

Background: Most hepatic triacylglycerols are believed to originate from fatty acids released from adipose tissue.
Results: Lipoprotein phosphatidylcholine is taken up by the liver and converted into triacylglycerols in vivo.
Conclusion: Lipoprotein-associated phosphatidylcholine is a quantitatively significant source of hepatic triacylglycerols.
Significance: The role of lipoprotein phosphatidylcholine should now be factored into our thinking about the development of hepatic steatosis.
Keywords: Biosynthesis, High Density Lipoprotein (HDL), Liver, Phosphatidylcholine, Triacylglycerol
Abstract
The increased prevalence of obesity and diabetes in human populations can induce the deposition of fat (triacylglycerol) in the liver (steatosis). The current view is that most hepatic triacylglycerols are derived from fatty acids released from adipose tissue. In this study, we show that phosphatidylcholine (PC), an important structural component of cell membranes and plasma lipoproteins, can be a precursor of ∼65% of the triacylglycerols in liver. Mice were injected with [3H]PC-labeled high density lipoproteins (HDLs). Hepatic uptake of HDL-PC was ∼10 μmol/day, similar to the rate of hepatic de novo PC synthesis. Consistent with this finding, measurement of the specific radioactivity of PC in plasma and liver indicated that 50% of hepatic PC is derived from the circulation. Moreover, one-third of HDL-derived PC was converted into triacylglycerols. Importantly, ∼65% of the total hepatic pool of triacylglycerol appears to be derived from hepatic PC, half of which is derived from HDL. Thus, lipoprotein-associated PC should be considered a quantitatively significant source of triacylglycerol for the etiology of hepatic steatosis.
Introduction
All nucleated mammalian cells are capable of synthesizing PC3 via the CDP-choline pathway (1). The rate-limiting enzyme for this pathway is CTP:phosphocholine cytidylyltransferase (CT) (2). In mice, two isoforms of CT exist, CTα and CTβ, of which CTα is the predominant isoform in liver (3, 4). The liver is the only organ that has an alternative pathway for synthesizing quantitatively significant amounts of PC. Hepatocytes can sequentially methylate phosphatidylethanolamine through the action of phosphatidylethanolamine N-methyltransferase (PEMT). The PEMT pathway is responsible for 30% of hepatic PC synthesis, whereas the remaining 70% is generated via the CDP-choline pathway (5).
Besides synthesizing PC, the liver can also obtain PC from the plasma through uptake of circulating lipoproteins. PC is the predominant phospholipid in the circulation. Among plasma lipoproteins, HDL particles have the highest concentration of PC (6). The lipids in these particles are mainly cholesteryl esters (24–45%) and phospholipids (42–51%) of which the majority is PC. Because mice have high levels of HDL, most of their circulating PC is associated with HDL (7). High plasma levels of HDL are associated with protection against the development of atherosclerosis (8). HDL particles are known to exhibit anti-inflammatory, anticoagulant, and antioxidant properties, all of which contribute to its antiatherogenic effect (for a review, see Nofer et al. (9)). Moreover, the major atheroprotective effect of HDL has been ascribed to its capacity to transport cholesterol from peripheral tissues and vascular macrophages toward the liver for subsequent elimination via the feces (10, 11). Therefore, extensive research has been performed on the hepatic uptake of HDL-cholesterol as part of this reverse cholesterol transport pathway (for a review, see Trigatti et al. (12)). Although phospholipids are the most abundant lipid in HDL, very little is known about the hepatic uptake of HDL-associated PC. Previous studies have shown that cultured hepatocytes efficiently take up HDL-PC by receptor-mediated uptake via the scavenger receptor class B, type I (SR-BI), by HDL particle endocytosis as well as by extracellular remodeling of HDL-PC by secretory phospholipase A2 (13). Furthermore, the intracellular fate of lipoprotein-associated PC has been investigated for both HDL and low density lipoproteins (LDLs). In isolated hepatocytes, PC associated with both LDL and HDL can serve as a precursor for the synthesis of triacylglycerols (TGs) (13, 14) and can therefore be involved in the development of fatty liver. Fatty liver (steatosis) can lead to non-alcoholic fatty liver disease, which is an accumulation of fat in the liver unrelated to alcohol consumption or chronic liver disease. Non-alcoholic fatty liver disease encompasses steatosis and non-alcoholic steatohepatitis in which inflammation and fibrosis can occur, leading to cirrhosis and in some instances hepatocellular carcinoma. The prevalence of non-alcoholic fatty liver disease is 10–24% in adult populations and 25–75% in the obese and diabetic populations and is thus one of the major health concerns in the population. Our previous studies suggest that uptake of lipoprotein-associated PC into hepatocytes may contribute to cellular TG accumulation. However, little is known about the quantitative importance of HDL-PC as a source for hepatic TG in whole animals.
The present study provides insight into the quantitative significance of hepatic uptake of HDL-PC in vivo as well as its subsequent metabolism. Additionally, we investigated whether disturbances in hepatic PC biosynthesis influence these processes by using mouse models that are deficient in either PEMT or CTα specifically in the liver. We demonstrate that HDL particles deliver ∼10 μmol of PC to the liver each day, thereby contributing half of the total hepatic pool of PC. Unexpectedly, we also show that 65% of the total hepatic pool of TG appears to be derived from hepatic PC.
MATERIALS AND METHODS
Animals
Male C57Bl/6 Pemt+/+ (wild-type) and Pemt−/− mice (backcrossed >7 generations) (15), liver-specific Ctα−/− mice and their CtαFLOX littermates (16) (referred to as wild-type controls), and SR-BI−/− mice and their C57Bl/6 wild-type controls (17) were fed standard rodent chow (LabDiet, catalog number 5001) ad libitum and had free access to water. All experimental procedures were approved by the University of Alberta's Institutional Animal Care Committee in accordance with guidelines of the Canadian Council on Animal Care.
