

Background: Whether Btk is involved in the regulation of NK cell innate function remains unknown.
Results: Btk−/− murine and human NK cells have decreased innate immune responses to the TLR3 ligand.
Conclusion: Btk is required for activation of NK cells.
Significance: Our results provide mechanistic insight into TLR3-triggered NK cell activation and make Btk a considerable target of the anti-inflammatory drug.
Keywords: Cytokine, Inflammation, Innate Immunity, Toll-like Receptors (TLR), Protein-tyrosine Kinase (Tyrosine Kinase), Acute Hepatitis, Cytotoxicity, Interferon-γ, NK Cells, Tyrosine Kinase Btk
Abstract
Bruton tyrosine kinase (Btk) is not only critical for B cell development and differentiation but is also involved in the regulation of Toll-like receptor-triggered innate response of macrophages. However, whether Btk is involved in the regulation of natural killer (NK) cell innate function remains unknown. Here, we show that Btk expression is up-regulated during maturation and activation of mouse NK cells. Murine Btk−/− NK cells have decreased innate immune responses to the TLR3 ligand, with reduced expressions of IFN-γ, perforin, and granzyme-B and decreased cytotoxic activity. Furthermore, Btk is found to promote TLR3-triggered NK cell activation mainly by activating the NF-κB pathway. Poly(I:C)-induced NK cell-mediated acute hepatitis was observed to be attenuated in Btk−/− mice or the mice with in vivo administration of the Btk inhibitor. Correspondingly, liver damage was aggravated in Btk−/− mice after the adoptive transfer of Btk+/+ NK cells, further indicating that Btk-mediated NK cell activation contributes to TLR3-triggered acute liver injury. Importantly, reduced TLR3-triggered activation of human NK cells was observed in Btk-deficient patients with X-linked agammaglobulinemia, as evidenced by the reduced IFN-γ, CD69, and CD107a expression and cytotoxic activity. These results indicate that Btk is required for activation of NK cells, thus providing insight into the physiological significance of Btk in the regulation of immune cell functions and innate inflammatory response.
Introduction
Bruton tyrosine kinase (Btk),4 a cytoplasmic tyrosine kinase and the most studied member of Tec family kinases, is expressed in almost all hematopoietic lineages except T cells (1–3). Btk has been shown to be crucial for B cell development, differentiation and function because of its predominant expression in different developmental stages of B lymphocytes, from hematopoietic stem cells, common lymphoid progenitor, to pre-B, pro-B, immature, and mature B cells (4). For example, Btk deficiency is associated with lack of circulating mature B cells and low reactivity to TI antigens of B cells, revealing a profound block in central B cell development and responsiveness of B cells (5, 6). Moreover, individuals with mutation of the gene encoding Btk, diagnosed as X-linked agammaglobulinemia (XLA), suffer from the disabled generation of all classes of immunoglobulins and therefore fail to mount effective humoral immune responses (7). The mutation of the Btk gene in mice leads to X-linked immunodeficiency, with a condition similar to XLA (8).
Although a series of defective functions of B cells have been ascertained in Btk-deficient models, there are also many reports regarding the defects of other cell types, such as macrophages, platelets, and osteoclasts in Btk-deficient mice (8–11). Btk-deficient macrophages show the enhanced susceptibility to apoptotic death upon exposure to the microbial and immune inflammatory signals such as LPS and IFN-γ in vitro. In mixed bone marrow chimeras, Btk deficiency primarily leads to loss of peripheral macrophages without affecting bone marrow development, suggesting a role of Btk in inflammation-induced macrophage apoptosis and subsequently regulating macrophage life span (8). Furthermore, specific Btk inhibition abolishes FcγRIII-induced TNF-α, IL-1β, and IL-6 production and thus suppresses myeloid cell-mediated arthritis (9). Moreover, once Btk-mediated signal is inhibited, thrombin-induced platelet aggregation is abolished due to failure of microtubular polymerization but maintains their depolymerization (10). However, whether Btk is involved in the regulation of NK cell function remains unknown. Therefore, the physiological and pathophysiological significance of Btk expression in different cell types of the hematopoietic system, such as NK cells, needs to be further investigated.
Because both Toll-like receptors (TLRs) and Btk are constitutively expressed in B cells and myeloid cells, the cooperation of Btk and TLRs has attracted much attention recently. In B1 cells, TLR9 signaling is suppressed by dephosphorylation of Btk, leading to attenuated activation of nuclear factor-κB (NF-κB) p65RelA and consequently blocking the excessive production of autoantibody (12). Btk-dependent induction of heme oxygenase-1 (HO-1) gene expression is observed in the macrophages upon stimulation with ligands of TLR2, TLR6, TLR7, and TLR9 (13). Btk−/− mice also show much more susceptibility to the infection of Listeria monocytogenes, which are recognized and cause downstream effects through TLR2 (14). These findings suggest that Btk is required for major TLR pathways but exert different functions in TLR-mediated responses. Our recent work shows that Btk participates in intracellular MHC class II molecule-mediated full activation of the TLR pathway by interacting with MyD88 and TRIF, promoting TLR-triggered production of proinflammatory cytokines and type I interferon in macrophages and dendritic cells (15). Therefore, Btk could cross-talk with TLR2, TLR3, TLR4, TLR6, TLR7, and TLR9 pathways in B cells or myeloid cells. However, whether Btk can cooperate with TLRs in other kinds of innate cells, such as NK cells, is still unknown.
NK cells are major effectors of the innate immune system, serving as the first line defense against infection, the regulator of immune response, and the monitoring of malignant cells (16). Indeed, the roles of NK cells are diverse, and their functions are controlled by the balance of a series of activating and inhibitory receptors (17, 18). Besides IL-2, IL-18, IL-15, NKAT, and STAT4, which have been previously confirmed to regulate cytotoxicity and production of TNF-α and IFN-γ of NK cells, more regulators have been identified to control NK cell functions. For example, activating transcription factor 3 (ATF3) decreases the transcription and secretion of IFN-γ in NK cells through interacting with a cis-regulatory element of the IFN-γ gene (19). Transcription factor Hlx expression in activated NK cells temporally controls and limits the monokine-induced production of IFN-γ, partially through the targeted depletion of STAT4 (20). EWS/FLI1-activated transcript 2 (EAT-2)A and EAT-2B are recently known to act as positive regulators of signaling lymphocyte activation molecule family receptor-specific NK cell cytotoxicity by promoting phosphorylation of Vav-1, which is known to be implicated in NK cell killing (17), yet PRDM1/Blimp-1 has the capacity to coordinately suppress the release of IFN-γ, TNF-α, and TNF-β by NK cells through direct binding to multiple conserved regulatory regions at the IFNG and TNF loci (21). It is known that activation of protein-tyrosine kinases is involved in TLR signaling and immune cell activation. Whether tyrosine kinase Btk is involved in the regulation of NK cell activation after challenge with ligands of TLRs needs to be identified.
