

Background: Mutations in the POLB gene are present in 40% of human colorectal tumors.
Results: The G231D variant is a slow polymerase that induces genomic instability and cellular transformation.
Conclusion: The slow G231D variant induces cellular transformation due to its inability to fill in single nucleotide gaps.
Significance: Slow pol β variants may drive tumorigenesis.
Keywords: Base Excision Repair, Cancer, DNA Polymerase, DNA Repair, Genomic Instability
Abstract
Rapidly advancing technology has resulted in the generation of the genomic sequences of several human tumors. We have identified several mutations of the DNA polymerase β (pol β) gene in human colorectal cancer. We have demonstrated that the expression of the pol β G231D variant increased chromosomal aberrations and induced cellular transformation. The transformed phenotype persisted in the cells even once the expression of G231D was extinguished, suggesting that it resulted as a consequence of genomic instability. Biochemical analysis revealed that its catalytic rate was 140-fold slower than WT pol β, and this was a result of the decreased binding affinity of nucleotides by G231D. Residue 231 of pol β lies in close proximity to the template strand of the DNA. Molecular modeling demonstrated that the change from a small and nonpolar glycine to a negatively charged aspartate resulted in a repulsion between the template and residue 231 leading to the distortion of the dNTP binding pocket. In addition, expression of G231D was insufficient to rescue pol β-deficient cells treated with chemotherapeutic agents suggesting that these agents may be effectively used to treat tumors harboring this mutation. More importantly, this suggests that the G231D variant has impaired base excision repair. Together, these data indicate that the G231D variant plays a role in driving cancer.
Introduction
Faithful DNA repair is necessary to maintain genomic integrity. The base excision repair pathway is responsible for repairing at least 20,000 lesions per cell per day (1). If left unrepaired, the lesions can give rise to genomic instability and tumorigenesis. The BER2 pathway is a multistep process that involves the enzymatic activities and interactions of many proteins. DNA polymerase β (pol β) is the main polymerase involved in BER and has two catalytic activities, deoxyribose phosphate (dRP) lyase and polymerase activities. One or both of these activities are necessary for survival because pol β knock-out mice are embryonic lethal.
There are three subpathways of BER depending on the type of glycosylase that excises the damaged base (2). For monofunctional glycosylases, the glycosylase removes the damaged base leaving an abasic site. Ape1 incises the abasic site leaving a 3′-OH and 5′-dRP group. pol β removes the dRP group with its lyase activity and fills in the missing nucleotide (3). Bifunctional glycosylases remove the damaged base and nick the backbone leaving a 3′-dRP group and a 5′-phosphate (4). Ape1 modifies the end to generate a 3′-OH group, and pol β fills in the gap. In an Ape1-independent pathway, bifunctional glycosylases with β,δ-elimination remove the damaged base and incise the backbone leaving 3′- and 5′-phosphates. Polynucleotide kinase modifies the 3′ end to generate an OH group, and pol β fills the gaps (5). In the final step of the pathway, the DNA is ligated by DNA ligase IIIα (6).
The G231D variant of pol β was identified in a colon tumor diagnosed as a stage 3 adenocarcinoma as described in the accompanying article (24). This variant was predicted to be damaging (24) and shown to catalyze DNA synthesis with a rate that is much lower than that of WT pol β. The Gly-231 residue is in close proximity to DNA, and its alteration could therefore influence catalysis. In this study, we sought to determine whether the colon cancer-associated variant G231D could drive carcinogenesis. Expression of G231D in immortal but nontransformed cells induced cellular transformation by a mutagenic mechanism. The G231D variant exhibited a slow catalytic rate as a result of its low affinity for dNTP substrate. In combination, our results strongly suggest that the G231D variant of pol β is a driver of carcinogenesis.
EXPERIMENTAL PROCEDURES
Materials
Ultrapure deoxynucleoside triphosphates and T4 polynucleotide kinase were purchased from New England Biolabs. [γ-32P]ATP (5 mCi) and ATP were purchased from PerkinElmer Life Sciences and Sigma, respectively.
Plasmids and Cloning
The G231D variant of pol β was generated using the Stratagene QuikChange® site-directed mutagenesis kit according to manufacturer's instructions. pET28a-WT pol β containing an N-terminal His tag was used as a template for protein expression and biochemical studies. For cell culture experiments, the hemagglutinin (HA)-tagged WT pol β in the pRVYTet retroviral vector was used as a template. The primers used for these reactions were purchased from Invitrogen, and the sequences are available upon request. Positive clones were sequenced at the Keck DNA Sequencing Facility, Yale University School of Medicine.
Protein Expression and Purification
The N-terminal His-tagged WT and G231D pol β pET28a plasmids were transformed into the Escherichia coli strain BL21 (DE3)-competent cells and purified as described previously by FPLC (7). All proteins were purified with >90% homogeneity as confirmed by Coomassie Blue staining of 10% SDS-polyacrylamide gels. The final protein was aliquoted, flash-frozen in liquid nitrogen, and stored at −80 °C. The final concentration was determined using the absorbance at 280 nm and the extinction coefficient for pol β (ϵ = 21,200 m−1 cm−1).
Preparation of DNA Substrates
The substrates used are shown in Table 1. Oligonucleotides were synthesized by the Keck DNA Sequencing Facility and purified by PAGE. 45AG and 45GC were used for kinetic studies, active site titrations, and gel mobility shift assays. LPSD was used for the BER assay. All primers were radiolabeled at the 5′ end using T4 polynucleotide kinase and [γ-32P]ATP. The downstream oligonucleotides were 5′-phosphorylated with nonradiolabeled ATP. The reactions were purified using Micro Bio-Spin® 30 chromatography columns (Bio-Rad) to remove unincorporated ATP. The oligonucleotides were annealed by combining the radiolabeled primer, the nonradiolabeled downstream oligonucleotide, and the template in 50 mm Tris-HCl, pH 8.0, and 0.25 m NaCl. The mixture was incubated at 95 °C for 5 min, slowly cooled to 50 °C over 30 min, and incubated at 50 °C for additional 20 min. Reactions were placed on ice and resolved on 12% native polyacrylamide gel to verify complete annealing of the oligonucleotides.