Radiolabeling of Phosphatidylcholine and HDL
Huh-7 human hepatoma cells were incubated for 16 h with 50 μCi of [3H]glycerol (PerkinElmer Life Sciences). Lipids were extracted, and [3H]PC was isolated by thin-layer chromatography (18). Glycerol labeling was chosen to limit the influence of lipid remodeling in the vasculature, a process that would greatly influence the results if PC were labeled in the fatty acid moiety. HDL was isolated from pooled mouse plasma by sequential ultracentrifugation (1.063 < d < 1.215) and labeled with [3H]PC as described elsewhere (13). For the experiments with primary hepatocytes, human HDL was isolated by sequential ultracentrifugation (1.063 < d < 1.215). For the LDL uptake experiments, human LDL was used that was isolated by sequential ultracentrifugation (1.019 < d < 1.063) and labeled with [3H]PC similarly to HDL labeling.
Hepatic Uptake of HDL
[3H]PC-HDL (0.5 × 106 dpm) was injected into mice via the tail vein. Disappearance of radioactivity from the plasma was assessed by dividing radioactivity in plasma at each time point by radioactivity at the initial time point (2 min after [3H]HDL injection). Fractional catabolic rates of plasma HDL-PC were calculated from the area under the curve, representing the decay of plasma radioactivity, using SAAM II software (SAAM Institute, Inc., Seattle, WA). Because the liver is the major organ for HDL-PC uptake (see Fig. 1), the rate of hepatic uptake of [3H]PC-HDL was calculated as the fractional catabolic rate multiplied by the total plasma pool of PC, which was determined by multiplication of the plasma volume of each mouse (4.5% of body weight) by the plasma PC concentration. Twenty-four hours after injection of [3H]HDL, the mice were sacrificed by cardiac puncture, and livers were excised, weighed, and stored at −80 °C. Similar experiments were performed using [3H]PC-LDL (radiolabel in the glycerol moiety of PC) in wild-type mice only. For these experiments, livers were harvested 6 h after injection of [3H]LDL particles.
FIGURE 1.
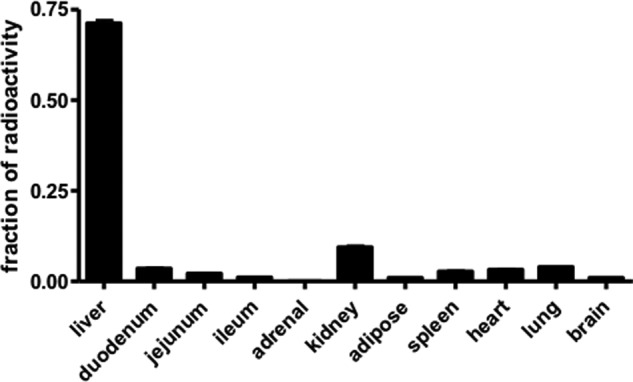
Radioactivity in mouse tissues after injection of [3H]PC-HDL. Mouse tissues were collected, and radioactivity was measured 15 min after injection of [3H]PC-HDL (0.5 × 106 dpm/mouse). Total recovery of radiolabel was 88% (61% in plasma, 19% in liver, and 8% in other organs). Data are expressed as the fraction of total radioactivity recovered in organs. Values represent means ± S.E. (error bars) (n = 8).
Analytical Procedures
Hepatic PC and phosphatidylethanolamine levels as well as plasma PC levels were determined by a phosphorous assay (19) after separation of the lipids by thin-layer chromatography. Hepatic and plasma TGs were quantified with an enzymatic kit (Roche/Hitachi; Roche Diagnostics GmbH). The amount of [3H] in 10 μl of plasma collected at the indicated times was measured by liquid scintillation counting. Radiolabel in the HDL fraction was determined after precipitation of apolipoprotein B-containing lipoproteins using phosphotungstic acid-MgCl2 (Sigma Diagnostics). To determine the proportion of PC that was hydrolyzed by a PC-phospholipase C (PLC) and subsequently re-esterified to TG, the radioactivity in TG was divided by the total radioactivity in TG and PC combined. The specific radioactivities of hepatic PC and TG were calculated, and the relative contribution of PC to the total hepatic TG pool was calculated by dividing the specific radioactivity of hepatic TG by the specific radioactivity of hepatic PC.
Incubation of Huh-7 Cells with HDL
Huh-7 cells were cultured in Dulbecco's modified Eagle's medium (DMEM) containing 10% fetal bovine serum until 80% confluence. Cells were then incubated in serum-free DMEM with or without human HDL (final concentration of PC, 2 mm) for 4 h. Cells were harvested, lipids were extracted, and cellular TG concentrations were determined.
Metabolic Labeling of Primary Mouse Hepatocytes
PC-PLC-mediated hydrolysis of PC was examined by treatment of primary mouse hepatocytes with the PC-PLC inhibitor Tricyclodecan-9-ylxanthogenate potassium salt (D609; Enzo Life Sciences International, Inc., Plymouth Meeting, PA). Hepatocytes were isolated from C57Bl/6 mice after perfusion of the liver with collagenase (20). The hepatocytes were cultured in DMEM containing 10% fetal bovine serum for 4–6 h followed by incubation in serum-free DMEM for 14 h. After incubation with or without 50 μg/ml D609 for 1 h, the cells were incubated for an additional 4 h in serum-free DMEM containing 0.4 mm [3H]oleic acid (PerkinElmer Life Sciences) (5 μCi/dish) complexed to 0.5% fatty acid-free bovine serum albumin and the indicated amounts of unlabeled human HDL (final concentration of PC, 0, 1, or 2 mm) in the presence or absence of 50 μg/ml D609. Cells were harvested, lipids were extracted, and the amounts of and radiolabel in PC and TG were measured.
Macrophage-to-liver Transport of Phosphatidylcholine
To determine whether there is net transport of PC from peripheral tissues to the liver, we adapted the experimental methods commonly used for measuring reverse cholesterol transport (21). J774 macrophages were cultured in DMEM containing 10% fetal bovine serum. Cellular PC was radiolabeled by incubating the cells with [3H]choline for 24 h followed by equilibration for 15 h in DMEM containing 2% fetal bovine serum. Cells were harvested and resuspended in serum-free DMEM. The [3H]PC-labeled macrophages (2 million cells/mouse) were injected intraperitoneally into C57Bl/6 mice. Blood was collected at 3, 6, 12, 24, and 30 h after injections, and after exsanguination at 30 h, livers were harvested and stored at −80 °C. The amount of [3H] in 30 μl of plasma collected at the indicated times was measured by liquid scintillation counting. Radioactivity in liver was determined after homogenizing the tissue and was related to total liver mass. All data were expressed as a percentage of the administered tracer dose.
