

The inclusion of fluorinated functional groups in small molecules has had a profound impact on the pharmaceutical, material, and agrochemical industries.[1, 2] In particular, the trifluoromethyl (CF3) substituent has emerged as an important functional group for the modulation of the physical properties in new pharmaceutical candidates as it has excellent metabolic stability, lipophilicity, and is electron-withdrawing in nature.[3] Myriad of fluorinated biologically active pharmaceutical compounds have been identified,[4] with an estimated 20% of drugs on the market containing fluorine.[1] On this basis, there has been a recent surge in the number of reports describing the formation of carbon–trifluoromethyl (C–CF3) bonds, demonstrating the continuing need for the development of efficient methods to incorporate these groups.
Early research into C–CF3 bond formation primarily focused on the exploration of nucleophilic and radical sources of the CF3 group.[5] These efforts resulted in the development of many trifluoromethylation reactions, including nucleophilic addition to carbonyl electrophiles,[6-7] halotrifluoromethylation of olefins,[8] enolate addition to the CF3 radical,[9] and formation of aryl–CF3 bonds.[10-11] While less extensively explored, electrophilic trifluoromethylating reagents have allowed for the trifluoromethylation of a range nucleophiles to be achieved.[12-13] In particular development of hypervalent iodine-based trifluoromethylating reagents by Togni has significantly broadened the scope of electrophilic trifluoromethylation methods.[14] Herein, we report our efforts in developing a new catalytic allylic trifluoromethylation of terminal olefins using the Togni electrophilic trifluoromethylating reagent 1 [Figure 1].[15]
Figure 1.

CuI-catalyzed oxidative trifluoromethylation of olefins.
Currently, only a limited number of methods are available to construct allylic–CF3 bonds from olefins. Research in this area has typically focused on perfluoroalkylations using iodonium salts, of which the trifluoromethyl variant is unstable and not synthetically viable.[16] The few methods that describe the preparation of molecules containing allylic–CF3 functional groups (e.g., 2) are not only limited in scope, but also require harsh reaction conditions, super-stoichiometric quantities of transition metal promoters, and toxic or expensive reagents.[17] An additional disadvantage of the reported methods is the required use of pre-functionalized starting materials such as allyl bromides or fluorosulfones.
We sought to develop a direct trifluoromethylation of unactivated olefins as a more convenient method to access 2. We hypothesized that this transformation might be achieved using a Cu-based strategy involving the generation of an allylic radical and a subsequent CF3• transfer [Scheme 1, Path A].[18] Alternatively, if reagent 1 could be used as an electrophilic CF3• equivalent, 2 may be generated via an atom transfer radical addition (ATRA)-type pathway (Path B).[19] Finally, an electrophilic trifluoromethylation proceeding through a cationic intermediate may also be viable (Path C).
Scheme 1.
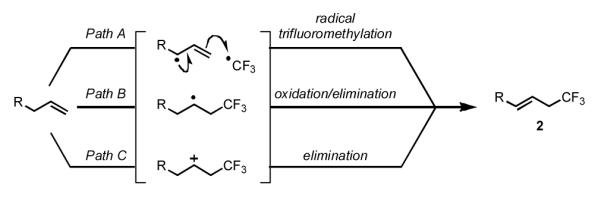
Plausible allylic trifluoromethylation mechanisms: allylic oxidation (Path A), atom transfer radical addition (Path B), electrophilic trifluoromethylation (Path C).
Commencing our studies, we examined the ability of various CuI/II salts to catalyze the trifluoromethylation of 4-phenyl-1-butene using electrophilic trifluoromethylating reagents.[12] Our most promising result was obtained using reagent 1 and CuCl as a catalyst, providing the corresponding linear allylic trifluoromethylation product 2a in good yield and high E/Z selectivity [Scheme 2]. We found the use of 1 to be convenient as it is easily prepared in three chromatography-free steps from inexpensive and recyclable starting materials.[14d] Mass spectral analysis indicated the desired product 2a was accompanied by chlorinated and other mono- and bis(trifluoromethylated) side products, complicating purification. Unfortunately conducting the reaction at 0 °C only suppressed side product formation to a minimal extent.
Scheme 2.
