

Abstract
Nitric oxide exerts ubiquitous signaling via post-translational modification of cysteine residues, a reaction termed S-nitrosylation. Important substrates of S-nitrosylation that influence cardiac function include receptors, enzymes, ion channels, transcription factors, and structural proteins. Cardiac ion channels subserving excitation-contraction coupling are potentially regulated by S-nitrosylation. Specificity is achieved in part by spatial co-localization of ion channels with nitric oxide synthases (NOS), enzymatic sources of NO in biologic systems, and by coupling of NOS activity to localized calcium/second messenger concentrations. Ion channels regulate cardiac excitability and contractility in millisecond timescales raising the possibility that NO-related species modulate heart function on a beat-to beat basis. This review focuses on recent advances in understanding of NO regulation of the cardiac action potential, and of the calcium release channel ryanodine receptor, which is crucial for the generation of force. S-nitroso (SNO) signaling is disrupted in pathological states in which the redox state of the cell is dysregulated, including ischemia, heart failure, and atrial fibrillation.
Keywords: Heart, nitrosylation, nitric oxide, ryanodine receptor, oxidation
Introduction
S-nitrosylation has emerged as an important and ubiquitous post-translational modification system, participating in cellular signaling throughout the phylogeny. S-nitrosylation is involved in cell signaling in bacteria1, plants2 and animals. In mammalian cells, S-nitrosylation affects crucial and diverse processes including neurotransmission and memory, gene expression, cellular excitability, mitochondrial energetics, control of blood flow, and respiration3, 4.
S-nitrosylation consists of the addition of a NO group to the thiol moiety of a cysteine residue. This covalent reaction can occur upon formal oxidation of NO or by trans-nitrosylation in which the NO group is exchanged between donor and acceptor thiol. The growing list of proteins that are targets for S-nitrosylated is vast 5, 6. Cysteine residues susceptible to S-nitrosylation are often located in an acid-base motif in which acidic and basic aminoacids surround the Cys-site of modification in primary and/or quaternary structure; an acid (D, E) in the +1 position appears to particularly important3, 7. In addition, hydrophobic motifs8 containing signature aromatic residues within a 6-7 angstrom sphere of influence have been shown to influence NO targeting 3, 9.
S-nitrosylation and denitrosyaltion reactions
Molecular mechanisms of S-nitrosylation and denitrosylation in biological systems are considered below.
Acidification of nitrite
At low pH, nitrite undergoes protonation to HNO2 (nitrous acid), which disproportionates to N2O3 (dinitrogen trioxide), a nitrosating agent. The reaction may be of relevance under ischemic conditions.
Trans-nitros(yl)ation
The transnitrosation reaction comprises the transfer of a NO+ group between two thiol groups (or between transition metal and thiol) The transnitrosation reaction involves nucleophilic attack of a thiolate on the nitrogen of SNO group. Acid-base catalysis is favored by the presence of basic amino acids surrounding the cysteine, which may lower thiol pKa (R-S-).
Metal-catalyzed
Accumulating evidence indicates that transition metals catalyze S-nitrosylation reactions in vivo 10, 11. These studies point to the possibility that enzymatic formation of SNO is an important and relevant signaling mechanism. Examples of metal catalyzed S-nitrosylation include the NO and nitrite dependent auto-S-nitrosylation of hemoglobin 12, 13,14 and the S-nitrosylation of glutathione by ceruloplasmin.
Denitrosylation
A number of enzymes have been shown to break down S-nitrosothiols; several meet stringent genetic criteria of being physiologically relevant. S-nitrosoglutathione reductase (GSNOR) and the thioredoxins (Trx1 and Trx2) have emerged as key denitrosylases. GSNOR metabolizes GSNO to GSNHOH, albeit a mixture of products will form depending on conditions, including GSSG, N-hydroxysulfenimide hydroxylamine, and NH315. The activity of GSNOR is critically dependent on glutathione and NADH. Mice deficient in GSNOR are protected from myocardial infarction16 but more susceptible to septic shock17. On the other hand, thioredoxin/thioredoxin reductase directly denitrosylates proteins (and potentially also low molecular weight S-nitrosothiols)18-21. Reduced thioredoxin operates with thioredoxin reductase and NADPH. Deficiency of thioredoxin/TrxR promotes Fas induced apoptosis. Xanthine oxidoreductase22, an enzyme involved in purine catabolism, is also able to degrade S-nitroglutathione and S-nitrosocysteine23, 24, but the physiological relevance of this activity has not been demonstrated.
Detection of S-nitrosylated proteins
The detection and quantification of S-nitrosylated proteins is technically challenging, in part due to the labile nature of the S-NO bond. In this section, we discuss the methods that have been most commonly used to evaluate the S-nitrosylation of ion channels.
Biotin switch protocol
The most widely used method for detecting and quantifying S-nitrosylated proteins is the “biotin switch” technique. This protocol developed by Jaffrey and colleagues25, 26, involves the formal reduction of S-nitrosylated proteins with ascorbate followed by tagging of the nascent thiols with a biotinylated compound N-[6-(biotinamido)hexyl]-3’-(2’-pyridyldithio)propionamide (biotin –HPDP), a thiol-specific biotinylating agent. If performed appropriately, the reaction is highly specific for S-nitrosylated vs. alternatively oxidized proteins27.
Photolysis-Chemiluminiscence
This assay involves homolysis of the SNO bond with UV light, followed by detection of NO by chemiluminescense28. Assays are performed in the presence and absence of HgCl2, which eliminates SNOs but not other NO complexes. ,This assay provided the first measurements of endogenous ion channel S-nitrosylation (RyR1 and RyR2)29-31, and remains a gold standard for SNO quantification, with limit of detection being in the range of pmol (the limit of detection for NO in the nitric oxide analyzer32). However, the assay is not available in many laboratories and is not amenable to preoteomic analysis.
Anti-S-nitrosocysteine antibodies
Antibodies have been raised against the SNO moiety in proteins (monoclonal and polyclonal), and are commercially available. These antibodies have demonstrated their usefulness in immunohistochemical studies of SNO-proteins33. In addition, anti-SNO antibodies have used been in Western blots of S-nitrosylated RyR1 34-37 and RyR238.
Impact of S-nitrosylation on the excitation-contraction coupling process
In the cardiac myocyte, contraction and relaxation is governed by excitation-contraction coupling39. During membrane depolarization triggered by voltage-gated Na+ channels, L-type Ca2+ channels (which are also voltage-sensitive) become activated. This inward Ca2+ current triggers massive release of stored Ca2+ from the sarcoplasmic reticulum (SR), through the Ca2+ release channel ryanodine receptor 2, a process termed calcium-induced calcium release. The SR Ca2+ release leads to Ca2+ binding to troponin C in the myofilaments and activation of ATPase activity of myosin, generating contraction. Relaxation of the muscle starts as Ca2+ dissociates from troponin C and is transported back into the SR by the pump SERCA2a and is extruded from the cell via a Na+/Ca2+ exchanger. NO appears to play a significant role in Ca2+ handling, principally by S-nitrosylation, as discussed below.
Voltage-gated sodium channels
In atrial and ventricular myocytes, the voltage-gated sodium (Nav) channels are responsible for fast depolarization. The main Nav expressed in the mammalian myocardium is encoded by the gene SCN5A. It consists of a single pore-forming α subunit of approximately 260 kDa. Nav channels typically inactivate very quickly at the more positive potentials. However, in cardiac myocytes, a late current is observed. Persistence of this current has been shown to be dependent on S-nitrosylation of Nav1.5. Nitrosylation is coupled to NOS1 activity40. Conversely, S-nitrosylation of sodium channels in sensory neurons blocks the current41-43.
