

Background: A class of analgesic α-conotoxins potently inhibits N-type calcium channels.
Results: The activity of α-conotoxins Vc1.1 and RgIA was reduced following knockdown of GABAB receptor expression in sensory neurons and could be reconstituted in HEK 293 cells expressing human GABAB receptors and Cav2.2.
Conclusion: GABAB receptors are needed for inhibition of Cav2.2 by Vc1.1 and RgIA.
Significance: These analgesic α-conotoxins activate human GABAB receptors.
Keywords: Antisense RNA, Calcium Channels, G Protein-coupled Receptors (GPCR), Immunochemistry, mRNA, Patch Clamp, GABA(B) Receptor, HEK 293 Cell, Conotoxin, Dorsal Root Ganglion Neuron
Abstract
α-Conotoxins Vc1.1 and RgIA are small peptides isolated from the venom of marine cone snails. They have effective anti-nociceptive actions in rat models of neuropathic pain. Pharmacological studies in rodent dorsal root ganglion (DRG) show their analgesic effect is mediated by inhibition of N-type (Cav2.2) calcium channels via a pathway involving γ-aminobutyric acid type B (GABAB) receptor. However, there is no direct demonstration that functional GABAB receptors are needed for inhibition of the Cav2.2 channel by analgesic α-conotoxins. This study examined the effect of the GABAB agonist baclofen and α-conotoxins Vc1.1 and RgIA on calcium channel currents after transient knockdown of the GABAB receptor using RNA interference. Isolated rat DRG neurons were transfected with small interfering RNAs (siRNA) targeting GABAB subunits R1 and R2. Efficient knockdown of GABAB receptor expression at mRNA and protein levels was confirmed by quantitative real time PCR (qRT-PCR) and immunocytochemical analysis, respectively. Whole-cell patch clamp recordings conducted 2–4 days after transfection showed that inhibition of N-type calcium channels in response to baclofen, Vc1.1 and RgIA was significantly reduced in GABAB receptor knockdown DRG neurons. In contrast, neurons transfected with a scrambled nontargeting siRNA were indistinguishable from untransfected neurons. In the HEK 293 cell heterologous expression system, Vc1.1 and RgIA inhibition of Cav2.2 channels needed functional expression of both human GABAB receptor subunits. Together, these results confirm that GABAB receptors must be activated for the modulation of N-type (Cav2.2) calcium channels by analgesic α-conotoxins Vc1.1 and RgIA.
Introduction
Neuropathic pain is caused by damage or disease to the central or peripheral nervous system. It is a major health burden and poorly managed due to a lack of efficacious analgesics (1–3). Current pharmacotherapies, including opioids, anti-epileptics, and antidepressants, have low efficacy and significant side effects (4, 5). Therefore, there is a clinical need to develop novel drugs leading to better treatment of neuropathic pain.
Conotoxins (toxins isolated from the venom of marine cone snails) target a wide variety of membrane receptors and ion channels and are a rich source of potential therapeutic agents. α-Conotoxins are small peptides that typically range in size from 12 to 19 amino acids, contain two disulfide bonds, and usually have an amidated C terminus. α-Conotoxin Vc1.1, a synthetic derivative of a peptide derived from the venom of the marine cone snail Conus victoriae, potently suppresses signs of pain when injected intramuscularly near the site of injury in rat models (6–8), demonstrating its potential as an analgesic therapeutic. RgIA, an α-conotoxin from Conus regius, has also been shown to significantly reduce chronic constriction nerve injury-induced hyperalgesia in rats (9, 10).
How α-conotoxins Vc1.1 and RgIA relieve neuropathic pain remains controversial. Both are potent antagonists of α9α10 nicotinic acetylcholine receptors (nAChR)4 (9–11), and Vc1.1 antagonizes the nicotine-induced increase in axonal excitability of unmyelinated C-fibers (12). Therefore, α9α10 nAChRs were proposed as the target for Vc1.1 and RgIA to mediate analgesia in chronic and neuropathic pain models (10). However, several structural analogs of Vc1.1 retain activity at α9α10 nAChRs but lose their anti-allodynic effect (13, 14). This suggests that a target other than α9α10 nAChR may contribute to the pain-relieving activity of α-conotoxins. Recent studies show that the analgesic α-conotoxins Vc1.1 and RgIA potently inhibit N-type calcium channel currents in rat sensory neurons (8, 15, 16). Pharmacological studies further reveal that Vc1.1 does not interact directly with voltage-gated calcium channels (VGCC) but instead via a voltage-independent G protein-coupled GABAB receptor-mediated mechanism (8, 15). Collectively, these findings indicate that several different membrane receptors, including the GABAB receptor, may contribute to the pain-relieving activity of a class of α-conotoxins.
To date, there is a lack of molecular studies confirming the interaction between α-conotoxins and GABAB receptor subunits and demonstrating the need for functional GABAB receptor dimers for Cav2.2 channel inhibition by α-conotoxins. This study examines whether or not a functional GABAB receptor is needed to link analgesic α-conotoxins and N-type VGCC current inhibition.
We took two different approaches to this study. First, we investigated VGCC currents in isolated rat DRG neurons following transient knockdown of the GABAB receptor subunit expression using small interfering RNA (siRNA). Second, we investigated Cav2.2 channel modulation by Vc1.1 in HEK 293 cells stably expressing Cav2.2 channels and transient transfection with both subunits of the human GABAB receptor. These approaches showed that GABAB receptor expression is necessary for the observed inhibition of N-type VGCCs by the α-conotoxins Vc1.1 and RgIA. A preliminary report of some of these results has been presented in abstract form (41).
EXPERIMENTAL PROCEDURES
DRG Neuron Preparation
DRG neurons were enzymatically dissociated from the ganglia of 4–14-day-old Wistar rats according to standard protocols. Briefly, rats were killed by cervical dislocation, as approved by the RMIT University Animal Ethics committee, and the spinal cords were removed. Ganglia of all areas of the spinal cord were collected in ice-cold Hanks' balanced salt solution (Invitrogen). They were incubated in 1 mg/ml collagenase (Sigma) in Hanks' balanced salt solution for 30 min at 37 °C, transferred to warm Neurobasal media (Invitrogen), and then triturated using a fire-polished Pasteur pipette to obtain a cell suspension. The suspension was centrifuged at 400 × g for 5 min and immediately used for transfection.
siRNA Knockdown of GABAB Receptor
Mission siRNA oligonucleotides (Sigma) for the rat gabbr1 gene (catalog no. SASI_Rn01_00121458) and gabbr2 gene (catalog no. SASI_Rn01_00107052) were used for transfection. Mission siRNA oligonucleotides comprising a scramble sequence with no homology to any known genes (siRNA universal negative control 1) were used as a negative control. Mock-transfected cells (without siRNA) served as an additional negative control. The siRNAs (100 nm final concentration of each siRNA duplex) were transfected into ∼5 × 104 dissociated DRG cells using the Amaxa Nucleofector II electroporation system in combination with the basic neuron SCN nucleofector kit (both Lonza, Cologne, Germany) following the manufacturer's protocol. To identify transfected cells during electrophysiological experiments, 200 nm fluorescein-labeled oligonucleotide (Block-iT Fluorescent Oligo, Sigma) was added to the transfection reaction mixture. After transfection, the cells were suspended again in Neurobasal media containing B27 supplement (both Invitrogen), 0.5 mm l-glutamine, and 1% penicillin/streptomycin and then seeded onto poly-d-lysine-coated multiwell plates or glass coverslips. The cells were incubated under humidified conditions in 95% air and 5% CO2 at 37 °C and used after 1–4 days.
