

Background: Previous small scale studies indicate that DNA polymerase β variants are present in 30% of human tumors.
Results: 40% of samples in a large human colorectal tumor collection harbor coding region variants, many of which exhibit altered function.
Conclusion: Aberrant activity or fidelity phenotypes exhibited by variants may contribute to tumorigenesis.
Significance: Expression of variants in human tumors plays a role in driving carcinogenesis.
Keywords: Cancer, DNA Damage, DNA Polymerase, DNA Repair, Tumor
Abstract
Previous small scale sequencing studies have indicated that DNA polymerase β (pol β) variants are present on average in 30% of human tumors of varying tissue origin. Many of these variants have been shown to have aberrant enzyme function in vitro and to induce cellular transformation and/or genomic instability in vivo, suggesting that their presence is associated with tumorigenesis or its progression. In this study, the human POLB gene was sequenced in a collection of 134 human colorectal tumors and was found to contain coding region mutations in 40% of the samples. The variants map to many different sites of the pol β protein and are not clustered. Many variants are nonsynonymous amino acid substitutions predicted to affect enzyme function. A subset of these variants was found to have reduced enzyme activity in vitro and failed to fully rescue pol β-deficient cells from methylmethane sulfonate-induced cytotoxicity. Tumors harboring variants with reduced enzyme activity may have compromised base excision repair function, as evidenced by our methylmethane sulfonate sensitivity studies. Such compromised base excision repair may drive tumorigenesis by leading to an increase in mutagenesis or genomic instability.
Introduction
Alterations of DNA repair pathways are associated with colorectal cancer. Their critical nature is evidenced by defects in mismatch repair (MMR),3 methylation reversal repair, and base excision repair (BER) being associated with this disease (1, 2). Tumor-specific silencing of the MMR gene MLH1 has been linked to microsatellite instability, a frequent alteration found in key genes implicated in hereditary nonpolyposis colon cancer (3). Additionally, in hereditary nonpolyposis colon cancer patients, germ line missense mutations have been identified in MMR pathway members, including MLH1, MSH2, and MSH6 (4–6). Aberrant or reduced levels of MMR can result in defects in apoptosis, leading to resistance to therapy (7, 8).
Reduced levels of O6-methylguanine DNA methyltransferase (MGMT) resulting from promoter hypermethylation have been observed in colorectal tumors (9, 10). Persistence of O6-methylguanine can lead to mispairing with thymine during DNA replication. An MGMT deficiency would be expected to increase spontaneous G→A transition mutations due to this mispairing, and such mutations have been observed in both p53 and K-ras within these MGMT-deficient colon tumors (9, 11). Alternatively, expression or overexpression of MGMT in tumors leads to resistance to alkylating agent-based chemotherapy (12).
The BER glycosylase MYH initiates repair of oxidative DNA damage in cells, and its absence may lead to accumulation of mutations related to tumorigenesis or tumor progression. MYH recognizes and removes the adenine that is mispaired with 8-oxoguanine, providing a substrate for BER. Biallelic germ line mutations that reduce MYH activity have been identified in colorectal tumor patients (13). As predicted, patients harboring mutation in the MYH gene also had a significant increase in tumor-specific G:C→T:A transversions within the adenomatous polyposis coli gene, leading to various truncations. Biallelic germ line MYH mutations have also been identified in conjunction with tumor-specific inactivating mutations in MLH1, indicating that BER defects may be linked to colorectal cancer (14).
The BER pathway is responsible for resolving up to 20,000 lesions per cell per day, which include oxidative and alkylation damage (15, 16). DNA pol β is the primary polymerase involved in BER, through its bifunctional deoxyribose phosphate lyase and polymerase activities, and it functions in all subpathways of BER (17).
Previous small scale studies have identified pol β variants on average in 30% of a variety of human cancers, including six out of eight colorectal tumors (18). The sample sizes from these studies ranged from 8 to 42 tumors depending on tumor type, and the described variants were not present in matched normal controls.
The majority of POLB variants identified in the previous small scale colorectal tumor study contained in-frame large deletions in the palm domain. Banerjee and co-workers (19, 20) characterized an 87-bp deletion variant (polβΔ) from this collection and showed that it does not support BER in cells. BER assays with whole cell extracts show that polβΔ acts as a dominant negative when expressed in the presence of wild type and that this effect is due to increased affinity for XRCC1 (21). Bound to XRCC1, polβΔ has a greater affinity for DNA than wild type but does not have polymerase activity in this context. HeLa cells expressing polβΔ in a wild-type background are also hypersensitive to both UV radiation and the alkylating agent methylnitronitrosoguanidine.
The only amino acid substitution identified in the previous small scale study was the nonsynonymous K289M variant. We have shown that K289M variant protein induces cellular transformation in immortalized mouse epithelial cells by increasing the mutation frequency of the cells (22, 23). We have also shown that this variant is a sequence context-dependent mutator, in that it induces mutations at a frequency 16 times higher than wild-type (WT) pol β within a nucleotide sequence context found in the adenomatous polyposis coli gene that is frequently mutated in colon cancer (22, 24).
The finding of POLB variants in six of eight colon tumors and the fact that most of them are functional pol β variants that could drive carcinogenesis led us to determine whether POLB was frequently mutated in colon cancer. In this study, we show that 40% of the colon tumors we characterized have mutations in the POLB gene, suggesting that normal pol β function is critical for the suppression of colon cancer. Importantly, many of the pol β variants we identified that were associated with late stage carcinomas were less active polymerases than WT pol β. Previous work from our laboratory (25) and work described in the accompanying paper (53) show that genomic instability arises in cells expressing low activity pol β variants and that this is likely to drive carcinogenesis.
EXPERIMENTAL PROCEDURES
Genomic DNA Extraction
Colon tumor cores were obtained from the YTMA8 cohort of the Yale University Pathology Tumor Collection; the YTMA8 cohort is described in Ref. 26. Patient data, including tumor stage, are available for most tumors in this collection. Formalin-fixed paraffin-embedded tissue blocks containing colorectal tumors were retrieved from the archives of the Department of Pathology, Yale University. Areas of invasive carcinoma were identified on corresponding hematoxylin and eosin-stained slides and tissue blocks, and 1.5 μm cores were isolated from formalin-fixed paraffin-embedded tumor tissue samples. Distinct normal tissue regions were identified by histological appearance and obtained from the same corresponding tumor block by microdissection if such a determination was possible by the presence of clear margins.
Genomic DNA was isolated from formalin-fixed paraffin-embedded cores using either the RecoverAll total nucleic acid isolation kit (Ambion) or the DNeasy kit (Qiagen) according to the manufacturer's instructions. DNA sample quality was evaluated by comparing the A260/A280 ratio obtained by UV spectroscopy (Nanodrop).
PCR Amplification and DNA Sequencing
Exons 1–14 as well as the 3′- and 5′-untranslated regions (UTRs) of the human POLB gene (NM_002690.1) were amplified using nested or semi-nested PCR with amplicon-specific primer sequences and conditions (supplemental Table S1). Exon 1 and the 5′UTR were present on the same amplicon. Consensus sequences were obtained from the AceView data base, Human 2007 build (www.ncbi.nlm.nih.gov). Primer sequences were aligned to the human genome using BLAST to confirm specificity (blast.ncbi.nlm.nih.gov). Direct sequencing of PCR products was performed at the Keck DNA Sequencing Facility, Yale University School of Medicine. Chromatogram files were visualized and aligned to consensus sequences using the Geneious software program, Version 3.8.5.