In this study, we found Btk expression is up-regulated during the maturation and activation of murine NK cells. Btk−/− NK cells, from both mice and human, show decreased expression of IFN-γ and reduced cytotoxicity in response to TLR3 ligand. Poly(I:C)-induced acute hepatitis is observed to be attenuated once the Btk gene is mutant or the Btk inhibitor is administered in vivo in mice, which can be reversed by adoptive transfer of Btk+/+ NK cells. In addition, Btk is found to promote TLR3-triggered NK cell activation mainly through the increased activation of the NF-κB pathway. Our study demonstrated for the first time that Btk is required for TLR3-triggered activation of NK cells, thus contributing to the better understanding of the physiological significance of Btk in the regulation of NK cell functions.
EXPERIMENTAL PROCEDURES
Mice
C57BL/6 mice (Joint Venture Sipper BK Experimental Animals Co., Shanghai, China) and Btk−/− mice (The Jackson Laboratory, Bar Harbor, ME) were maintained in a specific pathogen-free facility and used at 6–10 weeks of age. All animal experiments were performed in accordance with the Guide for the Care and Use of Laboratory Animals from the National Institutes of Health, with the approval of the Scientific Investigation Board of the Second Military Medical University, Shanghai.
XLA Patients
Five XLA patients (Btk−/−) and seven age-matched normal controls were recruited from Hong Kong, and the research protocol was approved by the Institutional Review Board of the University of Hong Kong/Hospital Authority Hong Kong West Cluster.
Purification of NK Cells from Mice and Patients
For isolation of mouse splenic NK cells, anti-CD3 and anti-DX5 microbeads were used as recommended by the manufacturer (Miltenyi). To purify mouse NK cells at different developmental stages, bone marrow lymphocytes or splenocytes from C57BL/6 mice were incubated with monoclonal antibodies (BD Biosciences), and then NK precursors (CD3− CD14− CD19− NK1.1− CD122+ NKG2D+), immature NK (CD3− NK1.1+ DX5−), and mature NK (CD3− NK1.1+ DX5+) were sorted with fluorescence-activated Dako MoFloTM XDP to a purity of >98% as described previously (18). For isolation of primary human NK cells, peripheral blood mononuclear cells were isolated from whole-blood samples of XLA patients (Btk−/−) and age-matched normal controls by Ficoll-Hypaque (GE Healthcare) gradient centrifugation. NK cells were magnetically separated from peripheral blood mononuclear cells with CD56 microbeads (Miltenyi Biotec). The purity of isolated CD56+ CD3− NK cells was consistently>97%, as determined by flow cytometry.
Activation of NK Cells
Mouse splenic NK cells were purified and then stimulated with poly(I:C) (20 μg/ml), PMA (50 ng/ml)/ionomycin (500 ng/ml), or IL-12 (20 ng/ml)/IL-18 (5 ng/ml), respectively, for the time indicated and then were collected for the phenotypic and functional analysis. Human NK cells were cultured in RPMI 1640 medium supplemented with 10% FBS (Invitrogen) at 1 × 106/ml with a supplement of poly(I:C) (Sigma) (final concentration 40 μg/ml) or an equal volume of PBS overnight in 37 °C, 5% CO2. For intracellular cytokine analysis, brefeldin A was added at a final concentration of 10 μg/ml at 4 h before the end of NK cell incubation as we described previously (22), and NK cells were washed with PBS twice before phenotype and function analysis to remove resident poly(I:C).
Quantitative PCR
Total RNA was prepared using the RNeasy kit (Qiagen, Valencia, CA). cDNA was generated with a cDNA archive kit (Applied Biosystems, Foster City, CA). Real time PCR was performed on a Light Cycler 1.5 PCR system (Roche Diagnostics) using SYBR Green real time PCR master mix reagents (Applied Biosystems, Foster City, CA) specific for mouse Btk and the internal control β-actin (15). Data were normalized to β-actin using the 2−ΔΔCt method.
Flow Cytometry
The following monoclonal antibodies were used in this study: anti-hCD56 (HCD56), anti-hCD3 (HIT3a), anti-hCD69 (FN50), anti-hCD107a (H4A3), anti-hIFN-γ (B27), anti-mCD3 (17A2), anti-mCD14 (Sa14-2), anti-mCD19 (6D5), anti-mNK1.1 (PK136), anti-mCD122 (5H4), anti-mNKG2D (C7), anti-mDX5 (DX5), anti-mCD69 (H1.2F3), anti-mLy-49A (YE1/48.10.6), and anti-mIFN-γ (XMG1.2) (Biolegend); anti-mLy-49C/I (5E6), anti-hNKG2D (1D11), anti-hNKp44 (p44–8.1), anti-hNKp46 (9E2), anti-h-granzyme-B (GB11), and anti-perforin (G9) (Pharmingen); anti-mperforin (eBioOMAK-D); and anti-mGranzyme-B (16G6), anti-mLy-49D (eBio4E5), and anti-mNKG2A/C/E (20d5) (eBioscience).
For surface staining, NK cells were stained with specific antibodies. For intracellular IFN-γ, perforin, and granzyme-B staining, cells were fixed, permeabilized, and then labeled with the antibodies indicated above. To avoid nonspecific staining, cells were incubated with FcR blocking reagent (Miltenyi Biotec) prior to the specific staining. All data were acquired on an LSR II (BD Biosciences) and analyzed by FlowJo software (TreeStar) as described previously (14, 23).
Immunoblot Assay
Cells were lysed with RIPA buffer (Cell Signaling Technology, Beverly, MA) supplemented with protease inhibitor mixture. Protein concentrations of the extracts were measured with BCA assay (Pierce). The immunoblot assay was performed as described previously (15).
Cytotoxicity Assay of NK Cells
For detection of mouse NK cell cytotoxicity, splenic NK cells were prepared and incubated at different E:T ratios with Yac-1 cells for 4 h at 37 °C. Then the cells were collected, stained with anti-NK1.1, stained with 7-amino-actinomycin D after washing, and analyzed by FACS. Live target cells were assayed by gating on the NK1.1− 7-AAD− population. Cytotoxicity (%) = (NK1.1− cells − NK1.1−7-AAD− cells)/(NK1.1− cells) × 100. For detection of human NK cell cytotoxicity, a live/dead cell-mediated cytotoxicity kit (Invitrogen) was used. Briefly, K562 target cells were stained with 3,3-dioctadecyloxacarbocyanine perchlorate and then cocultured with NK cells at an effector cell/target cell (E/T) ratio of 10:1 in the presence of propidium iodide for 2 h. After incubation, cytotoxicity was analyzed by flow cytometry and calculated as the percentage of DiO+PI+ cells in the total number of DiO+ cells as we described before (24).