TABLE 1.
DNA substratesa

a The template base is in boldface.
Cell Lines and Cell Culture
The pol β-deficient mouse embryonic fibroblasts (MEFs) (88 TAg) were a kind gift from Leona Samson (Massachusetts Institute of Technology). These cells were maintained in high glucose Dulbecco's modified Eagle's medium (DMEM) (Invitrogen) supplemented with 10% fetal bovine serum (FBS) (Gemini Bio-Products), 1% penicillin/streptomycin, 1% l-glutamine, and 220 μg/ml hygromycin B (Invitrogen) (used for selection) and grown at 37 °C in 5% CO2. GP2-293 cells (Clontech) were maintained in high glucose DMEM supplemented with 10% FBS, 1% penicillin/streptomycin, 1% l-glutamine, and 1 mm Hepes and grown at 37 °C in 5% CO2. C127 cells have been described (8). C127 cells were grown in high glucose DMEM supplemented with 10% FBS, 1% penicillin/streptomycin (Invitrogen), 1600 μg/ml G418, and 180 μg/ml hygromycin B at 37 °C in a 5% CO2-humidified incubator.
Transfection, Infection, and Expression Analysis
To obtain high titer retrovirus expressing WT and G231D pol β, the GP2-293 packaging cell line was transfected with pRVY pol β and pVSVG plasmids (9) using calcium phosphate. Low titer virus was removed 72 h post-transfection, and hygromycin B (200 μg/ml) was added to the cells to select spontaneous integrants. Stable integrants were transfected with pVSVG, and high titer virus was harvested after 72 h. pol β-deficient MEFs or C127 cells were seeded at 30% confluence and infected with high titer pol β virus in the presence of 4 μg/ml Polybrene. The following day, cells were split, and hygromycin B was added 2 days post-infection. Western blots of pol β confirmed expression in these cell lines. For the C127 cells, single clones were picked and screened for equal ratios of endogenous to exogenous expression.
Western Blotting
Cells were washed once with PBS, lysed in boiling 1× SDS buffer (50 mm Tris, pH 6.8, 100 mm DTT, 2% SDS, 10% glycerol), and boiled for an additional 5 min. Lysates were resolved on a 10% SDS-polyacrylamide gel and transferred to nitrocellulose membrane. pol β was detected by incubating the membrane with monoclonal mouse anti-pol β antibody (Abcam catalog no. 1831). Anti-tubulin (Cell Signaling Technologies) was used as a loading control. Blots were imaged using Bio Rad ChemiDocTM XRS+ and quantified using Image Lab Software.
Preparation of MEF Extract
pol β-deficient MEFs were rinsed three times in cold PBS, trypsinized, counted, and pelleted. The pellet was resuspended in Buffer I (10 mm Tris, pH 7.8, 200 mm KCl) to obtain 106 cells/20 μl. An equal volume of Buffer II (10 mm Tris, pH 7.8, 200 mm KCl, 2 mm EDTA, 40% glycerol, 0.2% IGEPAL, 2 mm DTT, 0.5 mm PMSF, supplemented with protease inhibitors) was added, and samples were rocked for 1 h at 4 °C. The extract was centrifuged at 16,000 × g for 10 min at 4 °C. The supernatant was collected, aliquoted, and flash-frozen in liquid nitrogen. Protein concentrations were determined by measuring the absorbance (562 nm) after the addition of BCA protein assay reagent (Thermo-Fisher Scientific) using BSA as a reference standard.
Chromosomal Aberrations
pol β-deficient MEFs expressing either WT or G231D pol β or empty vector were plated overnight in 10-cm dishes (106 cells/dish). Colcemid (Invitrogen) was added to the cells (0.1 μg/ml, 3 h) to arrest cells in mitosis. Cells were trypsinized, collected by centrifugation, washed one time with PBS, and resuspended in a hypertonic solution (0.56% KCl) for 30 min at 37 °C to lyse the cells. Cells were fixed using Carnoy's fixative (75% methanol, 25% acetic acid) and dropped onto slides. Cells were stained with Giemsa, and well spread metaphases were identified under ×100 objective (Zeiss). Images were taken using Spot Camera software (Diagnostic Instruments). Metaphase spreads were scored by eye for chromosomal fusions, breaks, and fragments.
Cellular Transformation
The focus formation assay was conducted as in Ref. 10. The presence of foci was also monitored by microscopic examination as described previously (10, 11). Anchorage-independent growth was assessed as described previously (11).
Proliferation
High passage C127 clones were plated at a density of 20,000 cells per 60-mm dish. Cells were counted every day for 5 consecutive days. Data were plotted as change in cell number per day.
Pre-steady State Burst Assay
Radiolabeled 1-bp gapped DNA (300 nm 45AG or 45GC) and pol β (100 nm) were combined with the correct dNTP and 10 mm MgCl2 in a rapid quench apparatus at 37 °C (KinTek Corp.). The reactions were quenched by the addition of 0.5 m EDTA. The reaction products were separated on a 20% denaturing polyacrylamide gels, visualized, and quantified using a Storm 860 Phosphorimager with ImageQuant software. Data were fitted to the burst in Equation 1,
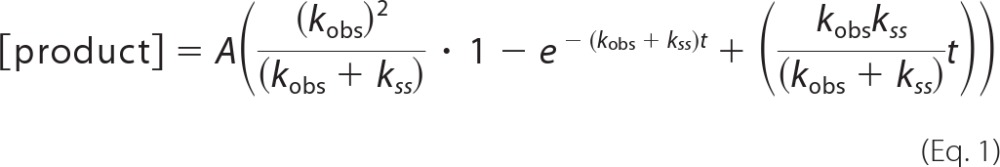 |
or to a single exponential as in Equation 2,
where A is the amplitude; kobs is the observed rate constant of the exponential phase; and kss is the rate constant of the linear phase (12). The single exponential equation was used when G231D did not display a biphasic burst.