TOPO TA Cloning
For the mutations producing S275N and E295K identified in the same tumor on the same amplicon, we used TOPO TA cloning to determine whether the mutations were present on the same DNA copy. 4 μl of PCR product was used with the TOPO TA cloning kit (Invitrogen) according to the manufacturer's instructions. Nine distinct clones were sequenced at the Keck DNA Sequencing Facility, Yale University, using primers provided by the TOPO TA cloning kit.
Variant Impact Predictions
The impact of each nonsynonymous amino acid substitution on enzyme function was assessed using both the PolyPhen (27) and SIFT (28) prediction algorithms. Default settings and thresholds were used for both programs. Variants that were predicted to affect protein function in either or both programs were selected for further analysis.
Plasmid DNA Constructs and Cloning
For protein purification, the wild-type pol β cDNA was cloned into the pET28a vector containing an N-terminal hexahistidine tag as described previously (29). pol β variant sequences were generated with the QuikChange site-directed mutagenesis protocol (Stratagene, primer sequences available upon request). Variant sequences were confirmed by direct sequencing at the Keck DNA Sequencing Facility, Yale University School of Medicine. For retroviral infection into mouse cells, the wild-type pol β cDNA with a C-terminal hemagglutinin (HA) tag was cloned into the pRVYtet retroviral vector as described previously (23). pol β variant sequences were generated and confirmed using the above protocol.
Protein Expression and Purification
The wild type and variant pol β pET28a constructs were transformed into Escherichia coli strain Rosetta(DE3) by electroporation. Luria Broth cultures supplemented with 50 μg/ml kanamycin and 34 μg/ml chloramphenicol were grown overnight at 37 °C, then diluted 1:100, and grown to an A600 of ∼0.5. Protein expression was then induced with 1 mm isopropyl β-d-thiogalactopyranoside for 2 h at 37 °C, and the cells were harvested by centrifugation. Induction of protein expression was evaluated by SDS-PAGE and Coomassie Blue stain. Pellets were stored at −80 °C and thawed overnight on ice at 4 °C immediately prior to purification. Proteins were purified either manually with nickel beads or by FPLC. The thawed pellet was resuspended in Buffer B (40 mm Tris-HCl, pH 8, 500 mm NaCl, 5 mm imidazole) for FPLC purification or lysis buffer (50 mm NaH2PO4, 300 mm NaCl, 10 mm imidazole) for nickel bead purification, supplemented with protease inhibitors (Roche Applied Science) and 1 mm PMSF, and sonicated on ice. The lysate was then clarified by centrifugation. For nickel bead purification, 1.5 ml of nickel beads (Qiagen) were equilibrated with lysis buffer. Clarified lysate was added to the beads and rocked for 90 min at 4 °C. Nickel beads were washed with lysis buffer, followed by two washes with wash buffer (50 mm NaH2PO4, 300 mm NaCl, 40 mm imidazole). His-tagged proteins were eluted from the beads by rocking with 250 μl of elution buffer (50 mm NaH2PO4, 300 mm NaCl, 250 mm imidazole) for 15 min at 4 °C. FPLC purification was performed as described previously (30). Briefly, a 5-ml HiTrap chelating HP column (GE Healthcare) was used with a linear imidazole elution gradient. Fractions were combined and concentrated to ∼1 ml and diluted to 10 ml. The resulting solution was applied to a 5-ml HiTrap SP HP column (GE Healthcare), and the final protein was eluted using a linear NaCl gradient. Fractions were combined and concentrated to 400–500 μl. For both purification protocols, glycerol was added to be 15% of the final volume, and preparations were stored at −80 °C. Purity was determined to be >85% for nickel bead-purified proteins and >90% for FPLC-purified proteins by SDS-PAGE and Coomassie Blue staining. Protein concentration was determined using the A280 (Nanodrop) and the extinction coefficient for pol β (ϵ = 21,200 m−1 cm−1).
DNA Substrate Preparation
The DNA oligonucleotides used in this study were prepared as described previously (29). Briefly, the oligonucleotides were synthesized by the Keck Oligo Synthesis Facility, Yale University School of Medicine, and purified by PAGE chromatography. Purified primer oligonucleotides were radiolabeled at the 5′ end with γ-32P, and purified downstream oligonucleotides were kinased with nonradioactive ATP, both using T4 polynucleotide kinase (New England BioLabs). Annealed products were assessed by resolving them on a 12% native polyacrylamide gel and visualized by autoradiography.
In Vitro Primer Extension Assays
Primer extension assays were performed using either 50 μm each of all four dNTPs or three, one, or zero dNTP(s) present in all combinations in buffer containing 50 mm Tris-HCl, pH 8, 10 mm MgCl2, 2 mm dithiothreitol (DTT), 20 mm NaCl, and 10% glycerol. These assays were performed using single turnover conditions with 750 nm pol β and 50 nm of 5-bp gapped DNA substrate. Reactions were carried out for 5 min at 37 °C, quenched with an equal volume of solution containing 1:1 90% formamide dye, 0.5 m EDTA, and placed on ice. Reaction samples were resolved on a 20% denaturing polyacrylamide gel containing 8 m urea, and the products were visualized using a Storm 860 Phosphorimager with ImageQuant software.
Pre-steady State Burst Kinetic Analysis
For comparatively faster variants, rapid chemical quench kinetics were performed using a KinTek apparatus (31) as described previously (29). Briefly, two reaction mixtures (600 nm DNA and 200 nm enzyme and 200 μm correct dNTP and 20 mm MgCl2) were separately prepared in Reaction Buffer (50 mm Tris, pH 8.0, 100 mm NaCl, 2 mm DTT, and 10% glycerol) at 2× concentrations and preincubated at 37 °C for 3 min. Mixtures were loaded onto the KinTek apparatus thermostated at 37 °C, and equal volumes of both solutions were reacted on a millisecond time scale from 0.02 to 3 s with 0.5 m EDTA added to quench the reactions. Extended products were resolved on a 20% polyacrylamide gel containing 8 m urea, visualized using a Storm 860 phosphorimager, and quantified with ImageQuant software. Extended product was plotted as a function of time using KaleidaGraph software (version 3.6.2) and fit by nonlinear regression to the burst equation, [product] = A (1 −exp (−kobst)) + ksst, where A is the amplitude of the burst; kobs is the observed rate constant for the exponential phase; kss is the observed steady state rate constant, and t is time. For comparatively slower variants, the observed rate constant was determined manually using the reaction conditions described above over a longer time course ranging from 30 s to 45 min as described previously (30). Extended product was plotted as a function of time and fit to a single exponential equation, [product] = A (1 −exp (−kobst), where A is the amplitude; kobs is the observed exponential phase rate constant, and t is time.
Cell Lines and Cell Culture
The mouse embryonic fibroblast (MEF) cell line 88TAg (pol β−/−) was a gift from Leona Samson (Massachusetts Institute of Technology). MEF cell lines were maintained in DMEM (Invitrogen) supplemented with 10% FBS (Gemini), 1% penicillin/streptomycin, and 1% l-glutamine (Invitrogen) and grown at 37 °C in a humidified 5% CO2 incubator. GP2-293 cells (Clontech) used for viral packaging were maintained in DMEM supplemented with 10% FBS, 1% penicillin/streptomycin, 1% l-glutamine, and 1 mm HEPES (Invitrogen).