The sodium channel is rich in cysteine: 33-40 Cys are present in various isoforms from brain, skeletal muscle, and heart. A full S-nitrosylation motif of the acid base type is found in the Nav of sensory neurons, but only partial in the heart 42. It is possible that beside S-nitrosylation, cardiac and non cardiac Nav channels also could be regulated by cysteine oxidation44, 45, perhaps under oxidative stress conditions.
Interestingly, a mutation in syntrophin associated with a form of the long QT syndrome (LQT3)46, results in aberrant S-nitrosylation of the sodium channel. Syntrophin, a member of the dystrophin-associated proteins normally serves as scaffold protein for NOS1 and the plasmalemmal calcium pump PMCA, an interaction that results in inhibition of NO production. Syntrophin mutation results in a disruption of the PMCA-NOS1 complex and favors interaction of NOS1 with the Na channel (subunit SCN5A). Release of PMCA increases NOS1 activity promoting S-nitrosylation of SCN5a and thereby increasing late Na+ currents. The impact of this process on the action potential duration was not investigated by the authors, but mutations of the sodium channel that lead to similar increased late currents prolong the duration of the action potential47.
L-type calcium channel
The L-type calcium channels consist of a major α1 subunit, which is pore-forming, and β, α2/δ and γ auxiliary subunits. In the heart the gene Cav1.2 encodes for the α1C, which subserves sensitivity to dihydropyridines48.
It has been reported that the L-type calcium channel can be activated by the NO donors SIN-1, nitrosocysteine (CysNO), and GSNO, in a manner that is independent of cGMP49. On the other hand, nitrosothiols apparently inhibit L-type channels that are expressed in heterologous systems50, 51. The mechanism of inhibition involves both reduction of open probability of single channels and a reduction of conductance. Although S-nitrosylation was not confirmed in these studies, the effects of S-nitrosothiols were independent of cGMP.
Recently, Sun and colleagues reported that the α1-subunit of the channel is constitutively S-nitrosylated in the mouse heart 52. They found that nitrosylation increases in ischemia and is associated with inhibition of the channel. Preconditioning of hearts with GSNO likewise increased the degree of nitrosylation53. In addition, GSNO reduced the damage after reperfusion and was associated with decreased Ca2+ influx through L-type calcium channels. Collectively, these data support the view that S-nitrosylation serves as a mechanism of cardioprotection.
In patients with atrial fibrillation (AF), it has been reported that the α1C subunit of the channel is hyper-nitrosylated, and an inverse relationship between nitrosylation and total glutathione cell content was observed 54. Hypernitrosylation of the channel may therefore contribute to the disease. The number and specific cysteine residues of L-type calcium channel that are S-nitrosylated remain unknown.
The L-type channel is also regulated by the cGMP-PKG pathway, probably through phosphorylation. This leads to decreased Ca2+ influx. L-type channel inhibition that is seen following stimulation of adrenergic receptors is mediated by NOS355, but when NOS1 is over expressed in the mouse heart, it may be responsible for the cGMP-mediated inhibition56. Reduced influx through the L-type would lead to a decreased load in the sarcoplasmic reticulum, reducing the amplitude of the Ca2+ transients. NO derived from both NOS3 and NOS1 may therefore serve as a protective mechanism to avoid Ca2+ overload after ischemia-reperfusion.
Voltage-gated potassium channels
Cardiac voltage-gated potassium channels determine the resting membrane potential and the duration of the cardiac action potential. Several channels participate, including the rapidly activating and inactivating transient outward current (Ito), the ultrarapid (IKur), rapid (IKr) and slow (IKs) components of the delayed rectifier and the inward rectifier (IK1)48.
The delayed rectifier K+ current is one of the major components that determine the timing of repolarization of cardiac myocytes. The delayed rectifier K+ current consists of two components: the rapidly activating (IKr) and the slowly activating component (IKs).
It has been reported that S-nitrosylation increases IKs in a manner dependent on NOS3 in guinea pig cardiomyocytes. This activation resulted in a shortening of the action potential duration57, 58. Cysteine 445 has been recently identified as the site S-nitrosylated in the pore-forming subunit KCNQ1 59. Interestingly, this cysteine is located within a consensus motif for nitrosylation (Lys,Arg,His)-Cys-(Asp,Glu), and mutation of the acid in the +1 position prevented S-nitrosylation. S-nitrosylation was also dependent on calmodulin, a situation analogous to that described for the RyR3. Also, in guinea pig cardiomyocytes, the overexpression of CAPON (also known as NOS1AP), a binding protein for NOS1, resulted in increased IKs60. This effect, along with a reduction in ICa,L, shortened action potential duration by 30%. NOS1 presumably translocated to the sarcolemma, although it was not confirmed if these effects were due to S-nitrosylation of the channels or cGMP-dependent events. The NOS1 effect on IKs was modest, probably due to co-existing regulation by NOS3.
In the atria, KV1.5 generates the ultrarapid component of the delayed rectifier (IKur), determining the duration and membrane potential of the plateau phase. S-nitrosylation inhibits this current61. A mechanism has been proposed based on three-dimensional modeling: nitrosylated Cys331 and Cys346 (which are located in the voltage-sensor region of the channel) are suggested to be stabilized by Ile262 and Arg342, effecting a conformational change.
Further, in the atria, KV4.3 which generates the transient outward K+ current (Ito) is also S-nitrosylated, although this has not been shown to change the gating properties62.
HERG channels (IKr) are reportedly inhibited by NO in a cGMP independent manner when expressed in X. laevis oocytes, although S-nitrosylation was not directly evaluated63.
Calcium release channel ryanodine receptor
Overview
The ryanodine receptor (RyR1 in skeletal muscle, RyR2 in the heart and RyR3 in brain) is a massive channel located in the membrane of the sarcoplasmic reticulum. In cardiac myocytes, RyR2 mediates SR Ca2+ release in response to the trans-sarcolemmal Ca2+ influx through L-type calcium channels, following membrane depolarization (calcium-induced calcium release). The RyR functions as a tetramer of 560 kDa subunits, and has a conductivity of around 150 pS for Ca2+. The channel is endogenously inhibited by Mg2+ and activated by Ca2+. The cardiac isoform RyR2 displays a sigmoid response to activating Ca2+ (Figure 3), whereas RyR1 displays a bell-shaped response, being inhibited by higher concentrations of cytosolic Ca2+. Additionally, the Ca2+ -dependent activity of the channel is modulated by Mg2+ and ATP64. Mg2+ inhibits the channel activity by competing with Ca2+ for high affinity binding sites. Redox modifications of RyR alter the effects of both Ca2+ and Mg2+.
Figure 3. Effect of oxidative and nitrosative modifications on the calcium dependence of the cardiac ryanodine receptor (RyR2).