Gel Mobility Shift Assay
The DNA binding constant was determined by gel electromobility shift assay as described previously (13) The dissociation constant for DNA (KD) was determined by fitting the fraction bound protein (Y) versus protein concentration with Equation 3,
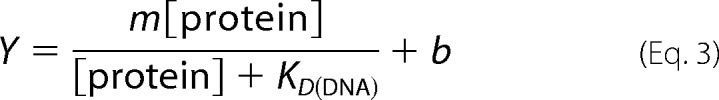 |
where Y is the amount of bound protein; m is a scaling factor, and b is the apparent minimum Y value (14).
Single Turnover Kinetic Assay
Radiolabeled gapped DNA (50 nm 45AG or 45GC) was incubated with WT pol β (500 nm active protein) as was determined by active site titration. The DNA/pol β mixture was incubated with varying concentrations of dNTP (5–2000 μm). For the correct dNTP, the reactions were run using a rapid quench apparatus at 37 °C for times from 0.1 to 30 s. For the incorrect dNTPs, the reactions were incubated at 37 °C for up to 90 min with samples being collected at multiple time points. All reactions were quenched with 0.5 m EDTA, and the extended product was analyzed as described with the pre-steady state burst assay. The concentration of extended product was plotted against time, and the data were fit to a single exponential curve to obtain the kobs for each dNTP concentration. A secondary plot of kobs for each dNTP concentration versus [dNTP] was fitted to the hyperbolic Equation 4,
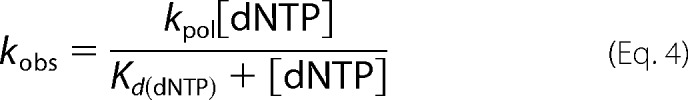 |
where kpol is the rate of polymerization, and Kd(dNTP) is the apparent dNTP binding constant. For G231D pol β, 500 nm protein was sufficient to bind 90% of DNA as was determined by the gel mobility shift assay.
Reconstituted Base Excision Repair Assay
Radiolabeled LPSD DNA (100 nm) was incubated with UDG (New England Biolabs) for 30 min at 37 °C. The UDG-treated DNA (5 nm) was incubated in pol β-deficient cell extract (5 μg) and WT or G231D pol β protein (10 nm) in BER buffer (45 mm Hepes, pH 7.8, 70 mm KCl, 2 mm DTT, 7.5 mm MgCl2, 0.5 mm EDTA, 2 mm ATP, 10 μl of dCTP) for 2 or 5 min at 37 °C. Reactions were quenched by the addition of an equal volume of 90% formamide dye and 0.5 m EDTA. Samples were resolved on 20% denaturing polyacrylamide gels, visualized using a Storm 860 Phosphorimager, and quantified using ImageQuant.
Clonogenic Survival Assays
2.2 × 105 cells were plated in 60-mm dishes. After 48 h, cells were treated with varying concentrations of either temozolomide (TMZ) (Sigma). Following treatment, cells were trypsinized and plated at several dilutions. Cells were allowed to grow for 9–11 days, and colonies were counted. Colonies were defined as having at least 50 cells. All experiments were performed 3–4 times.
Molecular Modeling and Molecular Dynamics Simulations
Modeling was carried out using the programs PyMOL, VMD, and NAMD2 (15, 16). The mutation from Gly-231 to Asp was introduced in PyMOL using a high resolution DNA co-crystal structure (PDB code 1bpx and 2fms) as starting models for the binary and ternary complexes, respectively. Initial bad contacts between the protein and the DNA backbone were analyzed and removed using features implemented in PyMOL. Subsequently, the initial model was further optimized by subjecting the structure to 500 steps of energy minimization in NAMD2, version 2.8 (16). Both the WT and G231D models were then subjected to periodic boundary molecular dynamics simulations performed for 6 ns (1-ns equilibration phase). NAMD2 uses the CHARMM27 force field to parameterize intramolecular interactions with long range nonbonding terms calculated up to a 12-Å cutoff for electrostatic and van der Waals interactions. Beyond this cutoff, a switching function turns off the calculation of nonbonding terms. All hydrogen bond lengths were held constant with the SHAKE-RATTLE-ROLL algorithm. The complex was embedded in a periodic boundary box of water molecules, which are parameterized by the TIP3P model. The ionic strength was adjusted to 0.2 mol/liter to allow for an appropriate screening of Coulomb forces. The simulation temperature during the dynamics run was maintained at 310 K using a temperature bath with a coupling constant of 5/ps. Root mean square deviation analysis of the entire trajectory was performed to evaluate systems mobility and to ensure proper convergence (data not shown). Trajectory analysis and molecular graphics images were generated using VMD and PyMOL (15, 16).
Circular Dichroism
Circular dichroism wavelength scans were performed with 1 μm pol β protein in 10 mm K2HPO4 at 23 °C using the Chirascan circular dichroism spectrometer (Applied Photosystems). Ellipticity was measured in 0.5 nm steps from 185 to 290 nm. Thermal scans were also performed at a set wavelength of 222 nm with a 5 °C/min step from 4.5 to 62 °C. Three measurements were taken for each enzyme and averaged.
Statistics
Two-tailed t tests and two-way analysis of variance were used as appropriate. Bonferroni's post hoc test was used to determine significant differences between the means of each group. All statistics were performed using GraphPad Prism version 5 (GraphPad Software, San Diego). Data are represented as means ± S.E.
