

Background: Aromatic binding proteins in R. palustris transport lignin degradation products and have the potential for bioremediation.
Results: The proteins bind aromatic compounds with high affinity and have dynamic elongated structures.
Conclusion: Ligand binding induces local changes of residues that stabilize the compounds through hydrophobic interactions.
Significance: The results provide thermodynamic and structural insights into solute-binding proteins for lignin degradation products.
Keywords: ABC Transporter, Isothermal Titration Calorimetry, Lignin Degradation, Protein Structure, X-ray Scattering, Small Angle X-ray Scattering
Abstract
Rhodopseudomonas palustris metabolizes aromatic compounds derived from lignin degradation products and has the potential for bioremediation of xenobiotic compounds. We recently identified four possible solute-binding proteins in R. palustris that demonstrated binding to aromatic lignin monomers. Characterization of these proteins in the absence and presence of the aromatic ligands will provide unprecedented insights into the specificity and mode of aromatic ligand binding in solute-binding proteins. Here, we report the thermodynamic and structural properties of the proteins with aromatic ligands using isothermal titration calorimetry, small/wide angle x-ray scattering, and theoretical predictions. The proteins exhibit high affinity for the aromatic substrates with dissociation constants in the low micromolar to nanomolar range. The global shapes of the proteins are characterized by flexible ellipsoid-like structures with maximum dimensions in the 80–90-Å range. The data demonstrate that the global shapes remained unaltered in the presence of the aromatic ligands. However, local structural changes were detected in the presence of some ligands, as judged by the observed features in the wide angle x-ray scattering regime at q ∼0.20–0.40 Å−1. The theoretical models confirmed the elongated nature of the proteins and showed that they consist of two domains linked by a hinge. Evaluation of the protein-binding sites showed that the ligands were found in the hinge region and that ligand stabilization was primarily driven by hydrophobic interactions. Taken together, this study shows the capability of identifying solute-binding proteins that interact with lignin degradation products using high throughput genomic and biophysical approaches, which can be extended to other organisms.
Introduction
Rhodopseudomonas palustris is among the most metabolically and environmentally versatile bacteria known, and it has been selected as a prime candidate for both bioremediation and biofuel studies (1). The microbe converts atmospheric carbon dioxide into biomass, degrades aromatic pollutants, breaks down aromatic compounds from lignin degradation products found in woody tissues, and generates energy by converting N2 into NH4 and H2. This versatility makes R. palustris suitable for cleaning up toxic aromatic compounds with a potential for biofuel production. However, the molecular mechanism underlying the uptake of aromatic compounds from the environment into R. palustris is still unclear. Understanding this mechanism is crucial for the development of innovative biotechnologies involving R. palustris and related organisms. Regarding this, the genome sequence of the R. palustris CGA009 strain revealed gene clusters associated with ABC transporter proteins that might be implicated in the uptake of aromatic compounds from the environment into the microbe (1).
In bacteria, ABC transporter proteins are the primary interfaces with the environment (2, 3). Bacterial ABC importers and exporters transport a variety of substrates across the cell membrane. ABC importers in particular are essential for the uptake of nutrients, including metals, small ions, mono- and oligosaccharides, peptides, amino acids, iron-siderophores, polyamines, and vitamins. A typical ABC importer generally consists of a periplasmic solute-binding protein, two integral membrane subunits, and two cytoplasmic ATPases (4, 5). It is the solute-binding proteins (SBPs)2 of the ABC superfamily that recognize and bind specific substrates in the cell wall and transfer them to the membrane subunits, and they are therefore responsible for the uptake of ligands from the environment.
Laboratory studies of growth requirements for environmentally derived R. palustris and other bacterial strains indicate that these organisms can metabolize a variety of aromatic compounds (6–8) and that the import of these compounds under anaerobic conditions likely occurs via ABC-type transporters implicating the SBPs in the uptake of these aromatic compounds (7, 9, 10). Indeed, based on genome sequence, annotation information, and differential scanning fluorimetry ligand binding screens, we recently identified four possible SBPs in R. palustris that showed binding to aromatic lignin monomers such as benzoate, 4-hydroxybenzoate, and p-coumarate (11). The results also showed that the proteins RPA0668, RPA0985, RPA4029, and RPA1789 exhibited aromatic ligand specificity. For instance, RPA0668 and RPA0985 showed specificity for both benzoate and 4-hydroxybenzoate, whereas RPA4029 and RPA1789 were specific for 4-hydroxybenzoate and p-coumarate, respectively. Interestingly, for RPA0668 and RPA1789, ligand specificity was consistent with gene clusters implicated in the biodegradation of the aromatic compounds. Although the compounds bound by this family of proteins have similar chemical properties, the proteins exhibit less than 40% sequence identity for any of the possible protein sequence alignments using the Blast algorithm (12). However, all the SBPs belong to the type I periplasmic binding fold superfamily (13), and comparison of the ABC transport protein sequences suggests that ligand affinity and specificity might be dictated by similar residues within their binding sites. Despite these findings, further structural studies are required to determine the properties, selectivity, and mode of aromatic ligand binding in these SBPs. Ultimately, determining which specific proteins and metabolic pathways are involved in the uptake of aromatic compounds could lead to utilization of R. palustris and other organisms in its genus for bioremediation and bioenergy objectives.
