

Abstract
Helicobacter hepaticus-infected Rag2-/- mice emulate many aspects of human inflammatory bowel disease, including the development of colitis and colon cancer. To elucidate mechanisms of inflammation-induced carcinogenesis, we undertook a comprehensive analysis of histopathology, molecular damage, and gene expression changes during disease progression in these mice. Infected mice developed severe colitis and hepatitis by 10 wk post-infection, progressing into colon carcinoma by 20 wk post-infection, with pronounced pathology in the cecum and proximal colon marked by infiltration of neutrophils and macrophages. Transcriptional profiling revealed decreased expression of DNA repair and oxidative stress response genes in colon, but not in liver. Mass spectrometric analysis revealed higher levels of DNA and RNA damage products in liver compared to colon and infection-induced increases in 5-chlorocytosine in DNA and RNA and hypoxanthine in DNA. Paradoxically, infection was associated with decreased levels of DNA etheno adducts. Levels of nucleic acid damage from the same chemical class were strongly correlated in both liver and colon. The results support a model of inflammation-mediated carcinogenesis involving infiltration of phagocytes and generation of reactive species that cause local molecular damage leading to cell dysfunction, mutation, and cell death. There are strong correlations among histopathology, phagocyte infiltration, and damage chemistry that suggest a major role for neutrophils in inflammation-associated cancer progression. Further, paradoxical changes in nucleic acid damage were observed in tissue- and chemistry-specific patterns. The results also reveal features of cell stress response that point to microbial pathophysiology and mechanisms of cell senescence as important mechanistic links to cancer.
Keywords: mass spectrometry, biomarkers
Chronic inflammation is a major risk factor for many human diseases, including cancer. Epidemiological studies suggest that at least 20% of all cancers are caused by chronic inflammatory conditions, such as the causative role of Heliobacter pylori and hepatitis B virus infections in gastric and liver cancer, respectively, and the strong association of inflammatory bowel disease (IBD) and colon cancer (1, 2). Although the underlying molecular mechanisms linking inflammation and cancer are poorly understood, many of the steps have been defined. In the case of infection, the inciting event is tissue injury, with local release of cytokines and other chemotactic factors causing infiltration and activation of macrophages and neutrophils to produce large quantities of other cytokines and chemokines as well as reactive chemical species that cause mutagenic and cytotoxic damage (3, 4). For example, up-regulation and activation of inducible NO synthase (iNOS) and NADPH oxidase in macrophages produces nitric oxide (NO) and superoxide ( ), respectively. Subsequent reactions yield a battery of reactive species, including the potent nitrosating agent nitrous anhydride (N2O3) and the oxidizing and nitrating agents nitrogen dioxide radical (
), respectively. Subsequent reactions yield a battery of reactive species, including the potent nitrosating agent nitrous anhydride (N2O3) and the oxidizing and nitrating agents nitrogen dioxide radical (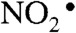 ), peroxynitrite (ONOO-), and nitrosoperoxycarbonate (
), peroxynitrite (ONOO-), and nitrosoperoxycarbonate (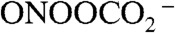 ) (5). Moreover, activated neutrophils are considered the primary source of the potent oxidant and halogenating agent hypochlorous acid (HOCl) via activation of myeloperoxidase (MPO) (6–8).
) (5). Moreover, activated neutrophils are considered the primary source of the potent oxidant and halogenating agent hypochlorous acid (HOCl) via activation of myeloperoxidase (MPO) (6–8).
These chemical mediators of inflammation can damage all classes of cellular molecules, including DNA, RNA, protein, lipids, and metabolites, by both direct and indirect mechanisms. For example, DNA damage caused by halogenation, deamination, and oxidation, as well as by nucleobase adduct formation caused by lipid peroxidation products, can potentially induce de novo mutations and epigenetic changes initiating tumor development (1, 2, 9). Several recent studies involving mouse models of inflammation have provided evidence supporting such a scenario (10–12). Similar pathways disrupt the function of RNA, as proposed for major human degenerative diseases such as Alzheimer’s disease, Parkinson’s disease, and atherosclerosis (13, 14), whereas protein damage caused by oxidation, nitration, and halogenation can impair function and stability, including those of tumor suppressors, oncogenes, and factors that maintain fidelity during DNA replication (4, 15, 16).
To better understand the complex pathophysiology of inflammation, we undertook a comprehensive analysis of chemical and biological end points in an established model of human IBD: Rag2-/- mice infected with Helicobacter hepaticus (17, 18). This Gram-negative spiral bacterium colonizes the liver and intestinal crypts of the cecum and the colon of several mouse strains, establishing a life-long infection (19, 20). H. hepaticus does not typically cause disease in immunocompetent mice, but infection in susceptible inbred strains can lead to hepatitis whereas immunodeficient mice develop chronic colitis (21–23). For example, mice lacking the recombinase-activating gene-2 (Rag2) do not possess functional lymphocytes, and H. hepaticus infection results in chronic colitis and colon cancer (17). H. hepaticus infection in Rag2-/- mice emulates many aspects of human IBD, with colonic and cecal infiltration of macrophages and neutrophils presumably causing increased production of reactive chemical species at sites of infection (18).
To test this hypothesis and identify potential biomarkers of the pathophysiology of inflammation, we characterized the time course of colon and liver pathology in H. hepaticus-infected Rag2-/- mice with a battery of quantitative molecular, histological, and bioanalytical assays. For example, we developed and applied an isotope-dilution liquid chromatography-coupled tandem mass spectrometry (LC-MS/MS) method (SI Appendix, Fig. S1) to quantify 14 different DNA and RNA damage products anticipated to represent the full spectrum of inflammation chemistries. This included the 2-deoxyribo- and ribonucleoside forms of base halogenation (5-chlorocytosine, 5-Cl-C), nitrosative deamination (xanthine, X; hypoxanthine, I), alkylation by lipid peroxidation products (1, N6-ethenoadenine, εA; 3,N4-ethenocytosine, εC), and oxidation (8-oxo-7,8-dihydroguanine, 8-oxo-G; spiroiminodihydantoin, Sp; guanidinohydantoin, Gh) (see SI Appendix, Fig. S2). Data from these chemical assays were correlated with quantitative scores from histopathological, immunohistochemical, and gene expression analyses. These analyses revealed a variety of unique features of infection-induced colitis and carcinogenesis.
Results
Histological Analysis of Colon and Liver from H. hepaticus-infected Rag2-/- Mice.
To monitor and characterize disease progression, colon and liver tissues were harvested at two different time-points following H. hepaticus infection: 10 and 20 weeks post-infection (w p.i.). Quantitative PCR analysis revealed that all infected Rag2-/- mice were positive for H. hepaticus, whereas the sham controls were negative. The tissue samples were subjected to a wide range of analyses, starting with histopathology and immunohistochemistry. Although there were minimal or no lesions in large intestine and liver in age-matched control groups, H. hepaticus-infected mice showed significant tissue pathology at both time-points (Fig. 1, SI Appendix, Fig. S3). At 10 w p.i., changes in the large intestine included inflammation, edema, epithelial defects, crypt atrophy, hyperplasia, and dysplasia, which were similar and most severe in the ileocecocolic junction and proximal colon (SI Appendix, Fig. S3 A and B). The most prominent histopathologic changes consisted of thickened colonic mucosa, infiltration of neutrophils and macrophages, and combinations of surface epithelial tattering or erosion and mucosal gland dilation or distortion with intraluminal cell debris. Where colonic dysplasia was a feature, mucosal glands were variably distorted and lined by crowded epithelial cells, often containing increased numbers of mitoses and reduced goblet cell differentiation. These changes were significant in the proximal and transverse colon but not statistically significant in the distal colon (SI Appendix, Fig. S3B). At 20 w p.i., lesions in the large intestine were most severe in the ileocecocolic junction and proximal colon and were similar to those observed at 10 w p.i. (Fig. 1A, SI Appendix, Fig. S3 A and B). However, progression of dysplasia to intramural carcinoma was evident in some animals at 20 w p.i., with carcinoma development characterized by invasion of neoplastic glands below the muscularis mucosae layer (SI Appendix, Fig. S3A). Glands were irregularly shaped and lined by atypical, tall columnar epithelial cells with indiscrete borders, cellular and nuclear pleomorphism, visible mitotic figures, and loss of goblet cells. In the liver, there was significant evidence of portal and lobular hepatitis, but not interface hepatitis, at both 10 and 20 w p.i. (SI Appendix, Fig. S3 C and D).
Fig. 1.

H. hepaticus-infected mice develop chronic colitis, colon carcinoma, and sporadic hepatitis. (A–C) HAIs of colitis were calculated by summing individual scores of inflammation, edema, epithelial defects, crypt atrophy, hyperplasia, and dysplasia for (A) cecum, (B) proximal colon, and (C) distal colon. (D) HIs were calculated by summing individual subfeature histopathology scores from lobular, portal, and interface inflammation, and the number of lobes that contained ≥5 inflammatory lesions. Statistical analysis for all panels is presented as box plots showing the median value (line), the interquartile range (box), and Tukey whiskers embracing data within 1.5-fold of the interquartile range; all data outside the range of the Tukey whiskers are presented as individual data points.