Transfection, Infection, and Expression Analysis
Retroviruses encoding variant or wild-type pol β proteins were generated by calcium phosphate co-transfection of GP2-293 cells with the pVSVG and pRVYtet plasmids. Following transfection, stable integrants were selected in the presence of 200 μg/ml hygromycin B (Invitrogen). These cell lines were used to generate high titer virus by transfection with pVSVG alone, and virus was harvested after 72 h post-transfection. MEF cell lines were grown to ∼30% confluence and infected with retrovirus supplemented with 4 μg of Polybrene. Stably expressing MEF pools were selected with 220 μg/ml hygromycin B. MEF pools were maintained in DMEM supplemented with 10% FBS, 1% penicillin/streptomycin, 1% glutamine, and 220 μg/ml hygromycin B and grown for no more than six passages. Expression of exogenous pol β was confirmed by Western blot as described previously using AbCam AB1831 (32) and normalized to tubulin (Cell Signaling Technology, catalog no. 2125S). Bands were visualized using an enhanced chemiluminescence kit according to the manufacturer's instructions (Bio-Rad) and a super-cooled high resolution CCD camera on the Bio-Rad ChemiDoc Imaging System. Bands were quantified using ImageLab software Version 3.0 (Bio-Rad).
MMS Sensitivity Assay
pol β-deficient MEFs expressing slow, damaging pol β variants were evaluated for sensitivity to the alkylating agent MMS as described previously (25). In brief, cells were seeded into 96-well plates at 1500 cells per well and incubated at 37 °C and 5% CO2 overnight to allow attachment. Cells were treated with various concentrations of MMS from 0 to 1.2 mm for 1 h at 37 °C and then washed in fresh media and incubated for 72 h at 37 °C and 5% CO2. Growth inhibition was determined by using the MTS CellTiter 96 AQueous One solution cell proliferation assay (Promega) according to the manufacturer's instructions. Four replicates of each cell line were averaged, and results were calculated as percentage growth of untreated control wells. Statistical analysis and graphing was performed using GraphPad Prism 5.0 software.
RESULTS
Forty Percent of Human Colorectal Tumors Harbor Mutations in Coding Regions of the POLB Gene
In our collection of 134 human colon tumors, we identified 75 tumors (56%) that harbor mutations in either the coding region or the UTR regions of POLB (Fig. 1A and supplemental Table S2). Of the 75 tumors with a mutation, 53 tumors (40%) contained at least one coding region mutation. Forty two tumors (31%) contain at least one nonsynonymous amino acid substitution (Table 1 and Fig. 1B), and 18 tumors (13%) contain at least one synonymous amino acid substitution (supplemental Table S3). Of these, two tumors contain early truncation mutations. One tumor has a single nucleotide deletion resulting in a frameshift. This frameshift affects all amino acids following Lys-168 and introduces a new stop codon at position 196. Coding region mutations map to all protein subdomains, and the majority of resulting amino acid variants has not been described previously (Fig. 2).
FIGURE 1.
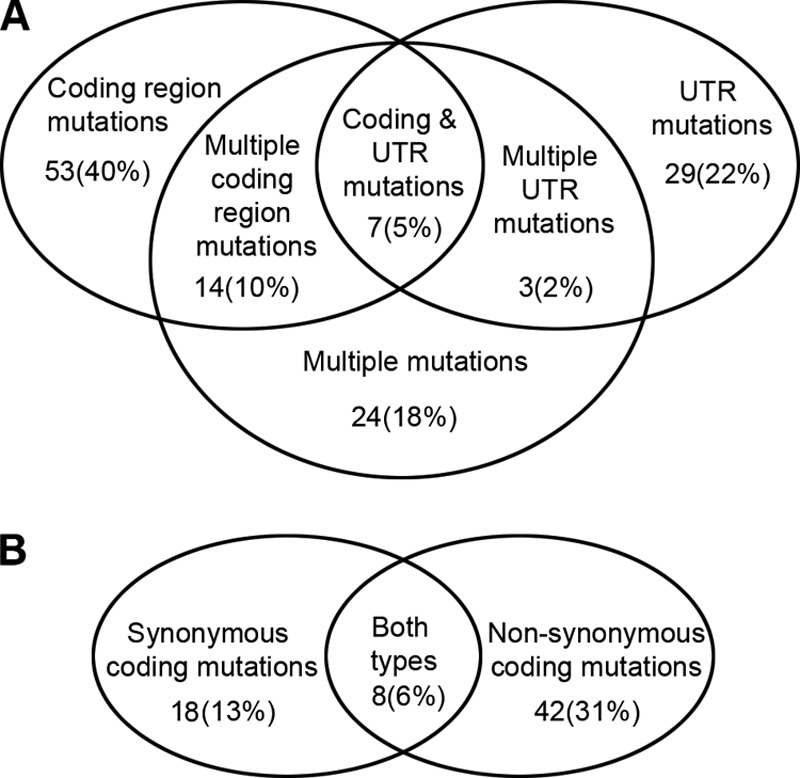
Schematic of identified mutations in tumors. A, number and percentage of tumors with the specific mutation category is indicated. B, number and percentage of tumors with synonymous and/or nonsynonymous coding mutations is indicated.
TABLE 1.
Nonsynonymous amino acid substitution variants identified in human colorectal tumors
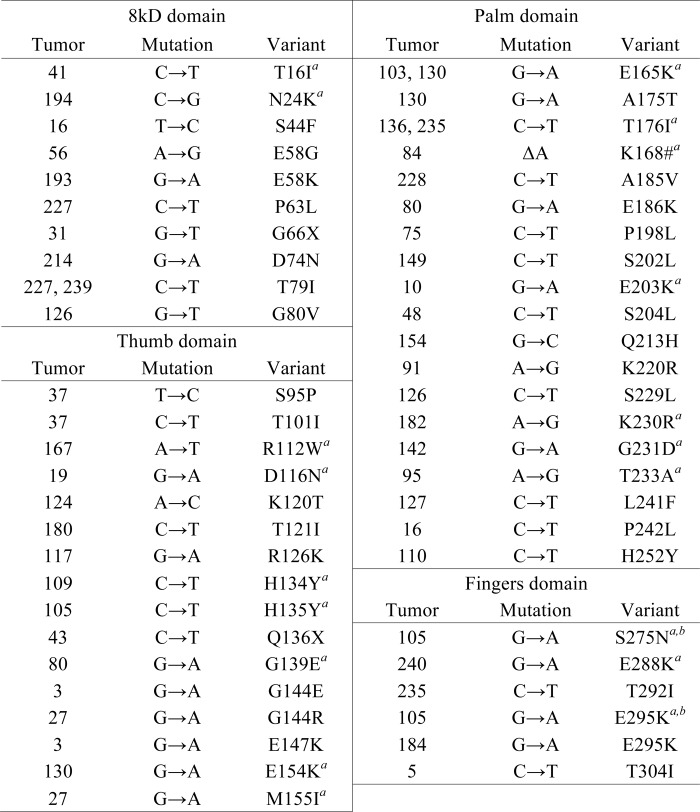
a Matched normal mucosa were sequenced for corresponding exon.
b Data were identified on the same DNA copy by TOPO-TA cloning.
FIGURE 2.

Nonsynonymous coding region variants are found in all domains of DNA pol β. A, pol β contains four subdomains: 8 kDa (red), thumb (green), palm (blue), and fingers (purple). A 1-bp gapped DNA substrate is indicated in gray (Protein Data Bank code 1bpy (52)). B, all nonsynonymous amino acid substitutions identified in this study are indicated as orange spheres.