Here, we report the thermodynamic and structural properties of RPA0668, RPA0985, RPA4029, and RPA1789 with aromatic ligands using isothermal calorimetry (ITC) and small/wide angle x-ray scattering (SAXS/WAXS), respectively. ITC was used to validate the binding specificity of the ABC transport proteins to different aromatic compounds. SAXS/WAXS of the proteins in solution yielded information about the global three-dimensional fold (SAXS) and the local conformational changes (WAXS) of the transporters in the presence and absence of benzoate, 4-hydroxybenzoate, and p-coumarate (14–17). Structural predictions, however, offered detailed three-dimensional structures of the proteins based on homology, threading, and ab initio methods, which were employed to generate different theoretical models of the ABC transport proteins from their primary sequences (18). Specifically, the approach was to compare the experimental SAXS/WAXS scattering profiles of the proteins with the calculated scattering curves of predicted models to determine the structural features of the R. palustris SBPs. Overall, the results provide the first structural insights into aromatic binding proteins and provide a model for aromatic ligand binding and selectivity, which can be extended to other organisms.
EXPERIMENTAL PROCEDURES
Protein Expression and Purification
RPA0668, RPA0985, RPA4029, and RPA1789 were produced as described previously (11). In brief, the genes were PCR-amplified from genomic DNA (ATCC, R. palustris CGA009 BAA-98D-5) using a KOD HiFi DNA polymerase reaction (Novagen) combined with a touchdown PCR program designed for the GeneAmp PCR System 9700 thermocycler machine (Applied Biosystems). The genes were cloned into two different vectors for both periplasmic and cytoplasmic expression (pBH31 and pMCSG7, respectively) and assessed for best solubility. Clones expressing soluble proteins were up-scaled, and His tag was purified using nickel-nitrilotriacetic acid (Qiagen) affinity chromatography columns. The purified proteins were dialyzed for buffer exchange, flash-frozen in liquid nitrogen, and stored at −80 °C until used.
Aromatic Ligand Stocks
Benzoate, 4-hydroxybenzoate, and tyrosine were dissolved in 50 mm HEPES, 150 mm NaCl buffer, pH 7.5. A stock solution of p-coumarate was prepared by dissolving the ligand in 100% DMSO and subsequent dilution in dialysis buffer (50 mm HEPES, 150 mm NaCl buffer, pH 7.5) for ITC measurements. Although tyrosine is not a direct product of lignin degradation, it was also used as a ligand for RPA1789 to compare the SAXS/WAXS results with the crystal structure of a tyrosine-bound homolog, recently deposited in the Protein Data Bank (PDB 3UK0). In ITC and SAXS/WAXS, a 10× ratio of ligand to protein concentration was used.
ITC Measurements
For ITC analysis of ligand binding, proteins were dialyzed for 18 h at 4 °C with exchanges of buffer (50 mm HEPES, 150 mm NaCl buffer, pH 7.5). Protein and ligand concentrations were determined spectrophotometrically. Isothermal titration experiments were performed at 25 °C using a VP-ITC calorimeter (Microcal, Inc.). In a typical experiment, 10-μl aliquots were injected ∼35 times at 4-min intervals from a 280-μl rotating stirrer-syringe to the reaction cell, containing 1.41 ml of ∼50 μm protein solution. Calorimetric data analysis was performed using the Origin 7.0 software provided by the instrument manufacturer (Microcal, Inc.). The binding stoichiometry (n), the enthalpy change (ΔH), and the binding constant (Ka) were directly obtained from the experimental titration curve.
SAXS/WAXS Data Acquisition and Processing
SAXS/WAXS data were recorded on the four ABC transport proteins in the absence and presence of the aromatic ligands. The 50 mm HEPES, 150 mm NaCl buffer was used as a background. In the absence of ligand, the scattering data were collected at 1, 2, 5, and 15 mg/ml. For the ligated protein complexes, the ligands were added to the proteins at a 10-fold excess to yield final protein concentrations of ∼5.0 and 14 mg/ml. The SAXS and WAXS data were collected simultaneously at the National Synchrotron Light Source (NSLS) beamline X9 using a PILATUS 300k SAXS detector and a Photonic Science CCD WAXS detector (19). In all SAXS/WAXS experiments, 15 μl of the protein and buffer solutions were continuously flowed through a 1-mm diameter capillary and exposed to the x-ray beam for 30 s. The measurements were carried out in triplicate. Data processing was performed using an automated Python-based package developed at X9. Overall, the two-dimensional scattering patterns recorded on the SAXS/WAXS detectors from protein solutions were first converted into one-dimensional scattering profiles. The SAXS/WAXS data were then merged, averaged, and buffer subtracted to obtain relative scattering intensity (I) as a function of momentum transfer vector, q (q = (4πsinθ)/λ), where λ is the beam wavelength, and θ is the scattering angle.
SAXS/WAXS Data Analysis
Data analysis and calculations were performed using the ATSAS software suite. The dimensions of the protein samples were first evaluated by estimating directly the radius of gyration (Rg) using the Guinier approximation. The Rg is the root mean square distance of an object from its center of mass. According to the Guinier approximation, for a monodisperse sample of globular protein, a plot of lnI(q) versus q2 should be linear for q < 1.3/Rg as described in Equation 1,
Deviations from linearity in the Guinier plots indicate aggregation or interparticle interactions and were also evaluated to confirm that no aggregation was present in any of the samples. All Guinier analyses were conducted using Primus (20).
The shape and maximum dimensions (Dmax) of each protein were then determined by the indirect Fourier transformation of their scattering data I(q), using GNOM (21). This process yields a pair distribution function P(r) (Equation 2) that describes the distribution of distances between pairs of elements within the entire volume of the scattering particle.
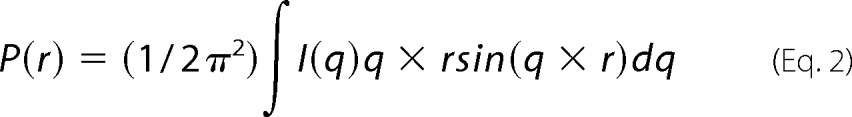 |
The three-dimensional scattering surfaces of the proteins were generated using the Dummy Atom Model Minimization (DAMMIN) method (22). The algorithm represents a protein as a collection of dummy atoms in a constrained volume with a maximum diameter defined experimentally by Dmax. It employs simulated annealing to generate the three-dimensional surface, and it calculates the scattering curve of the surface to evaluate its discrepancy (χ2) with the experimental data. For each protein, 10 surfaces were generated and averaged using DAMAVER (23). The averaged surfaces were then used as the final SAXS/WAXS three-dimensional structure.