In terms of disease progression, the histological activity indices (HAIs) of colitis (cumulative score of inflammation, edema, epithelial defects, crypt atrophy, hyperplasia, and dysplasia) showed significantly lower levels in the proximal colon at 20 w p.i. compared to 10 w p.i. but tended to increase in the distal colon at 20 w p.i. (P = 0.06) (Fig. 1 C and D). In agreement with the HAI in the proximal colon, the hepatitis index (HI) was significantly lower at 20 w p.i. than at 10 w p.i. (Fig. 1D). Consequently, the HAI of the proximal colon and the HI tended to correlate in H. hepaticus-infected mice (SI Appendix, Table S1).
To define the extent of the innate immune cell infiltrate in this infection model of colitis, we performed immunohistochemical analyses using F4/80 and MPO staining as markers for macrophage and neutrophil infiltration, respectively (Fig. 2, SI Appendix, Fig. S4). At both time-points, F4/80-positive and MPO-positive cells accumulated significantly and sustainably in the cecum and the proximal colon of H. hepaticus-infected Rag2-/- mice. However, between 10 and 20 w p.i., we observed a significant decrease in MPO-positive cells in the cecum, with a trend toward a decrease in the proximal colon (P = 0.07), whereas there was a trend towards increased macrophage infiltration at 20 w p.i. in the cecum and the proximal colon (P = 0.07). This is consistent with the view that neutrophils are recruited early during the inflammatory response, whereas macrophages persist longer at sites of inflammation (24).
Fig. 2.

H. hepaticus-induced colitis is associated tissue infiltration of neutrophils and macrophages. Cell counts (in situ) for MPO-positive neutrophils (A, B) and F4/80-positive macrophages (C, D) in the most affected regions of the lower bowel: cecum (A, C) and proximal colon (B, D). Values represent means ± SD.
LC-MS/MS Quantification of Inflammation-Related DNA and RNA Damage.
Toward the goal of defining the chemical mechanisms underlying inflammation-induced carcinogenesis, we used LC-MS/MS (SI Appendix, Fig. S1) to quantify the nucleoside forms of DNA and RNA damage products thought to represent the full spectrum of inflammation-related chemistries: nucleobase deamination (X and I), halogenation (5-Cl-C), oxidation (8-oxo-G, Sp, Gh), and alkylation (εA, εC) (SI Appendix, Fig. S2). This platform extends previously published DNA protocols (25, 26) to include the guanine secondary oxidation products Sp and Gh, the halogenated nucleobase 5-Cl-C, and a spectrum of RNA damage products (SI Appendix, Tables S2 and S3). Special attention was given to minimize adventitious damage by addition of deaminase inhibitors (coformycin, tetrahydrouridine), a metal chelator (deferoxamine), and an antioxidant (butylated hydroxytoluene) during sample preparation (SI Appendix, Fig. S1). LC-MS/MS parameters were optimized for maximal sensitivity resulting in limits of quantification (LOQ) of 1–10 fmol, which amounts to 0.4–8 lesions per 108 nucleotides (nt) in 100 μg of DNA or RNA, with basal levels of all DNA and RNA damage products above the LOQ in liver and colon from uninfected mice (Fig. 3, SI Appendix, Figs. S5 and S6; SI Appendix, Tables S2 and S3).
Fig. 3.

LC-MS/MS quantification of basal levels of nucleobase damage in colon and liver of uninfected mice. Basal levels of nucleobase damage products in DNA and RNA isolated from liver and colon from uninfected Rag2-/- mice. Analyses were performed as described in Materials and Methods. Statistical analysis is presented in box plots as described in Fig. 1.
This analytical platform was applied to liver and colon tissues from H. hepaticus-infected and control Rag2-/- mice at 10 and 20 w p.i. (SI Appendix, Tables S2 and S3). Analysis of DNA and RNA damage products in control mice revealed several important and unique findings. First, basal damage levels were significantly higher in RNA than in DNA for base deamination (X, I), oxidation (8-oxo-G), and chlorination (5-Cl-C) but not for lipid peroxidation-derived adducts (Fig. 3). Second, background levels of the guanine secondary oxidation products Sp and Gh were detected in the range of 1–7 lesions per 108 nt in DNA from the liver and colon, which is nearly 100 times lower than the parent 8-oxo-dG (Fig. 3). Sp and Gh have previously only been observed in vitro and in prokaryotes (reviewed in ref. 27). Third, the levels for all guanine-derived oxidation products were significantly lower in the colon compared to the liver, both in DNA and RNA (Fig. 3). The most prominent differences were observed for Sp and Gh, which were close to the LOQ in colon and significantly higher than the LOQ in the liver. Fourth, the levels of chemically related damage products showed strong and highly significant positive correlations, as demonstrated by correlations between Sp and Gh with their parent molecule 8-oxo-dG and between the lipid-peroxidation-derived etheno-adducts εA and εC in DNA and RNA, respectively (SI Appendix, Fig. S7). These observations satisfy assumptions about the chemical origins of these lesions.
The analytical methods were next used to define changes caused by H. hepaticus infection. Although no significant differences in damage levels were observed in the liver from infected mice, analyses of colon tissue revealed several significant findings, the most prominent of which was the increase in halogenated lesions. Statistically significant increases in 5-Cl-dC (50%) and 5-Cl-rC (33%) were observed at 20 w p.i., along with significant increases in dI (26%) in DNA (Fig. 4). Unexpectedly, we observed transient but significant decreases in εdA (35%), εdC (44%), and 8-oxo-G in RNA (25%) at 10 w p.i. (Fig. 4). The levels of other damage products in DNA and RNA remained unchanged in the infected mice compared to controls (SI Appendix, Tables S2 and S3).
Fig. 4.

H. hepaticus-induced DNA and RNA damage in colon. Rag2-/- mice were infected with H. hepaticus (H.h.) and colon tissue was isolated at 10 and 20 w p.i. for DNA and RNA damage analyses, as described in Materials and Methods. The figure contains data for damage products showing largest increases or decreases, with the complete dataset shown in SI Appendix, Tables S2 and S3. Statistical analysis is presented in box plots as described in Fig. 1.
Statistical analyses revealed several significant correlations between the nucleic acid damage and histopathology. Not surprisingly, simple correlation analysis of levels of DNA and RNA lesions with corresponding colonic HAI values revealed significant positive correlations for 5-Cl-dC, dI (Spearman coefficients r > 0.5, P < 0.05) (SI Appendix, Table S4), with weak tendencies for correlations of 5-Cl-rC (r = 0.45, P = 0.14) and Sp (r = 0.52, P = 0.087) with the colonic HAI.
Transcriptional Analysis of the DNA Repair and Oxidative Stress Responses in Inflammation.
Further insight into the mechanisms underlying tissue responses to inflammation was obtained by transcriptional profiling of genes involved in the generation of reactive chemical species (10 genes in the colon and 2 genes in the liver), stress response and glutathione metabolism (22 genes), DNA repair (53 genes), and cell cycle regulation (10 genes) (SI Appendix, Table S8). To determine which gene expression changes showed the strongest correlation with features of pathology, the data were subjected to principal component analysis (PCA). The PCA distinguished both tissue and infection status (SI Appendix, Fig. S8A), with liver and colon samples well distinguished in the first principal component and infection status separated in the second principal component. The genes contributing to these differences are shown in the corresponding loadings plots (SI Appendix, Fig. S8B), with associated volcano plots indicating the significance and fold-change in gene expression (Fig. 5, SI Appendix, Fig. S9). Interestingly, analysis of uninfected mice revealed higher expression of genes involved in the oxidative stress response in the liver compared to the colon, such as catalase (Cat), superoxide dismutases (Sod), thioredoxin reductase (Txnrd), glutathione peroxidases (Gpx), and glutathione S-transferases (Gst), which may be related to higher oxidative stress in the liver (SI Appendix, Fig. S9A). On the other hand, several genes involved in DNA repair showed higher expression in the uninfected colon compared to liver, including DNA glycosylases, such as NEIL3 and Ogg1, and ligase 1 (Lig1), which participate directly in the removal of the DNA base lesions such as 8-oxo-G, Sp, and Gh (28, 29). This may explain the lower levels of oxidatively derived nucleobase damage in the colon (Fig. 3).
Fig. 5.

Gene expression analysis of DNA repair and oxidative stress response factors in colon and liver tissues of H. hepaticus-infected Rag2-/- mice. QPCR-based gene expression profiling was performed on colon (A, C) and liver (B, D) tissues from control and H. hepaticus-infected mice (20 w p.i.), including 95 key genes involved in generation and detoxification of reactive chemical species (A, B) and DNA damage response (C, D) (SI Appendix, Table S8 lists all genes and gene ontology categories). The data are presented as volcano plots of changes in gene expression levels. Horizontal lines indicate P = 0.05 for N≥5. Vertical lines indicate no change in gene expression. Genes that are significantly up- or down-regulated are labeled with gene symbols.