qRT-PCR
RNA was isolated 24–48 h after transfection using the Absolutely RNA nanoprep kit (Agilent Technologies, Santa Clara, CA), and cDNA was synthesized from the RNA using the SuperScript III first-strand synthesis supermix (Invitrogen) for qRT-PCR. Expression levels of GABAB R1 and GABAB R2 mRNA were analyzed by qRT-PCR (Rotor-Gene 3000, Corbett Research, Sydney, Australia) using the SensiMix SYBR No-ROX kit (Bioline, London, UK) and the following primers: 5′-TCA AGA TCA TTC TCA TGC CTG-3′ and 5′-GTG AAC TGG AGC CAT ATG AG-3′ for GABAB R1, and 5′-GAA CGA GAC CAA CTT CTT CG-3′ and 5-CTC TGC TGT CTT GAA ATT GAG-3′ for GABAB R2. Additionally, in each sample, the cDNAs of the housekeeping genes succinate dehydrogenase complex, subunit A, ubiquitin C, and ribosomal protein L13a, were amplified using standard primer sets (Mouse Normalization Gene Panel, Bioline) to serve as internal references. Data were analyzed based on the comparative quantitation method (Rotor-Gene software, Corbett). For each sample, the relative expression level of GABAB receptor mRNA was calculated by comparing it with the geometric mean of the relative mRNA levels of the three housekeeping genes.
Antibodies
The primary antibodies used were mouse monoclonal anti-βIII tubulin (Promega, 1:2000), rabbit polyclonal anti-GABAB R1 (Abcam, Cambridge, UK, catalog no. ab75239, 1:800), and rabbit monoclonal anti-GABAB R2 (Abcam, catalog no. ab75838, 1:400) antibodies. The corresponding fluorescent secondary antibodies used were Alexa Fluor 488-conjugated goat polyclonal anti-rabbit IgG antibody (Invitrogen, 1:1000) and Alexa Fluor 555-conjugated goat polyclonal anti-mouse IgG antibody (Invitrogen, 1:1000).
Double Labeling Immunocytochemistry and Confocal Microscopy
Immunocytochemistry was performed on transfected DRG neurons 2–4 days after transfection. DRG neurons were fixed in 4% paraformaldehyde for 15 min at room temperature. After two washes with PBS, the cells were preincubated in blocking solution (10% goat serum, 1% Triton X-100 in PBS) for 30 min at room temperature, followed by overnight incubation at 4 °C with anti-β-III tubulin and either polyclonal anti-GABAB1 or monoclonal anti-GABAB2 antibodies in fresh blocking solution. After two washes with PBS, the fluorophore-conjugated secondary antibodies were applied for 1.5 h at room temperature. Cell nuclei were counterstained with DAPI. After four more washing steps with PBS, the cells were sealed with fluorescence mounting media (Dako, Glostrup, Denmark). Control experiments determining the specificity of the immunocytochemical procedures were validated by omitting the primary antibodies. Images were acquired using a Nikon A1 laser-scanning confocal microscope equipped with a Plan Apo VC ×60 water-immersion objective, an argon laser (488 nm), a solid state laser (561 nm), and NIS Elements software using the same settings for siRNA-transfected and control cells.
Quantitative Analysis of GABAB Receptor Protein Levels
DRG cells, immunolabeled as described above, were subjected to quantitative analysis of GABAB R1 receptor protein levels using a high-content imaging system (ImageXpressTM Micro, Molecular Devices, Sunnyvale, CA). Cells transfected with the siRNAs targeting the GABAB R1 and R2 subunits were compared with cells transfected with the nontargeting negative control siRNA. For each siRNA treatment group, 756 sites (each containing an average of 3.3 neurons) were scanned. Neurons were identified by βIII-tubulin expression. A threshold for R1 fluorescence was set above background or nonspecific staining levels. All βIII-tubulin-positive cells with above-threshold fluorescence were counted as high R1-expressing neurons. A difference in the percentage of high R1-expressing neurons between control and knockdown of GABABR1 and GABABR2 (R1+R2 KD) cell batches would indicate successful knockdown. Moreover, to detect subtle differences in expression between high R1-expressing neurons of the control and R1+R2 KD cell batches, we further evaluated R1 fluorescence intensities indicating protein expression levels by dividing the sum of the grayscale intensity of R1 fluorescence by the total βIII-tubulin-stained area.
DRG and HEK 293 Cell Electrophysiology
DRG neurons were transfected with either GABAB receptor-specific siRNAs or negative-control siRNA and grown on poly-d-lysine-coated glass coverslips for 1–4 days. Only DRG neurons exhibiting green fluorescence, due to co-transfection of the fluorescein-labeled oligonucleotide, were selected for electrophysiological recordings. Cells were transferred into a small volume (∼300 μl) recording chamber, constantly perfused with a solution containing (in mm) tetraethylammonium-Cl 140, BaCl2 2, d-glucose 10, and HEPES 10, pH 7.4 (with NaOH). The internal pipette solution contained (in mm) CsCl 140, MgCl2 1, MgATP 5, Na-GTP 0.1, bis(2-aminophenoxy)ethane-N,N,N′,N′-tetra-acetic acid-Cs4 5, and HEPES 10, pH 7.3 (with CsOH). Whole-cell Ba2+ currents were elicited from a holding potential of −80 mV, by 0.05 Hz, 200-ms step depolarizations to +10 mV. In each experiment, R1+R2 KD neurons and control neurons from the same batch of DRG preparations and transfections were tested successively. They were included for analysis only if the VGCC inward current inhibition in the R1+R2 KD neuron by baclofen was at least 10% less than the current inhibition by baclofen in the control neuron, indicating successful knockdown of the GABAB receptor.
HEK 293 cells stably expressing human Cav2.2 channels (α1B, α2δ1 and β3 subunits) (17) were transiently co-transfected with cDNAs encoding human GABAB R1 (10 μg), human GABAB R2 (10 μg) (OriGene Technologies, Inc., Rockville, MD), and enhanced green fluorescent protein (1 μg) (a reporter gene construct), using calcium phosphate precipitation (18). They were then cultured at 37 °C. In some experiments, cDNAs encoding the human dopamine receptor 2 (DRD2; OriGene Technologies) were used instead of GABAB R1 and R2. Patch clamp experiments were done 4–6 days after transfection, using the whole-cell recording configuration. HEK 293 cells were superfused with a solution containing (in mm) BaCl2 10, CsCl 85, tetraethylammonium-Cl 40, MgCl2 1, d-glucose 10, and HEPES 10, with pH 7.4 (with tetraethylammonium-OH). Fire-polished borosilicate patch pipettes (2–3 megohm tip resistances) were filled with a solution containing (in mm) cesium methane sulfonate 108, EGTA 9, MgCl2 4, MgATP 2, Na2GTP 0.5, and HEPES 9, pH 7.2 (with CsOH). Typically, cells were voltage-clamped at −80 mV, and membrane currents were elicited by 0.1 Hz, 150-ms step depolarizations to −10 mV.