Theoretical Predictions
Theoretical three-dimensional models of the ABC transport proteins were generated using SWISS-MODEL, PHYRE, TASSER, and ROBETTA algorithms. SWISS-MODEL (24) and PHYRE (25) predict protein structures by detecting closed and remote homologs, respectively, whereas I-TASSER (26) and ROBETTA (27) build protein models by combining remote homologs with ab initio calculations. The quality of the models was assessed by calculating their SAXS/WAXS scattering profiles using CRYSOL (28). For each protein, the best model was chosen by evaluating the χ2 between the computed scattering of the models and the experimental data. Superposition of the selected theoretical models with the SAXS/WAXS global surfaces was performed using SUPCOMB (29). The selected models were then employed for binding site prediction and ligand docking using BSP-SLIM. The method identifies ligand-binding sites by matching the predicted models to holo-structures in the Protein Data Bank (30). Ligand docking is then performed by pairing local shape and chemical features between ligand and the binding pockets. Interactions of the docked ligands with the proteins were also evaluated using PDBsum. CCP4MG was used for graphical analysis and figure generation.
RESULTS
Thermodynamic Properties
The affinity of the ABC transport proteins for the aromatic compounds identified by ligand screening was evaluated by ITC. Fig. 1 shows the ITC results obtained for RPA0668 with benzoate and 4-hydroxybenzoate. A previous ligand screening approach using a panel of 25 aromatic compounds indicated these ligands conferred the highest thermal stability. For these two ligands, the isotherms revealed normal sigmoidal titration curves (Table 1) with a similar binding affinity for benzoate and 4-hydroxybenzoate (dissociation constants (Kd) of 0.84 and 1.13 μm, respectively). Both interactions are enthalpy driven with an unfavorable entropic contribution. A calculated stoichiometry approximating 0.3 was consistently observed with multiple independent preparations of purified protein (Table 1). The binding fraction is dependent in part on the titration ligand as the sample purified protein preparation was utilized for benzoate and 4-hydroxybenzoate. In part, this is attributable to protein aggregation as some precipitate was consistently observed in the reaction cell at the conclusion of the experiment. Retained ligands may also contribute to lower stoichiometry, and some cellular aromatic compounds may not be removable by the purification and dialysis protocols in this study.
FIGURE 1.

Isothermal titration calorimetry of RPA0668 with benzoate (A) and 4-hydroxylbenzoate (B). The thermograms (top panel) and binding isotherms (bottom panel) are shown. The solid lines in the bottom panel are the fits to the experimental data.
TABLE 1.
ITC analysis of aromatic compound binding to solute-binding proteins from R. palustris
| Protein | Comment | Ligand | Na | Ka | Kd | ΔH | ΔS | ΔG |
|---|---|---|---|---|---|---|---|---|
| ×105m−1 | μm | kcal/mol | cal/mol/degree | cal/mol | ||||
| RPA 0668 | 25 °C | Benzoate | 0.16 ± 0.12 | 13.2 ± 6.0 | 0.84 ± 0.38 | −23.0 ± 1.41 | −49.2 ± 3.82 | −8323 ± 269 |
| Benzoate | 0.985a | 1.98 | 0.51 | 23.3 | −49.5 | −8589 | ||
| 25 °C | 4-Hydroxybenzoate | 0.284 ± 0.01 | 0.93 ± 0.28 | 1.13 ± 0.35 | −15.9 ± 3.96 | −0.82 ± 0.43 | −8137 ± 191 | |
| 4-Hydroxybenzoate | 0.999b | 1.13 | 0.88 | −13.1 | −1.61 | −8272 | ||
| RPA 0985 | 25 °C | Benzoate | 0.16 ± 0.026 | 1.95 ± 0.10 | 6.68 ± 4.62 | −1.59 ± 0.40 | 21.2 ± 2.7 | −7148 ± 349 |
| 37 °C | Benzoate | 0.18 ± 0.028 | 1.64 ± 0.07 | 7.06 ± 3.03 | −2.75 ± 3.96 | 14.9 ± 2.9 | −7353 ± 278 | |
| RPA 4029 | 4-Hydroxybenzoate | 0.62 ± 0.023 | 103 ± 20.9 | 0.095 ± 0.021 | −4.854 ± 0.14 | 15.8 ± 0.85 | −9562 ± 115 | |
| RPA 1789 | 25 °C | p-Coumarate | 0.93 ± 0.60 | 1.17 ± 0.077 | 8.61 ± 0.57 | 2.11 ± 3.96 | 30.3 ± 7.1 | −6904 ± 509 |
a Parameters were derived by assuming one binding site per molecule with the following thermodynamic parameters: n = 0.155, Kd = 0.57 μm, and ΔH = −24 kcal/mol.
b Parameters were derived by assuming one binding site per molecule with the following thermodynamic parameters: n = 0.29, Kd = 0.88 μm, and ΔH = −13 kcal/mol.
Two other proteins encoded by the RPA0985 and RPA4029 genes were also suggested to bind benzoate and/or 4-hydroxybenzoate based on the thermal stabilization screen. This functional assignment was confirmed by the ITC analysis as these proteins bound benzoate with observed Kd values in low micromolar or high nanomolar range (Table 1). The evolved heat obtained upon ligand titration of the RPA0985 protein was low, but similar kinetic and thermodynamic profiles were obtained at 25 and 37 °C (Table 1). Screening profiles obtained for the RPA1789 protein suggested a preference for propenoid ring substituents, which was confirmed by the observation of specific binding for p-coumarate with an observed Kd of 8.6 μm. Overall, the ITC results demonstrated that the four ABC transport proteins exhibited high affinity for the aromatic substrates with Kd values in the submicromolar range, similar to other SBPs (2, 3).
