

Polyketides produced by bacteria and fungi represent an extremely rich source of biologically active compounds that find wide-ranging applications as antibiotics, immunosuppresants, antitumor agents, and antiparasitic agents. The striking structural diversity of polyketides that gives rise to these wide-ranging applications is dictated both by the polyketide synthase (PKS) responsible for assembling the carbon backbone of the molecule, and by the tailoring enzymes (such as glycosyl transferases and hydroxylases) that modify the polyketide ring. Seminal work in our understanding of the complex polyketides came a number of years ago with the revelation that erythromycin produced by Saccharopolyspora erythraea is assembled by a modular PKS (1, 2). The first module is responsible for priming erythromycin with propionyl CoA and catalyzing a decarboxylative condensation with methylmalonyl CoA. The remaining five modules catalyze successive condensations with methylmalonyl CoA before a terminal thioesterase (TE) domain releases the aglycone, deoxyerythronolide B. The presence or absence of ketoreductase (KR), dehydratase (DH), and enoyl reductase (ER) catalytic domains within the module controls the extent to which the β-carbonyl of the growing polyketide chain is processed. In this issue of the Proceedings researchers at the University of Minnesota have revealed that a similar modular PKS is responsible for ketolide biosynthesis in Streptomyces venezulae (Fig. 1) (3).
Figure 1.
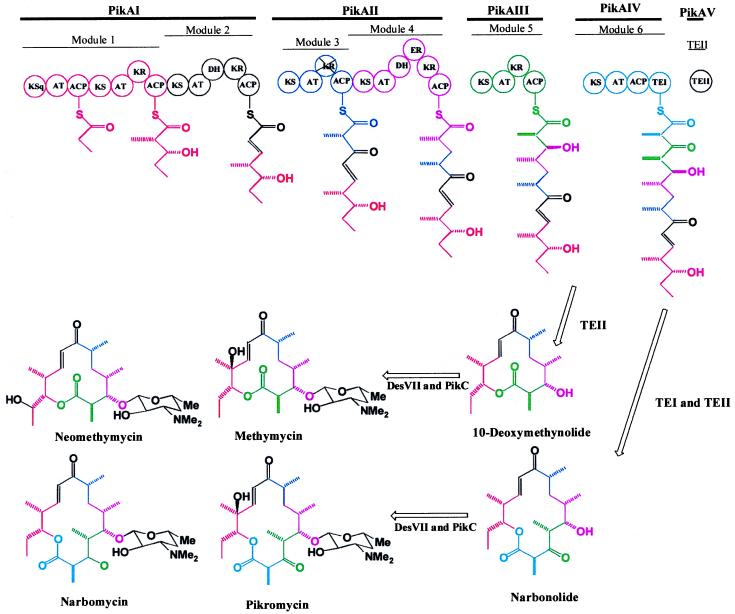
The role of the pik gene cluster in the biosynthesis of methymycin, neomethycin, narbomycin, and pikromycin (see text for details). Figure adapted from Xue et al. (3).
The discovery of the modular polyketide synthase has prompted two questions to be addressed: how is structural diversity achieved by using a modular type I PKS and can such a PKS be manipulated to access additional structural diversity? Recent analysis of additional modular PKS systems, including those involved in rapamycin, rifamycin, and niddamycin biosynthesis, has shown that a PKS polypeptide can contain 1–6 modules (4–6). The total number of modules controls the length of the polyketide chain that is formed before cyclization, whereas the catalytic domains of each module control the level of oxidation along the polyketide chain. The specificity of the acyl transferase domain within each module, which specifies for a malonyl CoA, methylmalonyl CoA, or ethylmalonyl CoA extender unit, controls the level of branching along polyketide chain (7, 8). Additional structural diversity is accomplished by the loading domains in the first module, which use a wide range of starter units such as 3-amino-5-hydroxybenzoic acid, isobutyryl CoA, and 3,4-dihydroxycyclohexanecarboxylic acid (4, 5, 9). Enzymes that catalyze the cyclization of the polyketide product also can contribute significantly to the structural diversity (10).
