

Abstract
Ferritins are ubiquitous and can be found in practically all organisms that utilize Fe. They are composed of 24 subunits forming a hollow sphere with an inner cavity of ~80 Å in diameter. The main function of ferritin is to oxidize the cytotoxic Fe2+ ions and store the oxidized Fe in the inner cavity. It has been established that the initial step of rapid oxidation of Fe2+ (ferroxidation) by H-type ferritins, found in vertebrates, occurs at a diiron binding center, termed ferroxidase center. In bacterial ferritins, however, X-ray crystallographic evidence and amino-acid sequence analysis revealed a trinuclear Fe binding center comprising a binuclear Fe binding center (sites A and B), homologous to the ferroxidase center of H-type ferritin, and an adjacent mononuclear Fe binding site (site C). In an effort to obtain further evidence supporting the presence of a trinuclear Fe binding center in bacterial ferritins and to gain information on the states of the iron bound to the trinuclear center, bacterial ferritin from Desulfovibrio vulgaris (DvFtn) and its E130A variant were loaded with sub-stoichiometric amounts of Fe2+ and products were characterized by Mössbauer and EPR spectroscopy. Four distinct Fe species were identified: a paramagnetic diferrous species, a diamagnetic diferrous species, a mixed valence Fe2+Fe3+ species and a mononuclear Fe2+ species. The latter three species were detected in the wild-type DvFtn, while the paramagnetic diferrous species was detected in the E130A variant. These observations can be rationally explained by the presence of a trinuclear Fe binding center, and the four Fe species can be properly assigned to the three Fe binding sites. Further, our spectroscopic data suggest that (1) the fully occupied trinuclear center supports an all ferrous state, (2) site B and C are bridged by a μ-OH group forming a diiron sub-center within the trinuclear center, and (3) this sub-center can afford both a mixed valence Fe2+Fe3+ state and a diferrous state. Mechanistic insights provided by these new findings are discussed and a minimal mechanistic scheme involving O-O bond cleavage is proposed.
Ferritin is a natural nano-protein cage that catalyzes the oxidation of the cytotoxic Fe2+ ions and stores the oxidized Fe in its inner protein cavity as a ferrihydrite phosphate mineral core.1–5 It is ubiquitous and may be found in all organisms that utilize Fe. Each ferritin molecule comprises 24 subunits of approximately 20 kDa subunit mass that are arranged into a hollow spherical protein shell with an outer diameter of ~12 nm and an inner diameter of ~8 nm.6–12 It can store up to thousands of Fe atoms in its cavity. Despite the lack of homology in their amino acid sequences, all ferritin subunits from different organisms share a common tertiary structure of a four-α-helix bundle. The 24-mer ferritin protein shells generally exhibit an octahedral 432 symmetry,13,14 with the exception of the Archaeoglobus fulgidus ferritin, which shows a tetrahedral 23 symmetry.11 Ferritin molecules from higher eukaryotic organisms, like vertebrates, are heteropolymers comprising two types of subunits designated as the H and L subunit. The H subunit catalyzes the initial step of rapid oxidation of Fe2+ (ferroxidation)15 and directs the oxidized Fe3+ into the inner cavity.16 The L subunit promotes the formation of the mineral core. The composition of H and L in a ferritin molecule depends on its source of origin. Ferritin molecules from brain and heart are rich in H subunit, whereas molecules from liver and spleen are low in H. A third type of subunit, designated as M, found in amphibians, is homologous to H. Prokaryotic ferritins, such as those from bacteria and archaea, are homopolymers composed of subunits more homologous to H than to L.
Extensive biochemical and physical investigations performed on human H ferritin (HuHF)5,6,15,17 and bull frog M ferritin (BfMF)18–23 firmly established that H-type subunits utilize molecular oxygen to rapidly oxidize Fe2+ ions, in a pair wise fashion, at a binuclear metal binding center, termed the ferroxidase center. X-ray crystallographic studies of HuHF6 and BfMF8 revealed that this substrate diiron binding center in H-type ferritins shares certain structural similarities with the cofactor site of the carboxylate-bridged diiron enzymes, such as the R2 subunit of the Escherichia coli ribonucleotide reductase,24 and the soluble methane monooxygenase.25 Reaction of O2 with the diiron enzymes yields H2O as the O2 reduction product and generates high-valent diiron intermediates for organic substrate oxidations.26,27 In contrast, reaction of O2 with H-type ferritins yields H2O219,28 and μ-oxo/μ-hydroxo diferric products as precursors to the mineral core.18 X-ray crystallographic studies and amino-acid sequence analysis indicated that L subunit lacks such a diiron binding ferroxidase center.7
In baceria, two classes of 24-meric ferritins have been identified: the canonical bacterial ferritin (Ftn) and the heme-containing bacterioferritin (Bfr).29 In E. coli, both classes of ferritins are found and have been studied extensively. The E. coli bacterioferritin (EcBfr) does have a diiron binding center30 that is structurally homologous to the diiron cofactor of the R2 subunit of E. coli ribonucleotide reductase. The Fe storage function of EcBfr, however, has not been clearly established. On the other hand, the Fe storage function of E. coli bacterial ferritin (EcFtnA) has been demonstrated both in vitro31 and in vivo.32 Amino-acid sequence comparisons and X-ray crystallographic studies9,33 showed that EcFtnA contains a substrate Fe binding site that is homologous to the diiron binding ferroxidase center of H-type ferritins (sites A and B). And, intriguingly, the X-ray crystallographic studies of EcFtnA7,9,33 further revealed the presence of a third Fe binding site (site C) in the vicinity of the H-type diiron ferroxidase center. Moreover, EcFtnA was found to oxidize approximately 3.5 Fe2+ ions per O2 molecule consumed,34 indicating that the O2 reduction product for EcFtnA ferroxidation may not be H2O2 and thus suggesting a ferroxidation mechanism that is distinct from that of H-type ferritins. Previous Mössbauer investigations on the initial Fe2+ oxidation by EcFtnA and its variants35,36 have provided data that suggest formation of μ-oxo/μ-hydroxo diferric complexes and mononuclear Fe3+ species as early products.
The presence of a trinuclear Fe binding center is not unique for EcFtnA and, in fact, may be a common feature for the Ftn-type ferritins of prokaryotes.29 Similar trinuclear Fe binding centers have been reported for the ferritins from Archaeoglobus fulgidus (AfFtn)11 and Pyrococcus furiosus (PfFtn).12 For the past decades, extensive investigations have been performed on eukaryotic H-type ferritins. A wealth of information concerning the mechanistic and functional aspects of the diiron ferroxidation center in H-type ferritins is known.37 In contrast, less is known for the trinuclear ferroxidase center of the prokaryotic Ftn.37 Despite their structural similarity, two distinct functions, gated iron pore versus catalytic oxidoreduction, have been proposed for the ferroxidase centers of EcFtn34 and PfFtn,38 respectively.
On the basis of its primary sequence, the ferritin from the sulfate reducer Desulfovibrio vulgaris (DvFtn) belongs to the prokaryotic Ftn class. Figure 1 compares the amino-acid sequence of DvFtn with those of EcFtnA and PfFtn. All residues (E17, E49, E50, H53, E94, E126, E129, and E130) (EcFtnA numbering) identified as ligands for the trinuclear Fe-binding center in EcFtnA33 are conserved in PfFtn and DvFtn. On the other hand, comparing the sequences of the bacterial ferritins with those of the two vertebrate H-type ferritins HuHF and BfMF (Figure 1), it is clear that residues E129 and E130 (EcFtnA numbering), which are ligands of site C in Ftn, are NOT conserved in the vertebrate H-type ferritins. The 3-dimentional structure of DvFtn is not known. But, for PfFtn, the X-ray crystallographic structure of the trinuclear Fe-binding center12 is practically super-imposable with that of EcFtnA.33 To provide a visual comparison of the structural differences and similarities between the ferroxidase centers of H-type feritin and prokaryotic Ftn, we overlaid the X-ray crystallographic structure of the binuclear ferroxidase center of HuHf 7 with that of the trinuclear ferroxidase center of PfFtn12 (Figure 2).
