

Abstract
Aims
Next-generation sequencing has opened the possibility of large-scale sequence-based disease association studies. A major challenge in interpreting whole-exome data is predicting which of the discovered variants are deleterious or neutral. To address this question in silico, we have developed a score called Combined Annotation scoRing toOL (CAROL), which combines information from 2 bioinformatics tools: PolyPhen-2 and SIFT, in order to improve the prediction of the effect of non-synonymous coding variants.
Methods
We used a weighted Z method that combines the probabilistic scores of PolyPhen-2 and SIFT. We defined 2 dataset pairs to train and test CAROL using information from the db-SNP: ‘HGMD-PUBLIC’ and 1000 Genomes Project databases. The training pair comprises a total of 980 positive control (disease-causing) and 4,845 negative control (non-disease-causing) variants. The test pair consists of 1,959 positive and 9,691 negative controls.
Results
CAROL has higher predictive power and accuracy for the effect of non-synonymous variants than each individual annotation tool (PolyPhen-2 and SIFT) and benefits from higher coverage.
Conclusion
The combination of annotation tools can help improve automated prediction of whole-genome/exome non-synonymous variant functional consequences.
Keywords: CAROL, PolyPhen-2, SIFT, Weighted Z method
Introduction
Advances in high-throughput technologies for next-generation sequencing have empowered the interrogation of thousands of individual genomes. Valuable emerging resources such as the 1000 Genomes [1] and UK10K Projects (http://www.uk10k.org/) provide reference panels for imputation and a deep catalogue of human sequence variation. Next-generation association studies are poised to survey most of the common and rare genetic variation of the human genome. Whole-exome and whole-genome sequence-based studies are being designed and increasingly carried out, both for common and rare diseases.
A major challenge in interpreting whole-exome/genome data is predicting which of the discovered variants are likely to be functional or disease-causing and which are neutral. To address this question in silico, several functional annotation tools focusing on the analysis of non-synonymous coding variants (ns variants) have been implemented. These tools are mainly based on sequence homology [2–4] ; empirically derived rules [5, 6] ; structural and functional features [7–12] ; artificial neural networks[13–16]; decision trees [17, 18]; random forests [19]; support vector machines [20–23] ; and Bayesian classifiers [24].
Two widely used functional annotation tools are the Polymorphism Phenotyping-2 (PolyPhen-2) and Sorting Intolerant From Tolerant (SIFT). Their common feature is the fact they both account for evolutionary protein patterns. PolyPhen-2 differs from SIFT in that it predicts the functional effects of mutations based on the posterior probability that a given mutation be deleterious, whilst SIFT directly computes the probability of an amino acid substitution occurring at each position of multiple aligned homologous sequences [2, 3, 24]. PolyPhen-2 also accounts for sequence and structural based-features, i.e. the specific site in which the substitution occurs and 3D structure of the protein [5, 6].
We developed a new algorithm called Combined Annotation scoRing toOL (CAROL), which combines information from PolyPhen-2 and SIFT. We use a weighted Z method to derive the combined score. We calibrate and validate CAROL using positive (known disease-causing) and negative (postulated non-disease-causing) control variants. CAROL has high predictive power for the effect of ns variants and has the distinct advantage of high coverage, i.e. low missing data rates.
Methods
Functional Annotation Tools
The profile scores of two typically used annotation tools were combined in order to obtain a score that better predicts the functional effect of amino acid substitutions. PolyPhen-2 performs multiple alignments with homologous sequences using BLAST and calculates position-specific independent count (PSIC) scores for each position (i) of the sequence in the profile matrix. The PSIC score is given by the log-likelihood of the given amino acid occurring at a particular position [25]. PolyPhen-2 then computes the absolute value of the difference between profile scores of both wild-type and variant amino acid residues for a specific position (dScore = Score1 − Score2), which corresponds approximately to the log-likelihood of substituting the wild-type amino acid for the mutant amino acid. Moreover, it also annotates the substitution site, e.g. active or binding, maps the substitution site to a known protein 3D structure, which indicates whether the substitution will affect any important feature of the protein, and checks for any contact with functional sites, e.g. contact with ligands, interaction between subunits of the protein alignment and contact with ‘critical’ residues [5, 24] by using a machine-learning approach. The prediction of the functional effect is calculated by naïve Bayes posterior probability and the qualitative prediction is based on cut-offs of the score.