In addition to these studies of natural PKSs, hybrid polyketides have been generated by a variety of techniques. Ring-contracted polyketides have been produced by movement of the terminal thioesterase to different modules of the PKS (11, 12), whereas analogues of the parent polyketide product have been produced by deletions, switches, and insertions of various catalytic domains and modules (13–16).
To date these studies have demonstrated that a single hybrid or natural polyketide synthase system typically is responsible for formation of a single product (thus S. hygroscopicus, which produces at least four complex polyketides, contains four separate PKS gene clusters; ref. 17). In addition, it has been shown that hybrid polyketide products are not always effective substrates for post-polyketide tailoring enzymes, as these often exhibit a strict substrate specificity (1, 15). In this issue of the Proceedings, Xue et al. (3) report a unique observation that a single PKS within a cluster of biosynthetic genes in S. venezulae is responsible for formation of both 12- and 14-membered macrolactones (3). In addition, the researchers have determined that two of the postpolyketide modifying enzymes have an unusually broad substrate and regiochemical specificity.
The first significant finding by Xue et al. is that four polypeptides, PikAI, PikAII, PikAIII and PikAIV, are involved in the production of the 14-membered ring macrolactone, narbonolide (the precursor to narbomycin and pikromycin), whereas just PikAI, PikAII, and PikAIII are required for production of the 12-membered macrolactone ring 10-deoxymethynolide (a precursor to methymycin and neomethymycin) (Fig. 1). In the pikromycin PKS the last two modules (modules 5 and 6) are encoded by separate ORFs, whereas previously identified PKS clusters that produce 14-membered macrolides contain both of these within a single ORF (1, 18). The researchers suggest that it is this feature that allows for early termination of the polyketide chain.
Xue et al. were faced with an intriguing question regarding the manner in which the nascent polyketide product is released from PikAIII and cyclized to give the 12-membered macrolide because, unlike PikAIV, this polypeptide does not contain a terminal TE domain. A hypothesis that a type II monofunctional thioesterase (Pik TEII encoded by pikAV) plays an important role was supported by the demonstration that inactivation of the corresponding pikAV gene led to a mutant that generated less than 5% of the 12-membered macrolide products. Xue et al. point out that various engineered polyketides of different chain lengths have been made by repositioning of the TE catalytic domain to different modules on a PKS (11, 12, 19), but that each positioning of the TE leads to only one discrete polyketide chain length product. The researchers raise an intriguing possibility that TEIIs along with novel PKS systems may allow for the simultaneous production of polyketides of different chain lengths. It should be noted, however, that many natural polyketide and nonribosomal polypeptide biosynthetic clusters (including the erythromycin PKS gene cluster) already contain homologues of Pik TEII (20), yet under most conditions produce predominantly one macrolide product. The utility of a Pik TEII in combinatorial biology technologies clearly will require further investigation.
Xue et al. also discovered that 5% of the narbonolide-derived polyketides were produced in the pikAV mutant and argue that Pik TEII plays a role in release of the nascent polyketide chain from both PikAIII and PikAIV. However, PikAIV already contains a TE domain, and a variety of in vitro studies (albeit with the erythromycin PKS TE) have clearly shown that this domain can function without a TEII (21). This observation, therefore, suggests that the exact role of Pik TEII is unclear. The observations of Xue et al. could equally implicate Pik TEII in having a role in editing (Heinz G. Floss, personal communication), possibly removing aberrant polyketides intermediates that for some reason have caused the catalytic machinery of pik PKS to stall. Such a role would explain why there is an abrogation of both 12-membered and 14-membered macrolides in a Pik TEII mutant. A similar conclusion has been drawn about the role of a TEII in tylosin biosynthesis in S. fradiae (Eric Cundliffe and Peter F. Leadley, personal communication), where it has been shown that only 10% of wild-type tylosin production is obtained in a TEII null mutant (A.R. Butler and E.C. Cundliffe, personal communication).
Regardless of its exact role, Xue et al. have clearly demonstrated that Pik TEII plays a major role in production of both 12- and 14-membered macrolide products in S. venezuelae. These observations undoubtedly will provide an impetus for more detailed investigations of the role of the class of enzymes in catalyzing the release of either the fully extended polyketide products, or earlier polyketide intermediates, from a PKS.