Figure 1.

Amino acid sequence alignment of EcFtnA, PfFtn, DvFtn, HuHf and BfMf. EcFtn numbering system is used throughout the manuscript. All three prokaryotic Ftn’s (EcFtn, PfFtn and DvFtn) share the same sequence numbers up to residue 165 with variation on the C-terminal extensions. The two eukaryotic H-type ferritins (HuHF and BfMF) have different N- and C-terminal extensions. The conserved residues are boxed in red. The residues that are conserved only in the bacterial ferritins are boxed in blue. The triangles indicates residues that are ligands to iron sites A and B. The stars indicates residues that are ligands to iron site C. Labels (A, B and C) for the three Fe binding sites are in accord with those designated by Stillman et al.9
Figure 2.

X-ray crystallographic structural comparison of the trinuclear iron binding center of PfFtnA (blue)12 and the binuclear ferroxidase site of HuHf (yellow).7 The residues are labeled according to the sequence number of EcFtnA (see Figure 1). The orange balls indicate the three bound Fe atoms, which are labeled as A, B and C in accord with Stillman et al.9 Fe – Fe distances are as indicated.
In order to obtain further evidence to support the presence of a trinuclear ferroxidase center in prokaryotic Ftn and to characterize such a ferroxidase center in detail prior to its reaction with oxygen, we have loaded apo wild-type DvFtn anaerobically with sub-stoichiometric amounts of Fe2+ (16, 31 and 62 Fe2+/24-mer) and characterized the Fe-loaded DvFtn’s by using Mössbauer and electron paramagnetic resonance (EPR) spectroscopy. As shown in Figure 2, the residue E130 bridges Fe sites B and C. A previous study34 showed that substitution of E130 in EcFtnA to alanine results in the detection of a transient blue (λmax = 600 nm) intermediate and reduces the Fe/O2 oxidation stoichiometry from 3–4 to 1.8,34 reminiscent of those properties that are characteristics of HuHf. Thus, it is important to investigate the effects of the E130A substitution on the state of the initially bound Fe ions. To this end, we have loaded the DvFtn E130A variant with 12 Fe2+ per 24-mer and characterized the Fe-loaded protein by using Mössbauer and EPR spectroscopy. The results not only provide evidence supporting the presence of a trinuclear ferroxidase center in DvFtn, but also reveal intricate spectroscopic properties of the three Fe sites, information that has a significant implication on the DvFtn ferroxidation mechanism.
Materials and methods
Sample preparation
Preparation of the overexpression vector and purification of recombinant DvFtn is described under the accompanying Supporting Information. All samples were prepared in anaerobic conditions in an anaerobic chamber. Protein samples were made free of oxygen by using a vacuum/argon manifold. The acidic 57FeSO4 solution was made from a 57Fe metal foil (95% plus enrichment) in an anaerobic chamber using the method described previously.39 Oxygen-free solution of purified DvFtn, in 0.2 M Tris-HCl pH 7.6 and 0.2 M NaCl buffer, were mixed with proper amount of the 57Fe-enriched ferrous solution for 30 min before transfer to EPR quartz tubes (350 μL) and Mössbauer sample holders (450 μL) for spectroscopic measurements.
Mössbauer and EPR spectroscopy
Mössbauer spectra were recorded in previously described spectrometers.39 The zero velocity of the spectra refers to the centroid of a room temperature spectrum of a metallic Fe foil. Analyses of the Mössbauer spectra were performed by using the WMOSS program (SEE Co., Edina, Minnesota), which was based on a spin Hamiltonian formalism conventionally used for Mössbauer analysis.40 EPR spectra were recorded on an X-band Brüker ER 200D-SRC spectrometer equipped with an Oxford Instrument ESR 910 continuous-flow cryostat.
Protein and metal determination
Protein concentration is expressed in moles of 24-subunit oligomer and was determined by using an extinction coefficient of (6.7 ± 0.05)×105 M−1 cm−1 at 280 nm. This value was calculated by using the method developed by Gill and von Hippel41 and is in accord with the experimental value of (6.5 ± 1.3)×105 M−1 cm−1 obtained by the BCA method (Sigma). The iron content was determined by Inductively Coupled Plasma-Atomic Emission Spectroscopy (Analytical Services Laboratory, Requimte) and by 2,4,6-tripyridyl-s-triazine assay as described by Fischer et al.42 The iron contents of the purified DvFtn from all preparations were found to be less than one Fe per 24-mer. No other metals were detected.
Results
EPR Spectroscopy
Low-temperature EPR spectra were recorded using a dual-mode resonance cavity. Under the parallel mode condition, no EPR resonances were detected for wild-type DvFtn loaded with Fe. When recorded under the perpendicular mode, wild-type DvFtn loaded with 16Fe/24-mer exhibits an axial-type EPR signal at the g ≤ 2.0 region (Figure 3), a type of EPR signal that is typically detected for spin S = 1/2 μ-oxo/μ-hydroxo carboxylate-bridged mixed valence Fe2+Fe3+ dinuclear centers in proteins, including hemerythrin,43 purple acid phosphatases,44,45 rubrerythin,46,47 R2 subunit of ribonucleotide reductases,48–50 and the hydroxylase component of soluble methane mono-oxygenases (MMOH).51,52 Detailed spectral analysis indicates that the experimental spectrum (shown in Figure 3) can be simulated as two overlapping S = 1/2 signals of comparable intensity having g values of 1.96, 1.84 and 1.82 (signal 1) and 1.92, 1.82 and 1.75 (signal 2) (Figure S1). The solid line shown in Figure 3 is the simulated composite spectrum. A similar EPR signal with two overlapping S = 1/2 components has also been observed in PfFtn.38 Consistent with the mixed valence Fe2+Fe3+ assignment, a previous redox titration study of PfFtn38 monitored by the EPR signal showed two mid-point redox potentials (in reference to the standard hydrogen electrode), +50 mV and +200 mV at pH 7.5, that can be assigned, respectively, to the Fe2+Fe2+/Fe2+Fe3+ and Fe2+Fe3+/Fe3+Fe3+ redox couples. The origin for the observation of two sets of principle g values is not known, however.
Figure 3.

EPR spectrum of Fe57-enriched DvFtn loaded with 16Fe/24-mer. The data (noisy gray line) were recorded at 12 K in a perpendicular mode. The smooth black line is a simulation assuming two sets of principle g values (see text and Figure S1). Other experimental conditions are: microwave frequency, 9.66 GHz; microwave power, 0.2 mW; modulation field, 0.5 mT.
Double integrations of the DvFtn EPR signal (Figure 3) were performed on four different samples, which yielded a total spin count of (0.34 ± 0.05) spin/Fe loaded. As the samples were loaded with an average of 16 Fe atoms per 24-mer, this translates to (5.4 ± 0.8) spin/24-mer. In other words, the DvFtn loaded with 16Fe/24-mer contains, on average, approximately 5 mixed valence Fe2+Fe3+ dinuclear centers per molecule. The intensity of this S = 1/2 signal depends strongly on the number of loaded Fe. It is most intense in the 16Fe/24-mer DvFtn and decreases with increasing Fe loading. In the 62Fe/24-mer DvFtn, the EPR signal disappears and the sample is EPR silent.