RESULTS
Expression of G231D Induces Cellular Transformation
Because the G231D variant was initially identified in a colon tumor, we queried whether its expression in mammalian cells resulted in a cancerous phenotype. We used the tetracycline off system to induce expression of G231D pol β in C127 cells. In this system, when tetracycline (Tet) is present in the medium, expression is turned off. Expression is induced once Tet is removed (17). Clones were selected that had equal levels of expression of exogenous to endogenous pol β protein (supplemental Fig. S1A). Expression of G231D in four of five independent clones induced cellular transformation (Fig. 1A, dashed lines), as indicated by a substantial increase in focus formation in the clones induced to express G231D compared with those not expressing G231D (solid lines). Note that expression of WT pol β in the C127 cells does not induce focus formation (Fig. 1B) (11). We also analyzed the ability of the cells to grow in soft agar as another measure of cellular transformation. Indeed, both G231D pol β clones 5 and 24 were capable of growing in an anchorage-independent manner when expression was induced (Fig. 1C) (p = 0.04 and 0.01, respectively). Finally, we measured the rate of proliferation in clones expressing either WT or G231D pol β. Under inducing conditions, G231D pol β clones 5 and 24 had a significant increase in proliferation compared with the noninduced counterparts (Fig. 1D) (p < 0.001 at day 5). There were no differences observed in the proliferation of WT pol β clones regardless of expression of WT pol β protein (9). Together, these data suggest that the expression of the variant G231D pol β induces cellular transformation.
FIGURE 1.

Expression of G231D in C127 cells induces cellular transformation. C127 clones either expressing (NT; open symbols, dashed lines) or not expressing (T; filled symbols, solid line) G231D (A) or WT pol β (B) were serially passaged up to passage 24 with 104 cells being plated, stained, and counted for foci every fourth passage. The numbers of foci are averages of the counts of two T25 flasks. C, high passage G231D clonal cell lines from the focus formation assay were plated in soft agar. Colonies were counted after 5 weeks of growth. Colonies in five fields were counted and averaged. Data are presented as the mean ± S.E. D, High passage G231D pol β clonal cell lines were plated in five dishes at a density of 20,000 cells per dish. Cells were trypsinized and counted each day. Data are plotted as the mean ± S.E. of the change in cell number per 24 h period. E, G231D clonal cell lines 5 and 24 were passaged in the presence (solid line) or absence (dashed lines) of Tet to induce expression. At passage 24 (indicated by arrow), Tet was added to the medium in the no Tet clones to extinguish expression. Cells were serially passaged to passage 32 and analyzed for the formation of foci.
To determine whether cellular transformation is dependent upon continuous expression of G231D, Tet was added to the medium of two of the transformed cell lines to extinguish expression (supplemental Fig. S1A). Foci continued to form even after expression was extinguished (Fig. 1E). This suggests that the transformation of cells by G231D resulted from a heritable change induced by the expression of G231D pol β.
G231D pol β Variant Does Not Display Biphasic Burst Kinetics
To provide a mechanism for G231D-induced cellular transformation, we characterized the biochemical properties of G231D pol β. In an initial activity screen it was shown that G231D pol β exhibits a slower rate of DNA synthesis compared with WT pol β in the primer extension assay (24). Here, we show that G231D exhibits a slower catalytic rate compared with WT using the 45AG DNA substrate in pre-steady state burst assays (Table 1). G231D pol β did not have the biphasic burst typical of pol β where the exponential phase is the polymerization and the linear phase is the rate-limiting step of product release. Instead, G231D fit to a single exponential indicating that the rate-limiting step is different from that of WT pol β and is now chemistry and not product release. The rate of G231D pol β was 0.1 s−1 compared with 14 s−1 for WT (Fig. 2). G231D pol β had the same overall structure and thermal stability as WT pol β (supplemental Fig. S2), so it is unlikely that its global structure is the reason for the decreased rate of catalysis.
FIGURE 2.

G231D pol β does not display biphasic burst kinetics. Pre-steady state kinetics was performed by incubating 100 μm 45AG DNA (Table 1) and 100 μm dTTP with 300 μm WT (A, filled circles) or G231D (B, open triangles) pol β. WT pol β reactions were on a time course ranging from 0.01 to 1 s, and G231D reactions ranged from 1 to 120 s. Products were resolved on 20% denaturing gels and imaged using a phosphorimager and quantified. Data were plotted as product formed versus time and fit to the full burst equation (WT) or single exponential (G231D). (WT, kobs = 14 ± 3 s−1; G231D, kobs = 0.11 ± 0.01 s−1).
G231D pol β Binds DNA Similar to WT pol β but Binds dNTPs with Less Affinity than WT pol β
To ascertain the biochemical mechanism underlying the slow catalytic rate of G231D pol β, we performed gel mobility shift assays to determine the KD(DNA) of WT and G231D pol β proteins. WT and G231D pol β bound DNA with a similar affinity (4.1 and 1.7 nm, respectively) (Fig. 3) for 45AG DNA. We next sought to determine the kinetic details of G231D pol β using single turnover kinetics. This allows us to obtain the rate of polymerization (kpol) and the equilibrium dissociation constant for dNTP (Kd(dNTP)) (Fig. 4). These studies revealed that G231D bound the correct dNTP (dTTP) with an affinity 15-fold less than WT pol β (Table 2) opposite template A. Interestingly, the rate of polymerization for the correct nucleotide is similar for G231D pol β compared with WT indicating that the slower burst/catalytic rate is mainly due to the impairment of nucleotide binding. Next, we performed pre-steady state burst kinetics with dNTP in a 2-fold excess of the Kd(dTTP) for G231D pol β (1.5 mm). Under these conditions, G231D pol β displayed the typical biphasic burst kinetics of WT pol β protein with a rate of 9 s−1 (Fig. 5). To ensure the excess dTTP did not alter the rate of WT pol β, we also analyzed the pre-steady state kinetics of WT pol β with 1.5 mm dTTP and observed no change in rate (14 s−1) (supplemental Fig. S3). This lower affinity for the correct nucleotide is not specific to the DNA sequence because G231D pol β binds dCTP with a 20-fold lower affinity compared with WT opposite templating base G (Table S1 and supplemental Figs. S4 and S5). Together, these data indicate that G231D pol β has a slower catalytic rate than WT pol β due to a defect in dNTP binding.