Recordings were carried out using a Multiclamp 700B amplifier (Molecular Devices), controlled by a Clampex9.2/Digidata 1332A acquisition system at room temperature (23–25 °C). Membrane currents were filtered at 2 kHz and sampled at 5 or 8 kHz. Leak and capacitative currents were subtracted using a −P/4 pulse protocol. Data were stored digitally on a computer for further analysis. Peak current amplitude in response to the depolarizing pulse was measured once a steady state was achieved (∼5–10 min). Current densities were calculated by dividing the normalized current amplitude by the cell capacitance measured immediately before the recording. Current amplitudes obtained in the presence of the drug were normalized by dividing the current amplitude obtained under control conditions.
Chemicals and Drugs
α-Conotoxins Vc1.1, RgIA, and AuIB were synthesized as described previously (19–22) and provided as a stock concentration in H2O of ∼1 mm by Dr. R. J. Clark (University of Queensland). The ω-conotoxins CVID and MVIIA were prepared as described previously (23). Baclofen and [d-Ala2,N-Me-Phe4,Gly5-ol]enkephalin acetate salt (DAMGO) were purchased from Sigma. (2S)-3-[[(1S)-1-(3,4-Dichlorophenyl)ethyl]amino-2-hydroxypropyl](phenylmethyl) phosphinic acid hydrochloride (CGP55845) was purchased from Tocris Bioscience (Bristol, UK). All drugs were diluted to the appropriate final concentration and applied via perfusion in the bath solution. Baclofen was usually tested following the application of the α-conotoxin.
Cell Surface Biotinylation and Western Blot
HEK 293 cells stably expressing human Cav2.2 channels were transfected as described above. They were grown in T175 flasks and treated with either 1 μm Vc1.1 or 30 μm baclofen. Immediately after treatment, biotinylation of cell surface proteins was done using the Pierce cell surface protein isolation kit (Thermo Fisher Scientific, Rockford, IL) according to the manufacturer instructions. Biotinylated Cav2.2 protein was detected by Western blot. Detailed methods can be found in the supplemental material.
Statistical Analysis
Numerical data are presented as means ± S.E. The statistical significance between the two groups was evaluated by Student's unpaired t test. The statistical significance between three or more groups was evaluated by one-way analysis of variance followed by the Bonferroni's post hoc test. Values of p < 0.05 were considered to be statistically significant. All statistical analyses were achieved using Prism Version 5.1 (GraphPad Software Inc., La Jolla, CA).
RESULTS
Knockdown of GABAB Receptor mRNA Expression in DRG Neurons
To effectively knockdown the expression of functional GABAB receptors (which are heterodimers of the two GABAB subunits R1 and R2), we tested siRNAs targeting either the R1 or R2 subunit in isolated DRG neurons. qRT-PCR showed that following 24–48 h post-transfection, the mRNA levels of R1 in cells transfected with an R1-specific siRNA significantly decreased to 32.6 ± 3.5% (n = 4) of control cells transfected with a scrambled, nontargeting siRNA (p < 0.0001) (Fig. 1A). R2 mRNA levels 24–48 h after transfection with the R2-specific siRNA were significantly reduced to 53.9 ± 8.0% (n = 4) of control (p = 0.007) (Fig. 1B). A further increase in siRNA concentration (300 nm) did not lead to a further knockdown of either GABAB receptor subunit (data not shown). The scrambled, nontargeting siRNA-transfected control cells exhibited R1 and R2 expression levels almost identical to mock-transfected (H2O transfected) cells (Fig. 1, A and B). This indicated that there was not a nonspecific effect due to the siRNA transfection process itself.
FIGURE 1.
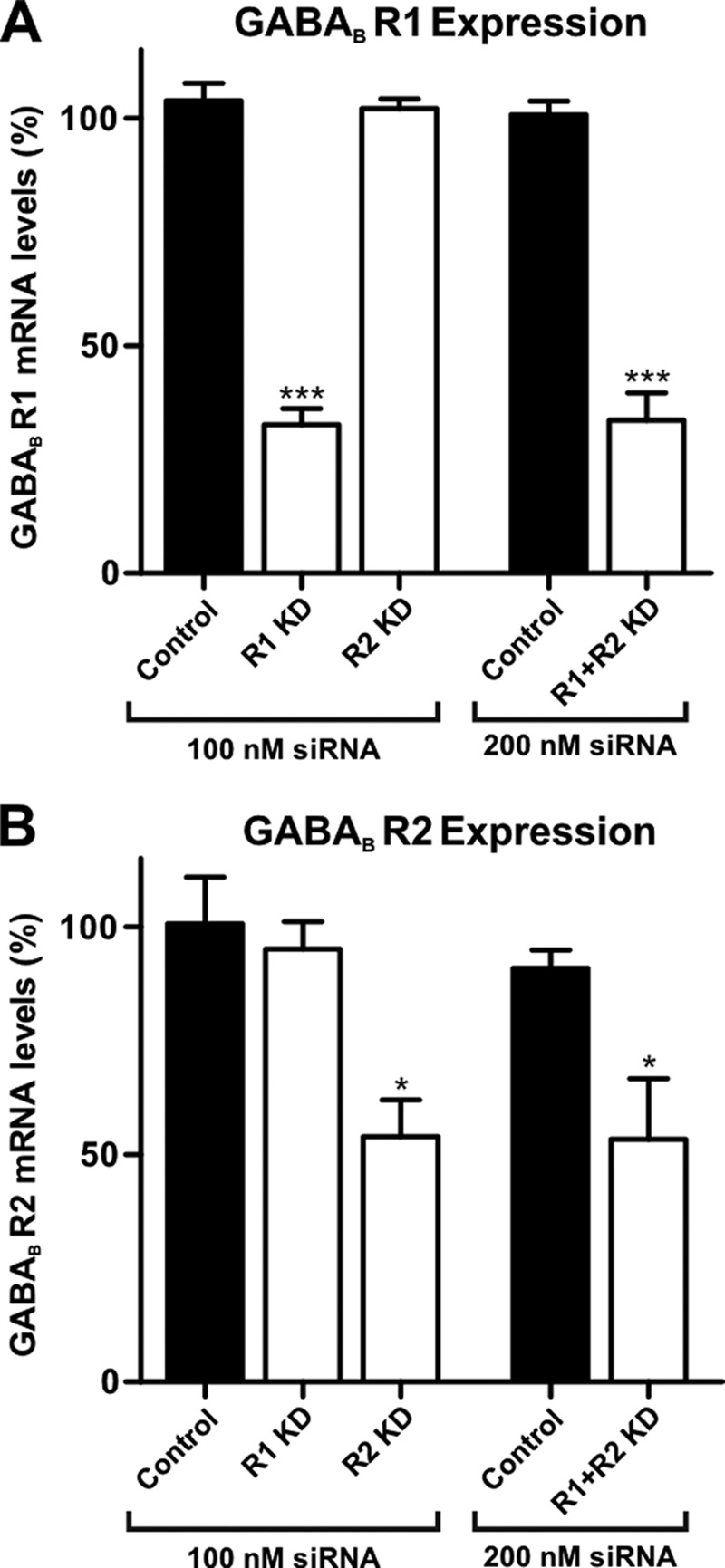
GABAB receptor mRNA knockdown by siRNA treatment in rat DRG neurons. Transfection of siRNA (100 nm) targeting GABAB receptor subunits GABAB R1 (R1 KD) and GABAB R2 (R2 KD) resulted in a knockdown by 67 ± 3% (n = 4) of R1 (A) and 46 ± 8% (n = 4) for R2 mRNA levels (B), respectively. This is compared with a negative control in which cells were transfected with an equal concentration of a scrambled, nontargeting siRNA. Co-transfection of R1- and R2-targeting siRNAs (R1+R2 KD) resulted in knockdown levels similar to single subunit knockdowns (66 ± 6 and 47 ± 13%, respectively; n = 5). mRNA levels were determined by qRT-PCR, calculated by comparative quantitation analysis, and normalized to the expression levels of water-transfected cells (*, p < 0.05; ***, p < 0.0001 versus control).