SIFT firstly searches for homologous sequences and chooses closely related sequences, which might share similar function. It then obtains multiple alignments for those sequences and calculates the normalized probabilities and conservation scores for each position in the multiple aligned sequences [2, 3, 26]. The normalized probability is determined by the ratio between the probability of observing the variant amino acid at position i in n homologous sequences and the maximum probability of the most frequent amino acid in position i, that is the wild-type amino acid most of the time. For SIFT scores > 0.05 the amino acid substitution is predicted to be tolerant, meaning that the variant amino acid has probabilities close to the wild-type amino acid and for SIFT scores < 0.05 the mutation is predicted to be deleterious (in this case the probability of the variant amino acid is much lower compared with the wild-type amino acid) [26].
As PolyPhen-2 and SIFT scores are not at comparable scales, we calculated the probability of the complement of the SIFT scores. The scaled scores (Pk) range between 0 and 1, in which scores closer to 1 indicate that the amino acid substitution is deleterious, and scores closer to 0 that it is neutral.
CAROL Algorithm
The CAROL algorithm is based on a weighted Z method, which combines the probabilistic score for each annotation tool (Pk):
where Zk is the standard normal deviates for each k-th annotation tool, and wk is the weight for the k -th tool, which is defined by the following log-likelihood:
According to wk, more weight will be given to bigger Pk, which reflects a higher probability that a mutation is deleterious. wk ranges between 0 and 6.9 as by default scores of 1 in SIFT and PolyPhen-2 are converted to 0.999. The derived probability of the CAROL score ranges between 0 and 1. As we are classifying a different dataset of that utilized by PolyPhen-2 and SIFT, we optimised PolyPhen-2, SIFT and CAROL’s power to predict deleterious and neutral effects at a threshold of 0.57, 0.96 and 0.98, respectively. The optimal threshold decision relied on the principle of obtaining the largest sensitivity whilst committing the smallest number of false positives, and it was determined by plotting a receiver operating characteristic (ROC) curve.
We investigated the incorporation of further functional annotation and conservation scores such as Protein ANalysis THrough Evolutionary Relationships (PANTHER) [4] and Genomic Evolutionary Rate Profiling (GERP) [27], but found that PolyPhen-2 and SIFT produced the most robust combination (online suppl. fig. S1; for all online supplementary material, see www.karger.com/doi/10.1159/000334984). PANTHER uses information from Hidden Markov Model (HMM) to classify both protein’s family and subfamily in order to predict protein function. In analogy to the other mentioned tools, PANTHER identifies and annotates conserved motifs in protein sequences. GERP test relies on the concept of rejected substitution to identify constrained elements. In other words, GERP identifies sequences that harbour fewer mutations than would be expected for neutral sequences reflecting the intensity of purifying past selection [27]. In addition to CAROL we explored three further combinations: CAROL+GERP, CAROL+PANTHER and CAROL+PANTHER+GERP (online suppl. fig. S1). The same weighted Z method was applied incorporating Pk given by PANTHER (corresponding to the probability of the complement of P deleterious). The weighting model was redefined when GERP (wG) was incorporated to:
We then compared the predictive power of CAROL with CONDEL (CONsensus DELeteriousness score of missense mutations) annotation tool. CONDEL is based on a weighted average of the normalized scores [28] that combines information from different annotation tools.
Calibration and Validation
To calibrate and validate CAROL, we defined two sets of positive and negative control variants selected from the Human Gene Mutation Database (HGMD-PUBLIC version) (http://www.hgmd.cf.ac.uk/ac/index.php), dbSNP Short Genetic Variations (http://www.ncbi.nlm.nih.gov/projects/SNP/) and 1000 Genomes Project [1]. HGMD is the most curated database of germline mutations in nuclear genes underpinning or associated with human inherited disorders [29]. dbSNP is a public catalogue of human genetic variation including both disease-causing clinical mutations, provided by locus-specific mutation databases (LSDBs), as well as neutral polymorphisms [30]. The 1000 Genomes Project aims to survey the majority of human genetic variation with allele frequency > 1% [1].