In terms of genes that distinguished H. hepaticus infection, 42% and 61% of the genes in the colon showed significantly different expression levels at 10 and 20 w p.i., respectively (Fig. 5, SI Appendix, Fig. S9B and Table S8), whereas 3.4% and 28% of the genes in the liver showed significantly different expression levels, respectively (Fig. 5, SI Appendix, Fig. S9C and Table S8). In infected colons at 20 w p.i., there was significant up-regulation of 80% of genes encoding proteins involved with the generation of reactive chemical species, including inducible nitric oxide synthase (Nos2), xanthine oxido-reductase (Xdh/XO), and several genes coding for NADPH oxidases, such as Nox1, Cybb, and Ncf2 (Fig. 5, SI Appendix, Table S8). Moreover, we observed up-regulation of genes thought to protect against oxidative stress, such as cell cycle checkpoint gene Cdkn2a, and epithelium-specific glutathione peroxidase 2 (Gpx2), thioredoxin reductase 1 (Txnrd1), and glutathione reductase (Gsr). Paradoxically, several genes of the stress response group (36%), such as Sod and Gst, and most of the genes involved in DNA repair (64%) showed significantly lower expression in H. hepaticus-infected colon compared to uninfected controls (Fig. 5, SI Appendix, Table S8). All major DNA repair pathways were represented in the down-regulated group: base excision repair (BER: Nthl1, NEIL3, and MUTYH), nucleotide excision repair (NER: Xpa, Xpc, Ercc6), mismatch repair (MMR: Mlh3, Msh3, Msh6), and double-strand break repair (DSBR: Prkdc, Mre11, Brca1). With the exception of Ercc3 and Mlh1, all genes of the NER and MMR groups were significantly down-regulated at 20 w p.i. Importantly, none of the housekeeping genes (i.e., 18S rRNA, Actb, Gapdh, Hprt1) were significantly altered by infection, which rules out a general suppression of gene expression in inflamed tissues. Interestingly, markers of cell proliferation, such as Pcna and Ki67, did not change in spite of histological evidence for cell proliferation in the infected colon. Ki67 expression was previously observed to increase only in H. hepaticus-infected mice in the infection-induced hepatocellular carcinoma tissue, with no increase in normal liver tissue (30).
Although some changes in gene expression were common to the liver and colon, such as up-regulation of Xdh/XO at 20 w p.i. (Fig. 5), there were several major infection-induced differences in the liver. Of note, expression of MPO was very highly induced in the liver, which may be related to inducible MPO expression in monocytes, because neutrophilic MPO is expressed only during early stages of neutrophil maturation in the bone marrow (7). Further, in direct contrast to the colon, H. hepaticus infection caused up-regulation of all major DNA repair pathways in the liver, including oxidative dealkylation of nucleobases (Alkbh1 and Alkbh3), BER (Lig1, NEIL3, and Ung), NER (Ercc6), MMR (Pms2), and DSBR (Brca1, Blm, and Exo1) (Fig. 5B). This is consistent with compensation for inflammation-induced DNA damage, which is further supported by up-regulation of several cell cycle checkpoint regulators, such as Trp53, Atr, Gadd45a, and Cdkn2a, in liver tissues from infected mice. On the other hand, several genes involved in stress response and glutathione metabolism, such as Cat, Gsta1, and Gsta4, were expressed at significantly lower levels in the liver of infected mice compared to controls (Fig. 5).
Discussion
IBD affects millions of people worldwide and significantly increases the risk of colon cancer, with > 20% of IBD patients developing colitis-associated cancers within 30 y of disease onset with a mortality rate > 50% (4). Although dysregulation of host-microbiome interactions, multifactorial genetic predisposition, and environmental factors are associated with IBD (4, 31), the molecular and chemical mechanisms linking colonic inflammation with cancer remain unclear, with potential involvement of tumor-initiating DNA damage, epigenetic changes, and interference with tumor-suppressive mechanisms (32). A central feature of current presumptive models of inflammation-mediated carcinogenesis involves infiltration of activated phagocytes at sites of infection, with subsequent generation of reactive oxygen, nitrogen, and halogen species that cause molecular damage leading to cell dysfunction, mutation, and cell death. Our comprehensive analysis of the H. hepaticus-infected Rag2-/- mouse model of colitis-associated carcinogenesis (17, 18) supports this model, with strong correlations among histopathology (Fig. 1), phagocyte infiltration (Fig. 2), and the increased level of HOCl-induced 5-Cl-dC (Fig. 4). The major results of these studies are summarized in Table 1 and Fig. 6, which reveals several unique features of the chemical and biological pathology linking inflammation and cancer in the mouse model of infection-induced colitis: strongly correlated histopathology and molecular damage suggestive of a major role for neutrophils in the inflammatory process, paradoxical changes in nucleic acid damage that are both tissue- and chemistry-specific, and features of cell stress response that are consistent with both known microbial pathophysiology and mechanisms of cell senescence.
Table 1.
Summary of significant results in H. hepaticus-infected Rag2-/- mice.
| Parameter |
10 wk p.i. |
20 wk p.i. |
||
| Histopathology | ↑ colitis, hepatitis | ↑ colitis, hepatitis | ||
| Immunohistochemistry (colon) | ↑ MØ, PMN | ↑ MØ, PMN | ||
| DNA damage | ↓ εdA, εdC | ↑ 5-Cl-dC, dI, Sp | ||
| RNA damage | ↓ 8-oxo-rG | ↑ 5-Cl-rC | ||
| Gene expression† | Reactive species generating | Colon | ↑ iNos, Nox1 | ↑ iNos, Xdh, Nox1, CYB, NCF |
| Liver | n.c.* | ↑ Mpo, Xdh | ||
| DNA repair | Colon | ↓ BER, NER, MMR, DSBR | ↓ BER, NER, MMR, DSBR | |
| Liver | ↑ Ung | ↑ BER, NER, MMR, DSBR | ||
| ↓ Apex2, Tdg | ↓ Polb | |||
| Cell cycle | Colon | ↓ Cdkn1a, Gadd45a, Hus1 | ↑ Cdkn2a | |
| ↓ Atm, Atr, Chek1Hus1 | ||||
| Liver | n.c. | ↑ Cdkn2a1, Chek2, Atr, Trp53 | ||
| Stress response | Colon | ↑ Gpx2 | ↑ Gpx2, Gsr, Txnrd1 | |
| ↓ Hspa1a, Cat | ↓ Cat, Txn1, Gpx4, SOD, GST, HSP | |||
| Liver | n.c. | ↓ Cat, Gsr, GST | ||
*n.c., no change from control; p.i., post-infection.
†Capitalized acronyms indicate groups of genes: BER base excision repair; CYB, cytochrome b; DSBR, double strand break repair; GST, glutathione S-transferase; HSP, heat shock protein; MMR mismatch repair; NCF, neutrophil cytosolic factor, NER, nucleotide excision repair, SOD, superoxide dismutase.
Fig. 6.

Model summarizing the potential mechanisms of inflammation-induced carcinogenesis.
Histopathological Correlates of Colitis and Hepatitis in H. hepaticus-Infected Rag2-/- Mice.
H. hepaticus colonizes the liver and colon of various mouse strains and is linked with the development of chronic colitis and hepatitis in several murine models (20). Although the complex population of immune cells in the intestinal lamina propria is thought to balance immune tolerance of luminal microbiota with pathogen defense (31), the lack of regulatory T cells in Rag2-/- mice disturbs the balance, with infection-induced inflammation leading to cancer (33). The present study demonstrated that H. hepaticus-infected Rag2-/- mice develop typhlocolitis (i.e., inflammation of the cecum and the colon) that evolved into colon cancer by 20 w p.i. (Fig. 1, SI Appendix, Fig. S3). Consistent with a diagnosis of typhlocolitis, the HAIs of the cecum and the proximal colon correlated moderately but with high statistical significance (r = 0.35, P < 0.01) in H. hepaticus-infected animals (SI Appendix, Table S1) and with infiltration of neutrophils and macrophages. Although we observed a progressive increase in the severity of disease in the cecum and the distal colon, histologic activity indices decreased significantly between 10 and 20 w p.i. in the proximal colon. This illustrates the spatiotemporal dynamic nature of colitis development in this model. Carcinomas were observed at the ileocecocolic junction in infected mice (10% of infected mice at 10 w p.i. and 20% at 20 w p.i.), which was one of the regions affected most by inflammation and histopathological changes (SI Appendix, Fig. S3A). This resembles neoplasia in human IBD patients, where carcinomas arise from colitis-associated dysplastic epithelial foci (20).
This study also demonstrates that H. hepaticus-infected Rag2-/- mice temporally develop hepatitis (Fig. 1B, SI Appendix, Fig. S3 C and D) that tended to correlate with the severity of colitis (r = 0.24, P = 0.07; HI versus HAI proximal colon) (SI Appendix, Table S1). In general, the hepatitis was mild to moderate at both time points, but sporadic incidents of severe hepatitis were consistently observed at 10 w p.i. The finding that the severity of colitis and hepatitis tended to correlate (SI Appendix, Table S1) suggests that both disease states are functionally and mechanistically linked, as had been observed in other models (e.g., ref. 34).
Determinants of the Spectrum of DNA and RNA Damage Products in Inflamed Colon and Liver.