Statistical Analysis
All values are means ± S.E. Statistical analysis was performed by Student's t test and one-way analysis of variance followed by post hoc Bonferroni correction for studies with primary hepatocytes. The level of significance was set at p < 0.05.
RESULTS
Hepatic Uptake of HDL-PC Is Quantitatively Important for Maintaining Hepatic PC Levels and Is Not Affected by Disturbances in PC Biosynthesis
Quantitatively, the liver is the major organ responsible for the uptake of HDL-cholesterol (10, 22). In mice injected with HDL in which the PC had been radiolabeled with [3H]glycerol, the liver accumulated ∼70% of the injected radioactivity recovered in tissues (Fig. 1). Hence, we quantified the daily uptake of HDL-PC by the liver based on the plasma fractional catabolic rate of HDL-PC under normal conditions as well as in response to disturbed hepatic PC synthesis, i.e. in Pemt−/− and Ctα−/− mice. Fig. 2, A and B, show the rate of decay of the radiolabel from plasma over a period of 24 h after injection. The fractional catabolic rate of HDL-PC was not affected by the lack of either PEMT or hepatic CTα (0.26 ± 0.01 versus 0.27 ± 0.01 pool/h for Pemt−/− mice and their wild-type controls, respectively, and 0.28 ± 0.02 versus 0.25 ± 0.02 pool/h for Ctα−/− mice and their CtαFLOX littermates, respectively). Although plasma PC levels were lower in both Pemt−/− and Ctα−/− mice compared with the respective control mice, the hepatic uptake of HDL-PC, estimated as the product of the fractional catabolic rate and the total pool of plasma PC, was similar for all genotypes (Fig. 2, C and D). Thus, although de novo synthesis of PC is disturbed in both Pemt−/− and liver-specific Ctα−/− mice, in neither of these mouse models was the uptake of HDL-PC from the circulation increased to maintain hepatic PC levels. Hepatic uptake of HDL-PC ranged from 7.3 to 11.4 μmol/day in these mouse models, similar to the amount of PC synthesized de novo in the liver each day (23). Twenty-four hours after injection, 87% of the radiolabel was still found in the HDL fraction of plasma, demonstrating that this uptake of [3H]PC is indeed mediated by HDL. Thus, uptake of HDL-associated PC into the liver is a quantitatively important source of PC for maintaining hepatic PC levels.
FIGURE 2.

Plasma decay and hepatic uptake of HDL-PC are not affected by PEMT deficiency or liver-specific elimination of CTα. After intravenous injection of [3H]PC-HDL into Pemt−/− and Ctα−/− mice and their wild-type controls, plasma radioactivity was measured over a period of 24 h. A and B represent the decay curves of 3H from plasma in Pemt−/− and Ctα−/− mice and their controls, respectively. C and D show hepatic uptake of HDL-PC calculated as the product of the HDL-PC plasma pool and the fractional catabolic rates derived from the decay curves. Values represent means ± S.E. (error bars) (n = 4–6 per group).
Phosphatidylcholine Is an Important Source of Hepatic Triacylglycerol
To investigate whether HDL-derived hepatic PC is converted into TG in vivo, we measured the 3H content of hepatic PC and TG. Twenty-four hours after an intravenous injection of [3H]HDL-PC, 67% of the 3H radiolabel in the liver was found in PC, whereas the remaining one-third of the 3H radiolabel was recovered in TG. Fig. 3, A and B, show the percentage of hepatic PC that was converted into TG under these conditions. In wild-type mice, one-third of hepatic PC was apparently hydrolyzed and re-esterified to generate TG. The absence of CTα in the liver did not influence the percentage of PC that was used for TG formation (Fig. 3B). However, mice lacking PEMT converted a larger proportion (49%) of hepatic PC into TG compared with their wild-type controls (Fig. 3A).
FIGURE 3.

Conversion of hepatic PC into TG in Pemt−/−, Ctα−/−, and their respective wild-type control mice. Twenty-four hours after intravenous injection of [3H]PC-HDL into Pemt−/− (A) and Ctα−/− (B) mice and their controls, livers were collected, and radioactivity in PC and TG was quantified. The percentages of PC converted into TG were calculated by dividing the radioactivity in TG by the total radioactivity in TG and PC combined. Values represent means ± S.E. (error bars) (n = 4–6 per group). *, p < 0.05.
To determine the quantitative importance of hepatic PC as a precursor for TG, the specific radioactivities of hepatic PC and TG were calculated. The fraction of hepatic TG that was generated from cellular PC was calculated by dividing the specific radioactivity of TG by the specific radioactivity of PC. According to this calculation, ∼65% of hepatic TG was derived from hepatic PC for wild-type and Pemt−/− mice (Fig. 4A). However, the contribution of hepatic PC to the total pool of hepatic TG was lower in mice lacking hepatic CTα compared with their controls (31 versus 67%) (Fig. 4B). To determine whether this process is specific for HDL-derived PC, we also injected [3H]PC-LDL into wild-type mice and measured radioactivity in hepatic PC and TG (Fig. 5). Similar to HDL-derived PC, 29% of PC taken up from LDL particles was converted into TG. These results together show that PC from both HDL and LDL particles is efficiently taken up by the liver and converted into TG. Thus, besides its importance in maintaining membrane integrity in hepatocytes, PC also has a major role as a direct precursor of hepatic TG.
FIGURE 4.

Hepatic PC is a quantitatively important source of hepatic TG. Twenty-four hours after intravenous injection of [3H]PC-HDL into Pemt−/− (A) or Ctα−/− (B) mice and their controls, livers were collected, and the specific activities (dpm/nmol) of hepatic PC and TG were calculated. The contribution of PC to the total hepatic TG pool was calculated by dividing the specific activity of hepatic TG by the specific activity of hepatic PC. Values represent means ± S.E. (error bars) (n = 4–6 per group). *, p < 0.05.
FIGURE 5.