Positive control variants were selected if they were annotated as ‘Clinical/LSDB variations’ present in the dbSNP and ‘HGMD-PUBLIC’ databases and if they were not found in the 1000 Genomes Project data. Negative control variants were selected if they had a 1000 Genomes Project frequency higher than 10% in all populations, if they were not annotated as ‘Clinical/LSDB variations’ in dbSNP, and if they were not included in the ‘HGMD-PUBLIC’ database. All positive and negative controls were ns variants, extracted using Ensembl API 62 (assembly GRCh37.p2) [31], having consequence ‘NON_SYNONYMOUS_CODING’ and excluding secondary consequences of ‘SPLICE_SITE’. The 1000 Genomes variants were obtained from dbSNP 132. The two sets were subsequently separated into training and testing sets in order to mitigate the risk of overtraining.
Running Parameters
PolyPhen-2, SIFT and CONDEL predictions were obtained in Ensembl API release 62 (more information is available at: http://www.ensembl.org/info/docs/variation/index.html). The version of CONDEL available in Ensembl only finds a weighted average between PolyPhen-2 and SIFT. PANTHER version 1.02 was installed and run in-house. We followed the standard parameters suggested by its authors (http://www.pantherdb.org/). We used the latest HMM library version 7.0. GERP scores for mammal species were also extracted using Ensembl API (for more information, see http://www.ensembl.org/Help/Faq?kw=compara). The CAROL algorithm was written in R language and can be accessed at: http://www.sanger.ac.uk/resources/software/carol/.
Results
HGMD-PUBLIC had 6,685 ns variant entries, the 1000 Genomes Project had 66,942 ns variant entries and db-SNP had 513,794 ns variant entries, of which 6,316 ns variants were classified as clinically related. In total, we compiled 2,939 positive and 14,536 negative control variants. We used a training set of 980 positive and 4,845 negative control variants to evaluate different versions of the combined annotation tool and to define optimal thresholds for each annotation tool. We subsequently tested CAROL on an independent set of 1,959 positive and 9,691 negative controls. Online suppl. figure S2 shows the probability distribution of Pk, for both positive and negative testing controls, for PolyPhen-2, SIFT and CAROL.
Performance statistics for the 3 different annotation tools are illustrated in table 1. CAROL correctly predicted 83% of the disease-causing substitutions as deleterious, corresponding to the most accurate prediction out of the 3 tools (80% for PolyPhen-2 and 81.9% for SIFT). CAROL also correctly classified 72.7% of the neutral variants as true negatives, which was similar to SIFT (72.7%) and higher than PolyPhen-2 (70.5%). CAROL was found to have the lowest type II error rate (17%) compared with PolyPhen-2 (20%) and SIFT (18.1%). CAROL’s type I error rate was 27.3%, similar to SIFT (27.3%) and lower that PolyPhen-2 (29.5%) (table 1). The lowest rate of missing data was observed for CAROL, it increased coverage compared to SIFT by 0.3% for the positive control set and 4.5% for the negative control set; and compared to PolyPhen-2, it increased coverage by 11.5% for the positive control set and 5.3% for the negative control set. Online suppl. table S1 illustrates the performance statistics for the training and the total datasets, in which similar results were obtained.
Table 1.
Sensitivity, specificity, type II error rate, type I error rate, missing values (NAs), total accuracy and area under ROC curve estimation for PolyPhen-2, SIFT and CAROL using an optimal threshold of 0.57, 0.96 and 0.98, respectively
| PolyPhen-2 | SIFT | CAROL | |
|---|---|---|---|
| Sensitivity | 0.800 | 0.819 | 0.830 |
| Specificity | 0.705 | 0.727 | 0.727 |
| Type II error | 0.200 | 0.181 | 0.170 |
| Type I error | 0.295 | 0.273 | 0.273 |
| NAs | 0.116–0.125 | 0.004–0.177 | 0.001–0.072 |
| Total accuracy | 0.721 | 0.744 | 0.745 |
| ROC area | 0.836 | 0.821 | 0.852 |
Numbers in bold denote where CAROL performs better than the other 2 annotation tools.
Sensitivity equals the number of true positives divided by the number of true positives plus false negatives; Specificity equals the number of true negatives divided by the number of true negatives plus false positives; Type II error equals 1 – sensitivity; Type II error equals 1 – specificity; Total accuracy equals the number of true positives plus negatives divided by the number of total positives plus negatives; ROC area equals the value of the Wilcoxon-Mann-Whitney test statistic [as in ref. 32].