In contrast to the wild-type DvFtn, the 16Fe/24-mer E130A variant exhibits no EPR resonance when the spectrum is recorded in the perpendicular mode, indicating that the protein does not contain half-integer spin systems. On the other hand, when recorded in the parallel mode, a resonance signal is detected in the low-field region (Figure 4), demonstrating the presence of a paramagnetic (i.e., S ≠ 0) integer-spin system.53 Integer-spin EPR signals have been reported for several Fe-containing proteins,53 including the fully reduced MMOH,54 which contains a carboxylate-bridged diferrous center. Taking into consideration the possible structural similarity between the diiron center in MMOH and in DvFtn-E130A, the observed integer-spin EPR signal (Figure 4) suggests strongly the presence of a fully reduced diferrous center in the 16Fe/24-mer loaded E130A variant, a suggestion that is confirmed by the Mössbauer data presented below.
Figure 4.

EPR spectrum of Fe57-enriched DvFtnA-E130A variant loaded with 16Fe/24-mer. The spectrum was recorded at 12 K in a parallel mode. Other experimental conditions are: microwave frequency, 9.39 GHz; microwave power, 20 mW; modulation field, 1.0 mT.
Mössbauer Results of E130A variant
To obtain more detailed information on the state of the loaded Fe, Mössbauer measurements were carried out in both weak (50 mT) and strong (8 T) applied-field conditions. Figure 5 shows the spectra of DvFtn-E130A variant loaded with 16Fe/24-mer recorded at 4.2 K in a parallel applied field of 50 mT (A) or 8 T (B). The 50-mT spectrum (Figure 5A) exhibits an intense asymmetric quadrupole doublet and some minor absorption that shows magnetic hyperfine structure. The intense quadrupole doublet can be simulated by using parameters (ΔEQ = (2.93 ± 0.05) mm/s and δ = (1.32 ± 0.03) mm/s) that are indicative of high-spin S = 2 Fe2+ with N/O ligands. Analysis of the data indicates that at least 90% of the Fe absorption can be attributed to this quadrupole doublet (solid line in Figure 5A), establishing that most, if not all, of the loaded Fe is in the S = 2 ferrous state. Under the influence of a strong applied field of 8 T, the Fe loaded E130A variant exhibits a paramagnetic (i.e., S ≠ 0) hyperfine structure (Figure 5B, vertical bars) that compares very well with the spectra of reduced Methylococus capsulatus MMOH (blue solid line) and BfMF (red solid line) loaded with 21Fe/24-mer recorded under the same experimental conditions (unpublished data). By visual inspection, the DvFtn-E130A 8-T spectrum also exhibits features very similar to those in the published high-field spectrum of reduced Methylosinus trichosporium MMOH.55 It is known that reduced MMOH contains an oxygen-activating carboxylate-bridged paramagnetic diferrous center25,27,52,54,55 and that BfMF loaded with substoichiometric Fe2+ also contains an oxygen-reactive carboxylate-bridged dinuclear ferroxidation center.8,18,22 The fact that most of the Fe atoms loaded into E130A variant are in the high-spin ferrous state, and that the high-field spectrum is similar to those of known diferrous centers in proteins suggest strongly that the Fe atoms in the 16Fe/24-mer E130A variant are organized as carboxylate-bridged diferrous centers. As commented previously by Fox et al.55 that it is impossible to obtain a unique set of parameters for the paramagnetic diferrous center by analyzing the high-field Mössbauer data alone because of the numerous number of unknown parameters (at least 15) involved, we did not attempt to analyze the high-field spectrum in more detail.
Figure 5.

Mössbauer spectra (vertical bars) of DvFtn-E130A variant loaded with 16Fe/24-mer. The spectra (vertical bars) were recorded at 4.2 K in a parallel field of 50 mT (A) or 8 T (B). The solid line in A is a simulation of a quadrupole doublet with parameters ΔEQ = 2.93 mm/s and δ = 1.32 mm/s. For comparison with known carboxylate-bridged diferrous proteins, the Mössbauer spectra of reduced Methylococus MMOH (blue line) and BfMf loaded with 21Fe/24-mer (red line) recorded under the same experimental conditions are overlaid with the spectrum of DvFtn in B.
Taking into consideration that (1) the Mössbauer spectra, presented above, are consistent with a carboxylate-bridged diferrous center, (2) the observed integer-spin EPR signal (Figure 4) may be assigned to a paramagnetic diferrous center, and (3) the Fe binding sites A and B of bacterial ferritins (which include EcFtnA, PfFtn and DvFtn) exhibit sequence homology and structural similarity (Figures 1 and 2) with that of the ferroxidase site of H-type ferritins (which include HuHF and BfMF), we conclude that the Fe atoms in the 16Fe/24-mer DvFtn-E130A must be occupying the iron-binding sites A and B, forming a carboxylate-bridged paramagnetic diferrous center (termed paramagnetic diferrous) similar to the diferrous ferroxidase center of H-type ferritins.
Mössbauer Results for Wild-type DvFtn
Figure 6 displays the spectra of DvFtn loaded with 62Fe (A and E), 31Fe (B and F) and 16Fe (C and G) per 24-mer. The spectra were recorded at 4.2 K in a parallel applied field of 50 mT (A, B, and C) or 8 T (E, F and G). From the 50-mT spectra, at least two spectral components corresponding to two distinct electronic systems can be clearly identified: an intense quadrupole doublet (absorption peaks at about 0 mm/s and 2.7 mm/s) arising from high-spin Fe2+ ions in integer spin electronic states, and a magnetically split spectrum with broad absorptions extending from −4 mm/s to +5 mm/s, indicative of half-integer spin states. In accord with the EPR spectrum of the Fe loaded DvFtn, which shows only two S = 1/2 mixed valence dinuclear Fe2+Fe3+ signals, the magnetic spectral component is attributed to the mixed valence Fe2+Fe3+ center. As the differences in the principle g values of these two overlapping EPR signals are small, Mössbauer spectroscopy does not have the sensitivity to distinguish them. Thus, in our analysis of the Mössbauer data, the magnetic spectral component is treated as arising from a single mixed valence Fe2+Fe3+ center.
Figure 6.

Mössbauer spectra (vertical bars) of wild-type DvFtn loaded with various substoichiometric amounts of Fe. The spectra are recorded at 4.2 K in a parallel field of 50 mT (left panel) or 8 T (right panel). The samples were loaded with 62Fe/24-mer (A and E), 31Fe/24-mer (B and F) and 16Fe/24-mer (C and G). The 16Fe/24-mer sample was reduced by sodium dithionite and spectra of the reduced sample are shown in D (50 mT) and H (8T). The color lines are simulations of the diamagnetic diferrous center (green), mixed valence Fe2+Fe3+ center (red) and mononuclear Fe2+ species (blue), using parameters listed in Table 1. They are plotted according to the percent absorptions listed in Table 2. The black solid lines overlaid with the experimental data are composite spectra of the simulated spectra shown.
Consistent with the EPR results, which show that the intensity of the S = 1/2 EPR signal decreases with increasing Fe loading and disappears in the 62Fe/24-mer sample, the percent absorption of the magnetic Mössbauer spectral component decreases substantially in the 31Fe/24-mer sample (Figure 6B) and becomes undetectable in the 62Fe/24-mer sample (Figure 6A). Only the high-spin Fe2+ quadrupole doublet is seen in the 62Fe/24-mer sample. The 8-T spectrum of this sample (Figure 6E) reveals further that this doublet represents at least two types of high-spin Fe2+ species in two distinct electronic environments. First, the intense absorption peaks, marked by arrows in Figure 6E, indicate that the doublet (observed in weak applied field) is magnetically split by an effective field that is exactly equal to the applied field, 8 T. In other words, most of the Fe2+ ions are present in a diamagnetic (S = 0) environment even though their intrinsic spins are 2. The most reasonable explanation for this observation is that the S = 2 Fe2+ ions are structured into an anti-ferromagnetically coupled diferrous center resulting in a diamagnetic (S = 0) ground state (termed the diamagnetic diferrous center). The remaining Fe2+ ions display a broad spectrum consistent with paramagnetic integer spin states but distinguishable from that of the paramagnetic diferrous center detected in E130A variant. This broad spectrum observed in high-field is tentatively assigned to a mononuclear Fe2+ center (see below).