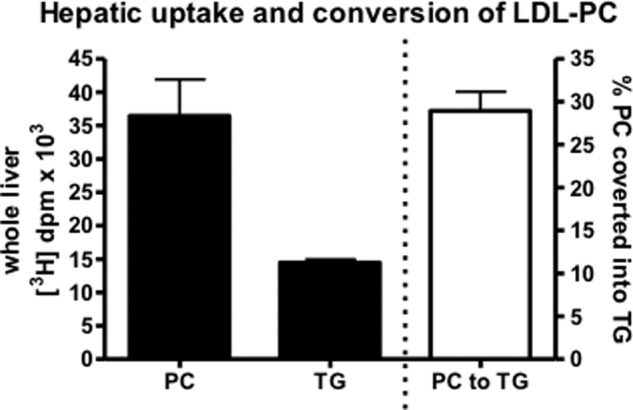
Conversion of LDL-derived PC into TG in livers of wild-type C57Bl/6 mice. Twenty-four hours after intravenous injection of [3H]PC-LDL into C57Bl/6 mice, livers were collected, and radioactivity in PC and TG was quantified. The percentages of PC converted into TG were calculated by dividing the radioactivity in TG by the total radioactivity in TG and PC combined. Values represent means ± S.E. (error bars) (n = 3).
Half of Newly Synthesized TG in Hepatocytes Is Derived from PC
The conversion of PC into TG involves a phospholipase C that hydrolyzes PC into diacylglycerol and phosphocholine (14) after which the diacylglycerol can be acylated to TG. As an additional approach for estimation of the quantitative significance of PC as a precursor of TG, mouse primary hepatocytes were incubated with various amounts of unlabeled human HDL (final concentration of PC, 0, 1, or 2 mm) along with 0.4 mm [3H]oleic acid (5 μCi/dish) complexed to 0.5% fatty acid-free bovine serum albumin, and after 4 h, the incorporation of [3H]oleic acid into PC and TG was measured. The mass of cellular PC and TG was not changed upon incubation with HDL (Fig. 6, A and B). However, incubation of hepatocytes with HDL reduced the specific radioactivity of cellular PC in a dose-dependent manner, indicating that a net uptake of HDL-PC into hepatocytes had occurred, resulting in dilution of the newly synthesized PC (Fig. 6C). Consequently, the specific radioactivity of TG was also reduced upon incubation with HDL (Fig. 6D), consistent with the conclusion that PC is a major contributor to hepatocyte TG and that HDL-PC is an important source of both hepatic PC and TG. To investigate further the quantitative importance of PC as a precursor of TG, we incubated hepatocytes with D609, an inhibitor of PC-specific PLC (24), to inhibit the hydrolysis of PC and thereby reduce the availability of diacylglycerol for acylation to TG. Cellular concentrations of PC and TG did not change upon incubation of hepatocytes with D609 (Fig. 6, A and B). In the absence of HDL, D609 reduced the specific radioactivity of TG by 50% (Fig. 5D), whereas the specific radioactivity of PC was unaffected (Fig. 6C), indicating that at least 50% of newly formed TG in primary mouse hepatocytes originates from the hydrolysis of PC. In the presence of D609, addition of HDL still reduced the specific activity of PC (Fig. 6C), whereas HDL did not further reduce the specific activity of TG (Fig. 6D). These data are consistent with those from the in vivo studies described above and show that uptake of HDL-PC contributes substantially to the total pool of PC in hepatocytes. Importantly, the data demonstrate the major importance of PC as a precursor of TG in hepatocytes both in vitro and in vivo.
FIGURE 6.

D609 and HDL reduce the specific radioactivity of TG in primary mouse hepatocytes labeled with [3H]oleic acid. Mouse primary hepatocytes were incubated for 1 h with or without the PC-PLC inhibitor D609 after which the cells were incubated for 4 h with [3H]oleic acid in the presence or absence of various concentrations of human HDL (final concentration of PC, 0, 1, or 2 mm). The concentrations of PC (A) and TG (B) as well as the radioactivity in these lipids were measured. C and D show the specific activity of PC and TG, respectively, relative to that in control cells incubated without HDL and/or D609. Values are means ± S.E. (error bars) of three independent hepatocyte preparations. *, significant difference versus no HDL with the same D609 treatment; #, significant difference versus no D609 treatment with the same HDL concentration determined using one-way analysis of variance tests (p < 0.05).
Uptake of HDL Increases TG Levels in Huh-7 Cells
To investigate the importance of HDL uptake for intracellular TG levels in another model, we treated human Huh-7 cells with human HDL. Compared with primary hepatocytes, these cells have lower basal TG levels (60 nmol/mg of protein; Fig. 7), which correspond more closely to hepatic TG levels in chow-fed mice in vivo (66 nmol/mg of protein in C57Bl/6 mice in this study; data not shown). After a 4-h incubation with HDL (final PC concentration was 2 mm), the amount of intracellular TG was 2-fold higher than in the cells that had not been incubated with HDL (Fig. 7). These data clearly demonstrate the quantitative importance of the uptake of HDL-derived PC for dictating the intracellular mass of TG in hepatocytes.
FIGURE 7.
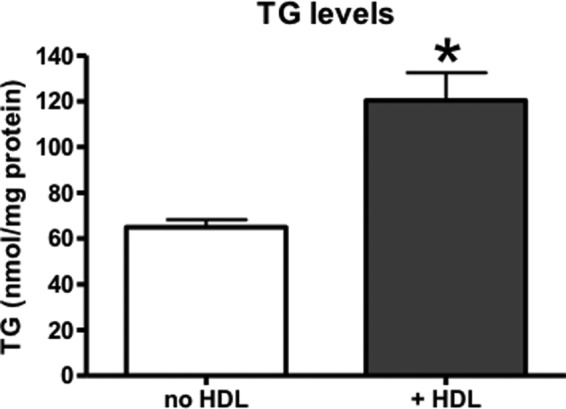
Cellular TG levels in Huh-7 cells increase after incubation with HDL-PC. Huh-7 cells were incubated for 4 h in serum-free DMEM with or without human HDL (final concentration of PC, 2 mm). Cells were harvested, lipids were extracted, and cellular TG concentrations were measured. Values are means ± S.E. (error bars) of three independent experiments, each performed in triplicate. *, p < 0.05.