No off-target effects of the GABAB subunit-targeting siRNAs were observed, because expression of the housekeeping genes, analyzed in parallel, was not affected by the addition of siRNAs. Furthermore, when mRNA expression of a single GABAB receptor subunit was disrupted by siRNA, the expression level of the other subunit was not affected (Fig. 1, A and B), indicating target-specific knockdown. Co-transfection of the R1- and R2-targeting siRNAs (R1+R2 KD) demonstrated that knockdown of each subunit was equally effective compared with single subunit knockdowns without interference (33.6 ± 6.1%, n = 5; p < 0.0001 mRNA levels of R1, and 53.4 ± 13.3%, n = 5; p = 0.026 mRNA levels of R2, respectively) (Fig. 1, A and B). Given that heterodimerization of R1 and R2 subunits is essential for functional GABAB receptors (24), we used the co-transfection protocol in the following experiments, expecting more effective knockdown.
GABAB Receptor Protein Knockdown by siRNA Treatment
Immunofluorescence labeling of GABAB receptors in rat DRG neurons and visual inspection by confocal microscopy revealed an apparent decrease in fluorescence intensities for GABAB R1 and GABAB R2 in R1+R2 KD neurons compared with scrambled, nontargeting siRNA-transfected DRG neurons (Fig. 2, A and B). The difference in intensity of R1 immunostaining was quantified using a high-content imaging system. In these experiments, we focused on R1 immunofluorescence because R2 immunofluorescence intensity and specificity were not as high as that of R1. Analysis revealed that the percentage of cells with R1-specific immunofluorescence above the set threshold was significantly lower in the R1+R2 KD group (35.1 ± 3.6%, n = 6) compared with control cells (57.3 ± 1.2%, n = 6) (p < 0.0001) (Fig. 3B). Furthermore, the fluorescence intensities of R1, which indicate expression levels, in cells with above-threshold R1 fluorescence were 20.2 ± 5.4% lower in knockdown cells compared with control cells (n = 6) (p = 0.021) (Fig. 3C). Taken together, the above data demonstrated that GABAB receptor knockdown by siRNA effectively decreases GABAB receptor protein expression in DRG neurons.
FIGURE 2.

GABAB receptor immunofluorescence is reduced in GABAB knockdown DRG neurons as observed using confocal microscopy. Immunolabeling of DRG neurons and visual inspection by confocal microscopy revealed an apparent decrease in fluorescence intensities for GABAB R1 and R2 in neurons transfected with siRNAs to disrupt GABAB R1 and R2 expression. This is compared with scrambled, nontargeting siRNA-transfected DRG neurons. Cells were immunostained 3 days after transfection, using antibodies directed against R1 (green) (A) or R2 (green) (B). Immunostaining of βIII-tubulin is shown in red. Representative images are shown. Scale bars, 50 μm.
FIGURE 3.

Quantitative evaluation of protein levels in GABAB R1 knockdown DRG neurons by high content imaging analysis revealed significantly reduced GABAB R1 expression. We compared DRG cells transfected with siRNAs targeting both GABAB receptor subunits (R1+R2 KD) with control cells transfected with scrambled, nontargeting siRNA (control). Quantitative analysis of immunofluorescence-labeled R1 proteins in DRG neurons 2–3 days after transfection revealed significantly reduced R1 protein expression in knockdown cells (R1+R2 KD). A, representative sites of the high content scan of a total of 756 sites per treatment group. DRG neurons showing weak green fluorescence, indicating low expression of R1 (white arrowheads), were counted as neurons with R1 expression below threshold in the statistical analysis. Scale bars, 50 μm. B, percentage of neurons exhibiting R1 immunofluorescence above threshold, which correlates to high R1 expression levels. C, R1 expression levels of neurons with R1 expression above threshold calculated from fluorescence intensities. Number of experiments indicated in parentheses (*, p < 0.05; ***, p < 0.0001 versus control).
Impairment of α-Conotoxin-mediated Inhibition of VGCC Currents by GABAB Receptor Knockdown
R1+R2 KD DRG neurons were used in patch clamp experiments to characterize the activity of the GABAB receptor agonist baclofen and α-conotoxins Vc1.1 and RgIA. Control and R1+R2 KD neurons exhibited comparable VGCC currents upon depolarization to +10 mV from a holding potential of −80 mV (Fig. 4, A and B). There was no significant difference in VGCC peak current density between control (−71.3 ± 7.7 pA/picofarads, n = 17) and R1+R2 KD (−59.0 ± 9.2 pA/picofarads, n = 17) (p = 0.312) DRG neurons. The effect of Vc1.1 on VGCC current amplitude in control and R1+R2 KD neurons was examined by bath application of Vc1.1 (100 nm), followed by the application of baclofen (30 μm).
FIGURE 4.

Inhibition of N-type calcium channels in response to baclofen and Vc1. 1 is lower in DRG neurons transfected with siRNAs that target the GABAB receptor. A and B show superimposed traces of depolarization-activated VGCC currents in scrambled, nontargeting siRNA-transfected (A) and GABABR knockdown siRNA-transfected (B) rat DRG neurons, in the absence (control) and presence of Vc1.1 (100 nm) and baclofen (30 μm). C and D, bar graph summary of data on peak VGCC current amplitude reduction in the presence of baclofen (C) and Vc1.1 (D). Vc1.1 inhibition of VGCC in R1+R2 KD neurons was significantly lower than in control neurons. Number of experiments is indicated in parentheses (**, p < 0.01 versus control).