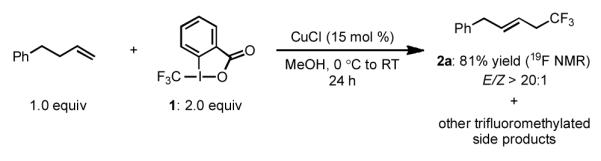
CuCl-catalyzed trifluoromethylation of 4-phenyl-1-butene: lead result.
With promising results obtained in our preliminary studies, we continued our efforts toward improving the efficiency of this reaction. Noting that the major side products contained two trifluoromethyl groups, we surmised that suppression of bis(trifluoromethylation) may be accomplished by the use of an excess of olefin. Thus, we evaluated various CuI/II catalysts in the trifluoromethylation of 4-phenyl-1-butene using an altered reaction stoichiometry of alkene:1 of 1.05:1 [Table 1]. Gratifyingly, the use of an excess of olefin reduced the amount of bis(trifluoromethylated) side products to ~5% independent of the identity of the CuI catalyst employed. The yields of these transformations were moderately lower than when 1 was used in excess, presumably due to Lewis acid-catalyzed decomposition of 1.[20] The modestly superior results obtained with (MeCN)4CuPF6 prompted us to continue optimization using this copper source. We found that reactions run in a range of solvents yielded a significant amount of desired product 2a. Interestingly, the E/Z ratio varied substantially depending on the identity of the alcoholic solvent examined [entries 5-9]. Methanol provided the best yield and E/Z ratio of the conditions studied [entry 5]. An additional increase in the alkene:1 ratio to 1.25:1.0 provided more consistent results and marginally higher yields [entry 6].
Table 1.
Selected optimization studies for the Cu-catalyzed trifluoromethylation of 4-phenyl-1-butene with 1.[[a]]
| |||||
|---|---|---|---|---|---|
|
| |||||
| Entry | CuI Source | Solvent | Conversion [%][[b]] |
Yield [%][[b]] |
E/Z [[c]] |
|
| |||||
| 1 | CuCl | MeOH | 100 | 63 | 96:4 |
| 2 | CuTC | MeOH | 100 | 68 | 97:3 |
| 3 | [Cu(OTf)]2PhH | MeOH | 93 | 61 | 86:14 |
| 4[[d]] | Cu(OTf)2 | CH2Cl2 | 81 | 0 | - |
| 5 | (MeCN)4CuPF6 | MeOH | 100 | 68 | 98:2 |
| 6 [[d],[e]] | (MeCN)4CuPF6 | MeOH | 100 | 71 | 98:2 |
| 7 | (MeCN)4CuPF6 | EtOH | 100 | 63 | 96:4 |
| 8 | (MeCN)4CuPF6 | i-PrOH | 100 | 43 | 95:5 |
| 9 | (MeCN)4CuPF6 | t-BuOH | 100 | 50 | 83:17 |
| 10 | (MeCN)4CuPF6 | Me2CO | 100 | 52 | 90:10 |
| 11 | (MeCN)4CuPF6 | MeCN | 24 | 0 | - |
| 12 | (MeCN)4CuPF6 | C6H6 | 100 | 27 | 89:11 |
| 13 | (MeCN)4CuPF6 | CH2Cl2 | 100 | 57 | 90:10 |
Conditions: alkene (0.205 mmol, 1.05 equiv), 1 (0.20 mmol, 1.0 equiv), Cu (0.030 mmol, 0.15 equiv) in MeOH (1.0 mL) at 0 °C for 15 min, then RT for 23 h.
Determined by 191F NMR spectroscopy using (trifluoromethoxy)benzene as an internal standard.
Determined by 19F NMR spectroscopy.
1.25 equiv of the alkene was used.
Average isolated yield of two independent runs on a 1.0 mmol scale (relative to 1). CuTC = copper(I) thiophene-2-carboxylate
We next proceeded to examine the scope of this reaction using our optimized protocol [Table 2]. The mild reaction conditions employed allowed for the trifluoromethylation of molecules containing a range of functional groups, including unprotected alcohols, protected amines, esters, amides, and alkyl bromides. Terminal epoxide-containing substrates required the use of a catalyst with lower Lewis acidity in order to avoid nucleophilic ring-opening by methanol; thus copper(I) thiophene-2-carboxylate (CuTC) was used for 2-(hex-5-en-1-yl)oxirane [entry 6]. In most cases, the E/Z selectivity was excellent, with an average ratio of 94:6 for the substrates examined. We found branched terminal olefins and 1,2-disubstituted olefins to be unsuitable substrates due to the formation of complex regioisomeric product mixtures. Furthermore, cyclic substrates furnished only trace amounts of product.[21]
 |
(1) |
Table 2.