HDL Mediates Net Transport of PC to the Liver
An important aspect of this hepatic uptake of HDL-PC that needs to be elucidated is to know from where the PC originates. To determine whether there is net transport of PC from peripheral tissues to the liver, we measured macrophage-to-liver transport of PC by adapting the widely used gold standard method for measuring reverse cholesterol transport (21). After intraperitoneal injection of radiolabeled macrophages, the appearance of the radiolabel in plasma increased over a period of 30 h (Fig. 8A). Twelve percent of the injected dose was recovered in the livers 30 h after injections (Fig. 8B). This clearly shows that there is indeed macrophage-to-liver transport of PC. At the 24-h time point, 80% of the radiolabel found in the circulation was associated with the non-apoB fraction of plasma (Fig. 8C), indicating that HDL is likely mediating this net transport of PC to the liver.
FIGURE 8.

Macrophage-to-liver transport of PC. C57Bl/6 mice were injected intravenously with [3H]PC-labeled J774 macrophages (2 × 106 cells containing 2 × 106 dpm in 0.6 ml of DMEM). A, 3H tracer in plasma over a 30-h period. B, 3H tracer recovered in liver 30 h after injections. C, percentage of plasma 3H in the HDL fraction 24 h after injections. Values are means ± S.E. (error bars) (n = 6).
Involvement of SR-BI in Hepatic Uptake of HDL-PC
Hepatic SR-BI is known to be the receptor responsible for selective uptake of HDL-cholesterol into the liver. Additionally, we have shown previously that in isolated hepatocytes SR-BI is responsible for 50% of the uptake of PC-oleic acid from HDL particles (13). To determine the involvement of SR-BI in hepatic uptake of HDL-PC in vivo, we injected wild-type and SR-BI−/− mice with human [3H]PC-HDL (labeled in the glycerol moiety of PC). Fig. 9A shows that there is no difference in the disappearance of radiolabel from the plasma between wild-type and SR-BI−/− mice. Moreover, the recovery of radiolabel in the liver 24 h after injections was similar in both mouse models (Fig. 9B). These results indicate that under these conditions SR-BI does not determine the rate of hepatic uptake of HDL-PC.
FIGURE 9.

Plasma decay and hepatic uptake of HDL-PC are not affected by SR-BI deficiency. After intravenous injection of [3H]PC-HDL into SR-BI−/− and wild-type C57Bl/6 mice, plasma radioactivity was measured over a period of 24 h. A, decay curves of 3H from plasma. B, recovery of radiolabel in liver 24 h after injections. Values represent means ± S.E. (error bars) (n = 7 per group).
DISCUSSION
Hepatic uptake of HDL has been studied extensively. Most of the previous studies were focused on the uptake of HDL-cholesterol as part of the atheroprotective, reverse cholesterol transport pathway. Only limited attention has been given to hepatic uptake of PC that is associated with HDL even though phospholipids are the most abundant lipid class in HDL particles. Previous studies from our laboratory showed that PC associated with either HDL or LDL is efficiently taken up by hepatocytes (13, 14). However, the quantitative importance of these processes was unknown. The current study reveals that the hepatic uptake of HDL-PC markedly contributes to hepatic PC levels in vivo by providing as much PC as is made in the liver each day. The quantitatively significant importance of PC as a source of hepatic TG in vivo that we observed in this study was completely unexpected.
The Source of HDL-PC
Each day, about 10 μmol of PC is delivered to the liver from HDL. Based on the specific radioactivities of PC in plasma and in the liver 24 h after the injection of [3H]HDL-PC, we calculated that approximately half of the hepatic PC pool is derived from the circulation. The uptake of HDL-PC was previously demonstrated in rats in which 38% of biliary PC originated from HDL (25). A question that arose from the findings of the present study is what is the source of HDL-PC? The liver is the major site for production of lipid-poor apoA-I, leading to ∼70% of the apoA-I in circulating HDL particles (26, 27). ApoA-I is secreted from the liver and lipidated with cholesterol and phospholipids in a process that requires hepatic ATP-binding cassette (ABC) protein A1 (28–30). It has been proposed that this initial lipidation of HDL accounts for the majority of PC associated with HDL, whereas most of the cholesterol is subsequently added to the HDL by ABCA1- or ABCG1-mediated efflux from peripheral tissues (31, 32). However, when HDL particles acquire cholesterol from peripheral tissues, the size of the particles increases. Consequently, HDL particles must also obtain PC from non-hepatic tissues to accommodate the increasing surface area of the particles possibly through either ABCA1- or ABCG1-mediated efflux from peripheral tissues or through transfer from other circulating lipoproteins. Our data clearly show that there is indeed transport of PC from macrophages to the liver, and in mice, this net transport of PC to the liver seems to be mediated by HDL. Because Pemt−/− and Ctα−/− mice have disturbed hepatic PC synthesis, we expected that these mice would have increased delivery of HDL-associated PC to the liver to maintain adequate hepatic PC levels. Surprisingly, however, hepatic uptake of HDL-PC was not different in these mice compared with their controls. This observation indicates that other processes, such as reduced secretion of PC into lipoproteins (7, 16, 33, 34) and compensatory up-regulation of the remaining PC biosynthetic pathway, i.e. the choline-pathway in Pemt−/− mice (35) and the PEMT pathway in Ctα−/− mice (16), are sufficient for maintaining hepatic PC levels in these mouse models.
HDL-PC as a Source of Hepatic TG
Most remarkable were the data on the metabolic fate of HDL-PC after its uptake by the liver. We observed that PC can be a quantitatively important source of hepatic TG because one-third of HDL-PC delivered to the liver appears to be hydrolyzed by PLC and subsequently re-esterified to form TG. That PC can serve as a source of TG was previously shown in rat hepatocytes for which incubation with [3H]oleic acid increased the amount of radiolabeled cellular and secreted TG with a concomitant loss of labeled PC (36). In addition, when cells were prelabeled with 1,2-di[1-14C]palmitoylphosphatidylcholine, part of the radiolabel was secreted as TG. More recently, we have shown that 50% of LDL-PC that is taken up by mouse hepatocytes is converted into TG (14). From that study, it became clear that the conversion of PC into TG involves the hydrolysis of PC by a PLC followed by esterification of the released diacylglycerol by acyl-coenzyme A:diacylglycerol acyltransferase 2. In addition, we showed that HDL-PC can also be converted into TG in primary mouse hepatocytes (13). We now demonstrate for the first time that HDL-PC is also converted into TG in vivo and that one-third of hepatic PC is converted into TG, thereby accounting for ∼65% of the TG in the liver.