FIGURE 3.

G231D has similar binding affinity for 45AG 1-bp gapped DNA as WT pol β. Various concentrations of WT (filled circles) or G231D (open triangles) protein were incubated with 0.1 nm 45AG DNA (Table 1) for 15 min at room temperature. Data were plotted as fraction product bound versus pol β concentration to obtain the KD(DNA). (WT, 4.1 ± 0.8 nm; G231D, 1.7 ± 0.2 nm).
FIGURE 4.

G231D pol β variant binds dNTP with less affinity than WT. The misincorporation of dCTP opposite template A for G231D is a representative plot of the single turnover misincorporation experiments. A, increasing concentrations of dCTP were incubated with 500 nm G231D pol β. The data were plotted as extended DNA formed versus time and fit to a single exponential equation to obtain the kobs for each dCTP concentration. B, secondary plot of the kobs versus the concentration of dCTP for G231D was fit to a hyperbolic equation to obtain kpol and the apparent Kd(dCTP) (0.00250 ± 0.00007 s−1 and 330 ± 30 μm, respectively).
TABLE 2.
Single turnover misincorporation opposite template A
Kinetic constants obtained from single turnover misincorporation experiments are listed for each enzyme (± S.E. of the fit).
| dNTP | Enzyme | kpol | Kd | kpolc/kpolia | Kdi/Kdca | Efficiencyb | Fold changec |
|---|---|---|---|---|---|---|---|
| s−1 | μm | μm−1 s−1 | |||||
| dTTP | WT | 46 ± 2 | 35 ± 5 | 1.3 | 55 | ||
| G231D | 13.1 ± 0.9 | 550 ± 70 | 2.4 × 10−2 | ||||
| dATP | WT | 0.025 ± 0.001 | 57 ± 10 | 1800 | 1.6 | 4.4 × 10−4 | 58 |
| G231D | 0.00250 ± 0.00007 | 330 ± 30 | 5200 | 0.60 | 7.6 × 10−6 | ||
| dCTP | WT | 0.079 ± 0.004 | 280 ± 40 | 580 | 8.0 | 2.8 × 10−4 | 110 |
| G231D | 0.0016 ± 0.0002 | 610 ± 110 | 8200 | 1.1 | 2.6 × 10−6 | ||
| dGTP | WT | 0.031 ± 0.002 | 84 ± 18 | 1500 | 2.4 | 3.7 × 10−4 | 93 |
| G231D | 0.00119 ± 0.00007 | 300 ± 50 | 11,000 | 0.55 | 4.0 × 10−6 |
a Subscript c denotes correct nucleotide; subscript i denotes incorrect nucleotide.
b Efficiency was calculated by dividing kpol by Kd for each condition.
c Fold change in efficiency (WT/G231D) is shown.
FIGURE 5.

G231D pol β displays biphasic burst kinetics with increased dTTP. Pre-steady state kinetics were performed by incubating 100 μm 45AG DNA (Table 1) and 1500 μm dTTP with 300 μm G231D pol β. The reactions were on a time course ranging from 0.01 to 1 s. Products were resolved on 20% denaturing gels and imaged using a phosphorimager and quantified. Data were plotted as product formed versus time and fit to the full burst equation. (kobs = 9 ± 1 s−1).
Expression of G231D Induces Chromosomal Aberrations
Cellular transformation by G231D appears to result from heritable changes induced by this protein upon expression in the C127 cells (Fig. 1E). In addition, the catalytic rate of G231D is significantly slower than that of WT and is due to G231D having a significantly decreased affinity for dNTP compared with WT pol β (Table 2 and supplemental Table S1). Previous studies by our laboratory showed that the E295K catalytically inactive pol β variant induced genomic instability in MEFs. Thus, we sought to determine whether the slower rate of G231D pol β resulted in an increase in genomic instability. Metaphase spreads of pol β−/− MEFs expressing either WT or G231D pol β were analyzed for chromosomal aberrations. The pol β−/− MEF cell line was used because the G231D mutation is homozygous and would not normally compete with WT pol β. Cells expressing G231D pol β had a significant increase in both fusions and breaks compared with WT (p < 0.001 and 0.5, respectively) (Fig. 6). The differences observed were not due to differences in the expression level as Western analysis indicated the cells expressed the proteins to similar amounts (supplemental Fig. S1B). We conclude that G231D induces cellular transformation by increasing genomic instability in cells.
FIGURE 6.

G231D pol β expression induces genomic instability in cells. Representative metaphase spread of pol β−/− MEFS expressing WT (A) or G231D pol β (B). Chromosomal fusions are shown with black arrows. C, number of aberrations per metaphase. A total of at least 50 metaphases were scored for each cell line. Data are plotted as mean ± S.E.
BER in the Presence of G231D Is Slow
To determine whether G231D participates in BER, we characterized BER using cell extracts. Radiolabeled double-stranded DNA with a uracil opposite the templating base G (LPSD substrate; Table 1) was incubated with UDG to excise the uracil. The substrate was then incubated with pol β-deficient MEF extract containing G231D pol β protein for 2 or 5 min. Although G231D pol β was capable of participating in the repair of a uracil lesion (23 and 47%, by 2 and 5 min, respectively; Fig. 7), it did so more slowly than WT pol β (35 and 59% by 2 and 5 min, respectively (9)).
FIGURE 7.