Chemical Inhibition of Btk in Vitro and in Vivo
Purified murine NK cells were pretreated with LFM-A13 (100 μm) for 30 min for in vitro inhibition of Btk. For in vivo experiments, WT mice were given 50 mg/kg LFM-A13 4h before poly(I:C) injection, and we then analyzed their serum ALT, AST, and IFN-γ at the indicated times.
Poly(I:C)-induced Acute Liver Injury
Mice were injected with poly(I:C), and liver histology, serology, and survival were assessed as we described previously (18).
Adoptive Transfer of WT NK Cells into Btk−/− Mice
Splenic NK cells from WT C57BL/6 mice were purified by magnetic activated cell sorting, and then intravenously injected into Btk−/− mice (5 × 106). After poly(I:C) was administrated to induce acute hepatitis, liver injury was assessed and evaluated as described previously (18).
Statistics
Data are expressed as means ± S.E. Statistical analysis was performed by Student's paired t test or two-way analysis of variance with a multiple-comparison test using Prism 5 (GraphPad Software). A value of p < 0.05 was considered statistically significant.
RESULTS
Btk Expression Increases Along with Differentiation and Activation of Murine NK Cells
To investigate whether Btk is involved in the regulation of NK cell function, we analyzed the expression of Btk in mature NK cells stimulated with PMA/ionomycin, poly(I:C), or IL-12/IL-18 (Fig. 1, A and B). Both mRNA expression and phosphorylated protein levels of Btk were significantly increased in NK cells after the stimulations. The expression of Btk protein gradually increased during the maturation of NK cells from small levels in NK cell precursors (Fig. 1C). These results suggest that Btk may be involved in the development and differentiation of NK cells.
FIGURE 1.
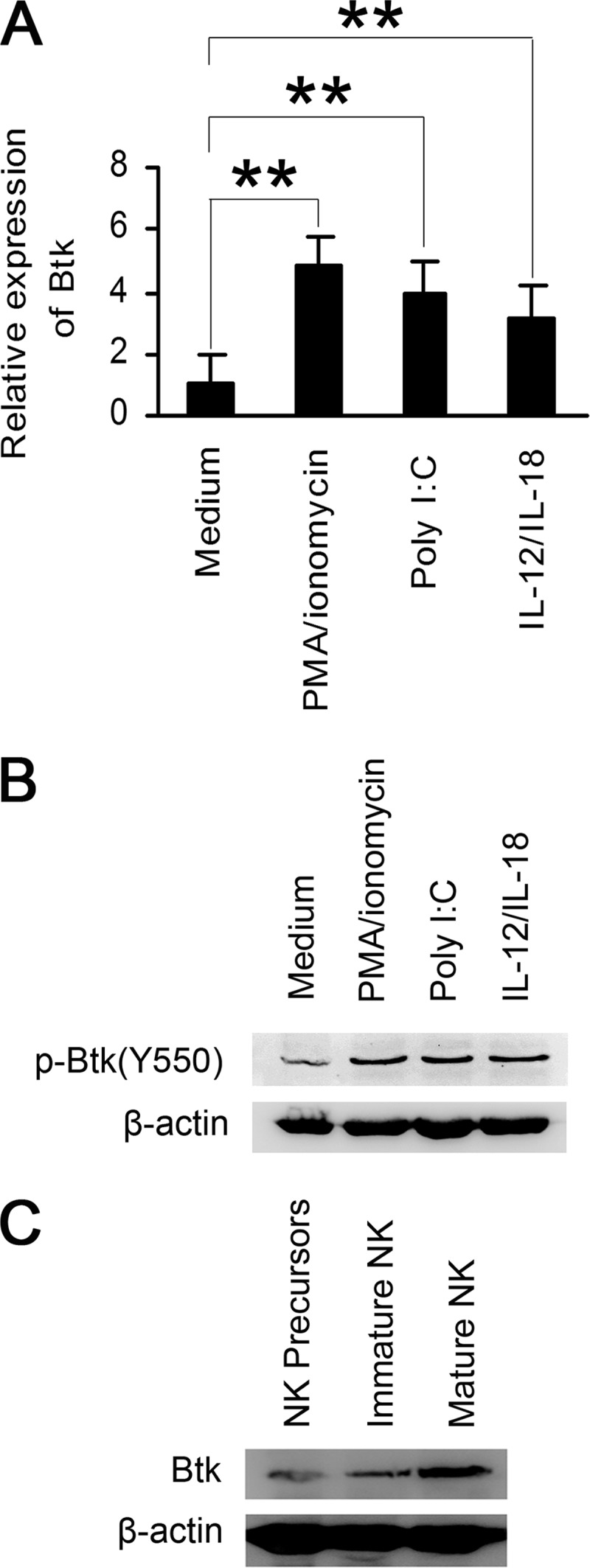
Btk expression is up-regulated during maturation and activation of murine NK cells. A and B, splenic NK cells were purified by magnetic activated cell sorting from C57BL/6 mice. Quantitative RT-PCR analyses of Btk (A) and Western blot analysis of p-Btk (B) were performed 8 h or 20 min after in vitro stimulation with PMA/ionomycin, poly(I:C), or IL-12/IL-18, respectively. C, expression of Btk protein was analyzed in different developmental stages of NK cells. Data are means ± S.E. of triplicate experiments. **, p < 0.01.
Btk Deficiency Leads to the Decreased Activation of NK Cells in Both Mice and Humans
By in vitro stimulation with poly(I:C), we found that NK cells isolated from Btk−/− mice (Btk−/− NK cells) had a lower CD69 expression compared with WT (Btk+/+) NK cells (Fig. 2A). The expressions of Ly49A, Ly49C/I, Ly49D, NKG2A/C/E, and NKG2D also decreased on unstimulated Btk−/− NK cells, compared with that in Btk+/+ NK cells (Fig. 2B). However, poly(I:C) stimulation significantly up-regulated the expression of NKG2D and Ly49D on mature Btk+/+ NK cells, although no differences in expression of NKG2D and Ly49D were observed on Btk−/− NK cells even after poly(I:C) treatment (Fig. 2B). Furthermore, intracellular expression of IFN-γ, perforin, and granzyme-B in Btk−/− NK cells was significantly decreased in response to the stimulation with poly(I:C) (Fig. 2C). Consequently, the cytotoxic activity of TLR3-activated Btk−/− NK cells was significantly reduced, compared with that in Btk+/+ NK cells (Fig. 2D).
FIGURE 2.