Structural Characterization
SAXS/WAXS offers relevant information about the dimension and shape of a protein in solution and was therefore used here to obtain insight into the structures of the free and ligand-bound ABC transport proteins. The SAXS/WAXS scattering curves of the proteins in the absence of aromatic ligands and their respective Guinier plots are presented in Fig. 2A. The scattering curves of the proteins at 1–5 mg/ml and the linearity of the Guinier plots at these concentrations indicated no detectable aggregation (Fig. 2A, inset). The Guinier analysis, which allowed the determination of the protein radius of gyration, revealed that RPA0668 had the largest dimension with an Rg of ∼27.15 ± 0.01 Å, followed by RPA4029, RPA0985, and RPA1789 with an Rg in the 23–24 Å range (Table 2). This was confirmed by the pair distribution functions, P(r), of the protein. The P(r) function is a real space representation of the scattering data and provides an approximation of the shape and dimension of a protein in solution. For the four proteins, the calculated P(r) functions (Fig. 2B) showed single peaks with Rg values similar to the ones obtained by the Guinier analysis (Table 2). In fact, a maximal dimension of ∼90 Å was obtained for RPA0668, whereas the other three ABC transport proteins showed Dmax of ∼80 Å. In addition, comparison of the protein P(r) functions with the P(r) of typical geometric shapes indicated that the ABC transport proteins were elongated with ellipsoid-like structures (15).
FIGURE 2.

SAXS/WAXS scattering (A) and P(r) functions of the ABC transport proteins (B). Guinier plots of the protein at concentrations of ∼2 mg/ml are shown in A, inset.
TABLE 2.
Structural parameters of the ABC transport proteins determined by Guinier approximation and indirect Fourier transformation of SAXS/WAXS data
| Protein | Rg (Guinier) | Rg (Gnom) | Dmax |
|---|---|---|---|
| Å | Å | Å | |
| RPA0668 | 27.15 (±0.01) | 27.17 (±0.06) | 92 |
| RPA0985 | 23.82 (±0.04) | 22.78 (±0.06) | 76 |
| RPA4029 | 24.46 (±0.49) | 23.83 (±0.07) | 80 |
| RPA1789 | 22.86 (±0.01) | 22.90 (±0.11) | 78 |
To corroborate this interpretation and to gain visual insights into the global architectures of the proteins, the three-dimensional surfaces from the SAXS/WAXS scattering data were generated. As shown in Fig. 3, the scattering curves of the three-dimensional surfaces fit the experimental data well, yielding a χ2 value in the range of 1.1–3.7. Moreover, the surfaces confirmed the elongated nature of the proteins and revealed differences in the ellipsoidal shapes of the proteins.
FIGURE 3.

A, SAXS/WAXS three-dimensional surfaces of the ABC transport proteins. B, comparison of the calculated scattering curves of the SAXS/WAXS surfaces (solid black lines) and the raw data (dotted line for RPA0668, solid line for RPA0985, dashed for RPA4029, and dashed-dotted for RPA1789). RPA0668 χ2 = 3.7 (R2 = 0.9996), RPA0985 χ2 = 2.4 (R2 = 0.9998), RPA4029 χ2 = 1.7 (R2 = 0.9998), and RPA1789 χ2 = 1.1 (R2 = 0.9996).
To examine the global and local structural changes associated with aromatic ligand binding, the SAXS/WAXS profiles of the liganded proteins were determined and compared with the free counterparts. Fig. 4 depicts the SAXS/WAXS scattering curves and the P(r) functions of RPA0668 in the absence and in the presence of added benzoate or 4-hydroxybenzoate ligands. The scattering patterns in the SAXS region (q ≤0.2) as well as the P(r) functions were quite similar, reflecting analogous topologies and global protein shapes in the unliganded and liganded states. Similar results were obtained for RPA0985 with benzoate and 4-hydroxybenzoate and for RPA4029 and RPA1789 in the presence of 4-hydroxybenzoate and p-coumarate, respectively. These results indicate that the global structure of the proteins remain unchanged in the presence of added ligand, implying that ligand binding only induces local changes in the proteins, which has also been observed in other SBPs (2, 3).
FIGURE 4.

A, SAXS/WAXS scattering curves, and B, P(r) functions of unliganded RPA0668 (solid line), liganded benzoate (dashed line), and liganded-4-hydroxylbenzoate (dotted line).
As a complement to SAXS, the WAXS region of the scattering curve has proven to be sensitive to local changes in protein structure induced by ligand binding. Therefore, to evaluate local protein changes in the presence of the aromatic ligands, we improved the quality of the WAXS region by increasing the concentration of the proteins to ∼15 mg/ml. Fig. 5, A and B, shows the WAXS regime for RPA0985 and RPA4029 in the absence and presence of benzoate and 4-hydroxybenzoate. For both proteins, the WAXS data did not change in the presence of benzoate as the aromatic ligand. Conversely, the WAXS data showed significant differences between the 4-hydroxybenzoate-bound and unbound states at q ∼0.20–0.40, suggesting local structural changes in the presence of this ligand. Likewise, fluctuations in the same WAXS region were observed for RPA1789 in the presence of tyrosine (Fig. 5C) and p-coumarate (data not shown). These differences were reproducible and 2–4 times larger than the error in the measurement. Taken together, these findings suggest that globally the ABC transport proteins retain their three-dimensional shapes in the presence of the aromatic ligands, but local movements occur upon ligand binding.
FIGURE 5.