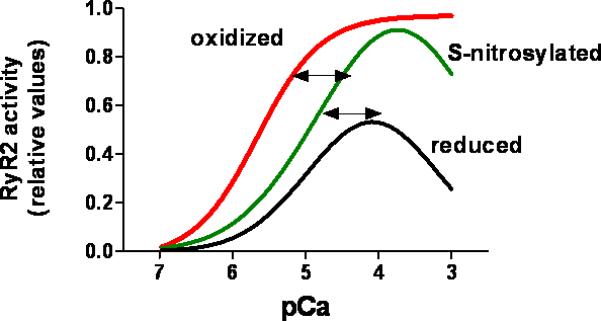
RyR is constitutively and reversibly S-nitrosylated, which increases RyR2 activity under normal conditions. Hyper- and hypo-nitrosylation may contribute to disease. Cysteine oxidation, likely a pathological modification, progressively increases RyR2 responsiveness to activating cytosolic Ca2+. Oxidative activation may be irreversible. The values for RyR2 activity are relative and were obtained from the literature as open probability and 3[H]ryanodine binding.
The activity of the channel can be modified by NO donors and molecules that affect the redox state of cysteine residues. Canine heart RyR and rabbit skeletal muscle RyR possess 85-100 Cys residues, about half as many of which are reactive thiols, that is, they can be S-nitrosylated, S-glutathionylated or oxidized to disulfide bonds.
In the case of RyR1, Cys3635 (which resides within a hydrophobic motif for S-nitrosylation), mediates regulation by NO65. NO nitrosylates Cys3635 and thereby displaces calcium-calmodulin34, activating the channel. A further dependence of S-nitrosylation on calmodulin has been observed previously29 and rationalized by a hydrophobic requirement for the NO effect. The corresponding cysteine in the RyR2 (rabbit heart) Cys3602 is also part of a putative hydrophobic site for S-nitrosylation, but is apparently not nitrosylated by NO. Instead, RyR2 is nitrosylated and activated by GSNO30.
For RyR, S-nitrosylation is principally activating, although very high supraphysiological concentrations of NO or other NO donors inhibit RyR1 and RyR2 in vitro66. More generally, the effects of NO may depend on the nature and concentration of the NO donor67, the redox state of the channel and the PO2. Studies performed in lipid bilayers have shown increase in open probability (Po) as well as a decrease. Generally, GSNO activates the channel independently of PO2 and redox state of the channel. Suko and colleagues showed that NOC-7 (1-hydroxy-2-oxo-3-(N-methyl-3-aminopropyl)-3 methyl-1-triazine) 0.1-0.3 mM and nitrosocysteine increased the PO while nitrosocysteine (4 mM) decrease it68. Hart et. al. found that S-nitroso-N-acetylpenicillamine (SNAP) at 10 μM increased the PO of the channel, while SNAP at higher concentration (1 mM) produced inactivation of the channel69. Nitroxyl anion, a reduced congener of NO (NO-), has been shown to activate the channel by cysteine oxidation70-72.
RyR may exist in different conformations depending on its redox state73 (Figure 3). The redox state of the channel is critical to RyR1 activity and its subsequent modulation by NO. Oxidants increase the activity and reducing agents inhibit it (GSH). High pO2 increases the open probability and a physiologic pO2 decreases it. At low (physiological) PO2, NO activates RyR1 through S-nitrosylation, whereas this effect is abrogated at high PO2. Given the intracellular concentrations of glutathione (~mM), it is possible that S-nitrosoglutathione acts as an intermediate or the direct species that nitrosylates the channel. In heart failure, the RyR is oxidized excessively74, 75 .
Both the cardiac and skeletal RyR are endogenously S-nitrosylated in vivo29, 31. In both cardiac and skeletal muscle, RyR nitrosylation is coupled to NOS1 activity76. In cardiac muscle, RyR2 co-localizes with NOS1 (Figure 1). NOS1 deficiency but not NOS3 deficiency impairs RyR2 nitrosylation indicating that NOS1 is the primary source of NO 76. However, S-nitrosylation of RyR2 in response to physiological stretch may be mediated by NOS377. Hypernitrosylation of RyR1 has been causally linked to pathologies of skeletal muscle, like malignant hyperthermia37 , muscular dystrophy36 and muscular fatigue35.
Figure 1. Ion channels regulated by S-nitrosylation in the cardiac myocyte.

So far, characterized: the sodium channel, which generates a rapid depolarization; the L-type calcium channel, which generates the inward Ca2+ current; the ryanodine receptor (RyR2) calcium release channel of the sarcoplasmic reticulum; the transient outward potassium channel ITo, which activates rapidly upon depolarization; IKur, IKr and IKs which generates the delayed rectifier. The figure also includes the source of NO, the NO synthases NOS1 and NOS3 and the oxidases that are present in the cardiac cell. XOR, xanthine oxidoreductase, NOS, nitric oxide synthase.
A number of ROS containing enzymes are found in or at the SR. Xanthine oxidoreducase co-precipitates with NOS178. Additionally, an NADH oxidase79and possibly an NADPH oxidase are found in the SR80 (Figure 1), altering RyR activity. Redox homeostasis, which becomes disturbed in pathological conditions like heart failure results in oxidation of RyR2 thiols, which may in turn impair S-nitrosylation.
Allosteric effectors
FK506-binding protein (FKBP12) is a small (12 kDa) protein that coordinates RyR gating. Removal of FKBP12 with the immunosuppressants FK506 or rapamycin increase the channel Po and promotes the emergence of subconductance states. In skeletal muscle triads, RyR1 S-nitrosylation with GSNO or NOR-3 leads to a four-fold increase in the Kd for FKBP1234. By contrast, oxidation with GSSG has no effect.
In cardiac SR vesicles, the oxidizing agents H2O2 and dioxide reduced the binding between RyR2 and FKBP12.6, the cardiac isoform of FKBP12. Use of a cysteine-null mutant FKBP12.6 did not prevent the redox-sensitive changes in binding. Interestingly, in a canine model of heart failure, RyR2 exists in an oxidized state (assessed by the content of free-cisterns) when FKBP12.6 binding is reduced75. Antioxidant treatment restores free cysteine content and FKP12.6 binding. The phosphorylation and nitrosylation states of the channel, which also affect FKP12.6 binding, were not assessed.
In vitro experiments using sarcoplasmic reticulum vesicles have shown that S-nitrosylation of RyR2 is enhanced at activating pCa values (around pCa 5) 31, 81, 82. In contrast, the reaction is inhibited by high Mg2+, which promotes channel closure (at low free Ca2+)31. This may have important physiological consequences: S-nitrosylation may enhance CICR under in conditions of high activity, increasing Ca2+ release in systole (high free Ca2+), but not in diastole (low free Ca2+ but high Mg2+). This is in contrast to the effect of oxidizing agents, which promote activity at low Ca2+ or high Mg2+ 83 .
Nitrosylation vs. glutathionylation
S-nitrosylation and glutathionylation have been shown to occur in RyR1 and RyR2 34, 80, 84. However, it is nitrosylation not glutathionylation that has been causally linked to physiological RyR activity. Skeletal muscle disorders exhibit increased nitrosylation that displaces FKBP12 from the complex. Both RyR1 and RyR2 show increased oxidation in disease but nature of the coincident oxidative modifications is not known.
Clinical Relevance
Since S-nitrosylation signaling is involved in multiple physiological processes, it is expected that aberrant nitrosylation could result in undesired behavior of the proteins involved. Consistent with this idea, arrhythmias and heart failure may be frequently associated with altered cellular redox state, which will tend to disrupt cellular S-nitrosylation and altered S-nitrosylation of specific ion channels or affiliate proteins may be relevant in the examples reviewed below.