Btk deficiency leads to the impaired activation of murine NK cells in response to TLR3 ligand. A, CD69 expression of splenic WT or Btk−/− NK cells with or without stimulation of poly(I:C). B, Ly49A, Ly49C/I, Ly49D, NKG2A/C/E, and NKG2D expression on splenic WT and Btk−/− NK cells with or without stimulation of poly(I:C). C, intracellular IFN-γ, perforin, and granzyme-B expression of WT and Btk−/− NK cells. D, cytotoxicity of WT and Btk−/− NK cells against YAC-1 cells. Data are mean ± S.D. of triplicate experiments. All experiments above were performed four times with similar results. Data are means ± S.E. of triplicate experiments.
We further investigated the function of primary NK cells isolated from XLA patients with poly(I:C) stimulation in vitro. Similarly, these human Btk−/− NK cells also exhibited the decreased expression of intracellular IFN-γ, CD69, CD107a, and reduced cytotoxic activity compared with normal NK cells (Fig. 3). Interestingly, expression of NKG2D, NKp44, NKp46, perforin, and granzyme-B in human Btk−/− NK cells was comparable with that in normal control cells regardless of whether it was stimulated with poly(I:C) or not (supplemental Fig. S1).
FIGURE 3.
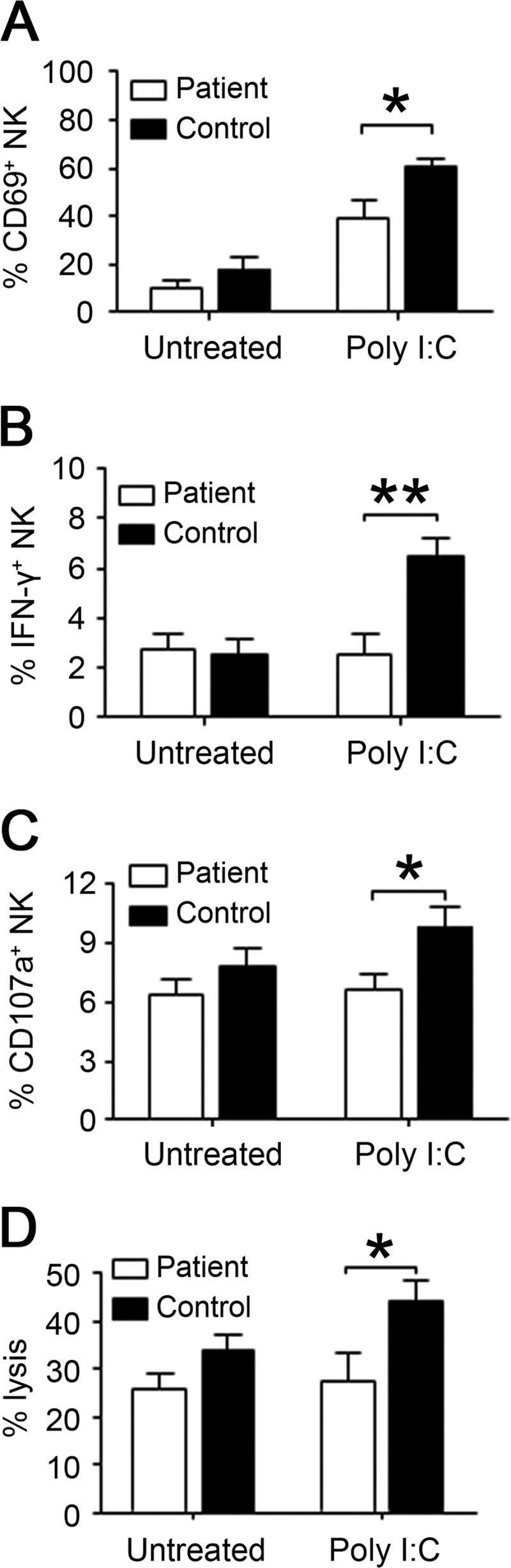
Impaired activation and cytotoxic activity of Btk−/− human NK cells after poly(I:C) stimulation. Highly purified human NK cells from XLA patients (Btk−/−) and age-matched normal controls were stimulated with poly(I:C) overnight and examined for the expressions of surface activation marker CD69 (A) and intracellular IFN-γ (B) by flow cytometry. C and D, treated NK cells were cocultured with K562 at E/T ratio of 10:1 in the presence of anti-CD107a antibody for 2 h. The surface expression of CD107a on NK cells (C) and specific lysis of target K562 cells (D) were then analyzed by flow cytometry. XLA patients (n = 5) and age-matched normal controls (n = 7) are shown. The data shown are represent for Mean±S.E., *, p < 0.05; **, p < 0.01.
To define the role of Btk in development and survival of NK cells, we also compared the percentages of NK cells in mononuclear cells between WT and Btk−/− mice, and we found that significantly less NK cells were contained in spleen, liver, and peripheral blood in Btk−/− mice (supplemental Fig. S2). However, no difference was found in the absolute number of NK cells in peripheral blood between the XLA patients and healthy controls, although there was a trend for a decrease in the percentage of NK cells in lymphocytes in XLA patients (supplemental Fig. S3). These data demonstrated that Btk is required for TLR3-triggered NK cell activation in both mice and human, although its role might be different in the development and survival of NK cells in mice and human.
TLR-triggered NK Cell Activation Is Suppressed by Btk Inhibitor
We then applied the chemical inhibitor of Btk, LFM-A13, to further evaluate the role of Btk in TLR3-triggered murine NK cell activation. As shown in Fig. 4A, expression of CD69 was decreased in TLR3-triggered NK cells after LFM-A13 treatment. Similarly, inhibition of Btk by LFM-A13 down-regulated the intracellular expressions of IFN-γ, perforin, and granzyme-B in TLR3-triggered NK cells (Fig. 4B). Consistently, LFM-A13-pretreated NK cells exhibited significantly decreased cytotoxicity (Fig. 4C). These data further confirmed that the inducible Btk is an important positive regulator in TLR3-triggered NK cell activation.
FIGURE 4.