The manner in which specific fermentation medium gives rise to differences in the ratio of the 12- and 14-membered macrolide products produced by S. venezuelae cannot be clearly determined from the current analysis of the pik cluster. Xue et al. consider that one possible explanation may be differential expression of PikAIV.
An additional significant finding from the analysis of the pik gene cluster is the observation that the glycosyl transferase DesVII is able to catalyze the transfer of desosamine onto both 12- and 14-membered polyketide aglycones. Furthermore, the researchers indicate that DesVII has an ability to use at least one alternate deoxysugar. An equally important finding is that there is only one cytochrome P450 hydroxylase (PikC) in the entire pik cluster. This enzyme thus is likely responsible for hydroxylation of both 12- and 14-membered macrolides and for catalyzing hydroxylation at two different carbons of the 12-membered macrolide to generate methymycin and neomethymycin. In the latter case, PikC is responsible for formation of both a secondary and a tertiary alcohol (Fig. 1). The broad substrate specificities of both DesVII and PikC, and the relaxed regiochemical specificity of PikC, clearly enable the relatively small cluster of genes (less than 60 kB) to be able to produce four different macrolide products and contrast the more stringent substrate specificity of hydroxylases and glycosyl transferases of other PKS systems.
A number of 6-deoxerythronolide B and rapamycin analogs that have been generated by a variety of approaches are not efficient substrates for the enzymes that catalyze the postpolyketide modification steps, which are essential for the biological activity of the polyketide (1, 15, 22). The work of Xue et al. raises the possibility that a variety of engineered polyketide structures may be glycosylated by using DesVII and hydroxlyated by using PikC. Although these enzymes most likely will not prove to be effective on all polyketide products generated from a combinatorial biology technology, they represent an important step toward finding postpolyketide modification enzymes with a broader substrate and regiochemical specificity. For instance, a gene shuffling approach using either desVII or pikC and their corresponding homologues from other PKS systems may allow for the directed evolution of such properties for glycosyl tranferases and hydroxylases, respectively (23).
The pik gene cluster is neither the first, nor likely the last, modular polyketide synthase gene cluster to be sequenced and analyzed. Nonetheless, as the work by Xue et al. elegantly demonstrates, the analyses have provided a tremendous insight into the manner in which nature capitalizes on the modular architecture of a complex polyketide synthase to obtain metabolic diversity. This information, together with a powerful new set of genetic tools, undoubtedly will be used in the combinatorial biology technologies that are actively and aggressively being pursued both in academic and industrial settings.
Acknowledgments
I thank Leonard Katz (Abbot Laboratories), Heinz Floss (University of Washington), and Eric Cunliffe (University of Leicester) for helpful conversations regarding modular polyketide synthases. My laboratory’s work on these systems is supported by the National Institutes of Health (GM50542) and the National Science Foundation (MCB9418581).
Footnotes
The companion to this commentary begins on page 12111 in issue 21 of volume 95.
