

Abstract
This study represents the first epidemiological study based on the national registry of primary immunodeficiencies (PID) in Korea. Patient data were collected from 23 major hospitals. A total of 152 patients with PID (under 19 yr of age), who were observed from 2001 to 2005, have been entered in this registry. The period prevalence of PID in Korea in 2005 is 11.25 per million children. The following frequencies were found: antibody deficiencies, 53.3% (n = 81), phagocytic disorders, 28.9% (n = 44); combined immunodeficiencies, 13.2% (n = 20); and T cell deficiencies, 4.6% (n = 7). Congenital agammaglobulinemia (n = 21) and selective IgA deficiency (n = 21) were the most frequently reported antibody deficiency. Other reported deficiencies were common variable immunodeficiencies (n = 16), X-linked agammaglobulinemia (n = 15), IgG subclass deficiency (n = 4). Phagocytic disorder was mostly chronic granulomatous disease. A small number of patients with Wiskott-Aldrich syndrome, hyper-IgE syndrome, and severe combined immunodeficiency were also registered. Overall, the most common first manifestation was pneumonia. This study provides data that permit a more accurate estimation PID patients in Korea.
Keywords: Primary Immunodeficiencies, Registry, Korea
INTRODUCTION
Primary immunodeficiencies (PID) are diverse entities which result from one or more abnormalities of the immune system. PID predisposes individuals to recurrent or unusual infections and has associations with other immune disorders including autoimmune diseases and lymphomas (1). Pediatricians and general practitioners are often poorly informed about the clinical presentation, diagnostic approach, and importance of primary immunodeficiencies. Thus, some patients may remain untreated for several years, which leads to many complications. With the novel progress in immunological and molecular techniques, including the development of diagnostic genetic tests (2), more than 150 of PIDs have been identified and characterized (3).
Reports on PID registries are currently available from different countries throughout Asia including Japan, Taiwan, Singapore, and Hong Kong (4-7). However, no national registry of PID has been established in Korea. This study provides, for the first time, data on the kinds and frequency of PID in Korea.
MATERIALS AND METHODS
Data collection
Patients were defined as individuals under 19 yr of age who were diagnosed with PID during the period of January 2001 to December 2005. Ten-page questionnaire was sent to all participating centers. All of the questionnaires were completed by the pediatricians involved in the care of the reported patients. The data were collected from 23 major university hospitals, distributed in 7 provinces of Korea.
Patient enrollment
The diagnosis of PID was based on standard criteria, which has been introduced by the Expert Committee of International Union of Immunological Societies (IUIS) on Primary Immunodeficiency (3). Patients with DiGeorge anomaly and a normal number and function of T cells were not included in the registry. Auto- and allo-immune neutropenias were also not included, also.
Registry questionnaire
A ten-page questionnaire was developed to determine: the patient's demographic information, including name, date of birth, and address, the diagnosis of PID, first clinical manifestation, age at the time of the onset of symptoms, age at the time of PID diagnosis, clinical manifestations (infectious, noninfectious), complications, family history of immunodeficiency, laboratory tests, follow-up information including the infections in the course of the illness, and kinds of treatment.
Laboratory analysis for the immunodeficient patients included complete blood count, blood smear, bone marrow biopsy, chemistry, immunoglobulin levels, isohemagglutinins, IgG subclasses titer, delayed cutaneous hypersensitivity reactions (Mantoux test, CMI skin test), antibody response to vaccine antigens (HBsAb), lymphocyte subpopulation (T cell and B cell), proliferation of T cells to mitogens, nitroblue tetrazolium (NBT) dye test, and dihydrorhodamine (DHR) assay. In recently diagnosed PID patients, DNA mutation analysis was usually performed when indicated.
Statistical analysis
Statistical analysis was performed at the Seoul National University College of Medicine, Medical Research Collaborating Center. The period prevalences were calculated using the population data registered between January and December 2005. The regional prevalence is based on the patients' home address. The data were analyzed with MS excel 2007 and SPSS Version 14.0.
Ethics statement
This study was approved by institutional board of the Catholic University of Korea, Daejeon St. Mary's Hospital (DC11RIMI-0077). Informed consent was waived by the board.
RESULTS
Frequency of different types of PID
A total of 152 patients, diagnosed with PID, was registered during the period from January 2001 to December 2005. Of the 152 patients, 119 were boys, giving a 3.6:1 sex ratio of boys to girls.