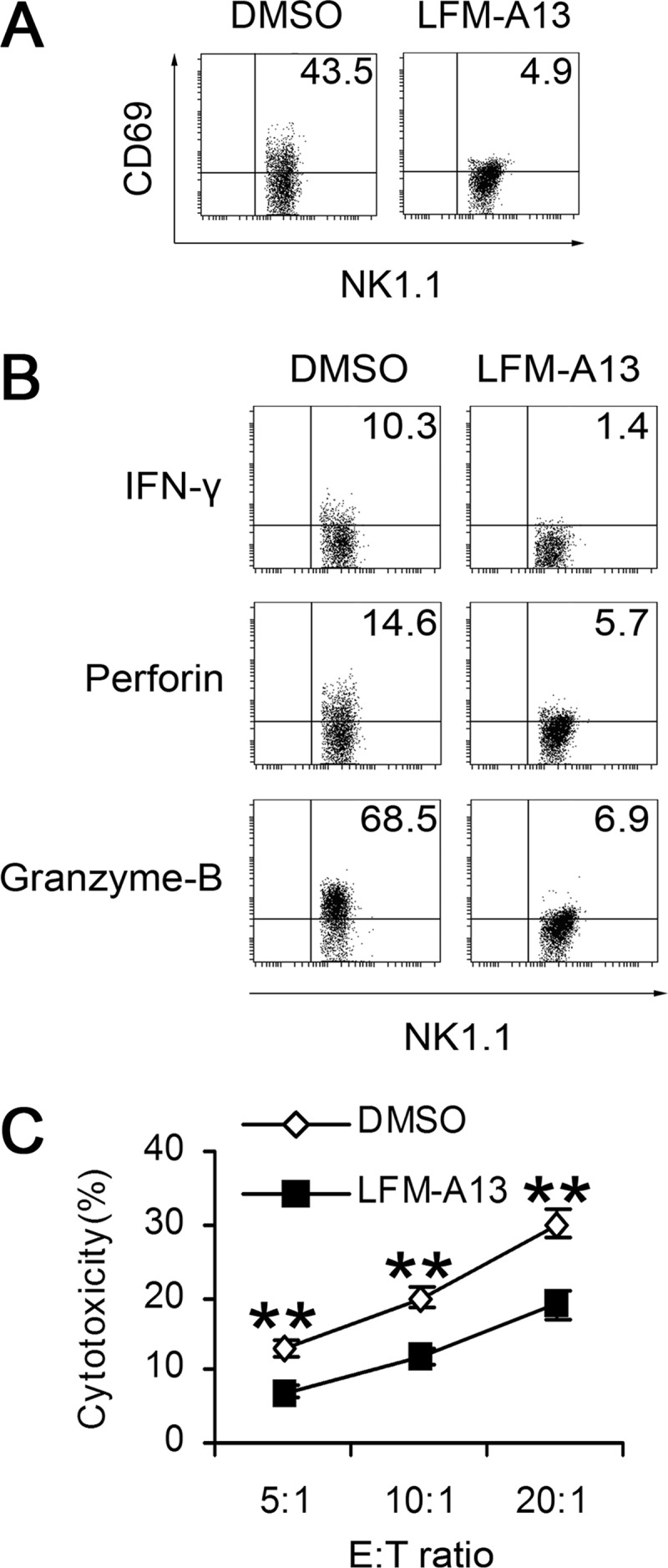
Suppression of TLR3-triggered NK cell activation by chemical inhibitor of Btk. CD69 (A), IFN-γ, perforin, granzyme-B expression (B), and cytotoxicity (C) of poly(I:C)-stimulated WT NK cells with or without pretreatment of Btk inhibitor LFM-A13 were analyzed. The data shown represent the means ± S.E., **, p < 0.01.
Btk Promotes TLR3-triggered NK Cell Activation Mainly through NF-κB Pathway
To illustrate the mechanism underlying the positive regulation of TLR3-triggered NK cell activation by Btk, we investigated the status of MAPK and NF-κB pathways in WT and Btk−/− NK cells. As shown in Fig. 5A, poly(I:C) stimulation failed to induce efficient phosphorylation of IKKα/β and degradation of IκBα in Btk−/− NK cells, resulting in the reduced phosphorylation of p65 and IRF3. Moreover, pretreatment with Btk inhibitor LFM-A13 suppressed TLR3-triggered activation of NF-κB in NK cells (Fig. 5B). Similarly the phosphorylations of ERK and JNK were also reduced in either Btk-deficient or LFM-A13-pretreated NK cells (Fig. 5, A and B). To investigate which signal pathway is responsible for the attenuated function of Btk−/− NK cells, we pretreated WT and Btk−/− NK cells with specific inhibitors for NF-κB, ERK, and JNK before poly(I:C) stimulation (Fig. 5C). The TLR3-induced IFN-γ production was significantly decreased by NF-κB inhibitor (pyrrolidine dithiocarbamate), although not in Btk−/− NK cells, in which NF-κB activation was already blunted. Inhibition of ERK and JNK did not significantly affect IFN-γ production in both WT and Btk−/− NK cells. These results indicated that Btk promotes TLR3-triggered NK cell activation, at least in part, through increased activation of the NF-κB pathway.
FIGURE 5.

Btk positively regulates TLR3-triggered NK cell functions by activating NF-κB pathway. Splenic NK cells were purified by magnetic activated cell sorting from WT or Btk−/− mice. A, Western blot analysis of MAPK and NF-κB pathways in TLR3-triggered Btk−/− and WT NK cells. B, MAPK and NF-κB pathways were analyzed in TLR3-triggered WT NK cells with or without LFM-A13 pretreatment. C, after pretreatment with pyrrolidine dithiocarbamate (PDTC) (NF-κB inhibitor, 50 μm), SP600125 (JNK inhibitor, 10 μm), or PD98059 (ERK inhibitor, 10 μm) for 30 min, intracellular IFN-γ expression of poly(I:C)-stimulated WT and Btk−/− NK cells was then analyzed.
Btk Contributes to the Poly(I:C)-induced NK Cell-mediated Acute Liver Injury
We further investigated the role of Btk in NK cell activation in vivo based on poly(I:C)-induced NK cell-mediated liver hepatitis model (18). After being challenged with poly(I:C), ALT and AST levels in the sera of WT mice increased significantly and reached the peak at 20 h after challenge, whereas IFN-γ production peaked at 8 h (Fig. 6, A and B). In Btk−/− mice, ALT and AST levels in the sera were significantly lower than the levels in WT mice, whereas IFN-γ production hardly increased at all (Fig. 6C). Crucially, Btk−/− mice achieved prolonged survival after being administered a lethal dose of poly(I:C) (Fig. 6D). Correspondingly, fewer necrotic areas and fewer infiltrated lymphocytes were observed in Btk−/− mice after poly(I:C) challenge by histological analysis (Fig. 6E). These data indicated that Btk might promote poly(I:C)-induced NK cell-mediated acute liver injury in mice.
FIGURE 6.

Btk deficiency attenuates poly(I:C)-induced acute liver injury in vivo. WT and Btk−/− mice were injected with poly(I:C) (20 μg/g body weight) intraperitoneally. Serum ALT (A), AST (B), IFN-γ (C) were assayed at the indicated times. Data are means ± S.E. of triplicate experiments. **, p < 0.01. D, survival of WT and Btk−/− mice was observed after injection of high dose of poly(I:C) (30 μg/g body weight). E, 18 h later, after fixed and embedded, sliced liver sections were stained with hematoxylin-eosin. Arrows indicated the liver necrosis and infiltration of lymphocytes.