In general, CAROL performed better than SIFT and PolyPhen-2, as shown by the total accuracy in table 1 and ROC curve (fig. 1). The ROC curve plots the proportion of variants correctly classified as deleterious (true positive rate or sensitivity) against the proportion of variants wrongly classified as so (false positive rate or type I error) for the 3 different prediction tools. In a ROC curve, an ideal prediction would give a vertical line of error noise 0, and a totally random prediction would give a line with a slope of 1. The estimated area under the curve was 83.6%, 82.1%, and 85.2%, for PolyPhen-2, SIFT and CAROL, respectively (table 1). CAROL achieved the highest accuracy of 74.5% compared with PolyPhen-2 (72.1%) and SIFT (74.4%), which is calculated by the number of true positives plus negatives divided by the number of total positives plus negatives. This indicates that CAROL gives relatively more accurate predictions compared with PolyPhen-2 and SIFT.
Fig. 1.
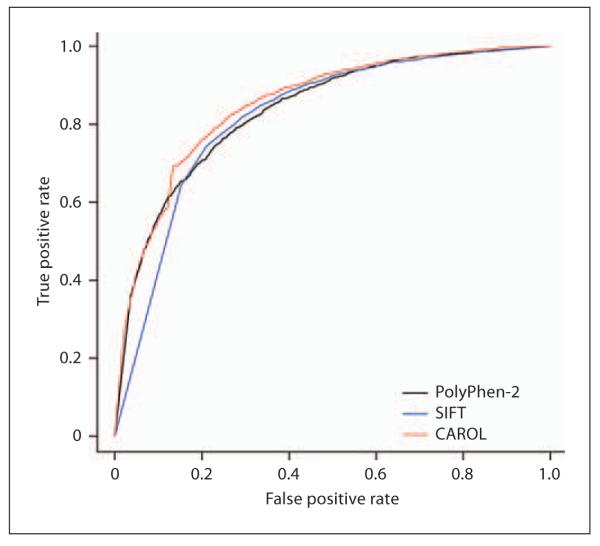
Receiver operating characteristic (ROC) curves of the 3 prediction tools. CAROL score makes a larger number of correct predictions (true positives, y-axis) for a given number of errors (false positives, x-axis) compared with SIFT and PolyPhen-2.
Importantly, CAROL has considerably increased coverage, i.e. lower rates of missing data, compared with the other tools. This is due to the fact that CAROL integrates the scores of the 2 component methods, and can still provide a score when one of the component methods does not. A common reason that might explain why PolyPhen-2 and SIFT did not make a prediction is because they were unable to find sufficient related protein sequences at the position of interest in their respective multiple sequence alignment pipelines. The addition of more predictive tools could in fact increase the coverage of CAROL, although it would not be considerable as its missing rate is already very low. CAROL’s performance was also slightly superior compared to that of CONDEL, as represented by its ROC curve, which has a predictive power of 0.849 (online suppl. fig. S3).
Discussion
Next-generation sequencing has opened the possibility of large-scale sequence-based disease association studies. A major challenge in interpreting whole-exome/ genome data is predicting the functional consequence of the discovered variants. Currently there are over 40 functional annotation tools, each one focusing particularly on different protein features. Combining all these tools is beyond the scope of this work. A recent approach based on the combination of different tools to improve the prediction of the effect on ns coding variants was proposed: CONDEL [28]. Here we introduce CAROL as a new method, which is an automated way of combining profile scores across the 2 most commonly used functional annotation tools: PolyPhen-2 and SIFT, by over-weighting sequence positions where the variant amino acid is most likely to be deleterious.
CAROL was found to have a higher predictive power and accuracy for the effect of ns variants than each of the 2 individual annotation tools. CAROL also has a lower type I error rate compared with SIFT and PolyPhen-2. A possible reason for the observed rate of type I error in the 3 annotation tools is that the negative set may contain a sizeable amount of mildly deleterious alleles. CAROL has the distinct advantage of higher coverage, i.e. less missing data, making it a well-suited approach for the automated prediction of whole-genome/exome ns variants and directly applicable to large-scale data generated by resequencing projects.
The assignment of functional scores has so far mainly focused on coding variants, although the majority of human genome sequence variation resides outside protein-coding regions. Non-coding variants demonstrably harbour important functional elements, and there are numerous examples of robust association between non-coding variants and complex diseases. Whilst challenging, developing functional annotation tools to comprehensively predict the effect of non-coding variants is an important research direction.