Scope of the CuI-catalyzed trifluoromethylation of terminal olefins with 1.[[a]]
| ||||
|---|---|---|---|---|
|
| ||||
| Entry | Product | Yield [%][[b]] | E/Z[[c]] | |
|
| ||||
| 1 | 2b [[d]] |

|
54 | 97:3 |
| 2 | 2c [[e]] |
|
67 | 97:3 |
| 3 | 2d [[e]] |
|
69 | 95:5 |
| 4 | 2e |
|
72 | 94:6 |
| 5 | 2f |
|
67 | 97:3 |
| 6 | 2g [[f]] |
|
70 | 93:7 |
| 7 | 2h |
|
78 | 96:4 |
| 8 | 2i |
|
79 | 95:5 |
| 9 | 2j |
|
75 | 94:6 |
| 10 | 2k |
|
73 | 94:6 |
| 11 | 2l [[e]] |

|
72 | 97:3 |
| 12 | 2m |

|
75 | 89:11 |
| 13 | 2n [[e]] |

|
80 | 95:5 |
Conditions: alkene (1.25 equiv), 1 (1.0 equiv), CuI (0.15 equiv) in MeOH (0.5 mL/0.10 mmol 1) at 0 °C for 15 min, then RT for 23 h. Reactions were run on a 0.50–1.00 mmol scale of 1.
Average isolated yield of two independent runs. Products contained approximately ≤ 5% other mono- and bis(trifluoromethylated) side products by 19F NMR spectroscopy.
Determined by 19F NMR spectroscopy.
1.0 equiv of the alkene was used.
(MeCN)4CuPF6 (0.25 equiv) was used.
CuTC (0.15 equiv) was used.
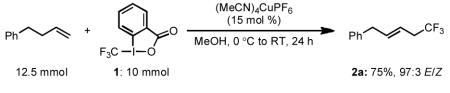
In order to demonstrate the robustness of this transformation, we conducted the trifluoromethylation of 4-phenyl-1-butene on a 10 mmol scale [Eq 1]. All reagents were weighed out on the bench top, open to the air, and the setup was conducted using standard Schlenk techniques. The results from this experiment indicate that the method described in this communication can be set up on the bench top without an accompanying sacrifice of the reaction efficiency.
 |
(2) |
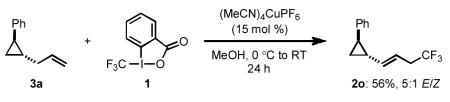
Similar to the proposed mechanism of the Kharasch-Sosnovsky CuI/II-catalyzed oxidation of olefins to generate allyl esters,[18] we wanted to probe whether this transformation proceeded through an allylic radical intermediate. We were intrigued, however, by the high selectivity for linear trifluoromethylated products obtained using the method described herein. This is in contrast to most reports of Kharasch-Sosnovsky-type oxidative alkene functionalizations, suggesting a possible divergence from this mechanistic pathway. In order to determine whether this transformation did indeed proceed through a free allylic radical, we conducted the trifluoromethylation of cyclopropane radical clock 3a [Eq 2]. Subjecting this substrate to our standard conditions provided the trifluoromethylated cyclopropane 2o in moderate yield, suggesting that a mechanism involving formation of an allylic radical is unlikely. However, we note that other trifluoromethylated side products were present (≤ 3 % yield each) that were not identifiable, which precludes us from conclusively stating that no ring-opened product was observed.
The results with cyclopropane 3a prompted us to consider an alternative mechanistic possibility, wherein the trifluoromethylation event occurs via an atom transfer radical addition-type pathway via homolytic cleavage of the alkene.[19] Data to support or refute this mechanism was sought by examining diethyl diallylmalonate as a cyclization radical clock [Scheme 3]. The major products obtained under these conditions were cyclopentane derivatives 4a and 4b. The presence of these species is explained by the occurrence of a 5-exo-trig cyclization that proceeds after the C–CF3 bond-forming event. It is unclear if the trifluoromethylation results in the generation a free-radical intermediate (5a) or an alkylcopper species (5b). After cyclization, termination occurs by a second trifluoromethylation or elimination to generate products 4a or 4b, respectively. Of note, we found that conducting the trifluoromethylation reaction in the presence of selected radical scavengers provided variable results that did not aid in our understanding of the reaction mechanism.[22] Further analysis will be necessary to more accurately elucidate the nature of this transformation.