Of 17 neurons that exhibited efficient functional knockdown of GABAB receptors, VGCC current amplitude was inhibited by 21.2 ± 2.9% in response to bath application of baclofen. However, control DRG neurons exhibited a baclofen-induced decrease of 36.6 ± 2.9% (n = 17) (p = 0.001) (Fig. 4, A and C, and Table 1). The remaining VGCC current was not inhibited more by bath application of the selective N-type VGCC antagonist, ω-conotoxin CVID (300 nm), as we observed previously (15). Bath application of 100 nm Vc1.1 to control neurons inhibited the VGCC current amplitude by 20 ± 2.6% (n = 10) (Fig. 4, A and D). However, in R1+R2 KD neurons, the effect of Vc1.1 on the VGCC current was significantly impaired, with the current amplitude reduced only by 5.1 ± 3.5% (n = 10) (p = 0.003) (Fig. 4, B and D, and Table 1). Note that in all neurons of the R1+R2 KD treatment group in which baclofen-mediated inhibition of VGCCs was impaired, Vc1.1-induced inhibition of VGCC currents decreased in all cases. The decrease in the number of functional GABAB receptors, caused by siRNA knockdown, resulted in a functional disruption of GABAB negative coupling to VGCCs. This confirms that Vc1.1, similar to baclofen, acts on N-type VGCCs via the GABAB receptor.
TABLE 1.
Relative GPCR-mediated decrease in VGCC peak current amplitude in control and R1+R2 KD DRG neurons
| Compound | Inhibitiona (control) | Inhibitiona (R1+R2 KD) | nb | pc | % siRNA attenuationd |
|---|---|---|---|---|---|
| % | % | ||||
| Vc1.1 | 20.0 ± 2.6 | 5.1 ± 3.5 | 10 | 0.003 | 74.5 |
| RgIA | 16.2 ± 2.3 | 4.6 ± 3.0 | 5 | 0.015 | 71.8 |
| Baclofen | 36.6 ± 2.9 | 21.2 ± 2.9 | 17 | 0.001 | 42.1 |
| DAMGO | 15.7 ± 2.6 | 22.3 ± 4.3 | 10 | 0.199 |
a Inhibition of VGCC peak current amplitude following addition of the compound indicated is calculated as 100 − I/I0 × 100.
b n is number of paired observations in control and R1 + R2 KD cells.
c p value indicates statistical significance of the difference in VGCC peak current amplitude inhibition in R1 + R2 neurons versus control neurons (% inhibition of control versus inhibition of R1 + R2 KD).
d Percentage of reduction of normalized current amplitude inhibition in knockdown cells compared with control cells is calculated as 100 − 100/inhibition(control) × inhibition(R1+R2 KD).
Disruption of VGCC current inhibition by GABAB receptor knockdown was more pronounced for Vc1.1-mediated N-type VGCC inhibition than for baclofen-mediated inhibition. In R1+R2 KD neurons, N-type VGCC current inhibition by Vc1.1 was 74.5% (n = 10) less than the current inhibition by Vc1.1 in control neurons, whereas baclofen-mediated current inhibition in R1+R2 KD neurons decreased by only 42.1% (n = 17), compared with the 17 corresponding control neurons (Table 1). Although the present findings substantiate previous reports that baclofen and Vc1.1 both act via the GABAB receptor (15, 16), the greater decrease in Vc1.1-mediated inhibition caused by siRNA-mediated GABAB receptor knockdown suggests that the two compounds may act via different downstream signaling cascades.
We also evaluated the activity of α-conotoxins RgIA and AuIB on the GABAB receptor in knockdown and control DRG neurons. RgIA (100 nm) reduced peak VGCC current amplitude in control neurons by 16.2 ± 2.3% (n = 5) (Table 1), which is consistent with previous studies showing RgIA activity on nontransfected DRG neurons (14, 15). In contrast, R1+R2 KD neurons considerably decreased RgIA-mediated inhibition, with a reduction of current amplitude by 4.6 ± 3% (n = 5) (p = 0.015) (Table 1). AuIB (100 nm), an analgesic α-conotoxin active at the α3β4 nAChR (20) and GABAB receptor (8), also inhibited VGCC current amplitude in control neurons (19.9 ± 3.8%; n = 3). However, in R1+R2 KD cells, inhibition of the VGCC current amplitude significantly decreased (7.6 ± 1.1%; n = 3) (p = 0.035). Together, these experiments show that the activity of these α-conotoxins on N-type VGCCs is dependent on GABAB receptor expression.
To show siRNA knockdown of the GABAB receptor was specific, control experiments examined the effect of μ-opioid receptor agonists on VGCC currents in transfected DRG neurons. μ-Opioid receptor activation by agonists, such as Met-enkephalin and DAMGO, has been shown to inhibit the N-type VGCC current via activation of a pertussis toxin-sensitive G protein-coupled receptor (GPCR) (25, 26). A recent study reported that different DRG neuron sensitivity to DAMGO corresponded with different DRG neuron soma sizes (27). We also observed a wide variation in individual DRG neuron sensitivity to DAMGO, with some neurons having minimal response because no specific neuron subtype was selected. Control DRG neurons showed 15.7 ± 2.6% (n = 10) inhibition of VGCC current amplitudes by 1 μm DAMGO (n = 10) when VGCC current amplitude was compared before and after adding DAMGO (p = 0.003). The neurons we studied were chosen at random, so any statistical difference in VGCC current inhibition between R1+R2 KD and control neurons would suggest the knockdown affected μ-opioid receptor signaling. However, we did not observe any statistically significant difference in VGCC current inhibition by 1 μm DAMGO between R1+R2 KD neurons (22.3 ± 4.3%; n = 10) and scrambled, nontargeting siRNA-transfected control neurons (15.7 ± 2.6%; n = 10) (p = 0.199) (Table 1). This confirmed that the impairment of VGCC current inhibition by baclofen, Vc1.1, RgIA, and AuIB in R1+R2 KD neurons was due to decreased GABAB receptor expression rather than the nonspecific effects of GABAB receptor-targeting siRNAs.
Reconstitution of the Human GABAB Receptor Signaling Pathway in HEK 293 Cells
To demonstrate that both subunits of the human GABAB receptor are needed to inhibit Cav2.2 channels with α-conotoxin Vc1.1, we reconstituted the GABAB receptor signaling pathway in HEK 293 cells stably expressing Cav2.2 channels (Cav2.2 cells). This was done by co-transfecting cDNAs of cloned human GABAB receptor subunits (R1 and R2) (Fig. 5).
FIGURE 5.

Inhibition of stably expressed Cav2. 2 channels in HEK 293 cells transiently co-transfected with human GABAB receptor subunits R1 + R2 by α-conotoxin Vc1.1 and baclofen. A–C, peak Ba2+ current amplitude elicited by step depolarization (0.1 Hz) to −10 mV from a holding potential of −80 mV plotted as a function of time. Bars indicate bath application of various compounds. Superimposed inward Ba2+ currents (top insets) are shown at the times marked by lowercase letters (see below). Dotted line indicates zero current level. A, in HEK 293 cells expressing Cav2.2 channels alone, depolarization-activated Ba2+ currents are not affected by α-conotoxin Vc1.1 (200 nm) but are almost completely inhibited by ω-conotoxin MVIIA (200 nm). B, in Cav2.2+R1+R2 cells, bath application of Vc1.1 (200 nm) inhibits inward Ba2+ current amplitude. C, in Cav2.2+R1+R2 cells, the effect of Vc1.1 was antagonized by bath application of the selective GABAB receptor antagonist CGP55845 (1 μm), whereas baclofen (50 μm), applied after the washout of CGP55845, inhibited the inward Ba2+ current. D, bar graph summary of the inhibition of peak Ba2+ current by Vc1.1 (200 nm) and baclofen (50 μm) in the absence of GABAB receptor subunits (Cav2.2 alone; control) or presence of Cav2.2 channels and R1, R2, or R1+R2. Number of experiments indicated in parentheses (*, p < 0.0001 versus control).