References
- 1. Donadio S, Staver M J, McAlpine J B, Swanson S J, Katz L. Science. 1991;252:675–679. doi: 10.1126/science.2024119. [DOI] [PubMed] [Google Scholar]
- 2.Cortes J, Haydock S F, Roberts G A, Bevitt D J, Leadley P F. Nature (London) 1990;348:176–178. doi: 10.1038/348176a0. [DOI] [PubMed] [Google Scholar]
- 3.Xue Y, Zhao L, Liu H-W, Sherman D H. Proc Natl Acad Sci USA. 1998;95:12111–12116. doi: 10.1073/pnas.95.21.12111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Schwecke T, Aparicio J F, Molner I, König A, Khaw L E, Haydock S F, Oliynyk M, Caffrey P, Cortes J, Lester J B, et al. Proc Natl Acad Sci USA. 1995;92:7839–7843. doi: 10.1073/pnas.92.17.7839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.August P R, Lang L T, Yoon Y J, Ning S, Müller R, Yu T-W, Taylor M, Hoffmann D, Kim C G, Zhang X, et al. Chem Biol. 1998;5:69–79. doi: 10.1016/s1074-5521(98)90141-7. [DOI] [PubMed] [Google Scholar]
- 6.Kakavas S J, Katz L, Stassi D. J Bacteriol. 1997;179:7515–7522. doi: 10.1128/jb.179.23.7515-7522.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Aparicio J F, Molnar I, Schwecke T, Konig A, Haydock S F, Khaw L E, Staunton J, Leadley P F. Gene. 1996;169:9–16. doi: 10.1016/0378-1119(95)00800-4. [DOI] [PubMed] [Google Scholar]
- 8.Stassi D L, Kakavas S J, Reynolds K A, Gunawardana G, Swanson S, Zeidner D, Jackson M, Liu H, Buko A, Katz L. Proc Natl Acad Sci USA. 1998;95:7305–7309. doi: 10.1073/pnas.95.13.7305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Denoya C D, Fedechko R W, Hafner E W, McArthur H A I, Morgenstern M R, Skinner D D, Stutzman-Engwall K, Wax R G, Wernau W C. J Bacteriol. 1995;177:3504–3511. doi: 10.1128/jb.177.12.3504-3511.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Konig A, Schwecke T, Molnar I, Böhm G A, Lowden P A S, Staunton J. Eur J Biochem. 1997;247:526–534. doi: 10.1111/j.1432-1033.1997.00526.x. [DOI] [PubMed] [Google Scholar]
- 11.Cortes J, Wiesmann K E H, Roberts G A, Brown M J, Staunton J, Leadley P F. Science. 1995;268:1487–1489. doi: 10.1126/science.7770773. [DOI] [PubMed] [Google Scholar]
- 12.Kao C M, Luo G, Katz L, Cane D E, Khosla C. J Am Chem Soc. 1995;117:9105–9106. [Google Scholar]
- 13.Oliynyk M, Brown M J B, Cortes J, Staunton J, Leadley P F. Chem Biol. 1996;3:833–839. doi: 10.1016/s1074-5521(96)90069-1. [DOI] [PubMed] [Google Scholar]
- 14.McDaniel R, Kao C M, Fu H, Hevezi P, Gustafsson C, Betlach M, Ashley G, Cane D E, Khosla C. J Am Chem Soc. 1997;119:4309–4310. [Google Scholar]
- 15.Ruan X, Pereda A, Stassi D L, Zeidner D, Summers R G, Jackson M, Shivakumar A, Kakavas S, Stavers M J, Donadio S, Katz L. J Bacteriol. 1997;179:6416–6425. doi: 10.1128/jb.179.20.6416-6425.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Marsden A F A, Wilkinson B, Cortes J, Dunster N J, Staunton J, Leadley P F. Science. 1998;279:199–202. doi: 10.1126/science.279.5348.199. [DOI] [PubMed] [Google Scholar]
- 17.Ruan X, Stassi D, Lax S, Katz L. Gene. 1997;203:1–9. doi: 10.1016/s0378-1119(97)00450-2. [DOI] [PubMed] [Google Scholar]
- 18.Swan D G, Rodriguez A M, Vilches C, Mendnez C, Salas J A. Mol Gen Genet. 1994;242:358–362. doi: 10.1007/BF00280426. [DOI] [PubMed] [Google Scholar]
- 19.Kao C M, Luo G, Katz L, Cane D E, Khosla C. J Am Chem Soc. 1996;118:9184–9185. [Google Scholar]
- 20.Marahiel M A, Stachehaus T, Mootz M D. Chem Rev. 1997;97:2651–2673. doi: 10.1021/cr960029e. [DOI] [PubMed] [Google Scholar]
- 21.Aggarwal, R., Caffrey, P., Leadley, P. F., Smith, C. J. & Staunton, J. (1995) J. Chem. Soc. Chem Commun., 1519–1520.
- 22.Khaw L E, Böhm G A, Metcalfe S, Staunton J, Leadlay P F. J Bacteriol. 1998;180:809–814. doi: 10.1128/jb.180.4.809-814.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Stemmer W P C. Proc Natl Acad Sci USA. 1994;91:10747–10751. doi: 10.1073/pnas.91.22.10747. [DOI] [PMC free article] [PubMed] [Google Scholar]