SAXS/WAXS of RPA0985 (A), RPA4029 (B), and RPA1789 (C) proteins at ∼15 mg/ml. Black curves are the scattering patterns obtained in the absences of ligands, and gray and light gray curves are in the presence of aromatic ligands. The insets show the WAXS regime from q = 0.2–0.45 Å−1.
By comparing the SAXS/WAXS data for RPA4029 and RPA1789 to the calculated scattering curves based on crystal structures from close homologs in the liganded states (Protein Data Bank codes 3TON and 3SNR), a similar conclusion can be drawn. As depicted in Fig. 6A, the theoretical scattering curve calculated from the crystal structure of a solute-binding protein (3TON) with 4-hydroxybenzoate, which shares 90% similarity with RPA4029, correlated poorly with the scattering curve of the unliganded ABC transport protein (χ2 of ∼11.2). But comparison of the same theoretical scattering data with the SAXS/WAXS data for RPA4029 bound to 4-hydroxybenzoate showed a substantial decrease in the χ2 (to ∼2.5), implying similarities of both proteins only in the ligated states and local structural fluctuations in the bound and unbound states. A similar behavior was observed for RPA1789 and a homolog with 92% similarity (3SNR) (Fig. 6B).
FIGURE 6.

Comparison of the experimental SAXS/WAXS scattering of RPA4029 (A) and RPA1789 (B) in the unbound (dashed line) and bound (dotted line) states with the calculated SAXS/WAXS curves of two close homologous (solid lines). Unbounded RPA4029 χ2 = 11.8 (R2 = 0.9974), bounded RPA4029 χ2 = 2.7 (R2 = 0.9983), unbounded RPA1789 χ2 = 4.2 (R2 = 0.9979), and bounded RPA1789 χ2 = 1.2 (R2 = 0.9991) are shown.
To further refine the structural details of the proteins, in silico structural models were predicted using four independent algorithms. A total of 21 models with different possible conformations was generated for each ABC transport protein. To select the most appropriate model for each protein, their SAXS/WAXS scattering profiles were calculated and compared with the experimental scattering curves. The best fit models are displayed in Fig. 7A along with the calculated scattering curves (solid lines in Fig. 7B). The calculated scattering curves of the models agree well with the SAXS/WAXS experimental data of the proteins with χ2 values ranging from 1.5 to 3.7 (Fig. 7B). This was also confirmed by the residual analysis of the experimental and calculated scattering curves (Fig. 7C). Moreover, superposition of the best theoretical models with the SAXS/WAXS global shapes of the proteins yielded a normalized partial discrepancy of ∼1, indicating high similarities (Fig. 7A). Overall, the structures of the validated models have an ellipsoidal three-dimensional fold consisting of two domains connected by a hinge region.
FIGURE 7.
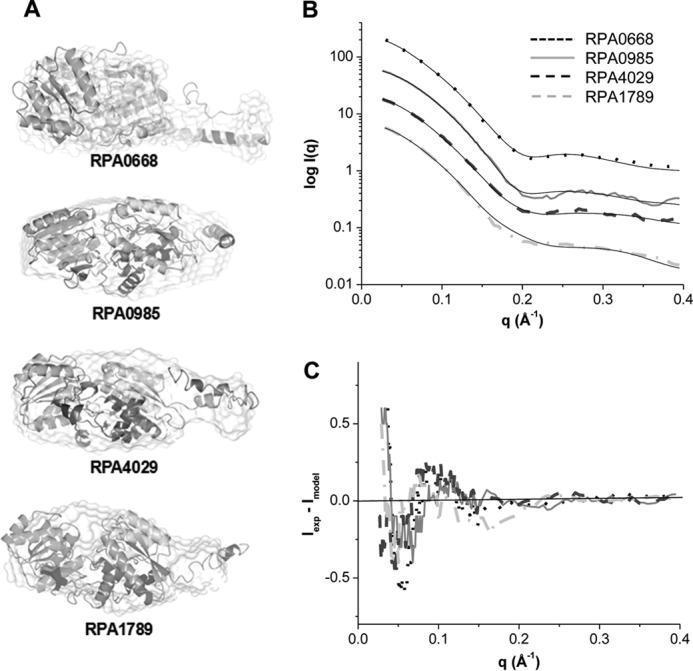
A, superposition of SAXS/WAXS surfaces with the best theoretical models obtained by I-Tasser. B, comparison of the calculated scattering curves of the theoretical models (solid black lines) and the experimental data of RPA0668 (dotted lines), RPA0985 (solid lines), RPA4029 (dashed lines), and RPA1789 (dashed-dotted lines). RPA0668 χ2 = 3.7 (R2 = 0.9992), RPA0985 χ2 = 2.5 (R2 = 0.9996), RPA4029 χ2 = 3.3 (R2 = 0.9996), and RPA1789 χ2 = 1.5 (R2 = 0.9998). C, residual plots of the experimental minus the calculated scattering curves of the theoretical models.
Finally, the best models were used for ligand-binding site predictions and docking to gain further information about the local structure and mechanism of aromatic ligand binding. For each protein, five different docked poses were predicted, and the ones with the highest docking scores were selected. The best docked structures along with representative aromatic ligands are depicted in Fig. 8. The results showed that, in the ABC transport proteins, the substrates bind in the hinge region formed by the two domains. As summarized in Table 3, in all cases, the aromatic compounds appear to be stabilized mainly through hydrophobic interactions. In RPA0985, RPA4029, and RPA1789, additional hydrogen bonding and van der Waals contacts between the hydroxyl and carboxyl groups of the compounds and nearby residues were also detected.
FIGURE 8.