To further confirm the contribution of Btk to the pathology of TLR-3-triggered liver damage, we applied LFM-A13 to inhibit the activity of Btk in vivo. As shown in Fig. 7, after intraperitoneal administration with LFM-A13, serum levels of ALT, AST, and IFN-γ in poly(I:C)-treated WT mice were markedly reduced. Histological analysis also showed attenuated liver damage, marked as less necrosis and fewer lymphocytes infiltration, in mice treated with LFM-A13 (Fig. 7D). These data emphasized the crucial role of Btk in TLR3-triggered NK cell activation in vivo.
FIGURE 7.

TLR3-triggered liver hepatitis is attenuated by inhibition of Btk in vivo. WT mice were administered with LFM-A13 (50 mg/kg) and then poly(I:C) (20 μg/g) intraperitoneally. Sera were collected for assays of ALT (A) and AST (B) 20 h later, and serum IFN-γ was examined 8 h after administration (C). Data are means ± S.E. of triplicate experiments. *, p < 0.05; **, p < 0.01. D, 18 h later, liver necrosis and infiltration of lymphocytes were observed after hematoxylin-eosin staining.
Replacement of Btk−/− NK Cells with WT NK Cells Rescues Poly(I:C)-induced Acute Liver Hepatitis in Mice
To determine whether the impaired NK cell function resulted in the attenuated experimental liver damage in Btk−/− mice observed above, we depleted NK cells in Btk−/− mice by PK136 monoclonal antibody and then transferred the purified WT NK cells 2 days later. One day post-cell transfer, poly(I:C) was administered. As shown in Fig. 8, the NK-deleted Btk−/− mice showed significantly declined levels of serum ALT, AST, and IFN-γ. In contrast, their serum ALT, AST, and IFN-γ levels dramatically increased after the infusion of WT NK cells. Moreover, more severe liver tissue damage was observed in Btk−/− mice receiving poly(I:C) administration after the transfer of WT NK cells (Fig. 8D). These results demonstrated that Btk-mediated NK cell activation is responsible for the pathogenesis of poly(I:C)-induced acute liver hepatitis, which further confirmed the indispensable role of Btk in TLR3-triggered NK cell activation.
FIGURE 8.

NK cell transfer aggravates TLR3-triggered acute liver injury in Btk−/− mice. NK cells were deleted in Btk−/− mice by intravenous injection of PK136 (200 μg/mouse) 2 days before transfer of purified WT NK cells, and then the mice were challenged with poly(I:C) (20 μg/g) intraperitoneally 24 h later. Serum ALT (A), AST (B), and IFN-γ (C) were examined as described in Fig. 6. Data are means ± S.E. of triplicate experiments. **, p < 0.01. D, liver necrosis and infiltration of lymphocytes were observed after hematoxylin-eosin staining 18 h after poly(I:C) injection.
DISCUSSION
In this report, we provide the functional description of Btk in NK cell activation. We show that Btk accumulates during maturation and activation of NK cells and acts as a positive regulator of NK cell activation by up-regulating expression of IFN-γ, granzyme-B, and perforin and enhancing cytotoxicity of NK cells in mice. The data strongly suggested Btk is responsible for poly(I:C)-induced NK cell activation, giving rise to the susceptibility of TLR3-triggered liver hepatitis. The observation that decreased TLR3-triggered activation of human NK cells from XLA patients is observed with the reduced IFN-γ, CD69, and CD107a expression and cytotoxicity, our results demonstrate that Btk is required for TLR3-triggered NK cell activation.
NK cells are important innate immune cells but they also bridge and influence adaptive immunity (25), whose activation relies on germ line-encoded receptors. Activated NK cells not only utilize polarized exocytosis of secretory lysosomes to kill infected or neoplastic cells (26, 27), but they also regulate and maintain the balance between positive and negative immune responses through secretion of cytokines and chemokines and killing of autologous immune cells, including activated T cells and dendritic cells (28, 29). Considering their enigmatic roles in different physiological processes, it is urgent to find critical regulators of NK cell function and to clarify their underlying mechanisms. On the basis of the observation of rapid Btk phosphorylation in NK cells after poly(I:C), PMA/ionomycin, or IL-12/IL-18 stimulation, we further investigated whether Btk regulates the activation of NK cells and their underlying mechanisms. As expected, Btk−/− NK cells exert decreased poly(I:C) up-regulated WT NK cells, along with the decreased expression of IFN-γ, perforin, and granzyme-B and consequently reduced cytotoxicity, which confirmed that Btk is a positive regulator of NK cell function upon poly(I:C) stimulation. Indeed, we also found Btk can also amplify the function of IL-12/IL-18-stimulated NK cells (data not shown). However, even though Btk deficiency led to a significantly reduced killing capacity of human NK cells with the decreased CD69, CD107a, and IFN-γ expression after poly(I:C) stimulation, NKG2D, NKp44, and NKp46 expression remained stable. Additionally, no statistical quantitative difference of NK cell number was observed in the peripheral blood between XLA patients and normal controls. These data suggested that although human and mice share essential developmental characteristics, some differences still exist. For example, X chromosome inactivation (XCI) allows dosage compensation of the expression from the sex chromosome in mammalian female cells (30). To clarify the corresponding mechanism, human embryonic stem cells and mouse embryonic stem cells were used and were supposed to shed light on the XCI process in early embryogenesis, but studies of embryonic stem cells from these two species largely indicated inconsistency in the status of XCI (31). Human embryonic stem cells showed the various states of XCI, which were proved to be caused by epigenic manipulations involving XIST (the crucial gene of XCI initiation) promoter methylation/demethylation as well as in vitro culture conditions (32). These findings suggest possible explanations for the partially inconsistent observations in Btk−/− NK cells of human and mouse, and epigenic regulation and environmental conditions may result in the inconsistent phenotype in different species. Recently, negative (ATF3, Hlx, and NK-κB p50) and positive regulators (STAT-4, EAT-2A, and -2B) have been reported one after another to modulate the activation progress and IFN-γ expression of NK cells (17, 20, 33), so we postulated it is probably that Btk cross-talks with them and initiates the downstream effects, and the underlying mechanism needs to be further studied.