In summary, preliminary visual analyses of the Mössbauer data reveal clearly that Fe loaded DvFtn contains at least three Fe species, namely, the mixed valence Fe2+Fe3+ dinuclear center, the diamagnetic diferrous center and the mononuclear Fe2+ species. The concentrations of these species vary with the Fe loading. On the basis of such an understanding, detailed analysis of the data was performed. The resulting characteristic Mössbauer parameters for each species are listed in Table 1 and their relative percent absorptions in each sample are listed in Table 2. The theoretical spectra simulated with these parameters are plotted in Figure 6 according to their corresponding percent absorptions. From Table 2, it can be seen that the decreases in the mixed valence species in the higher Fe loading samples is roughly compensated by the increases in the diamagnetic diferrous species. To provide direct evidence confirming the conversion of the mixed valence species to the diamagnetic diferrous species, the 16Fe/24-mer sample was reduced with sodium dithionite and Mössbauer spectra were recorded after the reduction (Figure 6, D and H). The data show clearly that dithionite converts all the mixed valence Fe2+Fe3+ species in the Fe loaded sample (60%) quantitatively into the diamagnetic diferrous species (from an initial 20% to a final 80%) and the amount of the mononuclear Fe2+ species (20%) remains constant (Table 2). This observation supports strongly that the mixed valence species and the diamagnetic diferous species represent two different oxidation states of a same dinuclear Fe center.
Table 1.
Mössbauer parameters of the Fe species detected in DvFtn and its E130A variant
| protein | species | Fe site | δ (mm/s) | ΔEQ (mm/s) | η | A/gnβn (T) |
|---|---|---|---|---|---|---|
| wild-type | diamagnetic diferrous | 1 | 1.22[3] | −2.83[5] | −0.82 | |
| 2 | 1.18[3] | 2.55[5] | 0 | |||
| mixed valence | Fe3+ | 0.52[3] | −1.18[5] | 0.3 | −(43.5, 53.4, 50.0) | |
| Fe2+ | 1.17[3] | 2.55[5] | 0 | +(28.9, 21.9 18.9) | ||
| mononuclear Fe2+ | 1.42[4] | 2.77[6] | ||||
| E103A | paramagnetic diferrous | 1.32[3] | 2.94[5] |
Values in the square parentheses are uncertainties of the last significant digits.
Table 2.
Percents Mössbauer absorption of the Fe species detected in DvFtn and its E130A variant loaded with various sub-stoichiometric amounts of Fe
| protein | Fe loading (Fe/24-mer) | % Mössbauer absorption of the Fe speciesa
|
|||
|---|---|---|---|---|---|
| diamagnetic diferrous | mixed valence Fe2+Fe3+ | mononuclear Fe2+ | paramagnetic diferrous | ||
| wild-type | 62b | 70 | 0 | 18 | 0 |
| 31 | 45 | 45 | 10 | 0 | |
| 16 | 20 | 60 | 20 | 0 | |
| 16+Na2S2O4 | 80 | 0 | 20 | 0 | |
| E130A | 16 | 0 | 0 | 0 | 100 |
A less than 5% uncertainty is estimated for the listed percent absorptions.
For the 62Fe/24-mer loading sample, approximately 12% of the absorption is attributed to small ferrihydrite clusters.
On the basis of the above analysis, the diamagnetic diferrous, mixed valence Fe2+Fe3+, and the mononuclear Fe2+ species contribute, respectively, 20%, 60% and 20% of the Mössbauer absorption of the 16Fe/24-mer sample. In an effort to obtain more information on the paramagnetic Fe2+ component, we use the simulated spectra of the diamagnetic diferrous center (green line in Figure 6G) and the mixed valence Fe2+Fe3+ (red line in Figure 6G) to remove their absorption contributions from the raw experimental spectrum (vertical bars in Figure 6G) and thus obtained a spectrum (Figure 7) representing closely the paramagnetic Fe2+ component. The pattern of this spectrum, which displays four sharp central absorption peaks and two broad outer peaks, resembles those observed for mononuclear Fe2+ proteins.56,57 Such a pattern is distinct from that of the paramagnetic diferrous spectrum (Figure 5), which displays absorptions arising from two distinct Fe sites as pointed out by Fox et al.55 To illustrate the point that the paramagnetic Fe2+ component is consistent with a mononuclear Fe2+ assignment, we have generated a theoretical spectrum by using a set of parameters (D = 6.0 cm−1, E/D = 0.25, δ = 1.38 mm/s, ΔEQ = 2.80 mm/s, η = 1.0 and A/gnβn = −(24, 24, 5.6) T) modified from that of reduced protocatechuate 3,4 dioxygenase57 and plotted the theoretical spectrum (solid line in Figure 7) against the experimental spectrum (vertical bars in Figure 7). It can be seen that, in regard to the overall patterns of the spectra, the theory compares reasonably well with the experimental data. It is important to comment, however, that the simulated spectrum is not a fit to the experimental data. It is presented here for the above mentioned illustration purpose only.
Figure 7.

Mössbauer spectrum (vertical bars) of the mononuclear Fe2+ species derived from the 8-T spectrum of the 62 Fe/24-mer loaded DvFtn (Figure 6G; see text for the derivation procedure). The solid line is a simulated spectrum using the parameters listed in text.
Discussion
X-ray crystallographic studies9,12 (see Figure 2) and sequence analysis (Figure 1) revealed the presence of a trinuclear Fe binding center in bacterial ferritins (e. g., EcFtnA, PfFtn and DvFtn). Mössbauer spectroscopic investigations36,58 of the ferroxidase reaction of EcFtnA showed formation of mononuclear and binuclear ferric products, suggesting involvement of a trinuclear Fe-binding site in the ferroxidase reaction. Here, to gain information on the states of the Fe atoms initially bound to the trinuclear center prior to ferroxidation, the bacterial ferritin DvFtn and its variant E130A were loaded with sub-stoichiometric amount of Fe2+ under anaerobic conditions, and Mössbauer and EPR spectroscopy were used to characterize these Fe loaded proteins. Four distinctive Fe species were identified (Table 1 and 2), namely, the paramagnetic diferrous species detected in the E130A variant, the diamagnetic diferrous species, the mixed valence Fe2+Fe3+ species and the mononuclear Fe2+ species observed in the wild-type DvFtn. For the wild-type DvFtn, effects of varying amounts of Fe loading were also investigated and percent Mössbauer absorptions of the various Fe species as a function of the amount of loaded Fe were determined (Table 2). To discuss the significance of our findings, we begin by making a reasonable and least confounding assignment of the observed Fe species in association with the Fe binding sites A, B and C.
Assignment for the paramagnetic diferrous center
We notice at first that the Mössbauer and EPR data of DvFtn-E130A variant loaded with 16Fe/24-mer shows only one Fe species, namely, the paramagnetic diferrous center. According to the X-ray crystallographic data of EcFtn,9 and PfFtn12, residue E130 (EcFtn numbering system) is one of the four ligands for site C, and bridges the Fe-binding sites B and C. Thus, substitution of E130 with alanine could significantly diminish the Fe-binding affinity of site C. On the other hand, even though E130 is also a ligand for site B, it is conserved only for bacterial Ftn-type ferritins and not for eukaryotic H-type ferritins. All residues (E17, E50, H53, E94, and E127) (EcFtn numbering system) that are identified as ligands to the diiron ferroxidation site of eukaryotic H-type ferritins (i.e., sites A and B)7,8 are retained in the E130A variant. It is therefore expected that the DvFtn-E130A variant would be able to assemble a diferrous center analogous to the ferroxidase centers of H-type ferritins.17,22 This expectation is consistent with the observation that DvFtn-E130A loaded with 16Fe/24-mer contains only the paramagnetic diferrous species, which exhibits Mössbauer and EPR spectroscopic properties resembling those of the carboxylate-bridged diferrous centers found in oxygen-activating non-heme enzymes24,25,52 and in H-type ferritins17,22 (see also Figure 5). Consequently, the paramagnetic diferrous species is assigned to a diferrous center occupying sites A and B (Figure 8).