Of the 152 registered patients, 16 died before 2005; therefore prevalence of PID at the end of 2005 was 11.25/million children under 19 yr old in Korea (Table 1). By age the highest prevalence (15.78/million) was observed between 5 and 9 yr of group (Table 2).
Table 1.
Frequency by the diagnosis, sex distribution and prevalence of primary immunodeficiencies in Korea

*Patients at the end of 2005. SCID, Severe combined immunodeficiency.
Table 2.
Prevalence of primary immunodeficiencies (at the end of 2005)
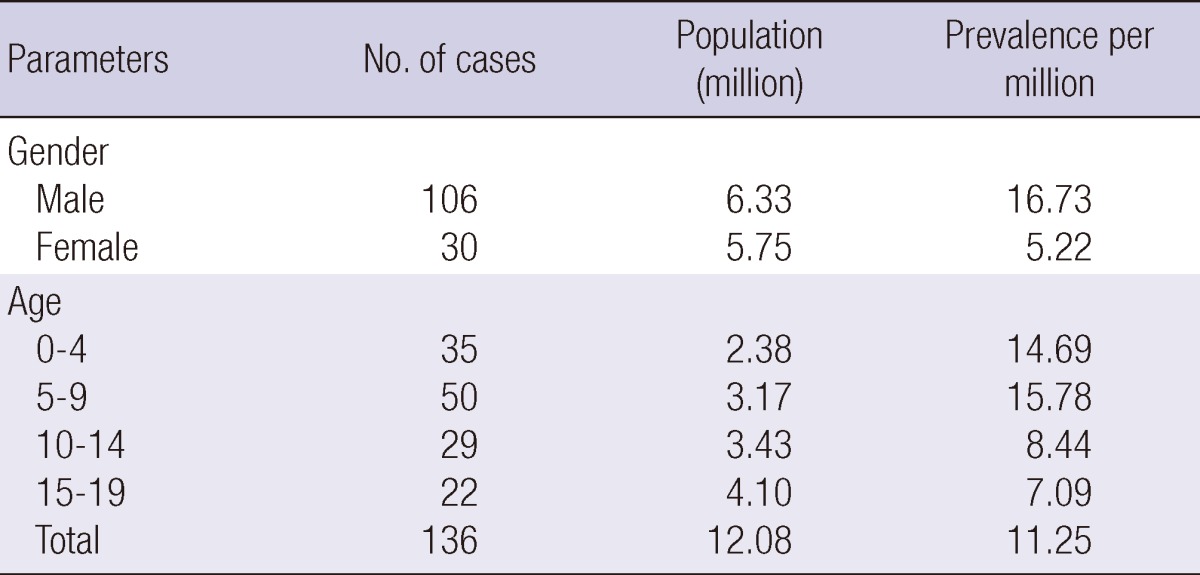
Of primary immunodeficiency, antibody deficiencies were the most common, constituting 53.3% of the patients (n = 81), followed by phagocytic disorders, (n = 44, 28.9%), combined immunodeficiencies, (n = 20, 13.2%), and T cell deficiencies (n = 7, 4.6%). Patients with complement deficiency were not found. Sex distribution, and prevalence rate are presented in Table 1 by diagnosis of primary immunodeficiency.
Congenital agammaglobulinemia (n = 21; 19 males and 2 females) and selective IgA deficiency (n = 21;11 males and 10 females) were the most frequently reported antibody deficiency. Other antibody deficiencies were common variable immunodeficiencies (CVID) (n = 16; 13 males and 3 females), X-linked agammaglobulinemia (XLA) (n = 15), IgG subclass deficiency (n = 4; 3 males and 1 female), transient hypogammaglobulinemia of infancy (n = 3; 2 males and 1 female), and non X-linked hyper-IgM syndrome (n = 1; 1 female). All T cell deficiencies were DiGeorge syndrome.
Phagocytic disorders were the second most frequent immunodeficiency disorder. Among the many neutrophil dysfunction syndromes, 44 cases (35 males and 9 females) of chronic granulomatous disease (CGD) was reported in this registry.
Wiskott-Aldrich syndrome was the most frequently reported type of combined immunodeficiencies (n = 6; 6 males). Other reported combined immunodeficiencies were hyper-IgE syndrome (n = 5; 3 males and 2 females), X-linked hyper-IgM syndrome (n = 4; 4 males), and severe combined immunodeficiencies (SCID) (n = 4; 4 males).