Atrial Fibrillation
Atrial fibrillation (AF) is the most common sustained arrhythmia observed in adults. In addition, it is a common complication of cardiac surgery. Among the electrophysiological alterations associated with AF, there is a decrease in ICa, which shortens the action potential and descreases atrial contractility85, 86.
A role for oxidative/nitrosative stress has recently been appreciated to contribute to this condition86. NO production has been shown to be reduced in a porcine model of AF87. NO blocks IKur both by S-nitrosylation and the cGMP pathway, accelerating repolarization. It has been hypothesized that a reduction in NO bioavailability (as is observed in experimental models of AF) would reduce the tonic inhibition of IKur, shortening the plateu phase of the AP. This remains to be tested in both experimental settings of AF and in human samples. Interestingly, and consistent with these ideas, the use of an nitrosylaing agent, sodium nitroprusside, has been tested for the prevention of AF after cardiac surgey88 with encouraging results. Whether this treatment resulted in S-nitrosylation or other redox modification of atrial ion channels is unknown.
Long QT syndrome
Long QT syndrome is a defect in cardiac repolarization identified by prolongation of the QT interval in the EKG. It is characterized by malignant ventricular arrhythmias and syncopal episodes. The syndrome has two main forms, acquired and congenital. Acquired long QT syndrome is often associated with heart failure and with the drugs such as antiarrhythmics, antidepressants, and phenothiazines. Heritable long QT is associated with mutations in genes that encode for ion channels (channelopathies)89.
A mutation in syntrophin associated with a form of the long QT syndrome (LQT3)46, results in aberrant S-nitrosylation of the sodium channel. Syntrophin mutation results in disruption of the PMCA-nNOS1 complex, which keeps NOS quiescent, and favors interaction of NOS1 with the Na channel (subunit SCN5A), promoting S-nitrosylation of SCN5a and thereby increasing late Na currents.
NOS1AP is a gene that encodes for nitric oxide synthase 1 adaptor protein, also known as CAPON (carboxy-terminal PDZ ligand of nNOS)90, a protein that regulates NOS1 subcellular location. A genome-wide study originally identified NOS1AP variants (single nucleotides polymorphisms, SNPs) with variations in the QT interval in a population of European origin91. Latter studies confirmed this observation and showed the association of NOS1AP SNPs with long QT92. Furthermore, some SNPs were associated with increased risk of cardiac sudden death93, and one particular SNP was associated with an increase in QT induced by the calcium channel blocker verapamil 94. The use of calcium channel blockers (verapamil and diltiazem) in carriers of a NOS1AP polymorphism was associated with increased cardiovascular mortality (not only cardiac sudden death) 95. Interestingly, non dihydropyridines blockers such as amilodipine and nifedipine did not increase the incidence of cardiovascular death in carriers of a NOS1AP polymorphism.
The mechanisms by which polymorphism of NOS1AP (CAPON) influence the QT period remain unknown, but it has been recently shown in an animal model that CAPON, through effects on NOS1, can reduce ICa and increase IKs, increasing the duration of the action potential60. In theory, if mutations of NOS1AP interfere with the proper location/activity of NOS1, this in turn will modify S-nitrosylation of the cardiac ion channels.
Heart failure
In canine models of heart failure, oxidation of RyR2 as measured as loss of free cysteines74, 75, 96 , has been observed. The oxidation is associated with diastolic Ca2+ leak, which is partially corrected by in vitro treatment with reductants agents like DTT, or by a variety of systemic treatments, including antioxidants (edaravone97), beta blockers96, and ACE inhibitors98. Heart failure is characterized by increased production of ROS that may derive from different sources. NADPH oxidase99, xanthine oxidase100 are up-regulated; uncoupled NOS3101, 102 and mitochondria4, 103 are additional sources of ROS, which may disrupt nitrosylation either by oxidation of cysteines that are target of nitrosylation or by elimination of NO. Adding to this complexity, nitric oxide synthase expression varies in heart failure. NOS1 has been reported to be up-regulated and translocated from the SR to the sarcolemma104, 105. Collectively, these changes will favor diminished S-nitrosylation of RyR by NO derived from nNOS in heart failure.
Conclusions
Cell signaling mediated by S-nitrosylation is ubiquitous in nature. Many cardiac ion channels are evidently regulated by S-nitrosylation. Specifically, S-nitrosylation modulates the major currents involved in the generation of the action potential and in the development of the calcium transient that is transduced into force generation. S-nitrosylation is regulated by the activity of the nitric oxide synthases and GSNO reductase, compartmentation of signals, and the redox state of the subcellular domain. Oxidative stress profoundly modifies NO effects especially in the case of the ryanodine receptor, which may function as a redox sensor.
The enzymatic systems that govern nitrosylation/denitrosylation reactions in the heart remain to be fully understood. Aberrant S-nitrosylation has been implicated in a variety of cardiac pathologies, including arrhythmias, reperfusion injury, preconditioning and heart failure. It is probable that many current pharmacological treatments including organic nitrates, beta blockers96, ACE inhibitors98, and statins106, mediate their actions, at least in a significant part, by restoring S-nitrosylation homeostasis.
Figure 2. Impact of S-nitrosylation on the cardiac action potential.
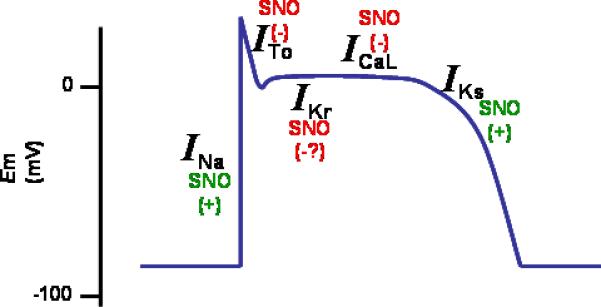
Between parenthesis, the effect of nitrosylation on the ionic currents.
The sodium current is increased by S-nitrosylation; the L-type calcium channel is inhibited, the transient-outward and the rapidly activating (IKr) potassium currents are inhibited; the slow component of the delayed rectifier is increased.
Acknowledgments
This work was supported by NIH grants RO1HL 65455, PO1-HL 75443, RO1-HL059130 and RO1 HL94849.