Scheme 3.
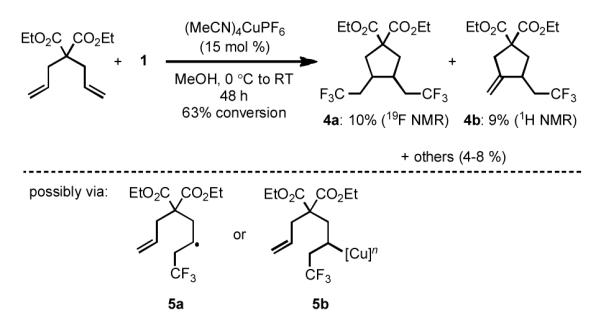
Examination of a diallylmalonate cyclization radical clock.
In conclusion, we have developed an allylic trifluoromethylation of unactivated terminal olefins. This method allows for the preparation of allyl–CF3 products previously difficult to access in a straightforward and efficient manner. The mild conditions for this transformation enable the trifluoromethylation of a range of substrates bearing numerous functional groups. A preliminary analysis suggests that the reaction mechanism is complex and multiple pathways leading to the desired allyl–CF3 products may be operating.[23] Future efforts will focus on examining the mechanistic details more extensively en route to expanding the generality and increasing the efficiency of this transformation.
Experimental Section
For 10.0 mmol scale reaction [Eq 1]: (E)-(5,5,5-trifluoropent-2-en-1-yl)benzene (2a). A 100 mL Schlenk flask was flame-dried under high vacuum and backfilled with argon. On the bench top, open to air, (MeCN)4CuPF6 (0.559 g, 1.50 mmol, 0.15 equiv) and 1 (3.16 g, 10.0 mmol, 1.0 equiv) were weighed out and added to the Schlenk flask. The flask was then sealed with a rubber septum, evacuated and backfilled with argon (this process was repeated for a total of three times) and cooled to 0 °C in an ice-water bath. The flask was charged with anhydrous methanol (50 mL) and 4-phenyl-1-butene (1.65 g, 1.88 mL, 12.50 mmol, 1.25 equiv) successively via syringe (a bright green-blue color is observed upon solvent addition). The reaction mixture was stirred for 30 min at 0 °C, after which the ice-water bath was removed and stirring was continued for an additional 23 h the reaction. The reaction mixture was partitioned between CH2Cl2 (75 mL) and sat. aq. NaHCO3 (75 mL). The aqueous layer was separated and extracted with CH2Cl2 (2 × 50 mL). The combined organic extracts were washed with sat. aq. NaHCO3 (75 mL), dried over Na2SO4 and concentrated in vacuo. The resultant oil was purified by silica gel chromatography (pentane) to afford 2a (1.503 g, 75%) as a clear and colorless oil (E/Z = 97:3) contaminated with 2.5 mol % of a bis(trifluoromethylated) side product. There was 3.5 mol % of a mono-trifluoromethylated side product with a 19F NMR shift consistent with the vinyl trifluoromethylation product (6).

Supplementary Material
Footnotes
Supporting information for this article is available on the WWW under http://www.angewandte.org or from the author.
We thank the National Institutes of Health (GM46059) for financial support of this project and for a postdoctoral fellowship to A.T.P. (F32GM093532). The Varian 300 MHz and Bruker 400 MHz NMR spectrometers used in this work were supported by grants from the National Science Foundation (CHE-9808061 and DBI-9729592) and National Institutes of Health (1S10RR13886-01), respectively.
Note: Reactions carried out on a 0.50–1.0 mmol scale of 1 [Table 2] were set up in an inert atmosphere glove box under a nitrogen atmosphere.