Geographical distribution
The patient's address was known in 152 cases and the geographic distribution and prevalence in each region were defined according to this information (Fig. 1). The results of the PID geographic distribution were heterogeneous. Significantly, Jeju province (region No. 7 in Fig. 1), which is the biggest island in Korea, showed the highest prevalence rate (100.47/million).
Fig. 1.

Map of Korea showing the provinces 1-7 and the prevalence of primary immunodeficiencies according to the regions.
Age distribution
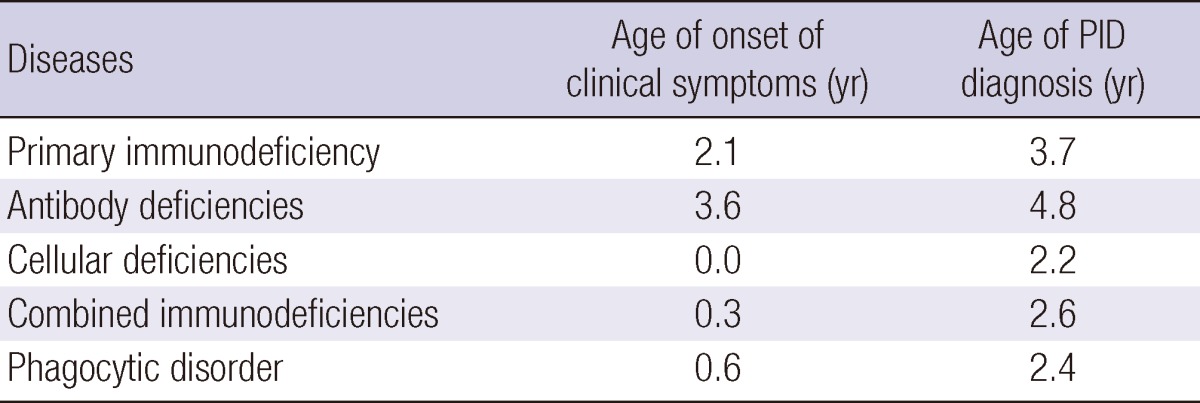
The highest prevalence (15.78/million) was observed between 5 and 9 yr of age (Table 2). The time elapsed between the onset of clinical symptoms and PID diagnosis was 19 months for PID; 14 months for antibody deficiencies, and 21 months for phagocytic disorders. Patients' clinical symptoms developed at 2.1 yr of age, and time of PID diagnosis was at 3.7 yr (Table 3).
Table 3.
Age of onset and diagnosis of primary immunodeficiencies
Family history
In this study, 23% of the patients had one or more family members with proven or suspected immunodeficiency. Most often a pair of siblings or cousins was affected.
Presenting manifestations
Overall, the most common first manifestation was pneumonia in PID patients. In antibody deficiencies, pneumonia was the most frequent initial manifestation (53.1%), and otitis media was the second most common presentation (11.1%). In T cell deficiencies, tetany was the most frequent symptom (71.4%). Congenital anomalies of the heart and great vessels were reported in 3 patients. The patients' remarkable susceptibility to infection was not always recognized in the cases of DiGeorge syndrome. In phagocytic disorders, lymphadenitis, perianal abscess, and pneumonia were the most common manifestations, being seen in 27.3%, 22.7%, and 15.9% of the cases, respectively (Table 4).
Table 4.
First manifestation of primary immunodeficiencies

FUO, Fever of unknown origin.
Bronchiectasis and respiratory failure were major complications in PID patients (n = 19, 11.5%). Short stature was observed in 16 patients (9.8%). Leukemia or lymphoma was noted in 7 patients (4.3%). Autoimmune diseases were uncommon; 5 cases (3.0%) were reported.
Wiskott-Aldrich syndrome was the most frequently reported type of combined immunodeficiencies (n = 6; 6 males). Four cases were diagnosed during the first year of life (66.6%). Triad symptoms for diagnosis of Wiskott-Aldrich syndrome (susceptibility to infection, thrombocytopenia, and eczema) were seen in all reported patients. Infections were generally severe and included pneumonia, skin infection, and lymphadenitis. In hyper-IgE syndrome, four cases (80%) were reported to have pneumatocele.