We expected that the fraction of HDL-PC converted into TG would have been reduced by PEMT and/or CTα deficiency to conserve PC for maintenance of hepatic PC levels. Thus, we were surprised that in neither Pemt−/− mice nor Ctα−/− mice was the fraction of PC that was converted into TG reduced. Unexpectedly, Pemt−/− mice converted an even larger fraction (49%) of their PC into TG than did their wild-type counterparts. We speculate that this increase in PC hydrolysis/re-esterification in Pemt−/− mice might be related to the need for choline-containing metabolites other than PC, such as choline, phosphocholine, or acetylcholine. The only mechanism for the net production of choline molecules endogenously in mammals is the PEMT-mediated methylation of phosphatidylethanolamine followed by catabolism of PC (37). Pemt−/− mice have completely lost this capability and consequently are fully dependent on dietary sources of choline. Therefore, we speculate that Pemt−/− mice might stimulate the generation of phosphocholine by increasing the PLC-mediated hydrolysis of PC to maintain sufficient amounts of choline-containing molecules.
PC Involvement in Hepatic TG Metabolism
It has been well established that the metabolism of PC and TG is inter-related. In general, a reduction in hepatic PC is associated with an increase in hepatic TG level. The provision of mice with a methionine-choline-deficient diet is a widely used model for studying PC deficiency (38, 39). Under these conditions, mice have markedly reduced hepatic PC synthesis and develop steatohepatitis (38, 39). Furthermore, Pemt−/− mice accumulate TG in their livers even when fed a chow diet. This increase in hepatic TG is more pronounced when the mice are fed a high fat diet in response to which they develop severe hepatic steatosis and hepatomegaly (15). Similarly, the liver-specific elimination of CTα reduces hepatic PC by 20% in chow-fed female Ctα−/− mice and increases hepatic TG levels by 40% (16). In all of these models, an underlying cause of TG accumulation in the liver is a reduction in hepatic VLDL secretion. For example, in primary hepatocytes from both Pemt−/− (20) and hepatic Ctα−/− mice (33) as well as in hepatocytes cultured in methionine-choline-deficient medium (40), the secretion of apoB48, apoB100, and TG was diminished. Moreover, plasma VLDL levels were markedly lower in Ctα−/− mice (16) as well as in Pemt−/− mice compared with control mice when fed a high fat/high cholesterol diet (7). Altogether, these findings clearly indicate that when hepatic PC levels are reduced VLDL secretion is diminished, resulting in hepatic accumulation of TG. In the data presented herein, we show that PC is also a quantitatively important direct precursor of hepatic TG and that this can provide as much as 65% of the TG in the liver. These findings illustrate another quantitatively important pathway by which PC is involved in hepatic TG metabolism.
Pathways Involved in Hepatic Uptake of HDL-PC
Half of hepatic PC is derived from the circulation, most of which is coming from HDL particles. Therefore, an important question that needs to be addressed is which receptors and pathways are involved in hepatic uptake of HDL-PC. The most likely receptor involved in this process would be SR-BI, the “HDL receptor” responsible for selective uptake of HDL-cholesteryl esters. In our previous studies (13), we have shown that besides extrahepatic remodeling and endocytosis of HDL particles SR-BI is responsible for 50% of uptake of PC in isolated hepatocytes. In the current study, however, we show that deficiency of SR-BI does not affect hepatic uptake of HDL-PC in vivo. This may indicate either that SR-BI is not involved in this process or that other pathways are compensating for the long term loss of SR-BI to enable adequate uptake of PC into the liver. On the other hand, in an acute model of adenoviral overexpression of SR-BI and thus increased hepatic uptake of HDL, hepatic TG levels increased 2-fold (41). This increase in TG might be due to increased uptake of HDL-associated PC into the liver followed by conversion of the PC into TG. Altogether, it seems that SR-BI may be partially involved in hepatic uptake of HDL-PC, but other compensatory pathways are also in place especially in the situation of long term SR-BI deficiency.
In summary, we have shown that delivery of HDL to the liver is not only important for its role in reverse cholesterol transport but is also an important component of hepatic metabolism of both phospholipids and triacylglycerols. It was very striking that such a large fraction of hepatic TG (65%) is made through the hydrolysis of PC. Thus, the origin of hepatic TG is much more complicated than was originally anticipated. This also implies that increasing HDL and reverse lipid transport to reduce the risk of atherosclerosis may cause hepatic steatosis as a side effect. The role of HDL-PC in the development of hepatic steatosis and non-alcoholic fatty liver disease should thus be factored into our thinking about obesity, diabetes, and liver diseases.
Acknowledgments
We are grateful to Drs. Jean Vance, Richard Lehner, and René Jacobs for helpful discussions and comments. We thank Dr. Bernardo Trigatti for supply of the SR-BI−/− mice and helpful discussions.
This work was supported in part by Canadian Institutes of Health Research Grant MOP 89793.
- PC
- phosphatidylcholine
- ABC
- ATP-binding cassette
- CT
- CTP:phosphocholine cytidylyltransferase
- PEMT
- phosphatidylethanolamine N-methyltransferase
- PLC
- phospholipase C
- TG
- triacylglycerol
- SR-BI
- scavenger receptor class B, type I.