G231D pol β participates in BER. LPSD DNA (Table 1) was treated with UDG and incubated with pol β-deficient cell extract in the presence of G231D pol β.
pol β-deficient Cells Expressing G231D pol β Have Increased Sensitivity to a Chemotherapeutic Agent
To determine whether the slower catalytic rate of G231D pol β would have an effect on the survival of cells upon exposure to chemotherapeutic agents, we expressed G231D pol β in pol β-deficient cells and assayed for cell survival in response to TMZ. We have previously showed that expressing WT pol β in the deficient cells rescues the cells from treatment with the alkylation agent, methylmethane sulfonate (10). Expression of G231D pol β only partially rescues the cells after treatment with methylmethane sulfonate (24) or TMZ (Fig. 8) indicating that the slower BER capacity of G231D pol β observed in vitro translates into less repair of the damage induced by these drugs. These data also suggest that people carrying this G231D mutation in their tumor would benefit from treatment with these BER drugs.
FIGURE 8.
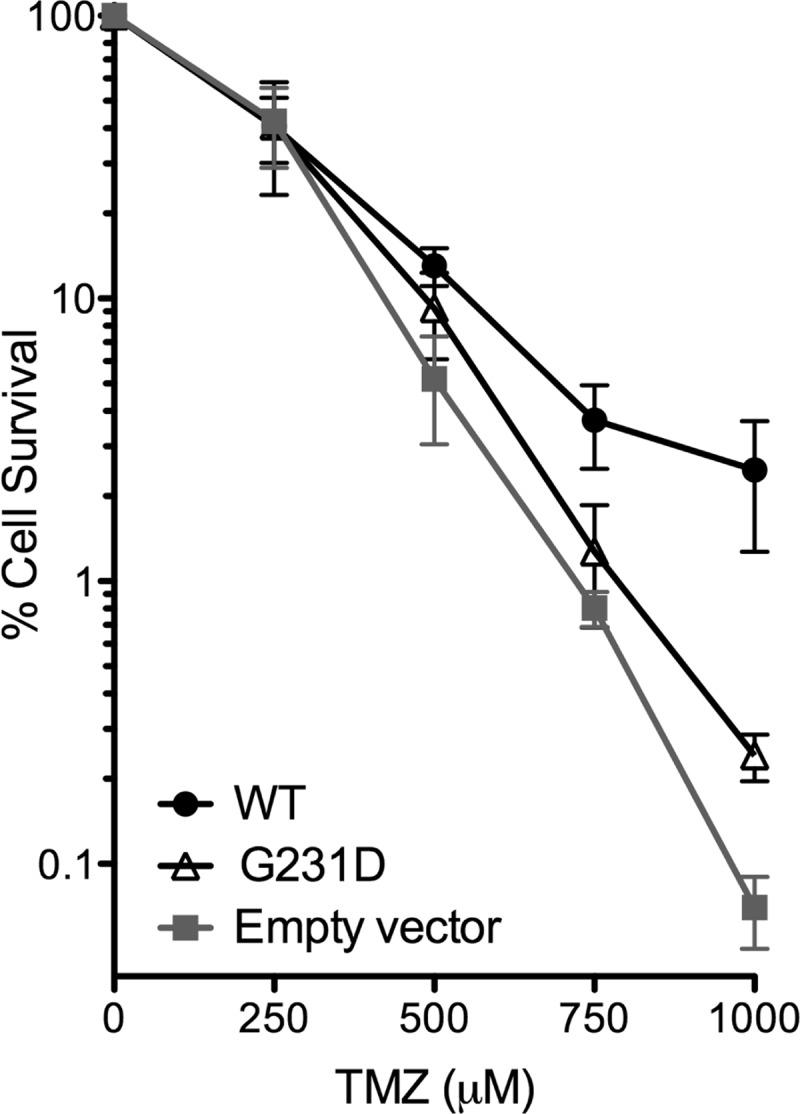
Expression of G231D sensitizes cells to chemotherapeutic agents. pol β-deficient cells expressing WT (filled circles), G231D (open triangles), or empty vector (filled squares) were treated with various concentrations of TMZ. Clonogenic survival assays were performed, and data are presented as mean ± S.E. of the percent survival (n = 3–4).
Substitution of Aspartate for Glycine at Position 231 Displaces the Template DNA in the Binary Complex
Gly-231 is distant from the active site of pol β so molecular dynamics simulations of the G231D variant were conducted on both the binary (pol β + DNA; Protein Data Bank code 1bpx) and ternary (pol β + DNA + dNTP; Protein Data Bank code 2fms) complexes to provide insight into how this residue impacts dNTP binding. In the binary complex, the change from a small noncharged residue (glycine) to the larger negatively charged aspartate appears to induce a displacement of both the protein and the DNA (Fig. 9A). The displacements are further exacerbated by the negative charges on the aspartate and the phosphate on the template strand between G9 and C10 (four base pairs upstream of the templating base). The distance between the α-carbon at position 231 of the protein and O2 of C10 on the template strand increases by 2.4 Å in the G231D variant compared with the wild-type polymerase (5.7 and 3.3 Å, respectively). Conversely, the model of the ternary complex of G231D shows fewer structural changes around position 231 (Fig. 9B). The distance between the α-carbon at position 231 of the protein and O2 of C10 on the template strand increases by only 0.6 Å when Gly-231 is replaced with aspartate (5.3 and 4.7 Å, respectively). As the only difference between the binary and ternary complexes is the presence of dNTP in the active site, this would suggest that binding of the nucleotide restores the G231D variant structure to a configuration more like that of the wild-type polymerase.
FIGURE 9.

G231D mutation disrupts template DNA and dNTP binding pocket. A and C, binary (open, no dNTP bound, Protein Data Bank code 1bpx) and B and D, ternary (closed, dNTP bound, Protein Data Bank code 2fms) crystal structures showing 231D pol β (blue) superimposed over WT pol β (red). A and B show residue 231. C and D show the dNTP binding pocket of 231D pol β (blue, cyan, and pink) superimposed over WT pol β (red, orange, and yellow).