Supplementary Material
Acknowledgements
E.Z. and J.A. are supported by the Wellcome Trust (WT088885/ Z/09/Z). M.L. is supported by Pfizer. The authors thank Aaron Day-Williams, Maricel Kann, Ivan Adzhubey, Pauline Ng and Emidio Capriotti for useful comments.
References
- 1.Durbin RM, Abecasis GR, Altshuler DL, Auton A, Brooks LD, Gibbs RA, Hurles ME, McVean GA. A map of human genome variation from population-scale sequencing. Nature. 2010;467:1061–1073. doi: 10.1038/nature09534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Ng PC, Henikoff S. Accounting for human polymorphisms predicted to affect protein function. Genome Res. 2002;12:436–446. doi: 10.1101/gr.212802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Ng PC, Henikoff S. SIFT: predicting amino acid changes that affect protein function. Nucleic Acids Res. 2003;31:3812–3814. doi: 10.1093/nar/gkg509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Thomas PD, Campbell MJ, Kejariwal A, Mi H, Karlak B, Daverman R, Diemer K, Muruganujan A, Narechania A. PANTHER: a library of protein families and subfamilies indexed by function. Genome Res. 2003;13:2129–2141. doi: 10.1101/gr.772403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Ramensky V, Bork P, Sunyaev S. Human non-synonymous SNPs: server and survey. Nucleic Acids Res. 2002;30:3894–3900. doi: 10.1093/nar/gkf493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Sunyaev S, Ramensky V, Koch I, Lathe W, 3rd, Kondrashov AS, Bork P. Prediction of deleterious human alleles. Hum Mol Genet. 2001;10:591–597. doi: 10.1093/hmg/10.6.591. [DOI] [PubMed] [Google Scholar]
- 7.Chasman D, Adams RM. Predicting the functional consequences of non-synonymous single nucleotide polymorphisms: structure-based assessment of amino acid variation. J Mol Biol. 2001;307:683–706. doi: 10.1006/jmbi.2001.4510. [DOI] [PubMed] [Google Scholar]
- 8.Cheng TM, Lu YE, Vendruscolo M, Lio P, Blundell TL. Prediction by graph theoretic measures of structural effects in proteins arising from non-synonymous single nucleotide polymorphisms. PLoS Comput Biol. 2008;4:e1000135. doi: 10.1371/journal.pcbi.1000135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Wang Z, Moult J. SNPs, protein structure, and disease. Hum Mutat. 2001;17:263–270. doi: 10.1002/humu.22. [DOI] [PubMed] [Google Scholar]
- 10.Worth CL, Bickerton GR, Schreyer A, Forman JR, Cheng TM, Lee S, Gong S, Burke DF, Blundell TL. A structural bioinformatics approach to the analysis of nonsynonymous single nucleotide polymorphisms (nsSNPs) and their relation to disease. J Bioinform Comput Biol. 2007;5:1297–1318. doi: 10.1142/s0219720007003120. [DOI] [PubMed] [Google Scholar]
- 11.Yue P, Li Z, Moult J. Loss of protein structure stability as a major causative factor in mono-genic disease. J Mol Biol. 2005;353:459–473. doi: 10.1016/j.jmb.2005.08.020. [DOI] [PubMed] [Google Scholar]
- 12.Yue P, Moult J. Identification and analysis of deleterious human SNPs. J Mol Biol. 2006;356:1263–1274. doi: 10.1016/j.jmb.2005.12.025. [DOI] [PubMed] [Google Scholar]
- 13.Bromberg Y, Rost B. SNAP: predict effect of non-synonymous polymorphisms on function. Nucleic Acids Res. 2007;35:3823–3835. doi: 10.1093/nar/gkm238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Ferrer-Costa C, Orozco M, de la Cruz X. Characterization of disease-associated single amino acid polymorphisms in terms of sequence and structure properties. J Mol Biol. 2002;315:771–786. doi: 10.1006/jmbi.2001.5255. [DOI] [PubMed] [Google Scholar]
- 15.Ferrer-Costa C, Orozco M, de la Cruz X. Sequence-based prediction of pathological mutations. Proteins. 2004;57:811–819. doi: 10.1002/prot.20252. [DOI] [PubMed] [Google Scholar]
- 16.Ferrer-Costa C, Orozco M, de la Cruz X. Use of bioinformatics tools for the annotation of disease-associated mutations in animal models. Proteins. 2005;61:878–887. doi: 10.1002/prot.20664. [DOI] [PubMed] [Google Scholar]