REFERENCES
- 1. Kennedy E. P., Weiss S. B. (1956) The function of cytidine coenzymes in the biosynthesis of phospholipids. J. Biol. Chem. 222, 193–214 [PubMed] [Google Scholar]
- 2. Vance D. E., Vance J. E. (2008) in Biochemistry of Lipids, Lipoproteins and Membranes (Vance D. E., Vance J. E., eds) 5th Ed., pp. 213–244, Elsevier, Amsterdam [Google Scholar]
- 3. Karim M., Jackson P., Jackowski S. (2003) Gene structure, expression and identification of a new CTP:phosphocholine cytidylyltransferase β isoform. Biochim. Biophys. Acta 1633, 1–12 [DOI] [PubMed] [Google Scholar]
- 4. Tang W., Keesler G. A., Tabas I. (1997) The structure of the gene for murine CTP:phosphocholine cytidylyltransferase, Ctpct. Relationship of exon structure to functional domains and identification of transcriptional start sites and potential upstream regulatory elements. J. Biol. Chem. 272, 13146–13151 [DOI] [PubMed] [Google Scholar]
- 5. DeLong C. J., Shen Y. J., Thomas M. J., Cui Z. (1999) Molecular distinction of phosphatidylcholine synthesis between the CDP-choline pathway and phosphatidylethanolamine methylation pathway. J. Biol. Chem. 274, 29683–29688 [DOI] [PubMed] [Google Scholar]
- 6. Jonas A., Philips M. C. (2008) in Biochemistry of Lipids, Lipoproteins and Membranes (Vance D. E., Vance J. E., eds) 5th Ed., pp. 485–506, Elsevier, Amsterdam [Google Scholar]
- 7. Noga A. A., Vance D. E. (2003) A gender-specific role for phosphatidylethanolamine N-methyltransferase-derived phosphatidylcholine in the regulation of plasma high density and very low density lipoproteins in mice. J. Biol. Chem. 278, 21851–21859 [DOI] [PubMed] [Google Scholar]
- 8. Assmann G., Schulte H., von Eckardstein A., Huang Y. (1996) High-density lipoprotein cholesterol as a predictor of coronary heart disease risk. The PROCAM experience and pathophysiological implications for reverse cholesterol transport. Atherosclerosis 124, (suppl.) S11–S20 [DOI] [PubMed] [Google Scholar]
- 9. Nofer J. R., Kehrel B., Fobker M., Levkau B., Assmann G., von Eckardstein A. (2002) HDL and arteriosclerosis: beyond reverse cholesterol transport. Atherosclerosis 161, 1–16 [DOI] [PubMed] [Google Scholar]
- 10. Fielding C. J., Fielding P. E. (1995) Molecular physiology of reverse cholesterol transport. J. Lipid Res. 36, 211–228 [PubMed] [Google Scholar]
- 11. Glomset J. A. (1980) High-density lipoproteins in human health and disease. Adv. Intern. Med. 25, 91–116 [PubMed] [Google Scholar]
- 12. Trigatti B., Covey S., Rizvi A. (2004) Scavenger receptor class B type I in high-density lipoprotein metabolism, atherosclerosis and heart disease: lessons from gene-targeted mice. Biochem. Soc. Trans. 32, 116–120 [DOI] [PubMed] [Google Scholar]
- 13. Robichaud J. C., van der Veen J. N., Yao Z., Trigatti B., Vance D. E. (2009) Hepatic uptake and metabolism of phosphatidylcholine associated with high density lipoproteins. Biochim. Biophys. Acta 1790, 538–551 [DOI] [PubMed] [Google Scholar]
- 14. Minahk C., Kim K. W., Nelson R., Trigatti B., Lehner R., Vance D. E. (2008) Conversion of low density lipoprotein-associated phosphatidylcholine to triacylglycerol by primary hepatocytes. J. Biol. Chem. 283, 6449–6458 [DOI] [PubMed] [Google Scholar]
- 15. Jacobs R. L., Zhao Y., Koonen D. P., Sletten T., Su B., Lingrell S., Cao G., Peake D. A., Kuo M. S., Proctor S. D., Kennedy B. P., Dyck J. R., Vance D. E. (2010) Impaired de novo choline synthesis explains why phosphatidylethanolamine N-methyltransferase-deficient mice are protected from diet-induced obesity. J. Biol. Chem. 285, 22403–22413 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Jacobs R. L., Devlin C., Tabas I., Vance D. E. (2004) Targeted deletion of hepatic CTP:phosphocholine cytidylyltransferase α in mice decreases plasma high density and very low density lipoproteins. J. Biol. Chem. 279, 47402–47410 [DOI] [PubMed] [Google Scholar]
- 17. Mardones P., Quiñones V., Amigo L., Moreno M., Miquel J. F., Schwarz M., Miettinen H. E., Trigatti B., Krieger M., VanPatten S., Cohen D. E., Rigotti A. (2001) Hepatic cholesterol and bile acid metabolism and intestinal cholesterol absorption in scavenger receptor class B type I-deficient mice. J. Lipid Res. 42, 170–180 [PubMed] [Google Scholar]
- 18. Jacobs R. L., Lingrell S., Dyck J. R., Vance D. E. (2007) Inhibition of hepatic phosphatidylcholine synthesis by 5-aminoimidazole-4-carboxamide-1-β-4-ribofuranoside is independent of AMP-activated protein kinase activation. J. Biol. Chem. 282, 4516–4523 [DOI] [PubMed] [Google Scholar]
- 19. Zhou X., Arthur G. (1992) Improved procedures for the determination of lipid phosphorus by malachite green. J. Lipid Res. 33, 1233–1236 [PubMed] [Google Scholar]
- 20. Noga A. A., Zhao Y., Vance D. E. (2002) An unexpected requirement for phosphatidylethanolamine N-methyltransferase in the secretion of very low density lipoproteins. J. Biol. Chem. 277, 42358–42365 [DOI] [PubMed] [Google Scholar]
- 21. Zhang Y., Zanotti I., Reilly M. P., Glick J. M., Rothblat G. H., Rader D. J. (2003) Overexpression of apolipoprotein A-I promotes reverse transport of cholesterol from macrophages to feces in vivo. Circulation 108, 661–663 [DOI] [PubMed] [Google Scholar]
- 22. Oram J. F., Yokoyama S. (1996) Apolipoprotein-mediated removal of cellular cholesterol and phospholipids. J. Lipid Res. 37, 2473–2491 [PubMed] [Google Scholar]