Figure 8.

Schematic representation of the Fe species detected in DvFtn and its E130A variant. The filled green or orange circles indicate that the sites are occupied by, respectively, Fe2+ or Fe3+ ions. The empty circles indicate unoccupied sites. The curves connecting two circles represent bridging carboxylate ligands.
Assignment for the diamagnetic diferrous center
The diamagnetic diferrous species is observed only in the wild-type DvFtn and not in the E130A variant, while the paramagnetic diferrous species is detected only in the E130A variant and not in the wild-type protein. For the wild-type DvFtn, there are three Fe-binding sites A, B and C, allowing for three different combinations of two Fe sites for a diferrous center. They are [A, B], [A, C] and [B, C]. Occupation of sites A and B would generate the paramagnetic diferrous center, which is not detected in the wild-type protein. Thus, a reasonable choice for the diamagnetic diferrous center would be either one of the latter two combinations, both involving site C. On the basis of the observations from the X-ray crystallographic structures of EcFtn9 and PfFtn12 that there exists a bridging carboxylate ligand (E130) for sites B and C, and that there is no candidate for bridging ligand for sites A and C, we postulate that the diamagnetic diferrous center occupies sites B and C (Figure 8). The association of site C with the diamagnetic diferrous center is consistent with the observation that the diamagnetic diferrous species is not detected in the E130A variant, because the E130A substitution presumably has significantly diminished the Fe-binding affinity of site C and thus prevented the formation of the diamagnetic diferrous center.
Presence of a μ-OH group in the diamagnetic diferrous center
Diferrous centers that display paramagnetic ground states are common for carboxylate-bridged non-heme diiron proteins, which include MMOH,54,55 ribonucleotide reductase R2 subunit,59–61 Δ9 desaturase,61 and H-type ferritins (see Figure 5). Carboxylate-bridged diferrous centers with diamagnetic (S = 0) ground states are rare. A well known example is the oxygen transfer protein, hemerythrin.62,63 This difference in the magnetic properties of the ground state is the result of distinct exchange coupling interactions between the two ferrous sites. A diamagnetic ground state is the outcome of significantly strong antiferromagnetic coupling between the two ferrous ions; A −J value of 15 cm−1 to 38 cm−1 (exchange coupling Hamiltonian
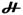 = −2J
S1•S2) was estimated for reduced hemerythrin.62,63 A paramagnetic ground state may result from weak exchange coupling (J less than 1 cm−1 in magnitude) together with relatively large zero-field splittings (D ~ 10 cm−1) of the ferrous ions.61 Since exchange interactions are mainly mediated by bridging ligands, this difference is likely to reflect distinct bridging ligand types and/or orientations. A unique structural feature of the hemerythrin diferrous center is the presence of a μ-OH group,64 which is absent in all other diferrous proteins of known structures.7,8,24,25 It is therefore likely that antiferromagnetic coupling in carboxylate-bridged diferrous centers may implicate the presence of a bridging OH group. Small molecule model-complex studies support such an assessment. A hydroxo-bridged diferrous complex65 with a (μ-OH)bis(μ-acetato)diiron(II) core structure exhibits antiferromagnetic coupling (−J ~ 13 cm−1) resembling that of the diferrous center in hemerythrin, while a water-bridged diferrous complex with a (μ-H2O)bis(μ-benzoate)diiron(II) core structure exhibits weak ferromagnetic interaction.66 On the basis of these considerations, we tentatively assign a bridging OH group for the diamagnetic diferrous species (Figure 8).
= −2J
S1•S2) was estimated for reduced hemerythrin.62,63 A paramagnetic ground state may result from weak exchange coupling (J less than 1 cm−1 in magnitude) together with relatively large zero-field splittings (D ~ 10 cm−1) of the ferrous ions.61 Since exchange interactions are mainly mediated by bridging ligands, this difference is likely to reflect distinct bridging ligand types and/or orientations. A unique structural feature of the hemerythrin diferrous center is the presence of a μ-OH group,64 which is absent in all other diferrous proteins of known structures.7,8,24,25 It is therefore likely that antiferromagnetic coupling in carboxylate-bridged diferrous centers may implicate the presence of a bridging OH group. Small molecule model-complex studies support such an assessment. A hydroxo-bridged diferrous complex65 with a (μ-OH)bis(μ-acetato)diiron(II) core structure exhibits antiferromagnetic coupling (−J ~ 13 cm−1) resembling that of the diferrous center in hemerythrin, while a water-bridged diferrous complex with a (μ-H2O)bis(μ-benzoate)diiron(II) core structure exhibits weak ferromagnetic interaction.66 On the basis of these considerations, we tentatively assign a bridging OH group for the diamagnetic diferrous species (Figure 8).
Assignment for the mixed valence Fe2+Fe3+ center
Our data show that reduction of the mixed valence Fe2+Fe3+ species by dithionite converts it into the diamagnetic diferrous species quantitatively. We thus conclude that the mixed valence Fe2+Fe3+ species is the one-electron oxidized state of the diamagnetic diferrous species. As it is not uncommon for mixed valence Fe2+Fe3+ diiron centers to have a bridging OH group,67,68 we expect that the μ-OH group in the diamagnetic diferrous state is retained in the Fe2+Fe3+ state. Furthermore, we tentatively assign the Fe3+ site to site B, and the Fe2+ site to site C (Figure 8), due to the following considerations.
Assignment for the mononuclear Fe2+ species
The Mössbauer results of the 62Fe/24-mer sample show that approximately 70% of the Fe absorptions can be attributed to the diamagnetic diferrous species, which accounts for 62×0.70 = 43.4 Fe2+ ions/24-mer. Thus, there are approximately 22 diamagnetic diferrous centers per 24-mer. With this same reasoning, the 18% Fe absorption arising from the mononuclear Fe2+ species means that there are ~11 mononuclear Fe2+ ions/24-mer. Because there are only 24 trinuclear Fe binding sites per 24-mer, and because 22 of these trinuclear sites contain the diamagnetic diferrous centers, it is inevitable that at least 9 (up to all) of the 11 mononuclear Fe2+ ions have to occupy same trinuclear sites with the diamagnetic diferrous centers. Thus, with the diamagnetic diferrous center occupying sites B and C, the mononuclear Fe2+ ion has to be at site A (Figure 8). The proximity between the diamagnetic diferrous center and the mononuclear Fe2+ ion is not expected to have a significant effect on their individual Mössbauer spectral properties, because the diamagnetic diferrous center is diamagnetic and, thus, would not interact magnetically with the adjacent mononuclear Fe2+ site. Also, exchange interaction between sites A and B is expected to be weak because there is no single-atom bridge between them.