Two-dimensional representation of RPA0668 (A), RPA0985 (B), and RPA4029 docked with 4-hydroxybenzoate (C), and RPA1789 with p-coumarate (D), generated by PDBsum. The server uses LigPlot to determine protein-ligand interactions. Accordingly, residues involved in hydrophobic contacts are represented by semicircles with spokes pointing toward the ligand atoms. The contacted atoms are also shown by spokes radiating back to the ligands. Hydrogen bonds are denoted by green dashed lines, and van der Waals interactions are indicated by solid lines.
TABLE 3.
Interaction of aromatic ligands with residues in the predicted binding sites
| Protein | Ligand | Hydrophobic Interactions | van der Waals interactions | H-bonding interactions |
|---|---|---|---|---|
| RPA0668 | Benzoate | Nine →Val131, Asn154, Gly156, Phe330 | None | None |
| 4-Hydroxybenzoate | Seven →Phe330, Asn154, Val131, Ala155 | None | None | |
| RPA0985 | 4-Hydroxybenzoate | Ten →Tyr61, Tyr123, Phe124, Thr125, Ala147, Ala148, Thr149, Pro194, Asp275 | Four →Tyr123, Ala148 | None |
| Benzoate | Nine →Pro60, Phe64Tyr61, Tyr123, Phe124, Thr125, Ala147, Ala247, Thr278, Asp275 | None | One →Tyr123 | |
| RPA4029 | 4-Hydroxybenzoate | Eight →Ser69, Thr70, Met281, Tyr197, Phe249, Pro251 | None | One → Ser69 |
| RPA1789 | p-Coumarate | Eight →Tyr168, Gln303, Phe304 | One →Gln303 | Two →Tyr168, Gln303 |
DISCUSSION
In this study, we have determined the structural properties of novel aromatic binding proteins from R. palustris and find similarities with other SBPs. The ITC analysis confirmed the inferred functional assignment as a binding protein for aromatic compounds with observed Kd values in a low micromolar or a high nanomolar range for all four binding proteins. The variance in thermodynamic parameters for the four SBPs suggests different binding site geometry and coordinating amino acids consistent with the observed sequence variance and sequence phylogeny for this protein family.
Bacterial SBPs have a conserved structure characterized by an elongated-ellipsoidal shape with two domains, both of which display an α/β-fold, connected by a hinge (2, 3). In all SBPs identified so far, the substrates are localized in the hinge between the two globular domains. The three-dimensional surfaces of the ABC transport proteins, derived from the SAXS/WAXS scattering data, demonstrated that indeed the proteins have elongated structures. Furthermore, the data showed that the proteins have different ellipsoidal shapes, indicating flexibility in their tertiary structures. Consistent with this interpretation is the fact that the theoretical models that best represented the ABC transport protein structures in solution also displayed different conformations in the N terminus (Fig. 7). Generally, SBPs are conformationally flexible, and the dynamics of the N-terminal region in the R. palustris proteins might reflect the inherent flexibility of the proteins in solution. Consistently, the lack of electron density around the N terminus in the crystal structures of some His tag-free SBPs, both in the liganded and unliganded states, confirms the disordered nature of this end terminal domain (31–33). It is important to emphasize that the functional relevance of this flexibility in SBPs has not been described thoroughly and warrants further investigation. Nevertheless, crystallographic, biochemical, and biophysical studies of several transporters have shown that the N-terminal domain of some SBPs interact with their cognate membrane proteins, and it is therefore reasonable to suggest that the flexibility of this terminus might be related to the mechanism of ligand transport (34).
In addition to the elongated and flexible nature of the ABC transport proteins in solution, the in silico studies also showed that they consist of two globular domains linked by three hinge regions. As with other SBPs, both domains in the ABC transport proteins are separated by a cleft and have similar super-secondary structures consisting of a central core of β-sheet flanked by α-helices. Likewise, evaluation of the ABC transport protein-computed binding sites showed that the docked ligands were found in the cleft region and that ligand stabilization is mostly driven by hydrophobic interactions.
Further stabilization of the aromatic ligands is achieved by hydrogen bonding and van der Waals contacts. Importantly, these interactions correlate well with the thermodynamic properties of the proteins. For instance, RPA0668 revealed similar affinity for both benzoate and 4-hydroxybenzoate, in line with the in silico observation that showed that both ligands were stabilized by analogous hydrophobic interactions. In RPA0985, 4-hydroxylbenzoate was further stabilized through van der Waals contacts between the hydroxyl and carboxyl groups of the ligand and adjacent residues (Table 3), and a 4000-fold increase in its affinity was also observed. Similarly, the additional van der Waals and hydrogen bond interactions in RPA1789 with p-coumarate explain the very high affinity observed for this aromatic ligand (K = 1.70 × 108 m−1). Taken together, the data suggest that ligand specificity is regulated by specific hydrophobic, van der Waals, and hydrogen bonding interactions. In fact, RPA4029 docked with 4-hydroxybenzoate showed eight hydrophobic contacts and one hydrogen bonding interaction, although docked benzoate only displayed two hydrophobic interactions. This explains in part the specificity of the RPA4029 protein for only 4-hydroxybenzoate.
Evaluation of the WAXS region provided additional insights into the mode of aromatic ligand binding in the ABC transport proteins. The observed changes in the 0.20–0.40 q region clearly show that the proteins undergo local structural rearrangement upon ligand binding. This region corresponds to features having length scales of about 20–30 Å (1/d (spacing) = q/2π). Fluctuations in this WAXS region of some proteins have been attributed to movement of whole domains from an open-unbound state to a closed-bound configuration (35). For example, in the maltose-binding protein, which is an SBP with the conserved two-domain structure, changes in this WAXS region were related to a 35° hinge bending along with an 8° rotational twisting of the N domain relative to the C domain induced by ligand binding (36). Similar fluctuations have been detected in the crystal structures of other bound and unbound SBPs (37, 38). On this basis, we propose that the ABC transport proteins undergo conformational changes upon aromatic ligand binding that involve rotation of both domains from an unliganded open state to a liganded closed conformation. Furthermore, the results showed that the WAXS region can also be used to assess ligand specificity of the ABC transport proteins toward the aromatic ligands. For RPA0985 and RPA4029 bound to benzoate, for example, the lack of noticeable changes in the WAXS pattern, as opposed to 4-hydroxybenzoate, indicates that this particular ligand does not induce closure of the two domains. Stabilization of the closed form, due to ligand binding, increases ligand affinity and selectivity. Thus, the inability of benzoate to stabilize the closed configuration in both proteins explains their specificity for 4-hydroxybenzoate.