References
- 1.Hausladen A, Privalle CT, Keng T, et al. Nitrosative stress: activation of the transcription factor OxyR. Cell. 1996;86:719–29. doi: 10.1016/s0092-8674(00)80147-6. [DOI] [PubMed] [Google Scholar]
- 2.Wang Y, Yun BW, Kwon E, et al. S-nitrosylation: an emerging redox-based post-translational modification in plants. J Exp Bot. 2006;57:1777–84. doi: 10.1093/jxb/erj211. [DOI] [PubMed] [Google Scholar]
- 3.Hess DT, Matsumoto A, Kim SO, et al. Protein S-nitrosylation: purview and parameters. Nat Rev Mol Cell Biol. 2005;6:150–66. doi: 10.1038/nrm1569. [DOI] [PubMed] [Google Scholar]
- 4.Hare JM, Stamler JS. NO/redox disequilibrium in the failing heart and cardiovascular system. J Clin Invest. 2005;115:509–17. doi: 10.1172/JCI200524459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Greco TM, Hodara R, Parastatidis I, et al. Identification of S-nitrosylation motifs by site-specific mapping of the S-nitrosocysteine proteome in human vascular smooth muscle cells. Proc Natl Acad Sci U S A. 2006;103:7420–5. doi: 10.1073/pnas.0600729103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Hao G, Derakhshan B, Shi L, et al. SNOSID, a proteomic method for identification of cysteine S-nitrosylation sites in complex protein mixtures. Proc Natl Acad Sci U S A. 2006;103:1012–7. doi: 10.1073/pnas.0508412103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Stamler JS, Toone EJ, Lipton SA, et al. (S)NO signals: translocation, regulation, and a consensus motif. Neuron. 1997;18:691–6. doi: 10.1016/s0896-6273(00)80310-4. [DOI] [PubMed] [Google Scholar]
- 8.Hess DT, Matsumoto A, Nudelman R, et al. S-nitrosylation: spectrum and specificity. Nat Cell Biol. 2001;3:E46–E49. doi: 10.1038/35055152. [DOI] [PubMed] [Google Scholar]
- 9.Derakhshan B, Hao G, Gross SS. Balancing reactivity against selectivity: the evolution of protein S-nitrosylation as an effector of cell signaling by nitric oxide. Cardiovasc Res. 2007;75:210–9. doi: 10.1016/j.cardiores.2007.04.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Foster MW, McMahon TJ, Stamler JS. S-nitrosylation in health and disease. Trends Mol Med. 2003;9:160–8. doi: 10.1016/s1471-4914(03)00028-5. [DOI] [PubMed] [Google Scholar]
- 11.Foster MW, Liu L, Zeng M, et al. A genetic analysis of nitrosative stress. Biochemistry. 2009;48:792–9. doi: 10.1021/bi801813n. [DOI] [PubMed] [Google Scholar]
- 12.Angelo M, Singel DJ, Stamler JS. An S-nitrosothiol (SNO) synthase function of hemoglobin that utilizes nitrite as a substrate. Proc Natl Acad Sci U S A. 2006;103:8366–71. doi: 10.1073/pnas.0600942103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Luchsinger BP, Rich EN, Gow AJ, et al. Routes to S-nitroso-hemoglobin formation with heme redox and preferential reactivity in the beta subunits. Proc Natl Acad Sci U S A. 2003;100:461–6. doi: 10.1073/pnas.0233287100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Basu S, Grubina R, Huang J, et al. Catalytic generation of N2O3 by the concerted nitrite reductase and anhydrase activity of hemoglobin. Nat Chem Biol. 2007;3:785–94. doi: 10.1038/nchembio.2007.46. [DOI] [PubMed] [Google Scholar]
- 15.Jensen DE, Belka GK, Du Bois GC. S-Nitrosoglutathione is a substrate for rat alcohol dehydrogenase class III isoenzyme. Biochem J. 1998;331:659–68. doi: 10.1042/bj3310659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Lima B, Lam GK, Xie L, et al. Endogenous S-nitrosothiols protect against myocardial injury. Proc Natl Acad Sci U S A. 2009;106:6297–302. doi: 10.1073/pnas.0901043106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Liu L, Hausladen A, Zeng M, et al. A metabolic enzyme for S-nitrosothiol conserved from bacteria to humans. Nature. 2001;410:490–4. doi: 10.1038/35068596. [DOI] [PubMed] [Google Scholar]
- 18.Benhar M, Forrester MT, Hess DT, et al. Regulated protein denitrosylation by cytosolic and mitochondrial thioredoxins. Science. 2008;320:1050–4. doi: 10.1126/science.1158265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Nikitovic D, Holmgren A. S-nitrosoglutathione is cleaved by the thioredoxin system with liberation of glutathione and redox regulating nitric oxide. J Biol Chem. 1996;271:19180–5. doi: 10.1074/jbc.271.32.19180. [DOI] [PubMed] [Google Scholar]
- 20.Sengupta R, Ryter SW, Zuckerbraun BS, et al. Thioredoxin catalyzes the denitrosation of low-molecular mass and protein S-nitrosothiols. Biochemistry. 2007;46:8472–83. doi: 10.1021/bi700449x. [DOI] [PubMed] [Google Scholar]
- 21.Stoyanovsky DA, Tyurina YY, Tyurin VA, et al. Thioredoxin and lipoic acid catalyze the denitrosation of low molecular weight and protein S-nitrosothiols. J Am Chem Soc. 2005;127:15815–23. doi: 10.1021/ja0529135. [DOI] [PubMed] [Google Scholar]
- 22.Trujillo M, Alvarez MN, Peluffo G, et al. Xanthine oxidase-mediated decomposition of S-nitrosothiols. J Biol Chem. 1998;273:7828–34. doi: 10.1074/jbc.273.14.7828. [DOI] [PubMed] [Google Scholar]
- 23.Aleryani S, Milo E, Rose Y, et al. Superoxide-mediated decomposition of biological S-nitrosothiols. J Biol Chem. 1998;273:6041–5. doi: 10.1074/jbc.273.11.6041. [DOI] [PubMed] [Google Scholar]
- 24.Jourd'heuil D, Mai CT, Laroux FS, et al. The reaction of S-nitrosoglutathione with superoxide. Biochem Biophys Res Commun. 1998;246:525–30. [PubMed] [Google Scholar]
- 25.Jaffrey SR, Erdjument-Bromage H, Ferris CD, et al. Protein S-nitrosylation: a physiological signal for neuronal nitric oxide. Nat Cell Biol. 2001;3:193–7. doi: 10.1038/35055104. [DOI] [PubMed] [Google Scholar]
- 26.Jaffrey SR, Snyder SH. The biotin switch method for the detection of S-nitrosylated proteins. Sci STKE. 2001;2001:L1. doi: 10.1126/stke.2001.86.pl1. [DOI] [PubMed] [Google Scholar]
- 27.Forrester MT, Foster MW, Stamler JS. Assessment and application of the biotin switch technique for examining protein S-nitrosylation under conditions of pharmacologically induced oxidative stress. J Biol Chem. 2007;282:13977–83. doi: 10.1074/jbc.M609684200. [DOI] [PubMed] [Google Scholar]
- 28.Stamler JS, Jaraki O, Osborne J, et al. Nitric oxide circulates in mammalian plasma primarily as an S-nitroso adduct of serum albumin. Proc Natl Acad Sci U S A. 1992;89:7674–7. doi: 10.1073/pnas.89.16.7674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Eu JP, Sun J, Xu L, et al. The skeletal muscle calcium release channel: coupled O2 sensor and NO signaling functions. Cell. 2000;102:499–509. doi: 10.1016/s0092-8674(00)00054-4. [DOI] [PubMed] [Google Scholar]