Although the role of Btk in TLR signaling is widely accepted, little is known about the relationship of Btk and TLR3 in the innate response. Previous observations of TLR3 ligand stimulation resulting in significantly lower production of TNF-α, but neither IL-6 nor IL-12p70, by dendritic cells from XLA patients in comparison with normal controls (35) lay the foundation for the exploration of the cooperation of the Btk and TLR3 signal pathway. In our experiments, the positive role of Btk on poly(I:C)-triggered NK cell activation was confirmed both in vitro and in vivo. Previous studies have shown that poly(I:C) could significantly induce NK cell accumulation and activation in the liver, as characterized by elevated CD69 expression (36). The activated NK cells could subsequently give rise to liver injury, showing local spotty necrosis in the livers and elevated serum levels of ALT and AST (37), whereas depletion of NK cells enhances liver regeneration and partially abolishes the negative effects of poly(I:C) on liver regeneration (34). Our data suggested that, as to NK cell-mediated TLR3-triggered acute liver injury, Btk deficiency leads to the limited local spotty necrosis, as well as decreased infiltration of inflammatory cells and reduced production of serum IFN-γ, ALT, and AST, contributing to reduced mortality induced by injection with lethal dose of poly(I:C). Accordingly, this consequent reduction in serum IFN-γ, ALT, AST, and histopathological processes was almost completely abrogated by adoptive transfer of WT NK cells into Btk−/− mice. So Btk may be, at least partially, responsible for the NK cell-mediated tissue damage during severe infections with bacteria and some viruses. But how Btk acts in the downstream signaling of TLR3 is still obscure. Previous reports showed that NK cells utilized TNF-related apoptosis-inducing ligand and FasL to eliminate primary hepatocytes in liver injury, and whether and how Btk regulated their downstream signals therefore deserve further research.
In conclusion, this is the first report about the positive regulatory role of Btk in NK cell function in vitro and in vivo. Btk, robustly increased during the maturation and activation of NK cells, promotes poly(I:C)-induced NK cell activation by enhancing the production of IFN-γ, perforin, and granzyme-B and cytotoxicity, consequently aggravating the TLR3-triggered liver damage mainly in an NF-κB-dependent manner. Our results provide new insight into the regulatory mechanism of TLR3-triggered NK cell functions and make Btk a considerable target of the anti-inflammatory drug in the future.

This article contains supplemental Figs. S1–S3.
- Btk
- Bruton tyrosine kinase
- TLR
- Toll-like receptor
- XLA
- X-linked agammaglobulinemia
- XCI
- X chromosome inactivation
- PMA
- phorbol 12-myristate 13-acetate
- NK
- natural killer
- ALT
- alanine aminotransferase
- AST
- aspartate aminotransferase.
REFERENCES
- 1. Mohamed A. J., Yu L., Bäckesjö C. M., Vargas L., Faryal R., Aints A., Christensson B., Berglöf A., Vihinen M., Nore B. F., Smith C. I. (2009) Bruton tyrosine kinase (Btk). Function, regulation, and transformation with special emphasis on the PH domain. Immunol. Rev. 228, 58–73 [DOI] [PubMed] [Google Scholar]
- 2. Liu Y., Wu Y., Lam K. T., Lee P. P., Tu W., Lau Y. L. (2012) Dendritic and T cell response to influenza is normal in the patients with X-linked agammaglobulinemia. J. Clin. Immunol., in press [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Ormsby T., Schlecker E., Ferdin J., Tessarz A. S., Angelisová P., Köprülü A. D., Borte M., Warnatz K., Schulze I., Ellmeier W., Horejsí V., Cerwenka A. (2011) Btk is a positive regulator in the TREM-1/DAP12 signaling pathway. Blood 118, 936–945 [DOI] [PubMed] [Google Scholar]
- 4. Nisitani S., Satterthwaite A. B., Akashi K., Weissman I. L., Witte O. N., Wahl M. I. (2000) Post-transcriptional regulation of Bruton tyrosine kinase expression in antigen receptor-stimulated splenic B cells. Proc. Natl. Acad. Sci. U.S.A. 97, 2737–2742 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Contreras C. M., Halcomb K. E., Randle L., Hinman R. M., Gutierrez T., Clarke S. H., Satterthwaite A. B. (2007) Btk regulates multiple stages in the development and survival of B-1 cells. Mol. Immunol. 44, 2719–2728 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Sharma S., Orlowski G., Song W. (2009) Btk regulates B cell receptor-mediated antigen processing and presentation by controlling actin cytoskeleton dynamics in B cells. J. Immunol. 182, 329–339 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Ng Y. Y., Baert M. R., Pike-Overzet K., Rodijk M., Brugman M. H., Schambach A., Baum C., Hendriks R. W., van Dongen J. J., Staal F. J. (2010) Correction of B-cell development in Btk-deficient mice using lentiviral vectors with codon-optimized human BTK. Leukemia 24, 1617–1630 [DOI] [PubMed] [Google Scholar]
- 8. Khare A., Viswanathan B., Gund R., Jain N., Ravindran B., George A., Rath S., Bal V. (2011) Role of Bruton tyrosine kinase in macrophage apoptosis. Apoptosis 16, 334–346 [DOI] [PubMed] [Google Scholar]
- 9. Di Paolo J. A., Huang T., Balazs M., Barbosa J., Barck K. H., Bravo B. J., Carano R. A., Darrow J., Davies D. R., DeForge L. E., Diehl L., Ferrando R., Gallion S. L., Giannetti A. M., Gribling P., Hurez V., Hymowitz S. G., Jones R., Kropf J. E., Lee W. P., Maciejewski P. M., Mitchell S. A., Rong H., Staker B. L., Whitney J. A., Yeh S., Young W. B., Yu C., Zhang J., Reif K., Currie K. S. (2011) Specific Btk inhibition suppresses B cell- and myeloid cell-mediated arthritis. Nat. Chem. Biol. 7, 41–50 [DOI] [PubMed] [Google Scholar]
- 10. Bouaziz A., Amor N. B., Woodard G. E., Zibidi H., López J. J., Bartegi A., Salido G. M., Rosado J. A. (2007) Tyrosine phosphorylation/dephosphorylation balance is involved in thrombin-evoked microtubular reorganization in human platelets. Thromb. Haemost. 98, 375–384 [PubMed] [Google Scholar]