The fully reduced tri-nuclear Fe2+ ferroxidase center
It is important to understand that the Mössbauer data also reveal that, in contrast to the diamagnetic diferrous species, the mixed valence Fe2+Fe3+ species does not occupy the same trinuclear site with the mononuclear Fe2+ ions. This is because the mixed valence species (S = 1/2) and the mononuclear Fe2+ species (S = 2) are both paramagnetic. Proximity of these two species would create spin-dipolar interactions between them, resulting in a half-integer spin system that would exhibit a magnetic Mössbauer spectrum comprising three nonequivalent magnetic spectral components, even in a weak applied field (e.g. 50 mT). The fact that such a spectrum is not detected in the Fe load DvFtn indicates unambiguously that the mixed valence diiron centers and the mononuclear Fe2+ species are occupying separate trinuclear Fe centers. Thus, the only allowed oxidation state for the fully occupied trinuclear center is the fully reduced state (i.e., all three sites are occupied by Fe2+). These observations suggest that binding of Fe2+ at site A must affect the redox properties of the diiron center at sites B and C so that the diamagnetic diferrous state is favored over the mixed valence Fe2+Fe3+ state. This suggestion may be rationalized if we assume that the Fe3+ ion of the mixed valence center is occupying site B; binding of Fe2+ to site A shifts the electron density of the bridging carboxylate, E50, toward site A and thus stabilizes Fe2+ over Fe3+ at site B.
Possible inter-subunit interaction
With the various Fe species properly assigned to the trinuclear Fe-binding site, and their corresponding percents Fe absorption determined (Table 2), it becomes possible to estimate the Fe occupancy of the trinuclear centers in a 24-meric molecule as a function of Fe loading. The results are presented in Table 3. In formulating Table 3, the occupancy of the fully occupied trinuclear centers is assumed to have its maximum possible value. For example, for the 16Fe/24-mer DvFtn the Mössbauer data show that there are, on average, 3 mononuclear Fe2+ species and 1.5 diamagnetic diferrous centers per 24-mer. Therefore, the possible presence of fully occupied trinuclear centers could vary from 0 to 1.5 per 24-mer. The maximum value 1.5 is listed in Table 3. With this assumption, the results (Table 3) show that the number of fully occupied trinuclear centers increases (from 1.5 to 11 per 24-mer) with increasing Fe loading (from 16 Fe to 62 Fe/24-mer, respectively), which is obviously as expected. But, interestingly, the results also show that increasing occupancy of the fully occupied trinuclear center appears to shift the FeB2+FeC2+/FeB2+FeC3+ ratio toward the reduced FeB2+FeC2+ state. Because the fully occupied trinuclear centers and the FeB2+FeC3+ centers have to be located in different subunits, this observation suggests inter-subunit cooperative effect that promotes binding of ferrous ions. Thus, in the absence of oxidizing agents, the trinuclear Fe binding sites in high Fe loading DvFtn may bind only ferrous ions.
Table 3.
Trinuclear ferroxidase site occupancy as a function of Fe loading
| Fe loading (Fe/24-mer) | site occupancy of Fe species/24-mer
|
||||
|---|---|---|---|---|---|
| FeA2+ | FeB2+FeC3+ | FeB2+FeC2+ | FeA2+FeB2+FeC2+ | vacant | |
| 16 | 1.5 | 5 | 0 | 1.5 | 16 |
| 31 | 0 | 7 | 4 | 3 | 10 |
| 62 | 0 | 0 | 11 | 11 | 2 |
Mechanistic considerations
X-ray crystallographic measurements showed that in comparison with H-type ferritins, bacterial ferritins contain an additional Fe binding site (site C) adjacent to a diiron binding center (sites A and B) that is homologous to the binuclear ferroxidase center of H-type ferritins (Figure 2).9,12 Here, we have loaded the bacterial ferritin DvFtn with sub-stoichiometric amounts of Fe under anaerobic condition, and employed EPR and Mössbauer spectroscopy to investigate the states of the Fe assembled on DvFtn. The result is consistent with the X-ray crystallographic findings that bacterial ferritin contains a trinuclear Fe binding center. Moreover, the spectroscopic data show that (1) the fully occupied ferroxidase center supports an all ferrous state, (2) sites B and C are bridged by a μ-OH group forming a diiron sub-center within the trinuclear center and (3) this diiron sub-center can afford both the mixed valence Fe2+Fe3+ state and the diferrous state.
Because sites A and B exhibit sequence and structural homologies to those of the diiron ferroxidase center of H-type ferritins, it was natural for previous investigators to consider that the trinuclear ferroxidase center functions as an H-type diiron ferroxidase center, at sites A and B, with modulation coming from a nearby mononuclear Fe2+ center, at site C.34 However, our current study shows that while DvFtn-E130A variant, as expected, does assemble a diferrous center at sites A and B with spectroscopic properties resembling those of the diferrous ferroxidase center in H-type ferritins, the wild type DvFtn, in fact, does not form a diiron sub-center at sites A and B, but rather, assembles a diiron sub-center at sites B and C with spectroscopic and redox properties that are distinct from those of the H-type ferroxidase center. In the following section, we propose a minimal mechanistic scheme (Scheme 1) for the bacterial ferritin ferroxidase reaction that incorporates these new findings. In formulating this scheme, we have also taken into consideration the previous finding that bacterial ferritin, EcFtnA, consumes one O2 molecule to oxidize approximately 3.5 Fe2+ ions in its ferroxidase reaction,34 indicating that H2O is a ferroxidase product.
Scheme 1.

Proposed minimal mechanism for bacterial ferritin ferroxidase reaction
Proposed minimal mechanistic scheme
According to the X-ray crystallographic structures of EcFtn9 and PfFtn,12 all three Fe-binding sites of bacterial ferritin receive four coordinate bonds from protein ligands (Figure 2). With the bridging OH group, both Fe atoms at sites B and C (FeB and FeC, respectively) are five-coordinated (not counting possible water ligands). Thus, the Fe at site A (FeA) is less saturated and is assumed to be the O2 binding site (Scheme 1). Binding of O2 to FeA2+ results in a ferric superoxide, which after reduction by FeB2+ becomes a ferric FeA3+-peroxide (or a μ-peroxo diferric FeA3+FeB3+ center) hydrogen-bonded to the μ-OH group of the mixed valence FeB3+FeC2+ sub-center (Intermediate I). Shifting of the proton to the peroxo group promotes breaking of the O-O bond and produces concomitantly a water molecule and a tri-ferric bis(μ-oxo/μ-hydroxo) cluster as precursor of the mineral core (Scheme 1). The ultimate step would require an additional reducing equivalent that may be provided by a protein residue. A candidate could be the conserved Y24 at the vicinity of the ferroxidase site (Figure 2). Other investigators have previously suggested that the observed Fe2+/O2 ratio of 3.5 (instead of 3.0 as suggested by scheme 1) is consistent with a portion of the oxidized protein residues being reduced by Fe2+ ions in solution.34
The proposed scheme is verifiable experimentally by using rapid kinetic methods, such as rapid freeze quench (RFQ) Mössbauer and EPR spectroscopy.69 As presented above, the current study has established the trinuclear all ferrous state as the initial state prior to reaction with O2. The proposed reaction intermediate I is identifiable by a combined EPR and Mössbauer characterization. With a μ-OH group present, the exchange interaction between FeB and FeC is expected to be much stronger than that between FeA and FeB.62,63,70 Thus, the spin state of I can be considered as the result of a spin-spin coupling between the FeA3+-peroxide and the antiferromagnetically coupled μ-OH FeB3+FeC2+ sub-center, which has a ground spin state of 1/2. The resulting spin state of I would depend on the nature and strength of the coupling. But, regardless of the spin state of I, its Mössbauer spectra would comprise three spin-coupled equal-intensity sub-spectra with characteristic parameters corresponding to two Fe3+ and one Fe2+ ions. The magnetic-field dependence of the Mössbauer spectra will provide further information on the details of the exchange coupling among the three Fe sites A, B and C. Currently, we are performing RFQ EPR and Mössbauer measurements to investigate the reaction of O2 with DvFtn pre-loaded with Fe2+, in an effort to examine the validity of the above proposed mechanistic scheme.