- 30.Sun J, Yamaguchi N, Xu L, et al. Regulation of the cardiac muscle ryanodine receptor by O(2) tension and S-nitrosoglutathione. Biochemistry. 2008;47:13985–90. doi: 10.1021/bi8012627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Xu L, Eu JP, Meissner G, et al. Activation of the cardiac calcium release channel (ryanodine receptor) by poly-S-nitrosylation. Science. 1998;279:234–7. doi: 10.1126/science.279.5348.234. [DOI] [PubMed] [Google Scholar]
- 32.Figueroa XF, Gonzalez DR, Martinez AD, et al. ACh-induced endothelial NO synthase translocation, NO release and vasodilatation in the hamster microcirculation in vivo. J Physiol. 2002;544:883–96. doi: 10.1113/jphysiol.2002.021972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Gow AJ, Chen Q, Hess DT, et al. Basal and stimulated protein S-nitrosylation in multiple cell types and tissues. J Biol Chem. 2002;277:9637–40. doi: 10.1074/jbc.C100746200. [DOI] [PubMed] [Google Scholar]
- 34.Aracena P, Tang W, Hamilton SL, et al. Effects of S-glutathionylation and S-nitrosylation on calmodulin binding to triads and FKBP12 binding to type 1 calcium release channels. Antioxid Redox Signal. 2005;7:870–81. doi: 10.1089/ars.2005.7.870. [DOI] [PubMed] [Google Scholar]
- 35.Bellinger AM, Reiken S, Dura M, et al. Remodeling of ryanodine receptor complex causes “leaky” channels: a molecular mechanism for decreased exercise capacity. Proc Natl Acad Sci U S A. 2008;105:2198–202. doi: 10.1073/pnas.0711074105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Bellinger AM, Reiken S, Carlson C, et al. Hypernitrosylated ryanodine receptor calcium release channels are leaky in dystrophic muscle. Nat Med. 2009;15:325–30. doi: 10.1038/nm.1916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Durham WJ, racena-Parks P, Long C, et al. RyR1 S-nitrosylation underlies environmental heat stroke and sudden death in Y522S RyR1 knockin mice. Cell. 2008;133:53–65. doi: 10.1016/j.cell.2008.02.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Bull R, Finkelstein JP, Galvez J, et al. Ischemia enhances activation by Ca2+ and redox modification of ryanodine receptor channels from rat brain cortex. J Neurosci. 2008;28:9463–72. doi: 10.1523/JNEUROSCI.2286-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Bers DM. Cardiac excitation-contraction coupling. Nature. 2002;415:198–205. doi: 10.1038/415198a. [DOI] [PubMed] [Google Scholar]
- 40.Ahern GP, Hsu SF, Klyachko VA, et al. Induction of persistent sodium current by exogenous and endogenous nitric oxide. J Biol Chem. 2000;275:28810–5. doi: 10.1074/jbc.M003090200. [DOI] [PubMed] [Google Scholar]
- 41.Bielefeldt K, Whiteis CA, Chapleau MW, et al. Nitric oxide enhances slow inactivation of voltage-dependent sodium currents in rat nodose neurons. Neurosci Lett. 1999;271:159–62. doi: 10.1016/s0304-3940(99)00553-4. [DOI] [PubMed] [Google Scholar]
- 42.Li Z, Chapleau MW, Bates JN, et al. Nitric oxide as an autocrine regulator of sodium currents in baroreceptor neurons. Neuron. 1998;20:1039–49. doi: 10.1016/s0896-6273(00)80484-5. [DOI] [PubMed] [Google Scholar]
- 43.Renganathan M, Cummins TR, Waxman SG. Nitric oxide blocks fast, slow, and persistent Na+ channels in C-type DRG neurons by S-nitrosylation. J Neurophysiol. 2002;87:761–75. doi: 10.1152/jn.00369.2001. [DOI] [PubMed] [Google Scholar]
- 44.Evans JR, Bielefeldt K. Regulation of sodium currents through oxidation and reduction of thiol residues. Neuroscience. 2000;101:229–36. doi: 10.1016/s0306-4522(00)00367-5. [DOI] [PubMed] [Google Scholar]
- 45.Kurata Y, Hisatome I, Tsuboi M, et al. Effect of sulfhydryl oxidoreduction on permeability of cardiac tetrodotoxin-insensitive sodium channel. Life Sci. 1998;63:1023–35. doi: 10.1016/s0024-3205(98)00364-6. [DOI] [PubMed] [Google Scholar]
- 46.Ueda K, Valdivia C, Medeiros-Domingo A, et al. Syntrophin mutation associated with long QT syndrome through activation of the nNOS-SCN5A macromolecular complex. Proc Natl Acad Sci U S A. 2008;105:9355–60. doi: 10.1073/pnas.0801294105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.George AL., Jr Inherited disorders of voltage-gated sodium channels. J Clin Invest. 2005;115:1990–9. doi: 10.1172/JCI25505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Nerbonne JM, Kass RS. Molecular physiology of cardiac repolarization. Physiol Rev. 2005;85:1205–53. doi: 10.1152/physrev.00002.2005. [DOI] [PubMed] [Google Scholar]
- 49.Campbell DL, Stamler JS, Strauss HC. Redox modulation of L-type calcium channels in ferret ventricular myocytes. Dual mechanism regulation by nitric oxide and S-nitrosothiols. J Gen Physiol. 1996;108:277–93. doi: 10.1085/jgp.108.4.277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Hu H, Chiamvimonvat N, Yamagishi T, et al. Direct inhibition of expressed cardiac L-type Ca2+ channels by S-nitrosothiol nitric oxide donors. Circ Res. 1997;81:742–52. doi: 10.1161/01.res.81.5.742. [DOI] [PubMed] [Google Scholar]
- 51.Poteser M, Romanin C, Schreibmayer W, et al. S-nitrosation controls gating and conductance of the alpha 1 subunit of class C L-type Ca(2+) channels. J Biol Chem. 2001;276:14797–803. doi: 10.1074/jbc.M008244200. [DOI] [PubMed] [Google Scholar]
- 52.Sun J, Picht E, Ginsburg KS, et al. Hypercontractile female hearts exhibit increased S-nitrosylation of the L-type Ca2+ channel alpha1 subunit and reduced ischemia/reperfusion injury. Circ Res. 2006;98:403–11. doi: 10.1161/01.RES.0000202707.79018.0a. [DOI] [PubMed] [Google Scholar]
- 53.Sun J, Morgan M, Shen RF, et al. Preconditioning results in S-nitrosylation of proteins involved in regulation of mitochondrial energetics and calcium transport. Circ Res. 2007;101:1155–63. doi: 10.1161/CIRCRESAHA.107.155879. [DOI] [PubMed] [Google Scholar]
- 54.Carnes CA, Janssen PM, Ruehr ML, et al. Atrial glutathione content, calcium current, and contractility. J Biol Chem. 2007;282:28063–73. doi: 10.1074/jbc.M704893200. [DOI] [PubMed] [Google Scholar]
- 55.Wang H, Kohr MJ, Wheeler DG, et al. Endothelial nitric oxide synthase decreases beta-adrenergic responsiveness via inhibition of the L-type Ca2+ current. Am J Physiol Heart Circ Physiol. 2008;294:H1473–H1480. doi: 10.1152/ajpheart.01249.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Burkard N, Rokita AG, Kaufmann SG, et al. Conditional neuronal nitric oxide synthase overexpression impairs myocardial contractility. Circ Res. 2007;100:e32–e44. doi: 10.1161/01.RES.0000259042.04576.6a. [DOI] [PubMed] [Google Scholar]