One of the major conclusions of our analysis of 14 different DNA and RNA damage products is that the goal of developing biomarkers from damage products caused by reactive species generation at sites of inflammation is hampered by our lack of understanding of the factors that govern the steady-state levels of damage products in an inflamed tissue. These include the nature of the agent causing the inflammation, the tissue involved, and the response of the affected cells. Although the methods used in the present studies allow analysis of damage in the total population of cell types present in inflamed colon and liver tissues, the results of the DNA and RNA damage analyses provide some unique insights into the factors governing molecular damage in inflamed tissues.
Consider first the complicated local chemistry of molecular damage in inflammation, starting with reactive halogen species generated in vivo by MPO. Although our previous pathology-focused studies of H. hepaticus infection in the Rag2-/- revealed that the use of an iNOS inhibitor to block NO production caused a reduction in colonic inflammation, hyperplasia, and dysplasia, a more significant reduction in colon pathology was achieved by depleting Gr-1+ neutrophils (18). Coupled with the fact that the major source of MPO is neutrophils (7), these observations point to neutrophil-dependent MPO-activity as a major contributor, along with macrophage-generated NO, to the colitis and colon cancer arising in H. hepaticus-infected mice. Our observations provide immunohistochemical and chemical confirmation of the role of reactive halogen species, with higher levels of 5-Cl-dC and 5-Cl-rC in H. hepaticus-infected colons and a persistent and high frequency of MPO-positive cells at sites of inflammation (Figs. 2 and 4). These results are all consistent with other observations of major roles for neutrophils in colitis, including the protective effect of selective neutrophil depletion and scavengers of neutrophil-derived HOCl (7, 35, 36) and the observation of neutrophil accumulation and increased MPO activity in intestinal lesions in IBD patients (7, 37–39). Further, chlorination-derived damage products have also been associated with other inflammatory conditions, such as higher levels of 5-chlorouracil in human atherosclerotic tissue (40) and dihalogenated 2′-deoxycytidine in lungs and livers of mice treated with proinflammatory lipopolysaccharaide (41). It has been observed that 5-Cl-dC mimics 5-methyl-dC and induces inappropriate DNMT1 methylation within CpG sequences (9, 42, 43), which may result in altered cytosine methylation patterns that could affect the expression of tumor suppressor genes and initiate carcinogenesis (9, 42, 43).
Localized oxidation chemistry also appears to play an important role in the spectrum of DNA and RNA lesions. Oxidants generated by normal respiration and activated phagocytes are predicted to cause oxidation of guanine in DNA to form 8-oxo-dG. Many studies support this model, with measurements of 8-oxo-dG from 0.5 to 4 lesions per 106 nt (10, 44, 45) agreeing with our analyses in the liver and colon (Fig. 3). Indeed, Sipowicz et al. demonstrated a two-fold increase in 8-oxo-dG in DNA from livers of A/JNCr mice infected with H. hepaticus, with evidence for oxidative stress induced by increased cytochrome P450 activity (46). However, 8-oxo-dG itself is approximately 1,000-fold more susceptible to oxidation than dG and in vitro studies have demonstrated that 8-oxo-dG oxidation results in secondary oxidation products such as Sp and Gh (26, 27, 47–49). The only previous in vivo evidence for the formation of 8-oxo-dG secondary oxidation products comes from studies of oxidatively stressed Escherichia coli (50), in which Sp was detected at levels of approximately 10–100 lesions per 106 nucleotides. Using a more sensitive analytical method, we have now been able to detect background levels of both Sp and Gh in both the liver and colon in mice (Fig. 3), at levels some 100 times lower than those observed in E. coli. The in vivo derivation of Sp and Gh from 8-oxo-dG in the liver and colon is supported by the significant positive correlations between the adduct levels (SI Appendix, Fig. S7). The lack of significant infection-induced increases in the steady-state levels of 8-oxo-dG, Sp, or Gh suggests that either reactive chemical mediators of inflammation present in this study do not cause oxidative DNA damage, or that efficient DNA repair balances the rate of oxidant-induced formation. It is important to point out, however, that the background steady-state level of Sp and Gh can contribute to the mutagenic burden in cells given that Sp is at least an order of magnitude more mutagenic than 8-oxo-dG (51).
Now consider the complications of DNA repair. The fact that we observed either no change or down-regulation of most DNA repair gene expression in infected colon at 10 and 20 w p.i., respectively, does not preclude a significant DNA repair capacity with extant proteins or translational control of gene expression. Indeed, the significant reduction in the levels of the etheno adducts in DNA in infected colon at 10 w p.i. (Fig. 4), and the suggestion of decreased levels of etheno adducts in the liver, is consistent with up-regulated DNA repair activity. In the case of etheno adducts, this is possibly related to alkyladenine DNA glycosylase (Aag), the loss of which has been shown to increase etheno adduct levels in colon DNA (11). To illustrate the complexity of damage and repair in inflammation, several studies showed higher levels of etheno adducts in inflammatory conditions in mice and humans (10, 11, 52, 53), whereas other reports noted lower levels of etheno adducts under pathological conditions (54, 55). In peripheral blood lymphocytes of colorectal carcinoma patients, levels of etheno adducts as well as DNA repair rates were lower than in healthy subjects (54). Further, Kirkali et al. observed lower levels of the purine oxidation products 4,6-diamino-5-formamidopyrimidine, 2,6-diamino-4-hydroxy-5-formamidopyrimidine (FapyG), 8-oxo-dG, and (5’S)-8,5′-cyclo-2′-deoxyadenosine in tumor tissue compared to surrounding normal tissue in patients with colorectal cancer (56).
Finally, our studies reveal tissue-specific differences in the levels of molecular damage products (Fig. 3). Kirkali et al. observed higher levels of DNA damage products in rectum compared to the colon (56), which corroborates our observations of tissue-specific differences in formation or repair of DNA damage.
Colitis-Associated Changes in Gene Expression Reflect Bacterial Pathophysiology in H. hepaticus-Infected Rag2-/- Mice.
Our comprehensive analysis of histopathology and molecular damage was complemented by an analysis of infection-induced changes in gene expression in colon and liver for 95 genes involved in the generation of reactive chemical species, stress response and glutathione metabolism, DNA repair, and cell cycle regulation. These changes in gene expression serve as biomarker signatures for both H. hepaticus infection in general and for organ-specific effects of the infection, as suggested by principal component analysis (SI Appendix, Fig. S8). The scores plot clearly distinguishes normal and infected colon and liver, with the tissue-specific effects apparent in the first component and infection-specific changes distinguished in the second component (SI Appendix, Fig. S8A).
Whereas the loading plot in SI Appendix, Fig. S8B shows the genes that account for each of the four groupings apparent in the scores plot, several rather dramatic shifts in specific functional gene categories are more clearly illustrated in the volcano plots shown in Fig. 5 and SI Appendix, Fig. S9. For example, virtually all DNA repair pathway genes were significantly down-regulated in the infected colon at 10 and 20 w p.i., including genes for MMR, BER, NER, and DSBR (Fig. 5C). This is somewhat paradoxical given the absence of infection-induced changes in the levels of most DNA damage products and the reduction in etheno adducts. However, as noted earlier, reduced gene expression may not correlate with the levels of functional repair proteins. The interesting complication here is that the DNA repair genes suppressed in the colon all showed increased expression in the liver (Fig. 5D), though to a smaller extent.
Our observation of H. hepaticus-induced down-regulation of DNA repair transcripts is consistent with studies of H. pylori infection, which has been shown to suppress DNA repair gene expression in infected cells in culture and in infected mouse and human tissues (57, 58). Whereas Machado et al. observed that infection caused reduced expression of base excision and mismatch repair genes and concomitant increases in genomic instability (58), Kim et al. demonstrated that H. pylori coculture with gastric cancer cells lines caused reduced levels of both mRNA and protein for both MutS and MutL mismatch repair protein, with heat-sensitive bacterial products responsible for the reductions (57). Further, Toller et al. found that co-culture of H. pylori with mouse and human cell lines led to increased levels of DNA double-strand breaks (59). These observations of increased genomic instability and double-strand breaks are consistent with the DNA-cleaving activity of the cytolethal distending toxin (CDT) produced by H. hepaticus. In addition to inducing cell cycle arrest and cell distention, CDT is essential for the carcinogenic effects of H. hepaticus infection in liver cancer models (60), presumably by inducing single-strand breaks that lead to replication-dependent double-strand breaks and subsequent recombination-induced mutations.
Another category of genes showing differential expression in the liver and colon (Fig. 5, SI Appendix, Fig. S9) are stress response genes. Considering the normal, uninfected condition, the gene expression results shown in SI Appendix, Fig. S9A support the notion that oxidative stress is higher in the liver than in the colon, because several genes involved in oxidative defense pathways were expressed at significantly higher levels in the normal liver (e.g., Cat, Sod, Gst). On the other hand, several DNA repair genes, mainly involved in BER, homologous recombination, and MMR as well as genes involved in cell cycle checkpoint regulation, exhibited significantly higher expression levels in the normal colon, which may be related to the higher exposure of colon tissue to food-borne toxicants. Interestingly, MUTYH and NEIL3, which were expressed at higher levels in the normal colon, are directly involved in oxidative stress repair, as MUTYH removes dA mispaired with 8-oxo-dG form the DNA backbone and NEIL3 is potentially involved in the repair of Sp and Gh (28, 29). The latter may directly account for lower levels of Sp and Gh in colon tissue compared to liver, as part of the generally lower levels of DNA lesions in the colon.