References
- [1].Muller K, Faeh C, Diederich F. Science. 2007;317:1881. doi: 10.1126/science.1131943. [DOI] [PubMed] [Google Scholar]
- [2].Hiyama T. Fluorine Compounds: Chemistry and Applications. Springer; Berlin: 2000. [Google Scholar]
- [3].Yamazaki T, Taguchi T, Ojima I. In: Fluorine in Medicinal Chemistry and Chemical Biology. Ojima I, editor. Wiley-Blackwell; Chichester: 2009. p. 3. [Google Scholar]
- [4].Welch JT. Tetrahedron. 1987;43:3123. [Google Scholar]
- [5].Dolbier WR. Chem. Rev. 1996;96:1557. doi: 10.1021/cr941142c. [DOI] [PubMed] [Google Scholar]
- [6].For reviews, see: Prakash GKS, Yudin AK. Chem. Rev. 1997;97:757. doi: 10.1021/cr9408991. Ma J-A, Cahard D. Chem. Rev. 2008;108:PR1. doi: 10.1021/cr800221v.
- [7].a) Billard T, Bruns S, Langlois BR. Org. Lett. 2000;2:2101. doi: 10.1021/ol005987o. [DOI] [PubMed] [Google Scholar]; b) Billard T, Langlois Bernard R., Blond G. Eur. J. Org. Chem. 2001;1467 doi: 10.1021/jo015587u. [DOI] [PubMed] [Google Scholar]; c) Pooput C, Dolbier WR, Médebielle M. J. Org. Chem. 2006;71:3564. doi: 10.1021/jo060250j. [DOI] [PubMed] [Google Scholar]
- [8].a) Kamigata N, Fukushima T, Yoshida M. J. Chem. Soc. Chem. Commun. 1989;1559 [Google Scholar]; b) Kamigata N, Fukushima T, Terakawa Y, Yoshida M, Sawada H. J. Chem. Soc. Perkin Trans. 1. 1991;627 [Google Scholar]; c) Ignatowska J, Dmowski W. J. Fluorine Chem. 2007;128:997. [Google Scholar]
- [9].a) Cantacuzène D, Dorme R. Tetrahedron Lett. 1975;16:2031. [Google Scholar]; b) Iseki K, Nagai T, Kobayashi Y. Tetrahedron Lett. 1993;34:2169. [Google Scholar]; c) Miura K, Taniguchi M, Nozaki K, Oshima K, Utimoto K. Tetrahedron Lett. 1990;31:6391. [Google Scholar]; d) Itoh Y, Mikami K. Org. Lett. 2005;7:649. doi: 10.1021/ol047565a. [DOI] [PubMed] [Google Scholar]; e) Nagib DA, Scott ME, MacMillan DWC. J. Am. Chem. Soc. 2009;131:10875. doi: 10.1021/ja9053338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].For a recent review, see: Tomashenko OA, Grushin VV. Chem. Rev. 2011 doi: 10.1021/cr1004293. ASAP, DOI: 10.1021/cr1004293.
- [11].a) Grushin VV, Marshall WJ. J. Am. Chem. Soc. 2006;128:12644. doi: 10.1021/ja064935c. [DOI] [PubMed] [Google Scholar]; b) Ball ND, Kampf JW, Sanford MS. J. Am. Chem. Soc. 2010;132:2878. doi: 10.1021/ja100955x. [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Wang X, Truesdale L, Yu J-Q. J. Am. Chem. Soc. 2010;132:3648. doi: 10.1021/ja909522s. [DOI] [PubMed] [Google Scholar]; d) Cho EJ, Senecal TD, Kinzel T, Zhang Y, Watson DA, Buchwald SL. Science. 2010;328:1679. doi: 10.1126/science.1190524. [DOI] [PMC free article] [PubMed] [Google Scholar]; e) Chu L, Qing F-L. Org. Lett. 2010;12:5060. doi: 10.1021/ol1023135. [DOI] [PubMed] [Google Scholar]; f) Senecal TD, Parsons AT, Buchwald SL. J. Org. Chem. 2011;76:1174. doi: 10.1021/jo1023377. [DOI] [PMC free article] [PubMed] [Google Scholar]; g) Knauber T, Arikan F, Röschenthaler G-V, Gooßen LJ. Chem. – Eur. J. 2011;17:2689. doi: 10.1002/chem.201002749. [DOI] [PubMed] [Google Scholar]
- [12].For a recent review, see: Shibata N, Matsnev A, Cahard D. Beilstein J. Org. Chem. 2010;6:65. doi: 10.3762/bjoc.6.65.