In summary, the affinity of the ABC transport proteins for aromatic ligands was validated by ITC. The global shape of the ABC transport proteins was determined, and local changes in their tertiary structures were detected in the presence of aromatic ligands. As with other transporters, binding of the aromatic ligands appears to induce movements of the amino acid residues in the hinge region. Theoretical models of the proteins were also obtained and validated with their experimental three-dimensional shapes. These models have enabled us to evaluate the mode of binding of the aromatic ligands in the ABC transport proteins. Taken together, the study has shown the capability of identifying solute-binding proteins that interact with lignin degradation products using high throughput genomic and biophysical approaches, which can be extended to other organisms. This study will enable the identification of other proteins with similar ligand-binding profiles and characteristics and enable refinement of sequence-based methods for extension to other organisms and systems.
Acknowledgments
We thank Dr. Lin Yang for scientific and technical support at beamline X9. The National Synchrotron Light Source is supported by United States Department of Energy Contract DE-AC02-98CH10886.
This work was supported by the United States Department of Energy, Office of Biological and Environmental Research, as part of the BER Genomic Science Program. This contribution originates from the “Environment Sensing and Response” Scientific Focus Area Program at Argonne National Laboratory.
- SBP
- solute-binding protein
- ITC
- isothermal calorimetry
- SAXS/WAXS
- small/wide angle x-ray scattering.
REFERENCES
- 1. Larimer F. W., Chain P., Hauser L., Lamerdin J., Malfatti S., Do L., Land M. L., Pelletier D. A., Beatty J. T., Lang A. S., Tabita F. R., Gibson J. L., Hanson T. E., Bobst C., Torres J. L., Peres C., Harrison F. H., Gibson J., Harwood C. S. (2004) Complete genome sequence of the metabolically versatile photosynthetic bacterium Rhodopseudomonas palustris. Nat. Biotechnol. 22, 55–61 [DOI] [PubMed] [Google Scholar]
- 2. Davidson A. L., Dassa E., Orelle C., Chen J. (2008) Structure, function, and evolution of bacterial ATP-binding cassette systems. Microbiol. Mol. Biol. Rev. 72, 317–364 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Eitinger T., Rodionov D. A., Grote M., Schneider E. (2011) Canonical and ECF-type ATP-binding cassette importers in prokaryotes. Diversity in modular organization and cellular functions. FEMS Microbiol. Rev. 35, 3–67 [DOI] [PubMed] [Google Scholar]
- 4. Locher K. P. (2009) Review. Structure and mechanism of ATP-binding cassette transporters. Philos. Trans. R. Soc. Lond. B Biol. Sci. 364, 239–245 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Rees D. C., Johnson E., Lewinson O. (2009) ABC transporters. The power to change. Nat. Rev. Mol. Cell Biol. 10, 218–227 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Elder D. J., Morgan P., Kelly D. J. (1992) Anaerobic degradation of trans-cinnamate and ω-phenylalkane carboxylic acids by the photosynthetic bacterium Rhodopseudomonas palustris. Evidence for a β-oxidation mechanism. Arch. Microbiol. 157, 148–154 [DOI] [PubMed] [Google Scholar]
- 7. Rahalkar S. B., Joshi S. R., Shivaraman N. (1993) Photometabolism of aromatic compounds by Rhodopseudomonas palustris. Curr. Microbiol. 26, 1–9 [Google Scholar]
- 8. Shoreit A. A. M., Shabeb M. S. A. (1994) Utilization of aromatic compounds by phototrophic purple nonsulfur bacteria. Biodegradation 5, 71–76 [Google Scholar]
- 9. Harwood C. S., Parales R. E. (1996) The β-ketoadipate pathway and the biology of self-identity. Annu. Rev. Microbiol. 50, 553–590 [DOI] [PubMed] [Google Scholar]
- 10. Pan C., Oda Y., Lankford P. K., Zhang B., Samatova N. F., Pelletier D. A., Harwood C. S., Hettich R. L. (2008) Characterization of anaerobic catabolism of p-coumarate in Rhodopseudomonas palustris by integrating transcriptomics and quantitative proteomics. Mol. Cell. Proteomics 7, 938–948 [DOI] [PubMed] [Google Scholar]
- 11. Giuliani S. E., Frank A. M., Corgliano D. M., Seifert C., Hauser L., Collart F. R. (2011) Environment sensing and response mediated by ABC transporters. BMC Genomics 12, Suppl. 1, S8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Altschul S. F., Gish W., Miller W., Myers E. W., Lipman D. J. (1990) Basic local alignment search tool. J. Mol. Biol. 215, 403–410 [DOI] [PubMed] [Google Scholar]
- 13. Marchler-Bauer A., Lu S., Anderson J. B., Chitsaz F., Derbyshire M. K., DeWeese-Scott C., Fong J. H., Geer L. Y., Geer R. C., Gonzales N. R., Gwadz M., Hurwitz D. I., Jackson J. D., Ke Z., Lanczycki C. J., Lu F., Marchler G. H., Mullokandov M., Omelchenko M. V., Robertson C. L., Song J. S., Thanki N., Yamashita R. A., Zhang D., Zhang N., Zheng C., Bryant S. H. (2011) CDD. A conserved domain database for the functional annotation of proteins. Nucleic Acids Res. 39, D225–D229 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Jacques D. A., Trewhella J. (2010) Small-angle scattering for structural biology. Expanding the frontier while avoiding the pitfalls. Protein Sci. 19, 642–657 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Mertens H. D., Svergun D. I. (2010) Structural characterization of proteins and complexes using small-angle x-ray solution scattering. J. Struct. Biol. 172, 128–141 [DOI] [PubMed] [Google Scholar]