In terms of changes associated with H. hepaticus infection, several transcripts involved in DSBR and BER were up-regulated in the liver at 20 w p.i, (Fig. 5), which would be consistent with either hepatic infection by H. hepaticus or hepatic DNA damage caused by humoral mediators from the infected colon. Another example involves expression of GPx2, an intestinal epithelium-specific glutathione peroxidase that reduces hydroperoxides by oxidation of glutathione, which was significantly up-regulated in H. hepaticus-infected colons at both time points (Fig. 5A, SI Appendix, Fig. S9B). This may be functionally important in that the stability of glutathione peroxidases is mainly regulated by their mRNA stability (61). GPx2 was implicated previously in the prevention of inflammatory colitis in the mouse (61, 62). In accordance with our results, it was shown that GPx2 is up-regulated in human colorectal cancer tissue (63, 64) and consequently may serve as a marker for inflammatory bowel disease (65).
Colitis-Associated Changes in Gene Expression Reflect Inflammation-Induced Senescence in H. hepaticus-Infected Rag2-/- Mice.
Many of the changes in gene expression observed in H. hepaticus-infected Rag2-/- mice are consistent with an infection-induced cell senescence, the growth arrest occurring when mitotically capable cells are subjected to any of a variety of stresses, such as the oncogenic stress of inflammation (66). One of the hallmarks of senescence is increased expression of apoptosis-inducing p16 (Cdkn2a) (66), the expression of which is increased significantly in both liver and colon at 20 w p.i (Fig. 5). p16 is up-regulated in premalignant lesions and lost in invasive colon carcinomas by Cdkn2a hypermethylation (67). The fact that H. hepaticus-generated CDT also induces p16 expression (68) suggests that a senescence response in the colon epithelium could result from both the infection itself and the resulting genotoxic stress of the inflammatory reaction. Associated with the increased p16 in these studies is reduced expression of DNA damage response activators ATM and ATR and their downstream partners Chek1 and Hus1. Loss of ATM/ATR could result in senescence and genomic instability leading to cell death (69, 70). Furthermore, both the hyperplasia associated with infection (SI Appendix, Fig. S3A) and the infiltration to activated phagocytes (Fig. 2) leads to a predominance of terminally differentiated cells, which may resemble a state of senescence. Our results support the notion that up-regulation of p16 is an inflammation-induced tumor suppressor mechanism that causes cell cycle arrest and senescence in heavily damaged cells.
In summary, the results of our comprehensive analysis of the biological and chemical pathology of H. hepaticus-induced colitis and colon cancer and H. hepaticus-induced hepatitis in the Rag2-/- mouse model provides a rigorous confirmation of a model of inflammation-mediated carcinogenesis involving infiltration of activated phagocytes at sites of infection, with subsequent generation of reactive oxygen, nitrogen, and halogen species that cause molecular damage leading to cell dysfunction, mutation, and cell death (Fig. 6). We have demonstrated strong correlations among histopathology, phagocyte infiltration, and molecular damage chemistry that suggest a major role for neutrophils in the inflammatory process and that reveal dynamic and unexpected changes in nucleic acid damage that are both tissue- and chemistry-specific. The results also reveal features of the cell stress response that point to microbial pathophysiology and mechanisms of cell senescence as important mechanistic links to cancer.
Materials and Methods
Chemicals.
All chemicals were of the highest available quality and purchased from Sigma-Aldrich unless indicated otherwise. LC-MS grade acetonitrile was obtained from EMD Chemicals. Water purified through a Milli-Q system was used throughout the study.
Animal Husbandry, H. hepaticus Infection, and Tissue Collection.
All experiments were performed in accordance with protocols approved by the Massachusetts Institute of Technology Committee on Animal Care and with the National Institutes of Health Guide for the Care and Use of Laboratory Animals. Rag2-/- mice (129S6/SvEvTac-Rag2tm1Fwa) were used throughout the study and housed in a specific pathogen-free barrier facility. Animals were infected with H. hepaticus (strain 3B1, ATCC51449) grown and confirmed as pure culture as described previously (17, 19). Two groups of mice aged 4–6 wk received 0.2 mL of fresh inoculums (infected group) or sterile media (control group) by gavage every other day for a total of three doses. Tissues from the first group of mice was harvested at 10 w p.i., whereas the second group of mice was euthanized at 20 w p.i. and tissue collected at necropsy. Tissues were removed by standard necropsy procedures. For histopathological evaluation the liver and entire large intestine, including the ileocecocolic junction and colon, were harvested from each mouse. For DNA, RNA, and protein damage analyses, separate groups of mice (n = 5–7 animals per group) were analyzed, respectively, in order to obtain sufficient amounts of tissue. For DNA and RNA analyses, liver lobes and whole colons were immediately submerged in RNAlater solution (Qiagen), placed on dry ice, and stored at -80 °C until further processing. Cecum and colon samples were analyzed by quantitative PCR (qPCR) using H. hepaticus-specific primers to confirm infection status (71).
Histopathologic Evaluation.
All tissues were fixed in 10% formalin, paraffin-embedded, sectioned at 5 μm, and stained with hematoxylin and eosin. Tissue sections were evaluated by a board-certified veterinary pathologist who was blinded to sample identity. The severity of lesions in the liver and large intestine was scored, using an ascending scale from 0 to 4, based on the degree of lesion severity: 0 (absent), 1 (mild), 2 (moderate), 3 (marked), and 4 (severe). For the liver, an HI was calculated by combining individual scores for lobular, portal, and interface hepatitis, as well as the number of lobes (out of a total of four) that contained ≥5 inflammatory lesions. Hepatitis was defined by a hepatitis index equal to or greater than 4. For the large intestine, lesions of inflammation, edema, epithelial defects, crypt atrophy, hyperplasia, and dysplasia were scored at separate locations (ileocecocolic junction, proximal colon, and distal colon). A HAI was calculated by combining individual scores of inflammation, edema, epithelial defects, crypt atrophy, hyperplasia, and dysplasia.
Immunohistochemistry.
Macrophages and neutrophils were identified in intestinal tissue sections by avidin-biotin complex immunohistochemistry using antibodies specific for myeloperoxidase (1:75; MPO; RB-373-A; Thermo Scientific), a neutrophil-specific marker, and F4/80 (1:150; MF48015; Caltag Laboratories), a macrophage-specific marker using previously established protocols (72). Briefly, ileo-cecal and colonic sections from 10 randomly selected animals (five males, five females) per group for each different time point were analyzed. Five randomly selected, well-oriented, full thickness mucosal sections at 40× objective field per mouse were screened to quantify the total number immunopositive cells in the mucosa and submucosa. The data were analyzed using an unpaired t-test and the values were plotted as the average number of positive cells/40X objective field.
Synthesis of Isotope-Labeled Internal DNA and RNA Standards.
Uniformly 15N-labeled internal standards of the DNA damage adducts dI, dX, 8-oxo-dG, εdA, and εdC were synthesized as described previously (25). RNA damage products rI, rX, 8-oxo-rG, εrA, and εrC were synthesized accordingly, using uniformly 15N-labeled ribonucleosides (rA, rG, rC) as starting materials (Cambridge Isotope Laboratories). Uniformly 13C, 15N-labeled internal standards of Sp and Gh were synthesized as described previously (26). Internal standards of 5-chloro-2′-deoxycytidine and 5-chlorocytidine were synthesized according to a protocol as previously described (73). Standards were purified by HPLC, characterized by LC-MS, and quantified by UV absorbance using published extinction coefficients (25, 26).
DNA and RNA Extraction.
To avoid adventitious nucleobase damage, deaminase inhibitors coformycin and tetrahydrouridine, and antioxidants deferoxamine and butylated hydroxytoluene were added throughout sample workup, as described previuosly (25, 74). DNA from approximately 200 mg of colon and liver tissue was extracted using the DNA Isolation Kit for Cells and Tissues (Roche) with modifications as published previously (25). RNA from approximately 100 mg of liver and colon tissue was extracted using the RNeasy Midi Kit (Qiagen) following the manufacturer’s instructions with the following specifications: homogenization was performed for 1 min using a Tissue Ruptor homogenizer (Qiagen) on a medium setting. RNA yields were improved by passing the homogenized sample 5 to 10 times through a blunt 20-gauge needle. An on-column DNase digestion was performed to remove genomic DNA contamination in preparation for gene expression analyses as described below. DNA and RNA concentrations were quantified by UV spectroscopy and samples stored at -80 °C.
Enzymatic Hydrolysis of DNA and RNA.
DNA (200 μg) was hydrolyzed to nucleosides by a combination of nuclease P1, DNase I, phosphodiesterase I (USB-Affymetrix), and alkaline phosphatase in the presence of deaminase inhibitors and antioxidants as described previously (25). The following amounts of internal standards were added to the digestion mixture: 1 pmol [15N]-dX, 1 pmol [15N]-dI, 1 pmol [15N]-8-oxo-dG, 100 fmol [15N]-1-N6-εdA, 100 fmol [15N]-3-N4-εdC, 1 pmol 13C, [15N]-5-Cl-dC, 5 pmol 13C, [15N]-Gh, and 2.9 pmol 13C, [15N]-Sp.