One tumor also has the E295K variant, which has been previously identified in a gastric tumor and has been shown to be an inactive polymerase that induces genomic instability and cellular transformation (25, 33). Interestingly, 82% of the missense mutations identified in this study are CG to TA transitions and are suggestive of mutagenesis occurring by deamination of 5-methylcytosine. A similar mutational bias toward CG to TA mutations has been reported previously in colon tumor studies (34–36).
The ratio of nonsynonymous to synonymous mutations is informative regarding selection. The idea is that mutants with nonsynonymous substitutions are able to be selected because the mutation alters protein structure and function. Synonymous mutations are thought to be silent and are therefore unlikely to undergo biological selection. A higher ratio of nonsynonymous/synonymous mutations compared with what is expected by chance indicates that the mutations are selected. Therefore, we estimated the selection pressure, φ, as described previously (37), for our observed 19 synonymous and 51 nonsynonymous mutations. A selection pressure greater than 1 indicates positive selection, whereas less than 1 is consistent with negative selection. We estimated that φ = 1.68 (95% confidence interval, 0.997–2.363; p = 0.025), showing that we have an excess of nonsynonymous mutations than what would be expected by chance. This estimate strongly suggests that cancer driver mutations exist among the mutations we observed.
Mutations Present in POLB-untranslated Regions May Be Functional
In sequencing the 3′- and 5′UTR of POLB, we found 20 tumors (15%) with 3′UTR mutations and 12 tumors (9%) with 5′UTR mutations (supplemental Table S4). Six mutations identified in the 3′UTR region are found in predicted miRNA-binding sites (TargetScan, Version 5.1) that are poorly conserved. These mutations may result in reduced binding of these miRNAs, leading to altered post-transcriptional regulation of pol β expression levels. Alternatively, mutations present in the 3′UTR may create novel miRNA-binding sites. For the identification of novel sites, we focused on miRNAs hsa-miR-(548–663) identified in human colorectal cells from tumors, normal tissue, and tumor cell lines (38). Wild-type and variant 3′UTR sequences were input into RNA hybrid (39) using a helix constraint from 2 to 8, with all other parameters set to defaults. 3′UTR sequences were input such that the position of the mutation would be included in the seed sequence, a 2–8-nucleotide region critical for miRNA binding (40). The same sequence region used for each variant was used for the wild-type sequence. Wild-type and variant 3′UTR sequences were compared against human miRNA residues 548–663. We compared the minimum free energy of hybridization values between wild-type and variant 3′UTR sequences. We identified decreases in minimum free energy binding to multiple 3′UTR variant sequences relative to wild type for miR-570, miR-583, miR-615–5p, miR-637, and miR-638 (supplemental Table S4). Given that these miRNAs are known to be present in human colorectal tissue, mutations identified in the POLB 3′UTR in colorectal tumors may result in aberrant translational repression. The 3′UTR of the pol β mRNA has also been shown to form a complex regulatory hairpin structure that interacts with Hax-1, an anti-apoptotic factor (41). Disruption of the conserved M2 hairpin by site-directed mutagenesis of a GGG motif was shown to affect mRNA stability and protein expression levels in FTO-2B cells. Here, we have identified mutations in this same GGG motif, which may exert a similar effect on hairpin formation and thus affect post-translational regulation of pol β (supplemental Table S4). One mutation identified in the 5′UTR region is present in a known binding site for the Sp1 transcription factor (5′-GCCCCGCCCC) (42). Alteration of the Sp1-binding site may result in aberrant levels of POLB transcription.
Colon Tumors Harbor Multiple Mutations in the POLB Gene
Of 134 tumors sequenced, 24 tumors (18%) were found to contain multiple mutations in POLB (supplemental Table S5). The status of these mutations in cells is unknown, as within these tumors there may be multiple distinct cells with single mutations that express different single variant proteins. Alternatively, there may be multiple single variants present on different DNA copies within the same cell, leading to expression of multiple distinct variant proteins within a single cell. There may also be multiple mutations present on the same DNA copy within a cell, giving rise to a single protein with multiple variant sites. In the case of S275N and E295K identified from the same exon and amplicon, from the same tumor, we were able to determine that they are present on the same DNA copy.
No Mutations Are Identified in Normal Tissue
We were able to obtain microdissected tissue outside of the tumor margin for 30 tumors, constituting 40% of the 75 tumors that had POLB mutation(s). Tumors with corresponding matched normal tissues are indicated in supplemental Table S2. For 37 mutations present in these 30 tumors, no corresponding mutation was detected using the same PCR and sequencing methods used to detect the tumor-associated mutation. This comparison between 30 tumors and their corresponding matched normal tissues is significant by the McNemar test with continuity correction performed using R2.12.0 software (p = 1.192 × 10−7). These results indicate that identified variants are likely tumor-specific. None of the coding region mutations identified in this study are known to be germ line single nucleotide polymorphisms and are therefore likely somatic mutations that are associated with the tumor (43). Polymorphisms present in the UTR regions were also not observed in our samples (dbSNP, www.ncbi.nlm.nih.gov). Given that the minor allele frequencies of POLB single nucleotide polymorphisms are less 0.02, we would not expect to observe these single nucleotide polymorphisms with the size of our cohort.
A Subset of Mutations Is Predicted to Affect Protein Function
Fifty one coding region mutations resulting in nonsynonymous amino acid substitutions were identified in 42 (31%) tumors. All nonsynonymous amino acid substitutions were evaluated using the SIFT and PolyPhen prediction algorithms to assess their effect on protein function. Seventeen tumors (13%) harbored a total of 21 nonsynonymous coding region mutations that were predicted by one or both programs to impact protein function (Table 2), and these protein products are herein referred to as damaging variants. The use of such algorithms facilitated the prioritization of variants for subsequent in vitro and in vivo analysis, and it is not considered to be an absolute predictor of the consequences of identified amino acid substitutions. For example, the K289M pol β colon cancer variant was not predicted to be damaging by these algorithms.
TABLE 2.
Algorithm predictions and gap-filling activity of pol β variants
| Damaging variant | SIFT | PolyPhen | In vitro enzyme activity relative to WTa |
|---|---|---|---|
| N24K | Tolerated | Possibly damaging | Normal |
| S44F | Tolerated | Probably damaging | Normal |
| E58G | Affect function | Probably damaging | Normal |
| P63L | Affect function | Benign | Normal |
| T79I | Affect function | Probably damaging | Normal |
| G80V | Affect function | Probably damaging | Greatly reduced |
| T101I | Tolerated | Possibly damaging | Normal |
| R112W | Tolerated | Probably damaging | Normal |
| T121I | Affect function | Probably damaging | Reduced |
| H134Y | Affect function | Probably damaging | Normal |
| H135Y | Tolerated | Probably damaging | Normal |
| G139E | Affect function | Probably damaging | Greatly reduced |
| T176I | Tolerated | Possibly damaging | Normal |
| P198L | Affect function | Probably damaging | Normal |
| S229L | Tolerated | Possibly damaging | Reduced |
| G231D | Tolerated | Probably damaging | Greatly reduced |
| L241F | Affect function | Probably damaging | Normal |
| P242L | Affect function | Probably damaging | Normal |
| S275N/E295K | Affect function | Probably damaging | Undetectable |
| T292I | Tolerated | Probably damaging | Greatly reduced |
| E295K | Affect function | Probably damaging | Undetectableb |
Damaging Variants Are Associated with Advanced Tumor Stages 3 and 4
Patient information regarding tumor stage (stages 1–4) was available for the corresponding tumors for 94 of 107 mutations identified (Table 3). Sixteen mutations resulting in damaging variants were found in tumors staged 3 or 4, whereas only three such mutations were found in tumors staged 1 or 2. An increase in the proportion of mutations that give rise to damaging amino acid substitutions observed in stages 3 and 4 is statistically significant (p = 0.0016, Fisher's exact test).