- 16. Makowski L., Gore D., Mandava S., Minh D., Park S., Rodi D. J., Fischetti R. F. (2011) X-ray solution scattering studies of the structural diversity intrinsic to protein ensembles. Biopolymers 95, 531–542 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Makowski L., Rodi D. J., Mandava S., Minh D. D., Gore D. B., Fischetti R. F. (2008) Molecular crowding inhibits intramolecular breathing motions in proteins. J. Mol. Biol. 375, 529–546 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Roy A., Kucukural A., Zhang Y. (2010) I-TASSER. A unified platform for automated protein structure and function prediction. Nat. Protoc. 5, 725–738 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Allaire M., Yang L. (2011) Biomolecular solution x-ray scattering at the National Synchrotron Light Source. J. Synchrotron Radiat. 18, 41–44 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Konarev P. V., Sokolova V. V., Koch M. H. J., Svergun D. I. (2003) PRIMUS. A Windows PC-based system for small angle scattering drug analysis. J. Appl. Crystallogr. 36, 1277–1282 [Google Scholar]
- 21. Svergun D. I. (1992) Determination of the regularization parameter in indirect-transform methods using perceptual criteria. J. Appl. Crystallogr. 25, 495–503 [Google Scholar]
- 22. Svergun D. I. (1999) Restoring low resolution structure of biological macromolecules from solution scattering using simulated annealing. Biophys. J. 76, 2879–2886 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Volkov V. V., Svergun D. I. (2003) Uniqueness of ab initio shape determination in small angle x-ray scattering. J. Appl. Crystallogr. 36, 860–864 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Kiefer F., Arnold K., Künzli M., Bordoli L., Schwede T. (2009) The SWISS-MODEL Repository and associated resources. Nucleic Acids Res. 37, D387–D392 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Kelley L. A., Sternberg M. J. (2009) Protein structure prediction on the Web. A case study using the Phyre server. Nat. Protoc. 4, 363–371 [DOI] [PubMed] [Google Scholar]
- 26. Zhang Y. (2008) I-TASSER server for protein three-dimensional structure prediction. BMC Bioinformatics 9, 40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Kim D. E., Chivian D., Baker D. (2004) Protein structure prediction and analysis using the Robetta server. Nucleic Acids Res. 32, W526–W531 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Svergun D. I., Barberato C., Koch M. H. J. (1995) CRYSOL. A program to evaluate x-ray solution scattering of biological macromolecules from atomic coordinates. J. Appl. Crystallogr. 28, 768–773 [Google Scholar]
- 29. Kozin M. B., Svergun D. I. (2001) Automated matching of high and low resolution structural models. J. Appl. Crystallogr. 34, 33–41 [Google Scholar]
- 30. Lee H. S., Lee C. S., Kim J. S., Kim D. H., Choe H. (2009) Improving virtual screening performance against conformational variations of receptors by shape matching with ligand binding pocket. J. Chem. Inf. Model. 49, 2419–2428 [DOI] [PubMed] [Google Scholar]
- 31. Chan S., Giuroiu I., Chernishof I., Sawaya M. R., Chiang J., Gunsalus R. P., Arbing M. A., Perry L. J. (2010) Apo- and ligand-bound structures of ModA from the archaeon Methanosarcina acetivorans. Acta Crystallogr. Sect. F Struct. Biol. Cryst. Commun. 66, 242–250 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Suzuki R., Wada J., Katayama T., Fushinobu S., Wakagi T., Shoun H., Sugimoto H., Tanaka A., Kumagai H., Ashida H., Kitaoka M., Yamamoto K. (2008) Structural and thermodynamic analyses of solute-binding protein from Bifidobacterium longum specific for core 1 disaccharide and lacto-N-biose I. J. Biol. Chem. 283, 13165–13173 [DOI] [PubMed] [Google Scholar]
- 33. Spurlino J. C., Lu G. Y., Quiocho F. A. (1991) The 2.3-Å resolution structure of the maltose- or maltodextrin-binding protein, a primary receptor of bacterial active transport and chemotaxis. J. Biol. Chem. 266, 5202–5219 [DOI] [PubMed] [Google Scholar]
- 34. Oldham M. L., Chen J. (2011) Crystal structure of the maltose transporter in a pretranslocation intermediate state. Science 332, 1202–1205 [DOI] [PubMed] [Google Scholar]
- 35. Makowski L. (2010) Characterization of proteins with wide-angle x-ray solution scattering (WAXS). J. Struct. Funct. Genomics 11, 9–19 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Fischetti R. F., Rodi D. J., Gore D. B., Makowski L. (2004) Wide-angle x-ray solution scattering as a probe of ligand-induced conformational changes in proteins. Chem. Biol. 11, 1431–1443 [DOI] [PubMed] [Google Scholar]
- 37. Gerstein M., Lesk A. M., Chothia C. (1994) Structural mechanisms for domain movements in proteins. Biochemistry 33, 6739–6749 [DOI] [PubMed] [Google Scholar]
- 38. Pflugrath J. W., Quiocho F. A. (1988) The 2 Å resolution structure of the sulfate-binding protein involved in active transport in Salmonella typhimurium. J. Mol. Biol. 200, 163–180 [DOI] [PubMed] [Google Scholar]