In HEK 293 cells with Cav2.2 alone (control), peak Ba2+ currents were not affected by α-conotoxin Vc1.1 (200 nm) (n = 10). However, the selective N-type (Cav2.2) VGCC inhibitor, ω-conotoxin MVIIA (200 nm), almost completely inhibited the inward Ba2+ current (n = 6) (Fig. 5A). In cells expressing Cav2.2 and co-transfected with GABAB R1 and R2 (Cav2.2+R1+R2 cells), Vc1.1 and baclofen caused inward Ba2+ current inhibition, indicated by the decrease of peak current amplitude during the administration of these compounds (40 ± 7%, n = 5 for Vc1.1; 65 ± 5%, n = 7 for baclofen) (Fig. 5, B–D). In Cav2.2, Cav2.2+R1 or Cav2.2+R2 cells, we did not observe inhibition of the Ba2+ current by Vc1.1 or baclofen (n ≥ 6) (Fig. 5D). Similarly, α-conotoxin Rg1A (200 nm) inhibited inward Ba2+ current only in Cav2.2+R1+R2 cells (20 ± 7.3%; n = 4; supplemental Fig. S1). However, the effect of Vc1.1 (200 nm) or baclofen (100 μm) was absent in Cav2.2 cells transiently transfected with the dopamine receptor D2 (n = 6; data not shown). In Cav2.2+R1+R2 cells, inhibition of Cav2.2 channels by Vc1.1 could be blocked by the selective GABAB receptor antagonist CGP55845 (n = 6) (Fig. 5C).
In a series of experiments, Ca2+ currents were evoked by depolarizing test pulses (50 ms, 0.1 Hz) to +20 mV from a holding potential of −80 mV, with or without a prepulse (20 ms), to +120 mV before the test pulse. Bath application of GABA (1 μm) inhibited Ca2+ current amplitude by 64 ± 5% without a prepulse and by 3 ± 5% (n = 8) with a prepulse (p < 0.0001). Vc1.1 (300 nm) caused either a 50 ± 10% (without a prepulse) or 36 ± 6% (with a prepulse, n = 5) decrease in Ca2+ current amplitude (p = 0.075). These data indicate that modulation of Cav2.2 by GABA is voltage-dependent, whereas inhibition by Vc1.1 does not significantly differ with or without a prepulse, so it is voltage-independent.
In summary, these data demonstrate that functional expressions of both R1 and R2 subunits are needed for the inhibition of Cav2.2 channels by Vc1.1 in the heterologous HEK 293 cell expression system.
To determine whether prolonged GABAB receptor activation by Vc1.1 or baclofen internalizes the Cav2.2 channel and reduces N-type VGCC current amplitude, we investigated HEK 293 cells stably expressing Cav2.2 using surface biotinylation studies. A similar mechanism of Cav2.2 internalization has been proposed for ORL1 receptor activation (28).
Cav2.2+R1+R2 cells were exposed to 1 μm Vc1.1 for 30 min, and cell surface proteins were biotinylated immediately afterward. Vc1.1 did not appear to cause internalization of Cav2.2, with cell surface Cav2.2 protein levels detected by Western blot unchanged compared with cells treated with either 30 μm baclofen or control solution only (n = 4; supplemental Fig. S2). Furthermore, there was no difference in cell surface Cav2.2 protein levels between Cav2.2+R1+R2 cells and Cav2.2 cells alone (lacking GABABR) when both were treated with Vc1.1. These results indicate that the observed N-type VGCC current inhibition by α-conotoxin Vc1.1 via the GABAB receptor is not due to internalization of Cav2.2 channels.
DISCUSSION
This study demonstrates that functional GABAB receptors are needed to observe the analgesic α-conotoxins Vc1.1 and RgIA decrease N-type VGCC peak current amplitude. Although previous pharmacological studies showed that Vc1.1 and RgIA can inhibit VGCC currents in rodent DRG neurons via activation of the G protein-coupled GABAB receptor (8, 15, 16), there were no molecular studies showing the need for expression of both GABAB receptor subunits.
We chose a knockdown approach to demonstrate dependence of α-conotoxin activity on GABAB receptor expression, because RNAi is a powerful tool for the investigation of molecular signaling cascades, and siRNAs have already been successfully used for knockdown of GABAB receptor subunits (29–31) and other target receptors and ion channels in pain pathways (32, 33). In siRNA knockdown experiments, we show that the inhibition of N-type VGCC currents in rat DRG neurons by α-conotoxins decreased following a transient knockdown of GABAB receptors. The effectiveness of the siRNA knockdown was underscored by qRT-PCR and immunocytochemistry, showing a decrease in GABAB receptor mRNA and protein expression, respectively.
Results from the GABAB receptor knockdown study were reinforced by a complementary approach using HEK 293 cells. These cells represent an established expression system for various membrane proteins (receptors and ion channels) and have been used to study signaling of heterologously expressed GABAB and other G protein-coupled receptors (14, 34). By co-transfecting both human GABAB receptor subunits into HEK 293 cells stably expressing Cav2.2 channels, we could reconstitute the GABAB receptor signaling pathway. In these cells, baclofen and α-conotoxins Vc1.1 and RgIA decreased Cav2.2-mediated Ba2+ currents. However, cells expressing Cav2.2 channels alone, or only one of the two GABAB receptor subunits, were insensitive to baclofen and Vc1.1. Thus, we have demonstrated that functional GABAB receptors are the pharmacological target of α-conotoxins Vc1.1 and RgIA. We have also shown that functional GABAB receptor expression is needed for the inhibition of Cav2.2 channels.
Our electrophysiological findings on GABAB receptor knockdown show that in control DRG neurons, the effect of Vc1.1 and RgIA on N-type VGCC currents was substantially less than that of baclofen. Moreover, Vc1.1 and baclofen clearly differed in their susceptibility to GABAB receptor knockdown. Inhibition of N-type VGCC currents in GABAB knockdown DRG neurons by Vc1.1 was almost completely abolished. In contrast, even though the effect of baclofen was significantly impaired, inhibition of VGCC current was still observed. These observations corroborate previous findings, indicating that Vc1.1 and baclofen act on N-type VGCCs via different pathways, both involving the GABAB receptor (15). N-type VGCCs can be inhibited by two distinct G protein-dependent inhibitory pathways, i.e. voltage-dependent and voltage-independent (35). Voltage-dependent inhibition is the most common pathway, involving Gβγ binding directly to the α-subunit of the N-type VGCC (36, 37). The inhibition of N-type VGCCs in DRG neurons by α-conotoxin Vc1.1 is voltage-independent and activates a pathway that involves Src tyrosine kinase (15). In contrast, the GABAB receptor agonist baclofen and its inhibition of VGCC currents are well characterized (38–40) and appear to inhibit N-type VGCC currents predominantly via the voltage-dependent pathway (35). These results suggest that the Vc1.1-mediated voltage-independent pathway is more sensitive to GABAB receptor knockdown (partial knockdown) than the baclofen-mediated voltage-dependent pathway.