Treatment
Regular intravenous immunoglobulin (IVIG) was infused in patients with hypogammaglobulinemia and recurrent infections. Sixty-three patients (41.4%) with antibody or combined deficiencies received IVIG as the primary treatment modality. Prophylactic treatment was prescribed to patients with T-cell deficiency and phagocytic disorders to prevent opportunistic infections. Interferon-gamma (IFN-γ) and an anti-fungal agent were given to patients with chronic granulomatous disease.
Hematopoietic stem cell transplantation was performed in 3 patients, of whom 2 were Wiskott-Aldrich syndrome, the other was X-linked SCID.
Mortality
Between January 2001 and December 2005, 15 (9.8%) of the 152 patients died; 8 died of pneumonia, 4 died of sepsis, 2 died of bleeding, and 1 died of lymphoma. Pneumonia was associated with the death in 53% of the fatal cases. The cause of death in two bleeding cases was pontine hemorrhage and gastrointestinal tract bleeding in Wikott-Aldrich syndrome.
DISCUSSION
This is the first epidemiological study of PID in Korea. A total of 152 patients representing 15 different diagnoses are registered. The period prevalence of PID is 11.25 per million children under 19 yr old.
The authors have divided the registered patients into four groups, including antibody deficiencies, T cell deficiencies, phagocytic disorders, and combined immunodeficiencies. Antibody deficiencies were the most frequent (53.3%, n = 81), followed by phagocytic disorders (28.9%, n = 44), combined immunodeficiencies (13.2%, n = 20), and T cell deficiencies (4.6%, n = 7).
The spectrum of PID in Korea is similar to that of other Asian countries (Table 5) (4-7). In this study, antibody deficiency was seen in nearly one half of the patients, which is consistent with other studies (4-14). Compared with other countries, Korea had a higher frequency of phagocytic disorders, placing it as the second most common immunodeficiencies, instead of combined deficiencies found in other studies (8-14). All phagocytic disorders were chronic granulomatous disease (CGD). The higher frequency of CGD in the registry could be due to the genetic makeup of the Korean population.
Table 5.
Comparative frequency of primary immunodeficiencies in other registries

*Not reported.
The reported frequency in 7 provinces was compared to this projected figure. Region No. 7 (Jeju) had more than double the prevalence of PID as compared to the other six provinces (Fig. 1). This may be partly due to diverging occurrence of these hereditary diseases.
Of the 152 registered patients, 119 were boys, giving a 3.6:1 sex ratio of boys to girls. The difference between the sexes was largely due to an almost-complete male predominance in severe combined immunodeficiency, X-linked agammaglobulinemia, and Wiskott-Aldrich syndrome.
The most common symptom in these cases was recurrent bacterial infections, which included pneumonia, lymphadenitis, and otitis media. Therefore, patients who present with recurrent or opportunistic infections should be screened for possible immunodeficiency.
One of the mainstays of PID management is the prescription of antimicrobials for prophylaxis or therapy. Intravenous immunoglobulin replacement therapy is also administered to patients with antibody immunodeficiency or combined immunodeficiency. Bone marrow transplantation has enabled long-term survival for some patients, usually those with SCID or Wiskott-Aldrich syndrome.
Although these conditions are uncommon, failure to diagnose and treat early may lead to substantial morbidity and mortality (15, 16). Moreover, as most of these conditions are inherited, making a precise diagnosis is important for genetic counseling of the immediate and extended family (17).
This study provides data that permits a more accurate estimation of the resources needed for PID patients in Korea. The data may also form the basis for national plans for various treatment modalities in PID, such as immunoglobulin replacement and bone marrow transplantation. With continuous PID registration in Korea, it will be possible to obtain population-based knowledge of the natural course of PID patients as part of an increasing international research effort.
The constructed registry reveals that antibody deficiencies and phagocytic disorders are the most common. The role of the PID national registry is important. It will raise awareness in pediatricians and general practitioners and provide them with information about PID.
ACKNOWLEDGMENTS
We thank all contributors to the national registry who provided information on patients with primary immunodeficiencies including care pediatricians throughout Korea.
Footnotes
This study was supported by research fund (2006-E63004-00) from the National Institute of Health, Korea.