- 57.Bai CX, Takahashi K, Masumiya H, et al. Nitric oxide-dependent modulation of the delayed rectifier K+ current and the L-type Ca2+ current by ginsenoside Re, an ingredient of Panax ginseng, in guinea-pig cardiomyocytes. Br J Pharmacol. 2004;142:567–75. doi: 10.1038/sj.bjp.0705814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Bai CX, Namekata I, Kurokawa J, et al. Role of nitric oxide in Ca2+ sensitivity of the slowly activating delayed rectifier K+ current in cardiac myocytes. Circ Res. 2005;96:64–72. doi: 10.1161/01.RES.0000151846.19788.E0. [DOI] [PubMed] [Google Scholar]
- 59.Asada K, Kurokawa J, Furukawa T. Redox- and calmodulin-dependent S-nitrosylation of the KCNQ1 channel. J Biol Chem. 2009 doi: 10.1074/jbc.M807158200. [DOI] [PubMed] [Google Scholar]
- 60.Chang KC, Barth AS, Sasano T, et al. CAPON modulates cardiac repolarization via neuronal nitric oxide synthase signaling in the heart. Proc Natl Acad Sci U S A. 2008;105:4477–82. doi: 10.1073/pnas.0709118105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Nunez L, Vaquero M, Gomez R, et al. Nitric oxide blocks hKv1.5 channels by S- nitrosylation and by a cyclic GMP-dependent mechanism. Cardiovasc Res. 2006;72:80–9. doi: 10.1016/j.cardiores.2006.06.021. [DOI] [PubMed] [Google Scholar]
- 62.Gomez R, Nunez L, Vaquero M, et al. Nitric oxide inhibits Kv4.3 and human cardiac transient outward potassium current (Ito1). Cardiovasc Res. 2008;80:375–84. doi: 10.1093/cvr/cvn205. [DOI] [PubMed] [Google Scholar]
- 63.Taglialatela M, Pannaccione A, Iossa S, et al. Modulation of the K(+) channels encoded by the human ether-a-gogo-related gene-1 (hERG1) by nitric oxide. Mol Pharmacol. 1999;56:1298–308. doi: 10.1124/mol.56.6.1298. [DOI] [PubMed] [Google Scholar]
- 64.Sanchez G, Hidalgo C, Donoso P. Kinetic studies of calcium-induced calcium release in cardiac sarcoplasmic reticulum vesicles. Biophys J. 2003;84:2319–30. doi: 10.1016/S0006-3495(03)75037-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Sun J, Xin C, Eu JP, et al. Cysteine-3635 is responsible for skeletal muscle ryanodine receptor modulation by NO. Proc Natl Acad Sci U S A. 2001;98:11158–62. doi: 10.1073/pnas.201289098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Meszaros LG, Minarovic I, Zahradnikova A. Inhibition of the skeletal muscle ryanodine receptor calcium release channel by nitric oxide. FEBS Lett. 1996;380:49–52. doi: 10.1016/0014-5793(96)00003-8. [DOI] [PubMed] [Google Scholar]
- 67.Gonzalez DR, Fernandez IC, Ordenes PP, et al. Differential role of S-nitrosylation and the NO-cGMP-PKG pathway in cardiac contractility. Nitric Oxide. 2008;18:157–67. doi: 10.1016/j.niox.2007.09.086. [DOI] [PubMed] [Google Scholar]
- 68.Suko J, Drobny H, Hellmann G. Activation and inhibition of purified skeletal muscle calcium release channel by NO donors in single channel current recordings. Biochim Biophys Acta. 1999;1451:271–87. doi: 10.1016/s0167-4889(99)00098-1. [DOI] [PubMed] [Google Scholar]
- 69.Hart JD, Dulhunty AF. Nitric oxide activates or inhibits skeletal muscle ryanodine receptors depending on its concentration, membrane potential and ligand binding. J Membr Biol. 2000;173:227–36. doi: 10.1007/s002320001022. [DOI] [PubMed] [Google Scholar]
- 70.Cheong E, Tumbev V, Abramson J, et al. Nitroxyl triggers Ca2+ release from skeletal and cardiac sarcoplasmic reticulum by oxidizing ryanodine receptors. Cell Calcium. 2005;37:87–96. doi: 10.1016/j.ceca.2004.07.001. [DOI] [PubMed] [Google Scholar]
- 71.Dai T, Tian Y, Tocchetti CG, et al. Nitroxyl increases force development in rat cardiac muscle. J Physiol. 2007;580:951–60. doi: 10.1113/jphysiol.2007.129254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Tocchetti CG, Wang W, Froehlich JP, et al. Nitroxyl improves cellular heart function by directly enhancing cardiac sarcoplasmic reticulum Ca2+ cycling. Circ Res. 2007;100:96–104. doi: 10.1161/01.RES.0000253904.53601.c9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Marengo JJ, Hidalgo C, Bull R. Sulfhydryl oxidation modifies the calcium dependence of ryanodine-sensitive calcium channels of excitable cells. Biophys J. 1998;74:1263–77. doi: 10.1016/S0006-3495(98)77840-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Terentyev D, Gyorke I, Belevych AE, et al. Redox Modification of Ryanodine Receptors Contributes to Sarcoplasmic Reticulum Ca2+ Leak in Chronic Heart Failure. Circ Res. 2008 doi: 10.1161/CIRCRESAHA.108.184457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Yano M, Okuda S, Oda T, et al. Correction of defective interdomain interaction within ryanodine receptor by antioxidant is a new therapeutic strategy against heart failure. Circulation. 2005;112:3633–43. doi: 10.1161/CIRCULATIONAHA.105.555623. [DOI] [PubMed] [Google Scholar]
- 76.Gonzalez DR, Beigi F, Treuer AV, et al. Deficient ryanodine receptor S-nitrosylation increases sarcoplasmic reticulum calcium leak and arrhythmogenesis in cardiomyocytes. Proc Natl Acad Sci U S A. 2007;104:20612–7. doi: 10.1073/pnas.0706796104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Petroff MG, Kim SH, Pepe S, et al. Endogenous nitric oxide mechanisms mediate the stretch dependence of Ca2+ release in cardiomyocytes. Nat Cell Biol. 2001;3:867–73. doi: 10.1038/ncb1001-867. [DOI] [PubMed] [Google Scholar]
- 78.Khan SA, Lee K, Minhas KM, et al. Neuronal nitric oxide synthase negatively regulates xanthine oxidoreductase inhibition of cardiac excitation-contraction coupling. Proc Natl Acad Sci U S A. 2004;101:15944–8. doi: 10.1073/pnas.0404136101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Cherednichenko G, Zima AV, Feng W, et al. NADH oxidase activity of rat cardiac sarcoplasmic reticulum regulates calcium-induced calcium release. Circ Res. 2004;94:478–86. doi: 10.1161/01.RES.0000115554.65513.7C. [DOI] [PubMed] [Google Scholar]
- 80.Sanchez G, Escobar M, Pedrozo Z, et al. Exercise and tachycardia increase NADPH oxidase and ryanodine receptor-2 activity: possible role in cardioprotection. Cardiovasc Res. 2008;77:380–6. doi: 10.1093/cvr/cvm011. [DOI] [PubMed] [Google Scholar]
- 81.Aracena P, Sanchez G, Donoso P, et al. S-glutathionylation decreases Mg2+ inhibition and S-nitrosylation enhances Ca2+ activation of RyR1 channels. J Biol Chem. 2003;278:42927–35. doi: 10.1074/jbc.M306969200. [DOI] [PubMed] [Google Scholar]