A method for hydrolysis of RNA to ribonucleosides was adapted from a recently developed protocol. RNA (100 μg) was dissolved in 30 μL of sodium acetate buffer (30 mM, pH 6.8) containing 3.3 mM zinc chloride, 33 μg/mL coformycin, 167 μM tetrahydrouridine, 8.33 mM deferoxamine, 1.67 mM butylated hydroxytoluene and each of following internal standards: 10 pmol [15N]-rX, 1 pmol rI, 1 pmol of [15N]-8-oxo-rG, 1 pmol [15N]-1-N6-εrA, 1 pmol [15N]-3-N4-εrC; and 1 pmol [15N,13C]-5-Cl-rC. The RNA was hydrolyzed by incubation with 4 U of nuclease P1 (4 μL of 1 U/μL) at 37 °C for 3 h, followed by dephosphorylation by addition of 40 μL of sodium acetate buffer (30 mM, pH 7.8), 34 U of alkaline phosphatase (2 μL of 17 U/μL) and 0.2 U of phosphodiesterase I (2 μL of 100 U/ml) at 37 °C, overnight. Enzymes were subsequently removed by microfiltration (Nanosep K10 Omega, Pall) and the filtrates concentrated under vacuum.
HPLC Prepurification of DNA and RNA Products.
Hydrolyzed DNA preparations were dissolved in 100 μL water and individual 2′-deoxyribonucleosides were resolved and recollected using an Agilent 1100 series HPLC system at empirically determined retention times (SI Appendix, Fig. S5 and Table S5) as described previously (25). The nucleoside-containing fractions were desiccated under vacuum, redissolved in 45 μL water, and analyzed by LC-MS/MS.
Based on a recently developed protocol, hydrolyzed RNA preparations were dissolved in 100 μL water and individual ribonucleosides were resolved and recollected by HPLC using a Phenomenex Synergi C18 reversed-phase column (250 × 4.6 mm, 4 μm particle size, 80 Å pore size). Gradient elution was performed with acetonitrile in 8 mM sodium acetate buffer at a flow rate of 0.5 mL/ min with a column temperature gradient of 5–45 °C; details are given in the SI Appendix, Table S6. Elution times for damage products are shown in SI Appendix, Fig. S5 and Table S7. The nucleoside-containing fractions were dried under vacuum, redissolved in 45 μL water, and analyzed by LC-MS/MS.
LC-MS/MS Detection of DNA and RNA products.
All measurements other than of Sp and Gh were performed using an Agilent 1100 series HPLC system interfaced with an API3000 tandem quadropole MS (AB Sciex) with a turbo ion spray ion source. Based on our published (25) and recently developed protocols, DNA and RNA samples were resolved on a Hypersil Gold aQ C18 reverse-phase column (150 × 2.1 mm; 3 μm particle size; Thermo Fisher) using 0.1% acetic acid in water (A) and 0.1% acetic acid in acetonitrile (B) with chromatographic parameters as specified in SI Appendix, Tables S5 and S7. The MS was operated in positive ion mode with the first and third quadrupoles fixed to unit resolution. Multiple reaction monitoring (MRM) mode was used for sample detection with dwell times set to 200 ms. Mass spectrometric parameters were optimized for maximal sensitivity and are as described in SI Appendix, Tables S5 and S7. A volume of 20 μL was injected per measurement representing equivalents of 89 μg of DNA and 45 μg of RNA.
Sp and Gh were analyzed using an Agilent 6430 triple quadrupole mass spectrometer interfaced with an Agilent 1200 capillary pumping system. Samples were resolved on a Hypercarb column (100 × 1.0 mm, 5 μm particle size; Thermo Scientific), using 0.3% formic acid in water (A) and 0.3% formic acid in acetonitrile (B) delivered from 2% to 70% B over 15 min at 20 μL/ min. The effluent from the first 5 min from the LC system was diverted to waste to minimize the contamination of the electrospray ionization source. A microelectrospray ionization source was used and the MS was operated in the positive ion mode, with all instrument parameters optimized for maximal sensitivity. MRM mode was used for sample detection with dwell times set to 200 ms.
Linear calibration curves (r2 > 0.99) for the isotope-labeled and unlabeled forms of each of the DNA and RNA damage products were generated with each set of samples. The quantities of DNA and RNA were corrected for contaminating RNA and DNA, respectively, by normalizing signal intensities of canonical 2′-deoxy- and ribo-nucleosides during the HPLC prepurification steps.
Gene Expression.
Analyses were performed using customized qPCR arrays (Qiagen SABiosciensces), which included 95 DNA repair and stress response genes (SI Appendix, Table S8) on an ABI 7900HT qPCR system (384-well block; Life Technologies). All steps were performed according to the manufacturer instructions with the following specifications: RNA quality and quantity was verified using a RNA 6000 Nano LabChip on an Agilent 2100 Bioanalyzer. Reverse transcription was performed with 400 ng of total RNA. Genomic DNA and reverse transcription controls were included in each PCR run to check for DNA contamination and impurities in RNA samples that could affect reverse transcription, respectively. Ct values were normalized to four different control genes (Actb, Gapdh, Hprt1, and 18SrRNA) for comparison of control and infected samples. For comparison of gene expression of liver and colon, samples Ct values were normalized to Gapdh, Hprt1, and 18SrRNA, because Actb showed significant intertissue variation. Data were analyzed using the PCR Array Data Analysis Web Portal (http://www.sabiosciences.com/pcrarraydataanalysis.php) according to the ΔΔCt method.
Statistical Analysis.
Statistical analyses were performed using Graph Pad Prism software (GraphPad Software). Colitis scores were compared using Mann—Whitney U-test for nonparametric data. Because data of adduct levels were not normally distributed, nonparametric Mann—Whitney rank sum test was performed to assess differences between two groups. The Wilcoxon signed-rank test was used for statistical analysis when values in control groups were consistently below the limit of quantification or showed no variance. Correlation analyses were performed using Spearman’s rank order correlation analysis. Analysis of gene expression data was performed using two-tailed Student’s t-test, with P < 0.05 considered statistically significant. SIMCA-P+ (Umetrics, Kinnelon) was used for multivariate data analysis.
Supplementary Material
ACKNOWLEDGMENTS.
The authors thank Drs. Pallavi Lonkar and Bo Pang for synthesis of several internal standards. This work was supported by the National Cancer Institute (CA026731). Bioanalytical studies were performed in the Bioanalytical Facilities Core of the Massachusetts Institute of Technology (MIT) Center for Environmental Health Sciences, which is supported by a grant from the National Institute for Environmental Health Sciences (ES002109). We thank Agilent Technologies for access to the 6430 triple quadrupole mass spectrometer. A.M. was supported by a fellowship of the German Academic Exchange Service (DAAD). E.G.P. was supported by 5T32-ES007020-34 National Institute of Environmental Health Science Training Grant in Toxicology. C.G.K. was supported by a Merck-MIT Fellowship.
Footnotes
The authors declare no conflict of interest.
See Author Summary on page 10753 (volume 109, number 27).
This article contains supporting information online at www.pnas.org/lookup/suppl/doi:10.1073/pnas.1207829109/-/DCSupplemental.