Supplementary Material
Acknowledgments
This work is supported in part by Fundação para a Ciência e Tecnologia (FCT/MEC) grant PTDC/QUI/64248/2006 to ASP, and by National Institutes of Health grant GM47295 to BHH. Requimte is funded in part by grant PEst-C/EQB/LA0006/2011 from FCT/MEC. FF is supported by a postdoctoral fellowship (SFRH/BPD/48430/2008) and MG by a Ph.D. fellowship (SFRH/BD/18825/2004) from FCT/MEC.
Footnotes
Construction of DvFtn overexpression vector, expression and purification of wild-type DvFtn and DvFtn-E130A variant, spin-Halmiltonian formulism used for analyses of the Mössbauer data, decomposition of the EPR spectrum of DvFtn and the 50-mT Mössbauer spectrum of the mononuclear Fe2+ species. This information is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.Andrews SC. Biochimica Et Biophysica Acta-General Subjects. 1800:691–705. doi: 10.1016/j.bbagen.2010.05.010. [DOI] [PubMed] [Google Scholar]
- 2.Theil EC. Current Opinion in Chemical Biology. 2011;15:304–311. doi: 10.1016/j.cbpa.2011.01.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Theil EC. In: Handbook of Metalloproteins. Messerschmidt A, Huber R, Poulos T, Weighardt K, editors. John Wiley & Sons; Chichester, UK: 2000. pp. 771–781. [Google Scholar]
- 4.Chasteen ND, Harrison PM. J Struct Biol. 1999;126:182–194. doi: 10.1006/jsbi.1999.4118. [DOI] [PubMed] [Google Scholar]
- 5.Harrison PM, Arosio P. Biochim Biophys Acta-Bioenergetics. 1996;1275:161–203. doi: 10.1016/0005-2728(96)00022-9. [DOI] [PubMed] [Google Scholar]
- 6.Lawson DM, Artymiuk PJ, Yewdall SJ, Smith JMA, Livingstone JC, Treffry A, Luzzago A, Levi S, Arosio P, Cesareni G, Thomas CD, Shaw WV, Harrison PM. Nature. 1991;349:541–544. doi: 10.1038/349541a0. [DOI] [PubMed] [Google Scholar]
- 7.Hempstead PD, Yewdall SJ, Fernie AR, Lawson DM, Artymiuk PJ, Rice DW, Ford GC, Harrison PM. J Mol Biol. 1997;268:424–448. doi: 10.1006/jmbi.1997.0970. [DOI] [PubMed] [Google Scholar]
- 8.Ha Y, Shi DS, Small GW, Theil EC, Allewell NM. J Biol Inorg Chem. 1999;4:243–256. doi: 10.1007/s007750050310. [DOI] [PubMed] [Google Scholar]
- 9.Stillman TJ, Hempstead PD, Artymiuk PJ, Andrews SC, Hudson AJ, Treffry A, Guest JR, Harrison PM. J Mol Biol. 2001;307:587–603. doi: 10.1006/jmbi.2001.4475. [DOI] [PubMed] [Google Scholar]
- 10.Macedo S, Romao CV, Mitchell E, Matias PM, Liu MY, Xavier AV, LeGall J, Teixeira M, Lindley P, Carrondo MA. Nat Struct Biol. 2003;10:285–290. doi: 10.1038/nsb909. [DOI] [PubMed] [Google Scholar]
- 11.Johnson E, Cascio D, Sawaya MR, Gingery M, Schroder I. Structure. 2005;13:637–648. doi: 10.1016/j.str.2005.01.019. [DOI] [PubMed] [Google Scholar]
- 12.Tatur J, Hagen WR, Matias PM. J Biol Inorg Chem. 2007;12:615–630. doi: 10.1007/s00775-007-0212-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Nordlund P, Eklund H. Current Opinion in Structural Biology. 1995;5:758–766. doi: 10.1016/0959-440x(95)80008-5. [DOI] [PubMed] [Google Scholar]
- 14.Crichton RR, Declercq J-P. Biochimica Et Biophysica Acta-General Subjects. 2010;1800:706–718. doi: 10.1016/j.bbagen.2010.03.019. [DOI] [PubMed] [Google Scholar]
- 15.Treffry A, Zhao Z, Quail MA, Guest JR, Harrison PM. Biochemistry. 1995;34:15204–15213. doi: 10.1021/bi00046a028. [DOI] [PubMed] [Google Scholar]
- 16.Turano P, Lalli D, Felli IC, Theil EC, Bertini I. Proc Natl Acad Sci U S A. 2005;107:545–50. doi: 10.1073/pnas.0908082106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Bou-Abdallah F, Papaefthymiou GC, Scheswohl DM, Stanga SD, Arosio P, Chasteen ND. Biochem J. 2002;364:57–63. doi: 10.1042/bj3640057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Hwang J, Krebs C, Huynh BH, Edmondson DE, Theil EC, Penner-Hahn JE. Science. 2000;287:122–5. doi: 10.1126/science.287.5450.122. [DOI] [PubMed] [Google Scholar]
- 19.Jameson GN, Jin W, Krebs C, Perreira AS, Tavares P, Liu X, Theil EC, Huynh BH. Biochemistry. 2002;41:13435–43. doi: 10.1021/bi026478s. [DOI] [PubMed] [Google Scholar]
- 20.Krebs C, Bollinger JM, Jr, Theil EC, Huynh BH. J Biol Inorg Chem. 2002;7:863–9. doi: 10.1007/s00775-002-0371-1. [DOI] [PubMed] [Google Scholar]
- 21.Moenne-Loccoz P, Krebs C, Herlihy K, Edmondson DE, Theil EC, Huynh BH, Loehr TM. Biochemistry. 1999;38:5290–5. doi: 10.1021/bi990095l. [DOI] [PubMed] [Google Scholar]
- 22.Pereira AS, Small W, Krebs C, Tavares P, Edmondson DE, Theil EC, Huynh BH. Biochemistry. 1998;37:9871–6. doi: 10.1021/bi980847w. [DOI] [PubMed] [Google Scholar]
- 23.Schwartz JK, Liu XS, Tosha T, Theil EC, Solomon EI. J Am Chem Soc. 2008;130:9441–50. doi: 10.1021/ja801251q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Logan DT, Su XD, Aberg A, Regnstrom K, Hajdu J, Eklund H, Nordlund P. Structure. 1996;4:1053–64. doi: 10.1016/s0969-2126(96)00112-8. [DOI] [PubMed] [Google Scholar]
- 25.Rosenzweig AC, Nordlund P, Takahara PM, Frederick CA, Lippard SJ. Chem Biol. 1995;2:409–18. [PubMed] [Google Scholar]
- 26.Bollinger JM, Edmondson DE, Huynh BH, Filley J, Norton JR, Stubbe J. Science. 1991;253:292–298. doi: 10.1126/science.1650033. [DOI] [PubMed] [Google Scholar]
- 27.Liu KE, Valentine AM, Wang DL, Huynh BH, Edmondson DE, Salifoglou A, Lippard SJ. J Am Chem Soc. 1995;117:10174–10185. [Google Scholar]
- 28.Zhao G, Bou-Abdallah F, Yang X, Arosio P, Chasteen ND. Biochemistry. 2001;40:10832–8. doi: 10.1021/bi011052j. [DOI] [PubMed] [Google Scholar]
- 29.Le Brun NE, Crow A, Murphy MEP, Mauk AG, Moore GR. Biochimica Et Biophysica Acta-General Subjects. 2010;1800:732–744. doi: 10.1016/j.bbagen.2010.04.002. [DOI] [PubMed] [Google Scholar]