TABLE 3.
Staging information for mutations in tumors found to harbor damaging pol β variants
| Mutations in tumors with staging information | |||
|---|---|---|---|
| Damaging variant | Stage 1 or 2 | Stage 3 or 4 | Total |
| No | 43 (93.5%) | 32 (67%) | 75 |
| Yes | 3 (6.5%) | 16a (33%) | 19 |
| Total | 46 | 48 | 94 |
a p = 0.0016, Fisher's exact test.
Many Damaging Variants Have Altered Activity in Vitro
To evaluate general polymerase activity, a DNA gap-filling assay using proteins purified by the nickel bead method was used. In this assay, wild-type pol β fills in a 5-bp gap and performs strand-displacement synthesis, displacing the downstream sequence to synthesize the full-length 45-mer product. This assay is used as a preliminary qualitative assessment of polymerase activity to determine whether any of the 21 variants behave differently than wild type. Eight of 21 variants, including the previously described E295K variant, exhibited reduced gap-filling or strand-displacement activity when incubated with the DNA substrate and all four dNTPs (Fig. 3 and Table 2). Variants that had altered activity in the gap-filling assay, with the exception of the previously characterized E295K variant, were selected for further analysis.
FIGURE 3.
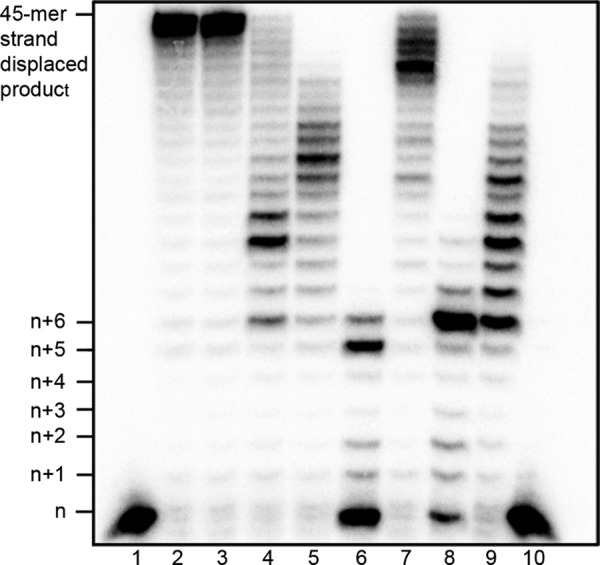
Damaging pol β variants exhibit altered gap-filling or strand displacement activity. A 5-bp gapped DNA substrate was incubated with pol β in the presence of all four dNTPs for 5 min as described under “Experimental Procedures.” Lane 1 is a wild-type pol β reaction carried out without dNTPs, as indicated by the unextended primer. Lane 2 is wild-type pol β. Lane 3 is S44F, representative of a damaging variant that is fully functional in this assay. The remaining lanes are as follows: lane 4, G80V; lane 5, T121I; lane 6, G139E; lane 7, S229L; lane 8, G231D; lane 9, T292I; and lane 10, S275N/E295K.
The variants G80V, T121I, G139E, S229L, G231D, T292I, and S275N/E295K were purified by FPLC. Pre-steady state burst kinetics of these variants were used to provide a quantitative comparison of burst rate and to determine whether the variants follow the same kinetic pathway as wild type. Under pre-steady state conditions, wild type exhibits a rapid burst of product formation followed by a slower linear phase corresponding to product release (Fig. 4A). The majority of variants that had an altered phenotype in the gap-filling assay exhibited reduced burst rates of varying degrees (Table 4). The two fastest variants, G80V and T121I, exhibited biphasic burst kinetics similar to wild type, however, with a reduced catalytic rate. The remaining variants exhibited significantly slower rates of product formation that were fit to the single exponential equation, rather than the biphasic burst, indicating that these variants follow an altered kinetic pathway compared with wild type (Fig. 4B).
FIGURE 4.

Representative pre-steady state burst kinetic plots of wild-type pol β (A) and G139E (B). A 1-bp gapped DNA substrate was incubated with pol β and dNTP on a millisecond or minute time scale as described under “Experimental Procedures.” Extended product formed was plotted as a function of time and fit to the biphasic burst equation for wild type with a kobs of 14 ± 3/s. Extended product formed by G139E was fit to a single exponential equation with a kobs of 0.004 ± 0.0001/s.
TABLE 4.
Burst rates and MMS rescue phenotypes for slow variants
| DNA pol β | kobs | MMS sensitivity rescue |
|---|---|---|
| s−1 | ||
| Wild type | 14 ± 3 | Complete |
| G80V | 14 ± 3 | Complete |
| T121I | 6 ± 1 | Partial |
| G139E | 0.004 ± 0.0001 | None |
| S229L | 0.3 ± 0.008 | Partial |
| G231D | 0.1 ± 0.005 | Partial |
| T292I | 1.9 ± 0.2 | Complete |
| S275N/E295K | Undetectable | None |
| E295Ka | Undetectable | None |
a Results were reported previously (25).
Most Damaging Variants Cannot Fully Complement the MMS Sensitivity of pol β-deficient Cells
To evaluate the consequences of reduced enzymatic rates on activity in a cellular context, we looked at the ability of slow variants to complement the MMS sensitivity of pol β-deficient cells. pol β-deficient cells are sensitive to the alkylating agent MMS, indicating that these cells have reduced BER capacity related to the absence of pol β (44). E295K has been previously shown to lack the ability to complement MMS sensitivity in pol β-deficient cells (25). Low passage pools of pol β-deficient MEFs expressing each slow variant, wild-type, or empty vector (pRVY) were generated as described under “Experimental Procedures.” Exogenous expression in our cell lines was confirmed by Western blot (supplemental Fig. S1). Expression of exogenous wild type rescues the MMS sensitivity of the pol β-deficient cells (Fig. 5A). The slow variants exhibit a variety of phenotypes, ranging from complete rescue to sensitivity on par with the empty vector. Representative data are shown for variants that exhibit complete, partial, or no rescue (Fig. 5, B–D). Observed rescue does not correlate to exogenous pol β expression levels. The degree of rescue appears to be related to the observed in vitro pre-steady state burst rate for most variants (Table 4), indicating that slower variants may not support BER in vivo. For example, the variant G231D has 140-fold reduction in rate relative to wild type and exhibits partial rescue in cells. The double variant S275N/E295K does not form any product under burst conditions and does not provide any rescue in cells in this context.
FIGURE 5.

Slow damaging variants exhibit varying degrees of rescue in a pol β-deficient MEFs to MMS. A, wild-type pol β rescues the sensitivity of pol β-deficient MEFs exposed to MMS compared with the empty vector control. B, representative example of complete rescue by a pol β variant, T292I. C, representative example of partial rescue by a pol β variant, G231D. D, representative example of a pol β variant that does not rescue, S275N/E295K.