References
- 1.Grivennikov SI, Greten FR, Karin M. Immunity, inflammation, and cancer. Cell. 2010;140:883–899. doi: 10.1016/j.cell.2010.01.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Lonkar P, Dedon PC. Reactive species and DNA damage in chronic inflammation: Reconciling chemical mechanisms and biological fates. Int J Cancer. 2011;128:1999–2009. doi: 10.1002/ijc.25815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Dale DC, Boxer L, Liles WC. The phagocytes: Neutrophils and monocytes. Blood. 2008;112:935–945. doi: 10.1182/blood-2007-12-077917. [DOI] [PubMed] [Google Scholar]
- 4.Terzic J, Grivennikov S, Karin E, Karin M. Inflammation and colon cancer. Gastroenterology. 2010;138:2101–2114. doi: 10.1053/j.gastro.2010.01.058. [DOI] [PubMed] [Google Scholar]
- 5.Dedon PC, Tannenbaum SR. Reactive nitrogen species in the chemical biology of inflammation. Arch Biochem Biophys. 2004;423:12–22. doi: 10.1016/j.abb.2003.12.017. [DOI] [PubMed] [Google Scholar]
- 6.Arnhold J, Flemmig J. Human myeloperoxidase in innate and acquired immunity. Arch Biochem Biophys. 2010;500:92–106. doi: 10.1016/j.abb.2010.04.008. [DOI] [PubMed] [Google Scholar]
- 7.van der Veen BS, de Winther MP, Heeringa P. Myeloperoxidase: Molecular mechanisms of action and their relevance to human health and disease. Antioxid Redox Signal. 2009;11:2899–2937. doi: 10.1089/ars.2009.2538. [DOI] [PubMed] [Google Scholar]
- 8.Winterbourn CC. Biological reactivity and biomarkers of the neutrophil oxidant, hypochlorous acid. Toxicology. 2002;181–182:223–227. doi: 10.1016/s0300-483x(02)00286-x. [DOI] [PubMed] [Google Scholar]
- 9.Valinluck V, Sowers LC. Endogenous cytosine damage products alter the site selectivity of human DNA maintenance methyltransferase DNMT1. Cancer Res. 2007;67:946–950. doi: 10.1158/0008-5472.CAN-06-3123. [DOI] [PubMed] [Google Scholar]
- 10.Pang B, et al. Lipid peroxidation dominates the chemistry of DNA adduct formation in a mouse model of inflammation. Carcinogenesis. 2007;28:1807–1813. doi: 10.1093/carcin/bgm037. [DOI] [PubMed] [Google Scholar]
- 11.Meira LB, et al. DNA damage induced by chronic inflammation contributes to colon carcinogenesis in mice. J Clin Invest. 2008;118:2516–2525. doi: 10.1172/JCI35073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Liao J, et al. Increased susceptibility of chronic ulcerative colitis-induced carcinoma development in DNA repair enzyme Ogg1 deficient mice. Mol Carcinog. 2008;47:638–646. doi: 10.1002/mc.20427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Li Z, Wu J, Deleo CJ. RNA damage and surveillance under oxidative stress. IUBMB Life. 2006;58:581–588. doi: 10.1080/15216540600946456. [DOI] [PubMed] [Google Scholar]
- 14.Feyzi E, et al. RNA base damage and repair. Curr Pharm Biotechnol. 2007;8:326–331. doi: 10.2174/138920107783018363. [DOI] [PubMed] [Google Scholar]
- 15.Laval F, Wink DA. Inhibition by nitric oxide of the repair protein, O6-methylguanine-DNA-methyltransferase. Carcinogenesis. 1994;15:443–447. doi: 10.1093/carcin/15.3.443. [DOI] [PubMed] [Google Scholar]
- 16.Graziewicz M, Wink DA, Laval F. Nitric oxide inhibits DNA ligase activity: Potential mechanisms for NO-mediated DNA damage. Carcinogenesis. 1996;17:2501–2505. doi: 10.1093/carcin/17.11.2501. [DOI] [PubMed] [Google Scholar]
- 17.Erdman SE, et al. CD4+ CD25+ regulatory T lymphocytes inhibit microbially induced colon cancer in Rag2-deficient mice. Am J Pathol. 2003;162:691–702. doi: 10.1016/S0002-9440(10)63863-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Erdman SE, et al. Nitric oxide and TNF-alpha trigger colonic inflammation and carcinogenesis in Helicobacter hepaticus-infected, Rag2-deficient mice. Proc Natl Acad Sci USA. 2009;106:1027–1032. doi: 10.1073/pnas.0812347106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Fox JG, et al. Helicobacter hepaticus sp. nov., a microaerophilic bacterium isolated from livers and intestinal mucosal scrapings from mice. J Clin Microbiol. 1994;32:1238–1245. doi: 10.1128/jcm.32.5.1238-1245.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Fox JG, Ge Z, Whary MT, Erdman SE, Horwitz BH. Helicobacter hepaticus infection in mice: Models for understanding lower bowel inflammation and cancer. Mucosal Immunol. 2011;4:22–30. doi: 10.1038/mi.2010.61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ward JM, et al. Chronic active hepatitis and associated liver tumors in mice caused by a persistent bacterial infection with a novel Helicobacter species. J Natl Cancer Inst. 1994;86:1222–1227. doi: 10.1093/jnci/86.16.1222. [DOI] [PubMed] [Google Scholar]
- 22.Ward JM, et al. Inflammatory large bowel disease in immunodeficient mice naturally infected with Helicobacter hepaticus. Lab Anim Sci. 1996;46:15–20. [PubMed] [Google Scholar]
- 23.Li X, et al. SCID/NCr mice naturally infected with Helicobacter hepaticus develop progressive hepatitis, proliferative typhlitis, and colitis. Infect Immun. 1998;66:5477–5484. doi: 10.1128/iai.66.11.5477-5484.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Soehnlein O, Lindbom L. Phagocyte partnership during the onset and resolution of inflammation. Nat Rev Immunol. 2010;10:427–439. doi: 10.1038/nri2779. [DOI] [PubMed] [Google Scholar]
- 25.Taghizadeh K, et al. Quantification of DNA damage products resulting from deamination, oxidation and reaction with products of lipid peroxidation by liquid chromatography isotope dilution tandem mass spectrometry. Nat Protoc. 2008;3:1287–1298. doi: 10.1038/nprot.2008.119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Yu H, Venkatarangan L, Wishnok JS, Tannenbaum SR. Quantitation of four guanine oxidation products from reaction of DNA with varying doses of peroxynitrite. Chem Res Toxicol. 2005;18:1849–1857. doi: 10.1021/tx050146h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Neeley WL, Essigmann JM. Mechanisms of formation, genotoxicity, and mutation of guanine oxidation products. Chem Res Toxicol. 2006;19:491–505. doi: 10.1021/tx0600043. [DOI] [PubMed] [Google Scholar]
- 28.Liu M, et al. The mouse ortholog of NEIL3 is a functional DNA glycosylase in vitro and in vivo. Proc Natl Acad Sci USA. 2010;107:4925–4930. doi: 10.1073/pnas.0908307107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.David SS, O’Shea VL, Kundu S. Base-excision repair of oxidative DNA damage. Nature. 2007;447:941–950. doi: 10.1038/nature05978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Rogers AB, et al. Hepatocellular carcinoma associated with liver-gender disruption in male mice. Cancer Res. 2007;67:11536–11546. doi: 10.1158/0008-5472.CAN-07-1479. [DOI] [PubMed] [Google Scholar]
- 31.Abraham C, Cho JH. Inflammatory bowel disease. N Engl J Med. 2009;361:2066–2078. doi: 10.1056/NEJMra0804647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Feagins LA, Souza RF, Spechler SJ. Carcinogenesis in IBD: Potential targets for the prevention of colorectal cancer. Nat Rev Gastroenterol Hepatol. 2009;6:297–305. doi: 10.1038/nrgastro.2009.44. [DOI] [PubMed] [Google Scholar]
- 33.Erdman SE, Poutahidis T. Roles for inflammation and regulatory T cells in colon cancer. Toxicol Pathol. 2010;38:76–87. doi: 10.1177/0192623309354110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Chen C, et al. Metabolomics reveals that hepatic stearoyl-CoA desaturase 1 downregulation exacerbates inflammation and acute colitis. Cell Metab. 2008;7:135–147. doi: 10.1016/j.cmet.2007.12.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Naito Y, Takagi T, Yoshikawa T. Molecular fingerprints of neutrophil-dependent oxidative stress in inflammatory bowel disease. J Gastroenterol. 2007;42:787–798. doi: 10.1007/s00535-007-2096-y. [DOI] [PubMed] [Google Scholar]
- 36.Natsui M, et al. Selective depletion of neutrophils by a monoclonal antibody, RP-3, suppresses dextran sulphate sodium-induced colitis in rats. J Gastroenterol Hepatol. 1997;12:801–808. doi: 10.1111/j.1440-1746.1997.tb00375.x. [DOI] [PubMed] [Google Scholar]
- 37.Kruidenier L, et al. Imbalanced secondary mucosal antioxidant response in inflammatory bowel disease. J Pathol. 2003;201:17–27. doi: 10.1002/path.1408. [DOI] [PubMed] [Google Scholar]
- 38.Kruidenier L, Kuiper I, Lamers CB, Verspaget HW. Intestinal oxidative damage in inflammatory bowel disease: Semi-quantification, localization, and association with mucosal antioxidants. J Pathol. 2003;201:28–36. doi: 10.1002/path.1409. [DOI] [PubMed] [Google Scholar]
- 39.Robinson CE, et al. Regulation of neutrophils in ulcerative colitis by colonic factors: A possible mechanism of neutrophil activation and tissue damage. J Lab Clin Med. 1997;130:590–602. doi: 10.1016/s0022-2143(97)90109-8. [DOI] [PubMed] [Google Scholar]