Nucleotide Binding Pocket of the G231D Variant Is Disrupted in the Binary Complex
The nucleotide binding pocket residues (Asp-276, Asn-279, Tyr-271, and Phe-272) are all misaligned in the G231D variant compared with wild type. Additionally, the templating base is rotated 130° out of the plane of the incoming nucleotide, which would prevent incorporation (Fig. 9C). As was the case in the area proximal to residue 231, the nucleotide binding pocket of the variant polymerase is not perturbed in the ternary complex (Fig. 9D). The magnesium ions are coordinated to the Asps; the DNA bases are aligned properly, and the binding pocket residues are no longer misaligned. This indicates that once the nucleotide binds in the binding pocket, the structure is the same as the binding pocket of the wild-type polymerase. This is in support of the kinetic data that indicate that nucleotide binding is impaired, but if given a large enough amount of nucleotide, the variant behaves like the wild-type polymerase.
DISCUSSION
The G231D variant was recently identified in a colon carcinoma (24). Here, we show that expression of the G231D colon carcinoma-associated variant in immortal but nontransformed epithelial cells results in cellular transformation as assessed by focus formation, anchorage-independent growth, and increased proliferation (Fig. 1). The mechanism of cellular transformation is likely genomic instability resulting from slow gap filling during BER by G231D. This slow gap filling leads to an increased frequency of chromosomal aberrations that have been shown to be associated with several different types of cancers, including colon carcinoma (18). Therefore, we conclude that the G231D colon cancer-associated pol β variant has the capacity to drive carcinogenesis.
G231D exhibits a significantly lower affinity for the correct dNTP substrate, resulting in a lower catalytic efficiency (Table 2 and Fig. 2). G231D binds to DNA with affinity similar to that of WT pol β (Fig. 3). However, our molecular modeling studies suggest that G231D repulses part of the template DNA in the binary complex and that this repulsion is propagated into the dNTP binding pocket, resulting in misalignment of the amino acid residues and the distortion of the templating base comprising this pocket (Fig. 9). This misalignment of amino acid side chains in the pocket results in a lower affinity of the protein for dNTP, but once it is able to bind, proper active site alignment occurs leading to catalysis.
Gap-filling by G231D is perturbed in BER reactions carried out in cell extracts (Fig. 7). We suggest that this is a direct result of the lower affinity of G231D for the dNTP substrate. The concentration of intracellular dNTP is estimated to be in the range of 2 to 7.6 μm and 6.9 to 79 μm (19) in mouse NIH3T3 and HeLa cells, respectively. The Kd(dTTP) of G231D is ∼550 μm, at least a 7-fold increase over the concentration in cells. Therefore, it is unlikely that a sufficient concentration of dNTP substrate is present in cells to support gap-filling by G231D. This is supported by our observation of the increased sensitivity of cells expressing G231D pol β to the alkylating agent TMZ (Fig. 8). Although G231D possesses some BER capacity and rescues cells more efficiently that empty vector, only a small sample size of cells survive treatment. These cells may have genomic instability through the accumulation of BER intermediates and chromosomal aberrations. We postulate that the slow filling of gaps during BER by G231D leads to the accumulation of single nucleotide gaps that could result in chromosomal aberrations we observe in the event of replication fork collapse. Chromosomal instability, as observed here with G231D (Fig. 6), has been shown to be associated with colon cancer (20).
We have characterized other tumor-associated variants of pol β, including E295K, I260M, D160N, and K289M. The E295K gastric tumor-associated variant also arose in one of the colon tumors we studied (24). We showed that this variant has no polymerase activity, and a recent study suggests that this is due to an inability of E295K to assume a closed conformation that is necessary for catalysis (21). Expression of E295K in mouse cells results in an increased frequency of sister chromatid exchanges and in cellular transformation. The I260M, D160N, and K289M pol β prostate, gastric, and colon carcinoma variants, respectively, also induce cellular transformation when expressed in cells. However, we have shown that these variants are sequence context-dependent mutators with catalytic rates similar to WT (9, 22, 23), and we suggested that they induce cellular transformation by inducing mutations in genes that are important for growth control. We have also shown that the L22P pol β gastric tumor-associated variant induces cellular transformation and exhibits substantially reduced dRP lyase activity (14). This deficient dRP lyase activity likely leads to genomic instability in cells, resulting in their transformation. Here, we have identified a fourth mechanism by which a pol β variant induces cellular transformation, low affinity for dNTP substrate, leading to genomic instability.
Several variants of pol β have been identified in tumors, and these variants have biochemical properties that lead to cellular transformation. The results of this study taken together with our previous data are consistent with the interpretation that pol β tumor-associated variants drive carcinogenesis.
This work was supported, in whole or in part, by National Institutes of Health Grants F32 CA156843 and T32 CA009259 (to A. A. N.) and 1 PO1 CA129186 (to J. B. S.).

This article contains supplemental Figs. S1–S5 and Table S1.
- BER
- base excision repair
- pol β
- DNA polymerase β
- MEF
- mouse embryonic fibroblast
- dRP
- deoxyribose phosphate
- TMZ
- temozolomide
- Tet
- tetracycline
- UDG
- uracil-DNA glycosylase.