DISCUSSION
The goal of this study was to determine whether the POLB gene is mutated in a large percentage of colorectal tumors, as suggested to be the case from a small scale study (45). We sequenced all exons, the 3′UTR and 5′UTR of the POLB gene. Here, we demonstrate that 75 (56%) of 134 human colorectal tumors studied harbor at least one mutation in POLB. We identified coding region mutations in 40% of tumors, most of which were not observed previously in other tumors and none of which are germ line single nucleotide polymorphisms. Our results strongly suggest that there are cancer driver mutations among the POLB nonsynonymous mutations. Therefore, our study suggests that normal functioning of pol β is critical for suppression of colon cancer. In combination with other studies of the POLB gene in tumors, our study demonstrates that there are no mutational hot spots in POLB in tumors. Many of the variants share the common phenotype of low polymerase activity, no matter where they map on the protein. Some of the tumor-associated variants we identified catalyze DNA synthesis with a rate slower than WT pol β. In our previous study of the E295K variant (25) and in the accompanying paper (53), we have shown that expression of low activity pol β variants induces genomic instability and cellular transformation, strongly suggesting that aberrant pol β function drives carcinogenesis.
Coding Region Mutations Exhibit Functional Phenotypes
Eight variants were found to exhibit functional phenotypes, including reduced polymerase activity and decreased BER function. Our selection of these eight variants was based on analysis using algorithms that are predictive of deficiencies in either polymerase or deoxyribose phosphate lyase activity. These predictions are likely imperfect and constitute an underestimate of variants with aberrant activity. Therefore, it is likely that there are additional colorectal cancer-associated variants we identified in the 134 tumors studied that result in aberrant BER that could drive carcinogenesis or impact therapeutic response.
All of the mutations identified, with the exception of E295K, appear to be novel tumor-associated somatic mutations. The previously characterized variant E295K, first identified in a gastric tumor and also identified here, does not possess any polymerase activity (25, 45). When E295K is expressed in cells, cellular transformation and genomic instability result. This variant also has been shown not only to fail to rescue MMS sensitivity in a pol β-deficient background but also to act as a dominant negative in the presence of wild-type pol β. Thus, E295K has a functional phenotype that is consistent with its acting as a driver of cancer. Interestingly, the colon tumor variant T304I, also not predicted to be damaging, has been previously identified as an 3′-azido-3′-thymidine-resistant variant (46). T304I has a pre-steady state burst rate similar to wild type, but it does not interact with XRCC1 in vivo (47), suggesting that it will not be scaffolded properly during BER, likely leading to a deficiency in gap filling. Threonine residue 79, found to be altered to isoleucine in our study, has been shown to contribute to polymerase fidelity (48).
In the accompanying paper (53), we show that the low activity G231D variant lacks discrimination at the dNTP binding step, and it induces genomic instability and cellular transformation when expressed in cells.
Mutations that result in synonymous amino acid substitutions may have an effect at the level of translation. The usage of a relatively rare codon during translation could lead to inappropriate ribosomal pausing followed by translation termination and degradation of the mRNA (49). For example, the substitution L195L identified here results from a codon change of CTG to CTA. In humans, the CTA codon is used 6-fold less frequently than CTG (50). In this case, the mutation encoding the synonymous L195L variant was present in homozygous form, suggesting that wild-type pol β may be expressed at a significantly lower level in this tumor, which could result in significantly fewer gaps being filled, and lead to genomic instability and cancer.
Alternatively, rare codons may be required to regulate the rate of translation to facilitate proper enzyme folding. A silent mutation that results in conversion of a rare codon to a more common codon could result in improper folding and subsequent peptide degradation due to a faster rate of translation. Here, the synonymous substitution L311L results from the TTG codon being altered to CTG, a nearly 4-fold more commonly used codon (50). Conversely, the L311L codon change could result in faster rates of translation without affecting protein folding, increasing overall pol β protein levels. Overexpression of pol β has previously been linked to various human cancers (51).
Noncoding Mutations May Affect Gene Regulation
Noncoding mutations identified in the 3′UTR may affect gene expression at the post-transcriptional level. Mutations identified that appear in putative miRNA-binding sites may disrupt targeting by miRNAs, leading to an increase in overall levels of pol β. Alternatively, 3′UTR mutations were found that create novel miRNA-binding sites that could result in inappropriate translational regulation in colorectal tissue. We also identified 3′UTR mutations within a key hairpin-forming motif previously shown to affect mRNA stability and protein levels. 5′UTR mutations may alter gene expression by preventing the binding of transcription machinery to the upstream promoter. In this study, we identified a mutation in the Sp1-binding site contained within the 5′UTR.
POLB Mutations Are Likely to Play a Role in Carcinogenesis
Many of the tumor-associated pol β variants identified in this study and predicted to be damaging exhibit low polymerase activity or, in the case of mutations in the untranslated regions, are predicted to result in a lower concentration of pol β in cells. Our previous results and those in the accompanying paper (53) show that these low activity variants induce genomic instability and cellular transformation. The demonstration of large numbers of pol β variants in colon tumors along with their functional phenotypes strongly suggests that aberrant pol β function drives carcinogenesis.
Acknowledgments
We thank Dr. David Rimm for data analysis tools and helpful discussions, Lori Charette for technical assistance, and Dr. Abhi Patel for helpful discussions regarding this work.
This work was supported, in whole or in part, by National Institutes of Health Grants CA120098 and 5-U19-CA105010 (to J. B. S.).

This article contains supplemental Fig. S1 and Tables S1–S5.
- MMR
- mismatch repair
- pol β
- DNA polymerase β
- MMS
- methylmethane sulfonate
- BER
- base excision repair
- MGMT
- O6-methylguanine DNA methyltransferase
- MEF
- mouse embryonic fibroblast
- miRNA
- microRNA.