- 23. Reo N. V., Adinehzadeh M., Foy B. D. (2002) Kinetic analyses of liver phosphatidylcholine and phosphatidylethanolamine biosynthesis using 13C NMR spectroscopy. Biochim. Biophys. Acta 1580, 171–188 [DOI] [PubMed] [Google Scholar]
- 24. Amtmann E. (1996) The antiviral, antitumoural xanthate D609 is a competitive inhibitor of phosphatidylcholine-specific phospholipase C. Drugs Exp. Clin. Res. 22, 287–294 [PubMed] [Google Scholar]
- 25. Portal I., Clerc T., Sbarra V., Portugal H., Pauli A. M., Lafont H., Tuchweber B., Yousef I., Chanussot F. (1993) Importance of high-density lipoprotein-phosphatidylcholine in secretion of phospholipid and cholesterol in bile. Am. J. Physiol. Gastrointest. Liver Physiol. 264, G1052–G1056 [DOI] [PubMed] [Google Scholar]
- 26. Brunham L. R., Kruit J. K., Iqbal J., Fievet C., Timmins J. M., Pape T. D., Coburn B. A., Bissada N., Staels B., Groen A. K., Hussain M. M., Parks J. S., Kuipers F., Hayden M. R. (2006) Intestinal ABCA1 directly contributes to HDL biogenesis in vivo. J. Clin. Investig. 116, 1052–1062 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Timmins J. M., Lee J. Y., Boudyguina E., Kluckman K. D., Brunham L. R., Mulya A., Gebre A. K., Coutinho J. M., Colvin P. L., Smith T. L., Hayden M. R., Maeda N., Parks J. S. (2005) Targeted inactivation of hepatic Abca1 causes profound hypoalphalipoproteinemia and kidney hypercatabolism of apoA-I. J. Clin. Investig. 115, 1333–1342 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Bodzioch M., Orsó E., Klucken J., Langmann T., Böttcher A., Diederich W., Drobnik W., Barlage S., Büchler C., Porsch-Ozcürümez M., Kaminski W. E., Hahmann H. W., Oette K., Rothe G., Aslanidis C., Lackner K. J., Schmitz G. (1999) The gene encoding ATP-binding cassette transporter 1 is mutated in Tangier disease. Nat. Genet. 22, 347–351 [DOI] [PubMed] [Google Scholar]
- 29. Brooks-Wilson A., Marcil M., Clee S. M., Zhang L. H., Roomp K., van Dam M., Yu L., Brewer C., Collins J. A., Molhuizen H. O., Loubser O., Ouelette B. F., Fichter K., Ashbourne-Excoffon K. J., Sensen C. W., Scherer S., Mott S., Denis M., Martindale D., Frohlich J., Morgan K., Koop B., Pimstone S., Kastelein J. J., Genest J., Jr., Hayden M. R. (1999) Mutations in ABC1 in Tangier disease and familial high-density lipoprotein deficiency. Nat. Genet. 22, 336–345 [DOI] [PubMed] [Google Scholar]
- 30. Rust S., Rosier M., Funke H., Real J., Amoura Z., Piette J. C., Deleuze J. F., Brewer H. B., Duverger N., Denèfle P., Assmann G. (1999) Tangier disease is caused by mutations in the gene encoding ATP-binding cassette transporter 1. Nat. Genet. 22, 352–355 [DOI] [PubMed] [Google Scholar]
- 31. Singaraja R. R., Stahmer B., Brundert M., Merkel M., Heeren J., Bissada N., Kang M., Timmins J. M., Ramakrishnan R., Parks J. S., Hayden M. R., Rinninger F. (2006) Hepatic ATP-binding cassette transporter A1 is a key molecule in high-density lipoprotein cholesteryl ester metabolism in mice. Arterioscler. Thromb. Vasc. Biol. 26, 1821–1827 [DOI] [PubMed] [Google Scholar]
- 32. Singaraja R. R., Van Eck M., Bissada N., Zimetti F., Collins H. L., Hildebrand R. B., Hayden A., Brunham L. R., Kang M. H., Fruchart J. C., Van Berkel T. J., Parks J. S., Staels B., Rothblat G. H., Fiévet C., Hayden M. R. (2006) Both hepatic and extrahepatic ABCA1 have discrete and essential functions in the maintenance of plasma high-density lipoprotein cholesterol levels in vivo. Circulation 114, 1301–1309 [DOI] [PubMed] [Google Scholar]
- 33. Jacobs R. L., Lingrell S., Zhao Y., Francis G. A., Vance D. E. (2008) Hepatic CTP:phosphocholine cytidylyltransferase-α is a critical predictor of plasma high density lipoprotein and very low density lipoprotein. J. Biol. Chem. 283, 2147–2155 [DOI] [PubMed] [Google Scholar]
- 34. Noga A. A., Vance D. E. (2003) Insights into the requirement of phosphatidylcholine synthesis for liver function in mice. J. Lipid Res. 44, 1998–2005 [DOI] [PubMed] [Google Scholar]
- 35. Walkey C. J., Donohue L. R., Bronson R., Agellon L. B., Vance D. E. (1997) Disruption of the murine gene encoding phosphatidylethanolamine N-methyltransferase. Proc. Natl. Acad. Sci. U.S.A. 94, 12880–12885 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Wiggins D., Gibbons G. F. (1996) Origin of hepatic very-low-density lipoprotein triacylglycerol: the contribution of cellular phospholipid. Biochem. J. 320, 673–679 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Zeisel S. H., Da Costa K. A., Franklin P. D., Alexander E. A., Lamont J. T., Sheard N. F., Beiser A. (1991) Choline, an essential nutrient for humans. FASEB J. 5, 2093–2098 [PubMed] [Google Scholar]
- 38. Rinella M. E., Elias M. S., Smolak R. R., Fu T., Borensztajn J., Green R. M. (2008) Mechanisms of hepatic steatosis in mice fed a lipogenic methionine choline-deficient diet. J. Lipid Res. 49, 1068–1076 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Rinella M. E., Green R. M. (2004) The methionine-choline deficient dietary model of steatohepatitis does not exhibit insulin resistance. J. Hepatol. 40, 47–51 [DOI] [PubMed] [Google Scholar]
- 40. Yao Z. M., Vance D. E. (1988) The active synthesis of phosphatidylcholine is required for very low density lipoprotein secretion from rat hepatocytes. J. Biol. Chem. 263, 2998–3004 [PubMed] [Google Scholar]
- 41. Wiersma H., Nijstad N., Gautier T., Iqbal J., Kuipers F., Hussain M. M., Tietge U. J. (2010) Scavenger receptor BI facilitates hepatic very low density lipoprotein production in mice. J. Lipid Res. 51, 544–553 [DOI] [PMC free article] [PubMed] [Google Scholar]