References
- 1.Primary immunodeficiency diseases: report of a WHO scientific group. Clin Exp Immunol. 1995;99:1–24. [PMC free article] [PubMed] [Google Scholar]
- 2.Gathmann B, Grimbacher B, Beauté J, Dudoit Y, Mahlaoui N, Fischer A, Knerr V, Kindle G ESID Registry Working Party. The European internet-based patient and research database for primary immunodeficiencies: results 2006-2008. Clin Exp Immunol. 2009;157:3–11. doi: 10.1111/j.1365-2249.2009.03954.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.International Union of Immunological Societies Expert Committee on Primary Immunodeficiencies. Notarangelo LD, Fischer A, Geha RS, Casanova JL, Chapel H, Conley ME, Cunningham-Rundles C, Etzioni A, Hammartröm L, Nonoyama S, et al. Primary immunodeficiencies: 2009 update. J Allergy Clin Immunol. 2009;124:1161–1178. doi: 10.1016/j.jaci.2009.10.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Hayakawa H, Iwata T, Yata J, Kobayashi N. Primary immunodeficiency syndrome in Japan. I. Overview of a nationwide survey on primary immunodeficiency syndrome. J Clin Immunol. 1981;1:31–39. doi: 10.1007/BF00915474. [DOI] [PubMed] [Google Scholar]
- 5.Lee WI, Kuo ML, Huang JL, Lin SJ, Wu CJ. Distribution and clinical aspects of primary immunodeficiencies in a Taiwan pediatric tertiary hospital during a 20-year period. J Clin Immunol. 2005;25:162–173. doi: 10.1007/s10875-005-2822-2. [DOI] [PubMed] [Google Scholar]
- 6.Lim DL, Thong BY, Ho SY, Shek LP, Lou J, Leong KP, Chng HH, Lee BW. Primary immunodeficiency diseases in Singapore: the last 11 years. Singapore Med J. 2003;44:579–586. [PubMed] [Google Scholar]
- 7.Lam DS, Lee TL, Chan KW, Ho HK, Lau YL. Primary immunodeficiency in Hong Kong and the use of genetic analysis for diagnosis. Hong Kong Med J. 2005;11:90–96. [PubMed] [Google Scholar]
- 8.Kirkpatrick P, Riminton S. Primary immunodeficiency diseases in Australia and New Zealand. J Clin Immunol. 2007;27:517–524. doi: 10.1007/s10875-007-9105-z. [DOI] [PubMed] [Google Scholar]
- 9.Stray-Pedersen A, Abrahamsen TG, Frøland SS. Primary immunodeficiency diseases in Norway. J Clin Immunol. 2000;20:477–485. doi: 10.1023/a:1026416017763. [DOI] [PubMed] [Google Scholar]
- 10.Matamoros Florí N, Mila Llambi J, Español Boren T, Raga Borja S, Fontan Casariego G. Primary immunodeficiency syndrome in Spain: first report of the National Registry in Children and Adults. J Clin Immunol. 1997;17:333–339. doi: 10.1023/a:1027382916924. [DOI] [PubMed] [Google Scholar]
- 11.Gooi HC. Primary immunodeficiency register, United Kingdom: update 1992. Immunodeficiency. 1993;4:191–192. [PubMed] [Google Scholar]
- 12.Affentranger P, Morell A, Spath P, Seger R. Registry of primary immunodeficiencies in Switzerland. Immunodeficiency. 1993;4:193–195. [PubMed] [Google Scholar]
- 13.Boyle JM, Buckley RH. Population prevalence of diagnosed primary immunodeficiency diseases in the United States. J Clin Immunol. 2007;27:497–502. doi: 10.1007/s10875-007-9103-1. [DOI] [PubMed] [Google Scholar]
- 14.Abuzakouk M, Feighery C. Primary immunodeficiency disorders in the Republic of Ireland: first report of the national registry in children and adults. J Clin Immunol. 2005;25:73–77. doi: 10.1007/s10875-005-0360-9. [DOI] [PubMed] [Google Scholar]
- 15.Geha RS, Notarangelo LD, Casanova JL, Chapel H, Conley ME, Fischer A, Hammarström L, Nonoyama S, Ochs HD, Puck JM, et al. Primary immunodeficiency diseases: an update from the International union of Immunological Societies Primary Immunodeficiency Diseases Classification Committee. J Allergy Clin Immunol. 2007;120:776–794. doi: 10.1016/j.jaci.2007.08.053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Rosen FS, Cooper MD, Wedgwood RJ. The primary immunodeficiencies. N Engl J Med. 1995;333:431–440. doi: 10.1056/NEJM199508173330707. [DOI] [PubMed] [Google Scholar]
- 17.Conley ME. Genetics of primary immunodeficiency disease. Rev Immunogenet. 2000;2:231–242. [PubMed] [Google Scholar]