REFERENCES
- 1. Jiricny J., Marra G. (2003) DNA repair defects in colon cancer. Curr. Opin. Genet. Dev. 13, 61–69 [DOI] [PubMed] [Google Scholar]
- 2. Parsons R., Li G. M., Longley M. J., Fang W. H., Papadopoulos N., Jen J., de la Chapelle A., Kinzler K. W., Vogelstein B., Modrich P. (1993) Hypermutability and mismatch repair deficiency in RER+ tumor cells. Cell 75, 1227–1236 [DOI] [PubMed] [Google Scholar]
- 3. Herman J. G., Umar A., Polyak K., Graff J. R., Ahuja N., Issa J. P., Markowitz S., Willson J. K., Hamilton S. R., Kinzler K. W., Kane M. F., Kolodner R. D., Vogelstein B., Kunkel T. A., Baylin S. B. (1998) Incidence and functional consequences of hMLH1 promoter hypermethylation in colorectal carcinoma. Proc. Natl. Acad. Sci. U.S.A. 95, 6870–6875 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Lynch H. T., de la Chapelle A. (1999) Genetic susceptibility to nonpolyposis colorectal cancer. J. Med. Genet. 36, 801–818 [PMC free article] [PubMed] [Google Scholar]
- 5. Børresen A. L., Lothe R. A., Meling G. I., Lystad S., Morrison P., Lipford J., Kane M. F., Rognum T. O., Kolodner R. D. (1995) Somatic mutations in the hMSH2 gene in microsatellite unstable colorectal carcinomas. Hum. Mol. Genet. 4, 2065–2072 [DOI] [PubMed] [Google Scholar]
- 6. Dunlop M. G., Farrington S. M., Carothers A. D., Wyllie A. H., Sharp L., Burn J., Liu B., Kinzler K. W., Vogelstein B. (1997) Cancer risk associated with germ line DNA mismatch repair gene mutations. Hum. Mol. Genet. 6, 105–110 [DOI] [PubMed] [Google Scholar]
- 7. Meyers M., Hwang A., Wagner M. W., Boothman D. A. (2004) Role of DNA mismatch repair in apoptotic responses to therapeutic agents. Environ. Mol. Mutagen 44, 249–264 [DOI] [PubMed] [Google Scholar]
- 8. Fink D., Aebi S., Howell S. B. (1998) The role of DNA mismatch repair in drug resistance. Clin. Cancer Res. 4, 1–6 [PubMed] [Google Scholar]
- 9. Esteller M., Toyota M., Sanchez-Cespedes M., Capella G., Peinado M. A., Watkins D. N., Issa J. P., Sidransky D., Baylin S. B., Herman J. G. (2000) Inactivation of the DNA repair gene O6-methylguanine-DNA methyltransferase by promoter hypermethylation is associated with G to A mutations in K-ras in colorectal tumorigenesis. Cancer Res. 60, 2368–2371 [PubMed] [Google Scholar]
- 10. Shima K., Morikawa T., Baba Y., Nosho K., Suzuki M., Yamauchi M., Hayashi M., Giovannucci E., Fuchs C. S., Ogino S. (2011) MGMT promoter methylation, loss of expression, and prognosis in 855 colorectal cancers. Cancer Causes Control 22, 301–309 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Esteller M., Risques R. A., Toyota M., Capella G., Moreno V., Peinado M. A., Baylin S. B., Herman J. G. (2001) Promoter hypermethylation of the DNA repair gene O6-methylguanine-DNA methyltransferase is associated with the presence of G:C to A:T transition mutations in p53 in human colorectal tumorigenesis. Cancer Res. 61, 4689–4692 [PubMed] [Google Scholar]
- 12. Gerson S. L. (2002) Clinical relevance of MGMT in the treatment of cancer. J. Clin. Oncol. 20, 2388–2399 [DOI] [PubMed] [Google Scholar]
- 13. Al-Tassan N., Chmiel N. H., Maynard J., Fleming N., Livingston A. L., Williams G. T., Hodges A. K., Davies D. R., David S. S., Sampson J. R., Cheadle J. P. (2002) Inherited variants of MYH associated with somatic G:C→T:A mutations in colorectal tumors. Nat. Genet. 30, 227–232 [DOI] [PubMed] [Google Scholar]
- 14. Lefevre J. H., Colas C., Coulet F., Bonilla C., Mourra N., Flejou J. F., Tiret E., Bodmer W., Soubrier F., Parc Y. (2010) MYH biallelic mutation can inactivate the two genetic pathways of colorectal cancer by APC or MLH1 transversions. Fam. Cancer 9, 589–594 [DOI] [PubMed] [Google Scholar]
- 15. Sung J. S., Demple B. (2006) Roles of base excision repair subpathways in correcting oxidized abasic sites in DNA. FEBS J. 273, 1620–1629 [DOI] [PubMed] [Google Scholar]
- 16. Lindahl T. (1993) Instability and decay of the primary structure of DNA. Nature 362, 709–715 [DOI] [PubMed] [Google Scholar]
- 17. Prasad R., Beard W. A., Strauss P. R., Wilson S. H. (1998) Human DNA polymerase β deoxyribose phosphate lyase. Substrate specificity and catalytic mechanism. J. Biol. Chem. 273, 15263–15270 [DOI] [PubMed] [Google Scholar]
- 18. Starcevic D., Dalal S., Sweasy J. B. (2004) Is there a link between DNA polymerase β and cancer? Cell Cycle 3, 998–1001 [PubMed] [Google Scholar]
- 19. Bhattacharyya N., Banerjee S. (1997) A variant of DNA polymerase β acts as a dominant negative mutant. Proc. Natl. Acad. Sci. U.S.A. 94, 10324–10329 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Bhattacharyya N., Banerjee T., Patel U., Banerjee S. (2001) Impaired repair activity of a truncated DNA polymerase β protein. Life Sci. 69, 271–280 [DOI] [PubMed] [Google Scholar]
- 21. Bhattacharyya N., Banerjee S. (2001) A novel role of XRCC1 in the functions of a DNA polymerase β variant. Biochemistry 40, 9005–9013 [DOI] [PubMed] [Google Scholar]
- 22. Lang T., Maitra M., Starcevic D., Li S. X., Sweasy J. B. (2004) A DNA polymerase β mutant from colon cancer cells induces mutations. Proc. Natl. Acad. Sci. U.S.A. 101, 6074–6079 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Sweasy J. B., Lang T., Starcevic D., Sun K. W., Lai C. C., Dimaio D., Dalal S. (2005) Expression of DNA polymerase β cancer-associated variants in mouse cells results in cellular transformation. Proc. Natl. Acad. Sci. U.S.A. 102, 14350–14355 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Muniappan B. P., Thilly W. G. (2002) The DNA polymerase β replication error spectrum in the adenomatous polyposis coli gene contains human colon tumor mutational hotspots. Cancer Res. 62, 3271–3275 [PubMed] [Google Scholar]
- 25. Lang T., Dalal S., Chikova A., DiMaio D., Sweasy J. B. (2007) The E295K DNA polymerase β gastric cancer-associated variant interferes with base excision repair and induces cellular transformation. Mol. Cell. Biol. 27, 5587–5596 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Ginty F., Adak S., Can A., Gerdes M., Larsen M., Cline H., Filkins R., Pang Z., Li Q., Montalto M. C. (2008) The relative distribution of membranous and cytoplasmic met is a prognostic indicator in stage I and II colon cancer. Clin. Cancer Res. 14, 3814–3822 [DOI] [PubMed] [Google Scholar]
- 27. Adzhubei I. A., Schmidt S., Peshkin L., Ramensky V. E., Gerasimova A., Bork P., Kondrashov A. S., Sunyaev S. R. (2010) A method and server for predicting damaging missense mutations. Nat. Methods 7, 248–249 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Kumar P., Henikoff S., Ng P. C. (2009) Predicting the effects of coding nonsynonymous variants on protein function using the SIFT algorithm. Nat. Protoc. 4, 1073–1081 [DOI] [PubMed] [Google Scholar]