REFERENCES
- 1. Barnes D. E., Lindahl T. (2004) Repair and genetic consequences of endogenous DNA base damage in mammalian cells. Annu. Rev. Genet. 38, 445–476 [DOI] [PubMed] [Google Scholar]
- 2. Fortini P., Parlanti E., Sidorkina O. M., Laval J., Dogliotti E. (1999) The type of DNA glycosylase determines the base excision repair pathway in mammalian cells. J. Biol. Chem. 274, 15230–15236 [DOI] [PubMed] [Google Scholar]
- 3. Prasad R., Beard W. A., Strauss P. R., Wilson S. H. (1998) Human DNA polymerase β deoxyribose phosphate lyase. Substrate specificity and catalytic mechanism. J. Biol. Chem. 273, 15263–15270 [DOI] [PubMed] [Google Scholar]
- 4. Svilar D., Goellner E. M., Almeida K. H., Sobol R. W. (2011) Base excision repair and lesion-dependent subpathways for repair of oxidative DNA damage. Antioxid. Redox. Signal. 14, 2491–2507 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Wiederhold L., Leppard J. B., Kedar P., Karimi-Busheri F., Rasouli-Nia A., Weinfeld M., Tomkinson A. E., Izumi T., Prasad R., Wilson S. H., Mitra S., Hazra T. K. (2004) AP endonuclease-independent DNA base excision repair in human cells. Mol. Cell 15, 209–220 [DOI] [PubMed] [Google Scholar]
- 6. Srivastava D. K., Berg B. J., Prasad R., Molina J. T., Beard W. A., Tomkinson A. E., Wilson S. H. (1998) Mammalian abasic site base excision repair. Identification of the reaction sequence and rate-determining steps. J. Biol. Chem. 273, 21203–21209 [DOI] [PubMed] [Google Scholar]
- 7. Murphy D. L., Jaeger J., Sweasy J. B. (2011) A triad interaction in the fingers subdomain of DNA polymerase β controls polymerase activity. J. Am. Chem. Soc. 133, 6279–6287 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Lowy D. R., Rands E., Scolnick E. M. (1978) Helper-independent transformation by unintegrated Harvey sarcoma virus DNA. J. Virol. 26, 291–298 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Donigan K. A., Hile S. E., Eckert K. A., Sweasy J. B. (2012) The human gastric cancer-associated DNA polymerase β variant D160N is a mutator that induces cellular transformation. DNA Repair 11, 381–390 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Lang T., Dalal S., Chikova A., DiMaio D., Sweasy J. B. (2007) The E295K DNA polymerase β gastric cancer-associated variant interferes with base excision repair and induces cellular transformation. Mol. Cell. Biol. 27, 5587–5596 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Sweasy J. B., Lang T., Starcevic D., Sun K. W., Lai C. C., Dimaio D., Dalal S. (2005) Expression of DNA polymerase β cancer-associated variants in mouse cells results in cellular transformation. Proc. Natl. Acad. Sci. U.S.A. 102, 14350–14355 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Yamtich J., Starcevic D., Lauper J., Smith E., Shi I., Rangarajan S., Jaeger J., Sweasy J. B. (2010) Hinge residue Ile-174 is critical for proper dNTP selection by DNA polymerase β. Biochemistry 49, 2326–2334 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Murphy D. L., Kosa J., Jaeger J., Sweasy J. B. (2008) The Asp-285 variant of DNA polymerase β extends mispaired primer termini via increased nucleotide binding. Biochemistry 47, 8048–8057 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Dalal S., Chikova A., Jaeger J., Sweasy J. B. (2008) The L22P tumor-associated variant of DNA polymerase β is dRP lyase-deficient. Nucleic Acids Res. 36, 411–422 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. DeLano W. L. (2002) The PyMOL Molecular Graphics System, DeLano Scientific, San Carlos, CA [Google Scholar]
- 16. Kale R. S., Bhandarkar M., Brunner R., Gursoy A., Krawetz N., Phillips J., Shinozaki A., Varadarajan A., Schulten K. J. (1999) J. Comput. Phys. 151, 283–312 [Google Scholar]
- 17. Lai C. C., Edwards A. P., DiMaio D. (2005) Productive interaction between transmembrane mutants of the bovine papillomavirus E5 protein and the platelet-derived growth factor β receptor. J. Virol. 79, 1924–1929 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Shih I. M., Zhou W., Goodman S. N., Lengauer C., Kinzler K. W., Vogelstein B. (2001) Evidence that genetic instability occurs at an early stage of colorectal tumorigenesis. Cancer Res. 61, 818–822 [PubMed] [Google Scholar]
- 19. Gandhi V. V., Samuels D. C. (2011) A review comparing deoxyribonucleoside triphosphate (dNTP) concentrations in the mitochondrial and cytoplasmic compartments of normal and transformed cells. Nucleosides Nucleotides Nucleic Acids 30, 317–339 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Herman J. G., Umar A., Polyak K., Graff J. R., Ahuja N., Issa J. P., Markowitz S., Willson J. K., Hamilton S. R., Kinzler K. W., Kane M. F., Kolodner R. D., Vogelstein B., Kunkel T. A., Baylin S. B. (1998) Incidence and functional consequences of hMLH1 promoter hypermethylation in colorectal carcinoma. Proc. Natl. Acad. Sci. U.S.A. 95, 6870–6875 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Kirby T. W., DeRose E. F., Cavanaugh N. A., Beard W. A., Shock D. D., Mueller G. A., Wilson S. H., London R. E. (2011) Metal-induced DNA translocation leads to DNA polymerase conformational activation. Nucleic Acids Res. 40, 2974–2983 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Dalal S., Hile S., Eckert K. A., Sun K. W., Starcevic D., Sweasy J. B. (2005) Prostate cancer-associated I260M variant of DNA polymerase β is a sequence-specific mutator. Biochemistry 44, 15664–15673 [DOI] [PubMed] [Google Scholar]
- 23. Lang T., Maitra M., Starcevic D., Li S. X., Sweasy J. B. (2004) A DNA polymerase β mutant from colon cancer cells induces mutations. Proc. Natl. Acad. Sci. U.S.A. 101, 6074–6079 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Donigan K. A., Sun K.-w., Nemec A. A., Murphy D. L., Cong X., Northrup V., Zelterman D., Sweasy J. B. (May 10, 2012) Human POLB gene is mutated in high percentage of colorectal tumors. J. Biol. Chem. 287, 23830–23839 [DOI] [PMC free article] [PubMed] [Google Scholar]