- 17.Dobson RJ, Munroe PB, Caulfield MJ, Saqi MA. Predicting deleterious nsSNPs: an analysis of sequence and structural attributes. BMC Bioinformatics. 2006;7:217. doi: 10.1186/1471-2105-7-217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Krishnan VG, Westhead DR. A comparative study of machine-learning methods to predict the effects of single nucleotide polymorphisms on protein function. Bioinformatics. 2003;19:2199–2209. doi: 10.1093/bioinformatics/btg297. [DOI] [PubMed] [Google Scholar]
- 19.Bao L, Zhou M, Cui Y. nsSNPAnalyzer: identifying disease-associated nonsynonymous single nucleotide polymorphisms. Nucleic Acids Res. 2005;33:W480–W482. doi: 10.1093/nar/gki372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Capriotti E, Calabrese R, Casadio R. Predicting the insurgence of human genetic diseases associated to single point protein mutations with support vector machines and evolutionary information. Bioinformatics. 2006;22:2729–2734. doi: 10.1093/bioinformatics/btl423. [DOI] [PubMed] [Google Scholar]
- 21.Kulkarni V, Errami M, Barber R, Garner HR. Exhaustive prediction of disease susceptibility to coding base changes in the human genome. BMC Bioinformatics. 2008;9(suppl 9):S3. doi: 10.1186/1471-2105-9-S9-S3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Tian J, Wu N, Guo X, Guo J, Zhang J, Fan Y. Predicting the phenotypic effects of non-synonymous single nucleotide polymorphisms based on support vector machines. BMC Bioinformatics. 2007;8:450. doi: 10.1186/1471-2105-8-450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Calabrese R, Capriotti E, Fariselli P, Martelli PL, Casadio R. Functional annotations improve the predictive score of human disease-related mutations in proteins. Hum Mutat. 2009;30:1237–1244. doi: 10.1002/humu.21047. [DOI] [PubMed] [Google Scholar]
- 24.Adzhubei IA, Schmidt S, Peshkin L, Ramensky VE, Gerasimova A, Bork P, Kondrashov AS, Sunyaev SR. A method and server for predicting damaging missense mutations. Nat Methods. 2010;7:248–249. doi: 10.1038/nmeth0410-248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Sunyaev SR, Eisenhaber F, Rodchenkov IV, Eisenhaber B, Tumanyan VG, Kuznetsov EN. PSIC: profile extraction from sequence alignments with position-specific counts of independent observations. Protein Eng. 1999;12:387–394. doi: 10.1093/protein/12.5.387. [DOI] [PubMed] [Google Scholar]
- 26.Ng PC, Henikoff S. Predicting deleterious amino acid substitutions. Genome Res. 2001;11:863–874. doi: 10.1101/gr.176601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Cooper GM, Stone EA, Asimenos G, Green ED, Batzoglou S, Sidow A. Distribution and intensity of constraint in mammalian genomic sequence. Genome Res. 2005;15:901–913. doi: 10.1101/gr.3577405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Gonzalez-Perez A, Lopez-Bigas N. Improving the assessment of the outcome of non-synonymous SNVs with a consensus deleteriousness score, Condel. Am J Hum Genet. 2011;88:440–449. doi: 10.1016/j.ajhg.2011.03.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Stenson PD, Ball EV, Mort M, Phillips AD, Shiel JA, Thomas NS, Abeysinghe S, Krawczak M, Cooper DN. Human gene mutation database (HGMD): 2003 update. Hum Mutat. 2003;21:577–581. doi: 10.1002/humu.10212. [DOI] [PubMed] [Google Scholar]
- 30.Sherry ST, Ward MH, Kholodov M, Baker J, Phan L, Smigielski EM, Sirotkin K. dbSNP: the NCBI database of genetic variation. Nucleic Acids Res. 2001;29:308–311. doi: 10.1093/nar/29.1.308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Rios D, McLaren WM, Chen Y, Birney E, Stabenau A, Flicek P, Cunningham F. A data-base and API for variation, dense genotyping and resequencing data. BMC Bioinformatics. 2010;11:238. doi: 10.1186/1471-2105-11-238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Sing T, Sander O, Beerenwinkel N, Lengauer T. ROCR: Visualizing classifier performance in R. Bioinformatics. 2005;21:3940–3941. doi: 10.1093/bioinformatics/bti623. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.