- 29. Murphy D. L., Kosa J., Jaeger J., Sweasy J. B. (2008) The Asp-285 variant of DNA polymerase β extends mispaired primer termini via increased nucleotide binding. Biochemistry 47, 8048–8057 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Murphy D. L., Jaeger J., Sweasy J. B. (2011) A triad interaction in the fingers subdomain of DNA polymerase β controls polymerase activity. J. Am. Chem. Soc. 133, 6279–6287 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Johnson K. A. (1993) Conformational coupling in DNA polymerase fidelity. Annu. Rev. Biochem. 62, 685–713 [DOI] [PubMed] [Google Scholar]
- 32. Dalal S., Chikova A., Jaeger J., Sweasy J. B. (2008) The L22P tumor-associated variant of DNA polymerase β is dRP lyase-deficient. Nucleic Acids Res. 36, 411–422 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Iwanaga A., Ouchida M., Miyazaki K., Hori K., Mukai T. (1999) Functional mutation of DNA polymerase β found in human gastric cancer. Inability of the base excision repair in vitro. Mutat. Res. 435, 121–128 [DOI] [PubMed] [Google Scholar]
- 34. Wang Z., Shen D., Parsons D. W., Bardelli A., Sager J., Szabo S., Ptak J., Silliman N., Peters B. A., van der Heijden M. S., Parmigiani G., Yan H., Wang T. L., Riggins G., Powell S. M., Willson J. K., Markowitz S., Kinzler K. W., Vogelstein B., Velculescu V. E. (2004) Mutational analysis of the tyrosine phosphatome in colorectal cancers. Science 304, 1164–1166 [DOI] [PubMed] [Google Scholar]
- 35. Sjöblom T., Jones S., Wood L. D., Parsons D. W., Lin J., Barber T. D., Mandelker D., Leary R. J., Ptak J., Silliman N., Szabo S., Buckhaults P., Farrell C., Meeh P., Markowitz S. D., Willis J., Dawson D., Willson J. K., Gazdar A. F., Hartigan J., Wu L., Liu C., Parmigiani G., Park B. H., Bachman K. E., Papadopoulos N., Vogelstein B., Kinzler K. W., Velculescu V. E. (2006) The consensus coding sequences of human breast and colorectal cancers. Science 314, 268–274 [DOI] [PubMed] [Google Scholar]
- 36. Timmermann B., Kerick M., Roehr C., Fischer A., Isau M., Boerno S. T., Wunderlich A., Barmeyer C., Seemann P., Koenig J., Lappe M., Kuss A. W., Garshasbi M., Bertram L., Trappe K., Werber M., Herrmann B. G., Zatloukal K., Lehrach H., Schweiger M. R. (2010) Somatic mutation profiles of MSI and MSS colorectal cancer identified by whole exome next generation sequencing and bioinformatics analysis. PLoS One 5, e15661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Greenman C., Stephens P., Smith R., Dalgliesh G. L., Hunter C., Bignell G., Davies H., Teague J., Butler A., Stevens C., Edkins S., O'Meara S., Vastrik I., Schmidt E. E., Avis T., Barthorpe S., Bhamra G., Buck G., Choudhury B., Clements J., Cole J., Dicks E., Forbes S., Gray K., Halliday K., Harrison R., Hills K., Hinton J., Jenkinson A., Jones D., Menzies A., Mironenko T., Perry J., Raine K., Richardson D., Shepherd R., Small A., Tofts C., Varian J., Webb T., West S., Widaa S., Yates A., Cahill D. P., Louis D. N., Goldstraw P., Nicholson A. G., Brasseur F., Looijenga L., Weber B. L., Chiew Y. E., DeFazio A., Greaves M. F., Green A. R., Campbell P., Birney E., Easton D. F., Chenevix-Trench G., Tan M. H., Khoo S. K., Teh B. T., Yuen S. T., Leung S. Y., Wooster R., Futreal P. A., Stratton M. R. (2007) Patterns of somatic mutation in human cancer genomes. Nature 446, 153–158 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Cummins J. M., He Y., Leary R. J., Pagliarini R., Diaz L. A., Jr., Sjoblom T., Barad O., Bentwich Z., Szafranska A. E., Labourier E., Raymond C. K., Roberts B. S., Juhl H., Kinzler K. W., Vogelstein B., Velculescu V. E. (2006) The colorectal microRNAome. Proc. Natl. Acad. Sci. U.S.A. 103, 3687–3692 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Rehmsmeier M., Steffen P., Hochsmann M., Giegerich R. (2004) Fast and effective prediction of microRNA/target duplexes. RNA 10, 1507–1517 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Lewis B. P., Shih I. H., Jones-Rhoades M. W., Bartel D. P., Burge C. B. (2003) Prediction of mammalian microRNA targets. Cell 115, 787–798 [DOI] [PubMed] [Google Scholar]
- 41. Sarnowska E., Grzybowska E. A., Sobczak K., Konopinski R., Wilczynska A., Szwarc M., Sarnowski T. J., Krzyzosiak W. J., Siedlecki J. A. (2007) Hairpin structure within the 3′UTR of DNA polymerase β mRNA acts as a post-transcriptional regulatory element and interacts with Hax-1. Nucleic Acids Res. 35, 5499–5510 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Widen S. G., Kedar P., Wilson S. H. (1988) Human β-polymerase gene. Structure of the 5′-flanking region and active promoter. J. Biol. Chem. 263, 16992–16998 [PubMed] [Google Scholar]
- 43. Yamtich J., Speed W. C., Straka E., Kidd J. R., Sweasy J. B., Kidd K. K. (2009) Population-specific variation in haplotype composition and heterozygosity at the POLB locus. DNA Repair 8, 579–584 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Sobol R. W., Horton J. K., Kühn R., Gu H., Singhal R. K., Prasad R., Rajewsky K., Wilson S. H. (1996) Requirement of mammalian DNA polymerase-β in base-excision repair. Nature 379, 183–186 [DOI] [PubMed] [Google Scholar]
- 45. Wang L., Patel U., Ghosh L., Banerjee S. (1992) DNA polymerase β mutations in human colorectal cancer. Cancer Res. 52, 4824–4827 [PubMed] [Google Scholar]
- 46. Kosa J. L. (1999) AZT-resistant Mutants of DNA Polymerase β Identified by in Vivo Selection. A Structure-Function Study of Polymerase Substrate Specificity. Ph.D. dissertation, Yale University [Google Scholar]
- 47. Odell I. D., Barbour J. E., Murphy D. L., Della Maria J. A., Sweasy J. B., Tomkinson A. E., Wallace S. S., Pederson D. S. (2011) Nucleosome disruption by DNA ligase III-XRCC1 promotes efficient base excision repair. Mol. Cell. Biol. 31, 4623–4632 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Maitra M., Gudzelak A., Jr., Li S. X., Matsumoto Y., Eckert K. A., Jager J., Sweasy J. B. (2002) Threonine 79 is a hinge residue that governs the fidelity of DNA polymerase β by helping to position the DNA within the active site. J. Biol. Chem. 277, 35550–35560 [DOI] [PubMed] [Google Scholar]
- 49. Buchan J. R., Stansfield I. (2007) Halting a cellular production line. Responses to ribosomal pausing during translation. Biol. Cell 99, 475–487 [DOI] [PubMed] [Google Scholar]
- 50. Nakamura Y., Gojobori T., Ikemura T. (2000) Codon usage tabulated from international DNA sequence databases. Status for the year 2000. Nucleic Acids Res. 28, 292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Srivastava D. K., Husain I., Arteaga C. L., Wilson S. H. (1999) DNA polymerase β expression differences in selected human tumors and cell lines. Carcinogenesis 20, 1049–1054 [DOI] [PubMed] [Google Scholar]
- 52. Pelletier H., Sawaya M. R., Wolfle W., Wilson S. H., Kraut J. (1996) Crystal structures of human DNA polymerase βcomplexed with DNA. Implications for catalytic mechanism, processivity, and fidelity. Biochemistry 35, 12742–12761 [DOI] [PubMed] [Google Scholar]
- 53. Nemec A. A., Donigan K. A., Murphy. L., Jaeger J., Sweasy J. B. (May 9, 2012) Colon cancer-associated DNA polymerase β variant induces genomic instability and cellular transformation. J. Biol. Chem. 287, 23840–23849 [DOI] [PMC free article] [PubMed] [Google Scholar]