- 30.Frolow F, Kalb AJ, Yariv J. Nat Struct Biol. 1994;1:453–60. doi: 10.1038/nsb0794-453. [DOI] [PubMed] [Google Scholar]
- 31.Hudson AJ, Andrews SC, Hawkins C, Williams JM, Izuhara M, Meldrum FC, Mann S, Harrison PM, Guest JR. Eur J Biochem. 1993;218:985–95. doi: 10.1111/j.1432-1033.1993.tb18457.x. [DOI] [PubMed] [Google Scholar]
- 32.Andrews SC. Adv Microb Physiol. 1998;40:281–351. doi: 10.1016/s0065-2911(08)60134-4. [DOI] [PubMed] [Google Scholar]
- 33.Hempstead PD, Hudson AJ, Artymiuk PJ, Andrews SC, Banfield MJ, Guest JR, Harrison PM. FEBS Lett. 1994;350:258–62. doi: 10.1016/0014-5793(94)00781-0. [DOI] [PubMed] [Google Scholar]
- 34.Treffry A, Zhao ZW, Quail MA, Guest JR, Harrison PM. FEBS Letters. 1998;432:213–218. doi: 10.1016/s0014-5793(98)00867-9. [DOI] [PubMed] [Google Scholar]
- 35.Bauminger ER, Treffry A, Quail MA, Zhao ZW, Nowik I, Harrison PM. Inorg Chim Acta. 2000;297:171–180. [Google Scholar]
- 36.Bauminger ER, Treffry A, Quail MA, Zhao Z, Nowik I, Harrison PM. Biochemistry. 1999;38:7791–802. doi: 10.1021/bi990377l. [DOI] [PubMed] [Google Scholar]
- 37.Bou-Abdallah F. Biochimica Et Biophysica Acta-General Subjects. 2010;1800:719–731. doi: 10.1016/j.bbagen.2010.03.021. [DOI] [PubMed] [Google Scholar]
- 38.Tatur J, Hagen WR. Febs Letters. 2005;579:4729–4732. doi: 10.1016/j.febslet.2005.07.045. [DOI] [PubMed] [Google Scholar]
- 39.Ravi N, Bollinger JM, Huynh BH, Edmondson DE, Stubbe J. J Am Chem Soc. 1994;116:8007–8014. [Google Scholar]
- 40.Huynh BH, Kent TA. Adv Inorg Biochem. 1984;6:163–223. [PubMed] [Google Scholar]
- 41.Gill SC, Vonhippel PH. Anal Biochem. 1989;182:319–326. doi: 10.1016/0003-2697(89)90602-7. [DOI] [PubMed] [Google Scholar]
- 42.Fischer DS, Price DC. Clin Chem. 1964;10:21. [PubMed] [Google Scholar]
- 43.Muhoberac BB, Wharton DC, Babcock LM, Harrington PC, Wilkins RG. Biochim Biophys Acta. 1980;626:337–345. doi: 10.1016/0005-2795(80)90128-2. [DOI] [PubMed] [Google Scholar]
- 44.Antanaitis BC, Aisen P, Lilienthal HR. J Biol Chem. 1983;258:3166–3172. [PubMed] [Google Scholar]
- 45.Averill BA, Davis JC, Burman S, Zirino T, Sandersloehr J, Loehr TM, Sage JT, Debrunner PG. J Am Chem Soc. 1987;109:3760–3767. [Google Scholar]
- 46.Legall J, Prickril BC, Moura I, Xavier AV, Moura JJG, Huynh BH. Biochemistry. 1988;27:1636–1642. doi: 10.1021/bi00405a037. [DOI] [PubMed] [Google Scholar]
- 47.Gupta N, Bonomi F, Kurtz DM, Ravi N, Wang DL, Huynh BH. Biochemistry. 1995;34:3310–3318. doi: 10.1021/bi00010a021. [DOI] [PubMed] [Google Scholar]
- 48.Hendrich MP, Elgren TE, Que L. Biochem Biophys Res Commun. 1991;176:705–710. doi: 10.1016/s0006-291x(05)80241-4. [DOI] [PubMed] [Google Scholar]
- 49.Davydov R, Kuprin S, Graslund A, Ehrenberg A. J Am Chem Soc. 1994;116:11120–11128. [Google Scholar]
- 50.Atta M, Andersson KK, Ingemarson R, Thelander L, Graslund A. J Am Chem Soc. 1994;116:6429–6430. [Google Scholar]
- 51.Woodland MP, Patil DS, Cammack R, Dalton H. Biochim Biophys Acta. 1986;873:237–242. [Google Scholar]
- 52.Fox BG, Surerus KK, Munck E, Lipscomb JD. J Biol Chem. 1988;263:10553–10556. [PubMed] [Google Scholar]
- 53.Hendrich MP, Debrunner PG. Biophysical Journal. 1989;56:489–506. doi: 10.1016/S0006-3495(89)82696-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Hendrich MP, Munck E, Fox BG, Lipscomb JD. J Am Chem Soc. 1990;112:5861–5865. [Google Scholar]
- 55.Fox BG, Hendrich MP, Surerus KK, Andersson KK, Froland WA, Lipscomb JD, Munck E. J Am Chem Soc. 1993;115:3688–3701. [Google Scholar]
- 56.Moura I, Huynh BH, Hausinger RP, Legall J, Xavier AV, Munck E. J Biol Chem. 1980;255:2493–2498. [PubMed] [Google Scholar]
- 57.Zimmermann R, Huynh BH, Munck E, Lipscomb JD. J Chem Phys. 1978;69:5463–5467. [Google Scholar]
- 58.Bauminger ER, Harrison PM, Nowik I, Treffry A. Biochemistry. 1989;28:5486–93. doi: 10.1021/bi00439a025. [DOI] [PubMed] [Google Scholar]
- 59.Pulver SC, Tong WH, Bollinger JM, Stubbe J, Solomon EI. J Am Chem Soc. 1995;117:12664–12678. [Google Scholar]
- 60.Atta M, Debaecker N, Andersson KK, Latour JM, Thelander L, Graslund A. J Biol Inorg Chem. 1996;1:210–220. [Google Scholar]
- 61.Strand KR, Yang YS, Andersson KK, Solomon EI. Biochemistry. 2003;42:12223–12234. doi: 10.1021/bi035248q. [DOI] [PubMed] [Google Scholar]
- 62.Maroney MJ, Kurtz DM, Nocek JM, Pearce LL, Que L. J Am Chem Soc. 1986;108:6871–6879. [Google Scholar]
- 63.Reem RC, Solomon EI. J Am Chem Soc. 1987;109:1216–1226. [Google Scholar]
- 64.Holmes MA, Letrong I, Turley S, Sieker LC, Stenkamp RE. J Mol Biol. 1991;218:583–593. doi: 10.1016/0022-2836(91)90703-9. [DOI] [PubMed] [Google Scholar]
- 65.Hartman JAR, Rardin RL, Chaudhuri P, Pohl K, Wieghardt K, Nuber B, Weiss J, Papaefthymiou GC, Frankel RB, Lippard SJ. J Am Chem Soc. 1987;109:7387–7396. [Google Scholar]
- 66.Hagen KS, Lachicotte R. J Am Chem Soc. 1992;114:8741–8742. [Google Scholar]
- 67.Thomann H, Bernardo M, McCormick JM, Pulver S, Andersson KK, Lipscomb JD, Solomon EI. Journal of the American Chemical Society. 1993;115:8881–8882. [Google Scholar]
- 68.Davydov RM, Davydov A, Ingemarson R, Thelander L, Ehrenberg A, Graslund A. Biochemistry. 1997;36:9093–9100. doi: 10.1021/bi9700375. [DOI] [PubMed] [Google Scholar]
- 69.Krebs C, Edmondson DE, Huynh BH. Meth Enzym. 2002;354:436–454. doi: 10.1016/s0076-6879(02)54034-9. [DOI] [PubMed] [Google Scholar]
- 70.Krebs C, Bollinger JM, Theil EC, Huynh BH. J Biol Inorg Chem. 2002;7:863–869. doi: 10.1007/s00775-002-0371-1. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.