- 40.Takeshita J, et al. Myeloperoxidase generates 5-chlorouracil in human atherosclerotic tissue: A potential pathway for somatic mutagenesis by macrophages. J Biol Chem. 2006;281:3096–3104. doi: 10.1074/jbc.M509236200. [DOI] [PubMed] [Google Scholar]
- 41.Kawai Y, et al. Endogenous formation of novel halogenated 2′-deoxycytidine. Hypohalous acid-mediated DNA modification at the site of inflammation. J Biol Chem. 2004;279:51241–51249. doi: 10.1074/jbc.M408210200. [DOI] [PubMed] [Google Scholar]
- 42.Lao VV, et al. Incorporation of 5-chlorocytosine into mammalian DNA results in heritable gene silencing and altered cytosine methylation patterns. Carcinogenesis. 2009;30:886–893. doi: 10.1093/carcin/bgp060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Valinluck V, Sowers LC. Inflammation-mediated cytosine damage: A mechanistic link between inflammation and the epigenetic alterations in human cancers. Cancer Res. 2007;67:5583–5586. doi: 10.1158/0008-5472.CAN-07-0846. [DOI] [PubMed] [Google Scholar]
- 44.Ravanat JL, et al. Cellular background level of 8-oxo-7,8-dihydro-2′-deoxyguanosine: An isotope based method to evaluate artefactual oxidation of DNA during its extraction and subsequent work-up. Carcinogenesis. 2002;23:1911–1918. doi: 10.1093/carcin/23.11.1911. [DOI] [PubMed] [Google Scholar]
- 45.Gedik CM, Collins A. Establishing the background level of base oxidation in human lymphocyte DNA: Results of an interlaboratory validation study. FASEB J. 2005;19:82–84. doi: 10.1096/fj.04-1767fje. [DOI] [PubMed] [Google Scholar]
- 46.Sipowicz MA, et al. Increased oxidative DNA damage and hepatocyte overexpression of specific cytochrome P450 isoforms in hepatitis of mice infected with Helicobacter hepaticus. Am J Pathol. 1997;151:933–941. [PMC free article] [PubMed] [Google Scholar]
- 47.Luo W, Muller JG, Rachlin EM, Burrows CJ. Characterization of spiroiminodihydantoin as a product of one-electron oxidation of 8-Oxo-7,8-dihydroguanosine. Org Lett. 2000;2:613–616. doi: 10.1021/ol9913643. [DOI] [PubMed] [Google Scholar]
- 48.Luo W, Muller JG, Rachlin EM, Burrows CJ. Characterization of hydantoin products from one-electron oxidation of 8-oxo-7,8-dihydroguanosine in a nucleoside model. Chem Res Toxicol. 2001;14:927–938. doi: 10.1021/tx010072j. [DOI] [PubMed] [Google Scholar]
- 49.Niles JC, Wishnok JS, Tannenbaum SR. Spiroiminodihydantoin and guanidinohydantoin are the dominant products of 8-oxoguanosine oxidation at low fluxes of peroxynitrite: Mechanistic studies with 18O. Chem Res Toxicol. 2004;17:1510–1519. doi: 10.1021/tx0400048. [DOI] [PubMed] [Google Scholar]
- 50.Hailer MK, Slade PG, Martin BD, Sugden KD. Nei deficient Escherichia coli are sensitive to chromate and accumulate the oxidized guanine lesion spiroiminodihydantoin. Chem Res Toxicol. 2005;18:1378–1383. doi: 10.1021/tx0501379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Henderson PT, et al. The hydantoin lesions formed from oxidation of 7,8-dihydro-8-oxoguanine are potent sources of replication errors in vivo. Biochemistry. 2003;42:9257–9262. doi: 10.1021/bi0347252. [DOI] [PubMed] [Google Scholar]
- 52.Nair J, De Flora S, Izzotti A, Bartsch H. Lipid peroxidation-derived etheno-DNA adducts in human atherosclerotic lesions. Mutat Res. 2007;621:95–105. doi: 10.1016/j.mrfmmm.2007.02.013. [DOI] [PubMed] [Google Scholar]
- 53.Nair J, et al. Increased etheno-DNA adducts in affected tissues of patients suffering from Crohn’s disease, ulcerative colitis, and chronic pancreatitis. Antioxid Redox Signal. 2006;8:1003–1010. doi: 10.1089/ars.2006.8.1003. [DOI] [PubMed] [Google Scholar]
- 54.Obtulowicz T, et al. Aberrant repair of etheno-DNA adducts in leukocytes and colon tissue of colon cancer patients. Free Radic Biol Med. 2010;49:1064–1071. doi: 10.1016/j.freeradbiomed.2010.06.027. [DOI] [PubMed] [Google Scholar]
- 55.Godschalk RW, et al. Decreased levels of lipid peroxidation-induced DNA damage in the onset of atherogenesis in apolipoprotein E deficient mice. Mutat Res. 2007;621:87–94. doi: 10.1016/j.mrfmmm.2007.02.012. [DOI] [PubMed] [Google Scholar]
- 56.Kirkali G, et al. Evidence for upregulated repair of oxidatively induced DNA damage in human colorectal cancer. DNA Repair. 2011;10:1114–1120. doi: 10.1016/j.dnarep.2011.08.008. [DOI] [PubMed] [Google Scholar]
- 57.Kim JJ, et al. Helicobacter pylori impairs DNA mismatch repair in gastric epithelial cells. Gastroenterology. 2002;123:542–553. doi: 10.1053/gast.2002.34751. [DOI] [PubMed] [Google Scholar]
- 58.Machado AM, et al. Helicobacter pylori infection induces genetic instability of nuclear and mitochondrial DNA in gastric cells. Clin Cancer Res. 2009;15:2995–3002. doi: 10.1158/1078-0432.CCR-08-2686. [DOI] [PubMed] [Google Scholar]
- 59.Toller IM, et al. Carcinogenic bacterial pathogen Helicobacter pylori triggers DNA double-strand breaks and a DNA damage response in its host cells. Proc Natl Acad Sci USA. 2011;108:14944–14949. doi: 10.1073/pnas.1100959108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Ge Z, et al. Bacterial cytolethal distending toxin promotes the development of dysplasia in a model of microbially induced hepatocarcinogenesis. Cell Microbiol. 2007;9:2070–2080. doi: 10.1111/j.1462-5822.2007.00939.x. [DOI] [PubMed] [Google Scholar]
- 61.Brigelius-Flohe R, Kipp A. Glutathione peroxidases in different stages of carcinogenesis. Biochim Biophys Acta. 2009;1790:1555–1568. doi: 10.1016/j.bbagen.2009.03.006. [DOI] [PubMed] [Google Scholar]
- 62.Esworthy RS, et al. Mice with combined disruption of Gpx1 and Gpx2 genes have colitis. Am J Physiol Gastrointest Liver Physiol. 2001;281:G848–G855. doi: 10.1152/ajpgi.2001.281.3.G848. [DOI] [PubMed] [Google Scholar]
- 63.Murawaki Y, et al. Aberrant expression of selenoproteins in the progression of colorectal cancer. Cancer Lett. 2008;259:218–230. doi: 10.1016/j.canlet.2007.10.019. [DOI] [PubMed] [Google Scholar]
- 64.Florian S, et al. Cellular and subcellular localization of gastrointestinal glutathione peroxidase in normal and malignant human intestinal tissue. Free Rad Res. 2001;35:655–663. doi: 10.1080/10715760100301181. [DOI] [PubMed] [Google Scholar]
- 65.Te Velde AA, Pronk I, de Kort F, Stokkers PC. Glutathione peroxidase 2 and aquaporin 8 as new markers for colonic inflammation in experimental colitis and inflammatory bowel diseases: An important role for H2O2? Eur J Gastroenterol Hepatol. 2008;20:555–560. doi: 10.1097/MEG.0b013e3282f45751. [DOI] [PubMed] [Google Scholar]
- 66.Rodier F, Campisi J. Four faces of cellular senescence. J Cell Biol. 2011;192:547–556. doi: 10.1083/jcb.201009094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Kriegl L, et al. Up and downregulation of p16(Ink4a) expression in BRAF-mutated polyps/adenomas indicates a senescence barrier in the serrated route to colon cancer. Mod Pathol. 2011;24:1015–1022. doi: 10.1038/modpathol.2011.43. [DOI] [PubMed] [Google Scholar]
- 68.Kosar M, et al. Senescence-associated heterochromatin foci are dispensable for cellular senescence, occur in a cell type- and insult-dependent manner and follow expression of p16(ink4a) Cell Cycle. 2011;10:457–468. doi: 10.4161/cc.10.3.14707. [DOI] [PubMed] [Google Scholar]
- 69.Wong KK, et al. Telomere dysfunction and Atm deficiency compromises organ homeostasis and accelerates ageing. Nature. 2003;421:643–648. doi: 10.1038/nature01385. [DOI] [PubMed] [Google Scholar]
- 70.Murga M, et al. A mouse model of ATR-Seckel shows embryonic replicative stress and accelerated aging. Nat Genet. 2009;41:891–898. doi: 10.1038/ng.420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Ge Z, White DA, Whary MT, Fox JG. Fluorogenic PCR-based quantitative detection of a murine pathogen, Helicobacter hepaticus. J Clin Microbiol. 2001;39:2598–2602. doi: 10.1128/JCM.39.7.2598-2602.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Sheh A, et al. 17beta-estradiol and tamoxifen prevent gastric cancer by modulating leukocyte recruitment and oncogenic pathways in Helicobacter pylori-infected INS-GAS male mice. Cancer Prev Res (Phila) 2011;4:1426–1435. doi: 10.1158/1940-6207.CAPR-11-0219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Ryu EK, MacCoss M. New procedure for the chlorination of pyrimidine and purine nucleosides. J Org Chem. 1981;46:2819–2823. [Google Scholar]
- 74.Dong M, Dedon PC. Relatively small increases in the steady-state levels of nucleobase deamination products in DNA from human TK6 cells exposed to toxic levels of nitric oxide. Chem Res Toxicol. 2006;19:50–57. doi: 10.1021/tx050252j. [DOI] [PMC free article] [PubMed] [Google Scholar]