- 82.Stoyanovsky D, Murphy T, Anno PR, et al. Nitric oxide activates skeletal and cardiac ryanodine receptors. Cell Calcium. 1997;21:19–29. doi: 10.1016/s0143-4160(97)90093-2. [DOI] [PubMed] [Google Scholar]
- 83.Donoso P, Aracena P, Hidalgo C. Sulfhydryl oxidation overrides Mg(2+) inhibition of calcium-induced calcium release in skeletal muscle triads. Biophys J. 2000;79:279–86. doi: 10.1016/S0006-3495(00)76290-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Sanchez G, Pedrozo Z, Domenech RJ, et al. Tachycardia increases NADPH oxidase activity and RyR2 S-glutathionylation in ventricular muscle. J Mol Cell Cardiol. 2005;39:982–91. doi: 10.1016/j.yjmcc.2005.08.010. [DOI] [PubMed] [Google Scholar]
- 85.Skasa M, Jungling E, Picht E, et al. L-type calcium currents in atrial myocytes from patients with persistent and non-persistent atrial fibrillation. Basic Res Cardiol. 2001;96:151–9. doi: 10.1007/s003950170065. [DOI] [PubMed] [Google Scholar]
- 86.Van Wagoner DR. Oxidative stress and inflammation in atrial fibrillation: role in pathogenesis and potential as a therapeutic target. J Cardiovasc Pharmacol. 2008;52:306–13. doi: 10.1097/FJC.0b013e31817f9398. [DOI] [PubMed] [Google Scholar]
- 87.Cai H, Li Z, Goette A, et al. Downregulation of endocardial nitric oxide synthase expression and nitric oxide production in atrial fibrillation: potential mechanisms for atrial thrombosis and stroke. Circulation. 2002;106:2854–8. doi: 10.1161/01.cir.0000039327.11661.16. [DOI] [PubMed] [Google Scholar]
- 88.Cavolli R, Kaya K, Aslan A, et al. Does sodium nitroprusside decrease the incidence of atrial fibrillation after myocardial revascularization?: a pilot study. Circulation. 2008;118:476–81. doi: 10.1161/CIRCULATIONAHA.107.719377. [DOI] [PubMed] [Google Scholar]
- 89.Marban E. Cardiac channelopathies. Nature. 2002;415:213–8. doi: 10.1038/415213a. [DOI] [PubMed] [Google Scholar]
- 90.Jaffrey SR, Snowman AM, Eliasson MJ, et al. CAPON: a protein associated with neuronal nitric oxide synthase that regulates its interactions with PSD95. Neuron. 1998;20:115–24. doi: 10.1016/s0896-6273(00)80439-0. [DOI] [PubMed] [Google Scholar]
- 91.Arking DE, Pfeufer A, Post W, et al. A common genetic variant in the NOS1 regulator NOS1AP modulates cardiac repolarization. Nat Genet. 2006;38:644–51. doi: 10.1038/ng1790. [DOI] [PubMed] [Google Scholar]
- 92.Aarnoudse AJ, Newton-Cheh C, de Bakker PI, et al. Common NOS1AP variants are associated with a prolonged QTc interval in the Rotterdam Study. Circulation. 2007;116:10–6. doi: 10.1161/CIRCULATIONAHA.106.676783. [DOI] [PubMed] [Google Scholar]
- 93.Kao WH, Arking DE, Post W, et al. Genetic variations in nitric oxide synthase 1 adaptor protein are associated with sudden cardiac death in US white community-based populations. Circulation. 2009;119:940–51. doi: 10.1161/CIRCULATIONAHA.108.791723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.van NC, Aarnoudse AJ, Eijgelsheim M, et al. Calcium channel blockers, NOS1AP, and heart-rate-corrected QT prolongation. Pharmacogenet Genomics. 2009;19:260–6. doi: 10.1097/FPC.0b013e328324e556. [DOI] [PubMed] [Google Scholar]
- 95.Becker ML, Visser LE, Newton-Cheh C, et al. A common NOS1AP genetic polymorphism is associated with increased cardiovascular mortality in users of dihydropyridine calcium channel blockers. Br J Clin Pharmacol. 2009;67:61–7. doi: 10.1111/j.1365-2125.2008.03325.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Mochizuki M, Yano M, Oda T, et al. Scavenging free radicals by low-dose carvedilol prevents redox-dependent Ca2+ leak via stabilization of ryanodine receptor in heart failure. J Am Coll Cardiol. 2007;49:1722–32. doi: 10.1016/j.jacc.2007.01.064. [DOI] [PubMed] [Google Scholar]
- 97.Yagi H, Horinaka S, Matsuoka H. Edaravone prevented deteriorated cardiac function after myocardial ischemia-reperfusion via inhibiting lipid peroxidation in rat. J Cardiovasc Pharmacol. 2005;46:46–51. doi: 10.1097/01.fjc.0000162772.16797.7f. [DOI] [PubMed] [Google Scholar]
- 98.Menshikova EV, Salama G. Cardiac ischemia oxidizes regulatory thiols on ryanodine receptors: captopril acts as a reducing agent to improve Ca2+ uptake by ischemic sarcoplasmic reticulum. J Cardiovasc Pharmacol. 2000;36:656–68. doi: 10.1097/00005344-200011000-00016. [DOI] [PubMed] [Google Scholar]
- 99.Heymes C, Bendall JK, Ratajczak P, et al. Increased myocardial NADPH oxidase activity in human heart failure. J Am Coll Cardiol. 2003;41:2164–71. doi: 10.1016/s0735-1097(03)00471-6. [DOI] [PubMed] [Google Scholar]
- 100.Minhas KM, Saraiva RM, Schuleri KH, et al. Xanthine oxidoreductase inhibition causes reverse remodeling in rats with dilated cardiomyopathy. Circ Res. 2006;98:271–9. doi: 10.1161/01.RES.0000200181.59551.71. [DOI] [PubMed] [Google Scholar]
- 101.Takimoto E, Champion HC, Li M, et al. Oxidant stress from nitric oxide synthase-3 uncoupling stimulates cardiac pathologic remodeling from chronic pressure load. J Clin Invest. 2005;115:1221–31. doi: 10.1172/JCI21968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Moens AL, Takimoto E, Tocchetti CG, et al. Reversal of cardiac hypertrophy and fibrosis from pressure overload by tetrahydrobiopterin: efficacy of recoupling nitric oxide synthase as a therapeutic strategy. Circulation. 2008;117:2626–36. doi: 10.1161/CIRCULATIONAHA.107.737031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Zimmet JM, Hare JM. Nitroso-redox interactions in the cardiovascular system. Circulation. 2006;114:1531–44. doi: 10.1161/CIRCULATIONAHA.105.605519. [DOI] [PubMed] [Google Scholar]
- 104.Bendall JK, Damy T, Ratajczak P, et al. Role of myocardial neuronal nitric oxide synthase-derived nitric oxide in beta-adrenergic hyporesponsiveness after myocardial infarction-induced heart failure in rat. Circulation. 2004;110:2368–75. doi: 10.1161/01.CIR.0000145160.04084.AC. [DOI] [PubMed] [Google Scholar]
- 105.Damy T, Ratajczak P, Shah AM, et al. Increased neuronal nitric oxide synthase-derived NO production in the failing human heart. Lancet. 2004;363:1365–7. doi: 10.1016/S0140-6736(04)16048-0. [DOI] [PubMed] [Google Scholar]
- 106.Haendeler J, Hoffmann J, Zeiher AM, et al. Antioxidant effects of statins via S-nitrosylation and activation of thioredoxin in endothelial cells: a novel vasculoprotective function of statins. Circulation. 2004;110:856–61. doi: 10.1161/01.CIR.0000138743.09012.93. [DOI] [PubMed] [Google Scholar]