- 11. Lee S. H., Kim T., Jeong D., Kim N., Choi Y. (2008) The tec family tyrosine kinase Btk regulates RANKL-induced osteoclast maturation. J. Biol. Chem. 283, 11526–11534 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Kubo T., Uchida Y., Watanabe Y., Abe M., Nakamura A., Ono M., Akira S., Takai T. (2009) Augmented TLR9-induced Btk activation in PIR-B-deficient B-1 cells provokes excessive autoantibody production and autoimmunity. J. Exp. Med. 206, 1971–1982 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Vijayan V., Baumgart-Vogt E., Naidu S., Qian G., Immenschuh S. (2011) Bruton tyrosine kinase is required for TLR-dependent heme oxygenase-1 gene activation via Nrf2 in macrophages. J. Immunol. 187, 817–827 [DOI] [PubMed] [Google Scholar]
- 14. Bao Y., Han Y., Chen Z., Xu S., Cao X. (2011) IFN-α-producing PDCA-1+ Siglec-H-B cells mediate innate immune defense by activating NK cells. Eur. J. Immunol. 41, 657–668 [DOI] [PubMed] [Google Scholar]
- 15. Liu X., Zhan Z., Li D., Xu L., Ma F., Zhang P., Yao H., Cao X. (2011) Intracellular MHC class II molecules promote TLR-triggered innate immune responses by maintaining activation of the kinase Btk. Nat. Immunol. 12, 416–424 [DOI] [PubMed] [Google Scholar]
- 16. Sun J. C., Lanier L. L. (2011) NK cell development, homeostasis, and function. Parallels with CD8+ T cells. Nat. Rev. Immunol. 11, 645–657 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Wang N., Calpe S., Westcott J., Castro W., Ma C., Engel P., Schatzle J. D., Terhorst C. (2010) Cutting edge. The adapters EAT-2A and -2B are positive regulators of CD244- and CD84-dependent NK cell functions in the C57BL/6 mouse. J. Immunol. 185, 5683–5687 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Zhang M., Han Y., Han C., Xu S., Bao Y., Chen Z., Gu Y., Xia D., Cao X. (2009) The β2 integrin CD11b attenuates polyinosinic:polycytidylic acid-induced hepatitis by negatively regulating natural killer cell functions. Hepatology 50, 1606–1616 [DOI] [PubMed] [Google Scholar]
- 19. Rosenberger C. M., Clark A. E., Treuting P. M., Johnson C. D., Aderem A. (2008) ATF3 regulates MCMV infection in mice by modulating IFN-γ expression in natural killer cells. Proc. Natl. Acad. Sci. U.S.A. 105, 2544–2549 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Becknell B., Hughes T. L., Freud A. G., Blaser B. W., Yu J., Trotta R., Mao H. C., Caligiuri de Jesús M. L., Alghothani M., Benson D. M., Jr., Lehman A., Jarjoura D., Perrotti D., Bates M. D., Caligiuri M. A. (2007) Hlx homeobox transcription factor negatively regulates interferon-γ production in monokine-activated natural killer cells. Blood 109, 2481–2487 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Smith M. A., Maurin M., Cho H. I., Becknell B., Freud A. G., Yu J., Wei S., Djeu J., Celis E., Caligiuri M. A., Wright K. L. (2010) PRDM1/Blimp-1 controls effector cytokine production in human NK cells. J. Immunol. 185, 6058–6067 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Tu W., Zheng J., Liu Y., Sia S. F., Liu M., Qin G., Ng I. H., Xiang Z., Lam K. T., Peiris J. S., Lau Y. L. (2011) The aminobisphosphonate pamidronate controls influenza pathogenesis by expanding a γδ T cell population in humanized mice. J. Exp. Med. 208, 1511–1522 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Zheng J., Liu Y., Qin G., Lam K. T., Guan J., Xiang Z., Lewis D. B., Lau Y. L., Tu W. (2011) Generation of human Th1-like regulatory CD4+ T cells by an intrinsic IFN-γ- and T-bet-dependent pathway. Eur. J. Immunol. 41, 128–139 [DOI] [PubMed] [Google Scholar]
- 24. Qin G., Mao H., Zheng J., Sia S. F., Liu Y., Chan P. L., Lam K. T., Peiris J. S., Lau Y. L., Tu W. (2009) Phosphoantigen-expanded γδ T cells display potent cytotoxicity against human and avian influenza virus-infected monocyte-derived macrophages. J. Infect. Dis. 200, 858–865 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Vivier E., Tomasello E., Baratin M., Walzer T., Ugolini S. (2008) Functions of natural killer cells. Nat. Immunol. 9, 503–510 [DOI] [PubMed] [Google Scholar]
- 26. Stinchcombe J. C., Griffiths G. M. (2007) Secretory mechanisms in cell-mediated cytotoxicity. Annu. Rev. Cell Dev. Biol. 23, 495–517 [DOI] [PubMed] [Google Scholar]
- 27. Dustin M. L., Long E. O. (2010) Cytotoxic immunological synapses. Immunol. Rev. 235, 24–34 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Strowig T., Brilot F., Münz C. (2008) Noncytotoxic functions of NK cells. Direct pathogen restriction and assistance to adaptive immunity. J. Immunol. 180, 7785–7791 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Vivier E., Raulet D. H., Moretta A., Caligiuri M. A., Zitvogel L., Lanier L. L., Yokoyama W. M., Ugolini S. (2011) Innate or adaptive immunity? The example of natural killer cells. Science 331, 44–49 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Erwin J. A., Lee J. T. (2008) New twists in X-chromosome inactivation. Curr. Opin. Cell Biol. 20, 349–355 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Chow J. C., Yen Z., Ziesche S. M., Brown C. J. (2005) Silencing of the mammalian X chromosome. Annu. Rev. Genomics Hum. Genet. 6, 69–92 [DOI] [PubMed] [Google Scholar]
- 32. Shen Y., Matsuno Y., Fouse S. D., Rao N., Root S., Xu R., Pellegrini M., Riggs A. D., Fan G. (2008) X-inactivation in female human embryonic stem cells is in a nonrandom pattern and prone to epigenetic alterations. Proc. Natl. Acad. Sci. U.S.A. 105, 4709–4714 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Tato C. M., Mason N., Artis D., Shapira S., Caamano J. C., Bream J. H., Liou H. C., Hunter C. A. (2006) Opposing roles of NF-κB family members in the regulation of NK cell proliferation and production of IFN-γ. Int. Immunol. 18, 505–513 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Sun R., Gao B. (2004) Negative regulation of liver regeneration by innate immunity (natural killer cells/interferon-γ). Gastroenterology 127, 1525–1539 [DOI] [PubMed] [Google Scholar]
- 35. Taneichi H., Kanegane H., Sira M. M., Futatani T., Agematsu K., Sako M., Kaneko H., Kondo N., Kaisho T., Miyawaki T. (2008) Toll-like receptor signaling is impaired in dendritic cells from patients with X-linked agammaglobulinemia. Clin. Immunol. 126, 148–154 [DOI] [PubMed] [Google Scholar]
- 36. Dong Z., Wei H., Sun R., Hu Z., Gao B., Tian Z. (2004) Involvement of natural killer cells in poly(I:C)-induced liver injury. J. Hepatol. 41, 966–973 [DOI] [PubMed] [Google Scholar]
- 37. Dong Z., Wei H., Sun R., Tian Z. (2007) The roles of innate immune cells in liver injury and regeneration. Cell. Mol. Immunol. 4, 241–252 [PubMed] [Google Scholar]