One possible way GPCR agonists could reduce VGCC current amplitude is through receptor-mediated internalization of the channels. This mechanism has been proposed for inhibition of N-type VGCC currents after activation of the ORL1 receptor (28). However, a recent study in rat DRG neurons found no functional loss of surface N-type calcium channels after prolonged ORL1 activation by nociceptin/orphanin FQ (27). Our surface biotinylation studies on HEK 293 cells expressing human Cav2.2 and GABAB receptor and exposed to either Vc1.1 or baclofen did not reveal any decrease in cell surface Cav2.2 protein levels. This suggests that Vc1.1 activates a GABAB receptor-mediated signaling cascade that inhibits N-type VGCC currents by channel inactivation rather than channel internalization.
Our findings not only strengthen the pharmacological significance of the observed N-type VGCC inhibition by α-conotoxins, Vc1.1 and RgIA, but extend them to show that these α-conotoxins are active at human GABAB receptors. In vivo studies have shown that Vc1.1, RgIA, and AuIB administered intramuscularly in rats can potently suppress neuropathic pain following partial nerve ligation (6–8). To date, there are at least five α-conotoxins that inhibit N-type VGCCs in DRG neurons, and the three (Vc1.1, RgIA, AuIB) that have been tested in animal pain models are analgesic. Further studies of the structure-activity relationships are likely to identify the specific residues that confer potency and selectivity for the GABAB receptor and will improve understanding of how α-conotoxins act on the GABAB receptor. In the future, other α-conotoxins and analogs activating the GABAB receptor may be discovered, making the human GABAB receptor a novel target for drugs used to treat neuropathic pain.
Acknowledgments
We are grateful to Dr. Richard Clark (University of Queensland) for kindly providing the synthesized α-conotoxins. We thank Professor MacDonald Christie for helpful discussions and Dr. Nadia Cerminara for comments on a draft of the manuscript.
This work was supported by National Health and Medical Research Council Program Grant 569927 and Australian Research Council Project Grant DP1093177.

This article contains supplemental Figs. S1 and S2.
- nAChR
- nicotinic acetylcholine receptor
- Cav2.2+R1+R2
- cells expressing Cav2.2 and co-transfected with GABAB R1 and R2 subunits
- DAMGO
- [d-Ala2,N-Me-Phe4, Gly5-ol]enkephalin acetate salt
- DRG
- dorsal root ganglion
- GABAB
- γ-aminobutyric acid type B
- GABAB R1 and R2
- γ-aminobutyric acid type B receptor subunits R1 and R2
- qRT-PCR
- quantitative real time-PCR
- R1+R2 KD
- knockdown of GABABR1 and GABABR2
- VGCC
- voltage-gated calcium channel.
REFERENCES
- 1. Cousins M. J., Brennan F., Carr D. B. (2004) Pain relief. A universal human right. Pain 112, 1–4 [DOI] [PubMed] [Google Scholar]
- 2. Zimmermann M. (2001) Pathobiology of neuropathic pain. Eur. J. Pharmacol. 429, 23–37 [DOI] [PubMed] [Google Scholar]
- 3. Baron R. (2006) Mechanisms of disease. Neuropathic pain, a clinical perspective. Nat. Clin. Pract. Neurol. 2, 95–106 [DOI] [PubMed] [Google Scholar]
- 4. Dworkin R. H., O'Connor A. B., Backonja M., Farrar J. T., Finnerup N. B., Jensen T. S., Kalso E. A., Loeser J. D., Miaskowski C., Nurmikko T. J., Portenoy R. K., Rice A. S., Stacey B. R., Treede R. D., Turk D. C., Wallace M. S. (2007) Pharmacological management of neuropathic pain. Evidence-based recommendations. Pain 132, 237–251 [DOI] [PubMed] [Google Scholar]
- 5. Jensen T. S., Gottrup H., Sindrup S. H., Bach F. W. (2001) The clinical picture of neuropathic pain. Eur. J. Pharmacol. 429, 1–11 [DOI] [PubMed] [Google Scholar]
- 6. Sandall D. W., Satkunanathan N., Keays D. A., Polidano M. A., Liping X., Pham V., Down J. G., Khalil Z., Livett B. G., Gayler K. R. (2003) A novel α-conotoxin identified by gene sequencing is active in suppressing the vascular response to selective stimulation of sensory nerves in vivo. Biochemistry 42, 6904–6911 [DOI] [PubMed] [Google Scholar]
- 7. Satkunanathan N., Livett B., Gayler K., Sandall D., Down J., Khalil Z. (2005) α-Conotoxin Vc1.1 alleviates neuropathic pain and accelerates functional recovery of injured neurones. Brain. Res. 1059, 149–158 [DOI] [PubMed] [Google Scholar]
- 8. Klimis H., Adams D. J., Callaghan B., Nevin S., Alewood P. F., Vaughan C. W., Mozar C. A., Christie M. J. (2011) A novel mechanism of inhibition of high voltage activated calcium channels by α-conotoxins contributes to relief of nerve injury-induced neuropathic pain. Pain 152, 259–266 [DOI] [PubMed] [Google Scholar]
- 9. Vincler M., Wittenauer S., Parker R., Ellison M., Olivera B. M., McIntosh J. M. (2006) Molecular mechanism for analgesia involving specific antagonism of α9α10 nicotinic acetylcholine receptors. Proc. Natl. Acad. Sci. U.S.A. 103, 17880–17884 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Vincler M., McIntosh J. M. (2007) Targeting the α9α10 nicotinic acetylcholine receptor to treat severe pain. Expert Opin. Ther. Targets 11, 891–897 [DOI] [PubMed] [Google Scholar]
- 11. Ellison M., Haberlandt C., Gomez-Casati M. E., Watkins M., Elgoyhen A. B., McIntosh J. M., Olivera B. M. (2006) α-RgIA. A novel conotoxin that specifically and potently blocks the α9α10 nAChR. Biochemistry 45, 1511–1517 [DOI] [PubMed] [Google Scholar]
- 12. Lang P. M., Burgstahler R., Haberberger R. V., Sippel W., Grafe P. (2005) A Conus peptide blocks nicotinic receptors of unmyelinated axons in human nerves. Neuroreport 16, 479–483 [DOI] [PubMed] [Google Scholar]
- 13. Nevin S. T., Clark R. J., Klimis H., Christie M. J., Craik D. J., Adams D. J. (2007) Are α9α10 nicotinic acetylcholine receptors a pain target for α-conotoxins? Mol. Pharmacol. 72, 1406–1410 [DOI] [PubMed] [Google Scholar]
- 14. Adams D. J., Callaghan B., Berecki G. (2012) Analgesic conotoxins: Block and G protein-coupled receptor modulation of N-type (Cav2.2) calcium channels. Br. J. Pharmacol. 166, 486–500 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Callaghan B., Haythornthwaite A., Berecki G., Clark R. J., Craik D. J., Adams D. J. (2008) Analgesic α-conotoxins Vc1.1 and RgIA inhibit N-type calcium channels in rat sensory neurons via GABAB receptor activation. J. Neurosci. 28, 10943–10951 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Callaghan B., Adams D. J. (2010) Analgesic α-conotoxins Vc1.1 and RgIA inhibit N-type calcium channels in sensory neurons of α9 nicotinic receptor knockout mice. Channels 4, 51–54 [DOI] [PubMed] [Google Scholar]
- 17. Dai G., Haedo R. J., Warren V. A., Ratliff K. S., Bugianesi R. M., Rush A., Williams M. E., Herrington J., Smith M. M., McManus O. B., Swensen A. M. (2008) A high throughput assay for evaluating state dependence and subtype selectivity of Cav2 calcium channel inhibitors. Assay Drug Dev. Technol. 6, 195–212 [DOI] [PubMed] [Google Scholar]
- 18. Jordan M., Schallhorn A., Wurm F. M. (1996) Transfecting mammalian cells. Optimization of critical parameters affecting calcium-phosphate precipitate formation. Nucleic Acids Res. 24, 596–601 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Clark R. J., Fischer H., Nevin S. T., Adams D. J., Craik D. J. (2006) The synthesis, structural characterization, and receptor specificity of the α-conotoxin Vc1.1. J. Biol. Chem. 281, 23254–23263 [DOI] [PubMed] [Google Scholar]
- 20. Clark R. J., Daly N. L., Halai R., Nevin S. T., Adams D. J., Craik D. J. (2008) The three-dimensional structure of the analgesic α-conotoxin, RgIA. FEBS Lett. 582, 597–602 [DOI] [PubMed] [Google Scholar]
- 21. Luo S., Kulak J. M., Cartier G. E., Jacobsen R. B., Yoshikami D., Olivera B. M., McIntosh J. M. (1998) α-Conotoxin AuIB selectively blocks α3β4 nicotinic acetylcholine receptors and nicotine-evoked norepinephrine release. J. Neurosci. 18, 8571–8579 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Schnölzer M., Alewood P., Jones A., Alewood D., Kent S. B. (1992) In situ neutralization in Boc-chemistry solid phase peptide synthesis. Rapid, high yield assembly of difficult sequences. Int. J. Pept. Protein Res. 40, 180–193 [DOI] [PubMed] [Google Scholar]
- 23. Lewis R. J., Nielsen K. J., Craik D. J., Loughnan M. L., Adams D. A., Sharpe I. A., Luchian T., Adams D. J., Bond T., Thomas L., Jones A., Matheson J. L., Drinkwater R., Andrews P. R., Alewood P. F. (2000) Novel ω-conotoxins from Conus catus discriminate among neuronal calcium channel subtypes. J. Biol. Chem. 275, 35335–35344 [DOI] [PubMed] [Google Scholar]
- 24. Bettler B., Kaupmann K., Mosbacher J., Gassmann M. (2004) Molecular structure and physiological functions of GABAB receptors. Physiol. Rev. 84, 835–867 [DOI] [PubMed] [Google Scholar]
- 25. Schroeder J. E., McCleskey E. W. (1993) Inhibition of Ca2+ currents by a μ-opioid in a defined subset of rat sensory neurons. J. Neurosci. 13, 867–873 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Nomura K., Reuveny E., Narahashi T. (1994) Opioid inhibition and desensitization of calcium channel currents in rat dorsal root ganglion neurons. J. Pharmacol. Exp. Ther. 270, 466–474 [PubMed] [Google Scholar]
- 27. Murali S. S., Napier I. A., Rycroft B. K., Christie M. J. (2012) Opioid-related (ORL1) receptors are enriched in a subpopulation of sensory neurons, and prolonged activation produces no functional loss of surface N-type calcium channels. J. Physiol. 590, 1655–1667 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Altier C., Khosravani H., Evans R. M., Hameed S., Peloquin J. B., Vartian B. A., Chen L., Beedle A. M., Ferguson S. S., Mezghrani A., Dubel S. J., Bourinet E., McRory J. E., Zamponi G. W. (2006) ORL1 receptor-mediated internalization of N-type calcium channels. Nat. Neurosci. 9, 31–40 [DOI] [PubMed] [Google Scholar]
- 29. Hand K. S., Harris N. C., Spruce A. E. (2000) An antisense investigation of the role of the γ-aminobutyric acidB1 receptor subunit in Ca2+ channel modulation in rat sensory neurons. Neurosci. Lett. 290, 49–52 [DOI] [PubMed] [Google Scholar]
- 30. Naseer M. I., Lee H. Y., Ullah N., Ullah I., Park M. S., Kim S. H., Kim M. O. (2010) Ethanol and PTZ effects on siRNA-mediated GABAB1 receptor. Down-regulation of intracellular signaling pathway in prenatal rat cortical and hippocampal neurons. Synapse 64, 181–190 [DOI] [PubMed] [Google Scholar]
- 31. Lee H. Y., Li S. P., Park M. S., Bahk Y. H., Chung B. C., Kim M. O. (2007) Ethanol's effect on intracellular signal pathways in prenatal rat cortical neurons is GABAB1-dependent. Synapse 61, 622–628 [DOI] [PubMed] [Google Scholar]
- 32. Röhl T., Kurreck J. (2006) RNA interference in pain research. J. Neurochem. 99, 371–380 [DOI] [PubMed] [Google Scholar]
- 33. Dray A. (2008) Neuropathic pain. Emerging treatments. Br. J. Anaesth. 101, 48–58 [DOI] [PubMed] [Google Scholar]
- 34. Tedford H. W., Zamponi G. W. (2006) Direct G protein modulation of Cav2 calcium channels. Pharmacol. Rev. 58, 837–862 [DOI] [PubMed] [Google Scholar]
- 35. Raingo J., Castiglioni A. J., Lipscombe D. (2007) Alternative splicing controls G protein-dependent inhibition of N-type calcium channels in nociceptors. Nat. Neurosci. 10, 285–292 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Ikeda S. R. (1996) Voltage-dependent modulation of N-type calcium channels by G-protein βγ subunits. Nature 380, 255–258 [DOI] [PubMed] [Google Scholar]
- 37. Herlitze S., Garcia D. E., Mackie K., Hille B., Scheuer T., Catterall W. A. (1996) Modulation of Ca2+ channels by G-protein βγ subunits. Nature 380, 258–262 [DOI] [PubMed] [Google Scholar]
- 38. Dolphin A. C., Scott R. H. (1987) Calcium channel currents and their inhibition by (−)-baclofen in rat sensory neurons. Modulation by guanine nucleotides. J. Physiol. 386, 1–17 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Tatebayashi H., Ogata N. (1992) Kinetic analysis of the GABAB-mediated inhibition of the high threshold Ca2+ current in cultured rat sensory neurons. J. Physiol. 447, 391–407 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Campbell V., Berrow N., Dolphin A. C. (1993) GABAB receptor modulation of Ca2+ currents in rat sensory neurones by the G protein Go. Antisense oligonucleotide studies. J. Physiol. 470, 1–11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Adams D. J., Cuny H., De Faoite A., Huynh T. G. (2011) Analgesic α-conotoxins. Inhibition of N-type calcium channels via GABAB receptor activation. Program No. 233.04, 2011 Neuroscience Meeting Planner Society for Neuroscience, Washington, D. C [Google Scholar]