- [13].Yagupolskii LM, Kondratenko NV, Timofeeva GN. J. Org. Chem. USSR. 1984;20:103. [Google Scholar]
- [14].a) Kieltsch I, Eisenberger P, Togni A. Angew. Chem. Int. Ed. 2007;46:754. doi: 10.1002/anie.200603497. [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2007;119:768. [Google Scholar]; b) Eisenberger P, Kieltsch I, Armanino N, Togni A. Chem. Commun. 2008;1575 doi: 10.1039/b801424h. [DOI] [PubMed] [Google Scholar]; c) Kieltsch I, Eisenberger P, Stanek K, Togni A. Chimia. 2008;62:260. [Google Scholar]; d) Stanek K, Koller R, Togni A. J. Org. Chem. 2008;73:7678. doi: 10.1021/jo8014825. [DOI] [PubMed] [Google Scholar]; e) Koller R, Huchet Q, Battaglia P, Welch JM, Togni A. Chem. Commun. 2009;5993 doi: 10.1039/b913962a. [DOI] [PubMed] [Google Scholar]; f) Koller R, Stanek K, Stolz D, Aardoom R, Niedermann K, Togni A. Angew. Chem. Int. Ed. 2009;48:4332. doi: 10.1002/anie.200900974. [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2009;121:4396. [Google Scholar]; g) Niedermann K, Früh N, Vinogradova E, Wiehn MS, Moreno A, Togni A. Angew. Chem. Int. Ed. 2011;50:1059. doi: 10.1002/anie.201006021. [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2011;123:5. [Google Scholar]; h) Shimizu R, Egami H, Nagi T, Chae J, Hamashima Y, Sodeoka M. Tetrahedron Lett. 2010;51:5947. [Google Scholar]; i) Liu T, Shen Q. Org. Lett. 2011;13:2342. doi: 10.1021/ol2005903. [DOI] [PubMed] [Google Scholar]; j) Allen AE, MacMillan DWC. J. Am. Chem. Soc. 2011;132:4986. doi: 10.1021/ja100748y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Eisenberger P, Gischig S, Togni A. Chem. – Eur. J. 2006;12:2579. doi: 10.1002/chem.200501052. [DOI] [PubMed] [Google Scholar]
- [16].a) Umemoto T, Kuriu Y, Nakayama S.-i. Tetrahedron Lett. 1982;23:1169. [Google Scholar]; b) Umemoto T. Chem. Rev. 1996;96:1757. doi: 10.1021/cr941149u. [DOI] [PubMed] [Google Scholar]; c) Umemoto T, Kuriu Y, Shuyama H, Miyano O, Nakayama S-I. J. Fluorine Chem. 1982;20:695. [Google Scholar]
- [17].a) Kobayashi Y, Yamamoto K, Kumadaki I. Tetrahedron Lett. 1979;20:4071. [Google Scholar]; b) Duan J-X, Chen Q-Y. Journal Chem. Soc. Perkin Trans 1. 1994;725 [Google Scholar]; c) Konno T, Takehana T, Mishima M, Ishihara T. J. Org. Chem. 2006;71:3545. doi: 10.1021/jo0602120. [DOI] [PubMed] [Google Scholar]; d) Kim J, Shreeve JM. Org. Biomol. Chem. 2004;2:2728. doi: 10.1039/B412480B. [DOI] [PubMed] [Google Scholar]
- [18].Kharasch MS, Sosnovsky G. J. Am. Chem. Soc. 1958;80:756. Kharasch MS, Sosnovsky G, Yang NC. J. Am. Chem. Soc. 1959;81:5819. For a review see: Rawlinson DJ, Sosnovsky G. Synthesis. 1972;1
- [19].Davies T, Haszeldine RN, Tipping AE. J. Chem. Soc. Perkin Trans. 1. 1980;927 For a review, see Eckenhoff WT, Pintauer T. Catalysis Rev. 2010;52:1.
- [20].Fantasia S, Welch JM, Togni A. J. Org. Chem. 2010;75:1779. doi: 10.1021/jo9025429. [DOI] [PubMed] [Google Scholar]
- [21].Subjecting cis-cyclodecene to the standard reaction conditions [Table 2] furnished only trace amounts of the expected allylic CF3 product despite complete consumption of 1, as determined by 19F NMR spectroscopy. Use of hex-4-en-1-ol produced a complex mixture of mono- and bis(trifluoromethylated) products in approximately 30% combined yield.
- [22].We conducted the trifluoromethylation of 4-phenyl-1-butene under the standard reaction conditions [Table 2] in the presence of varying amounts of several radical scavengers: galvinoxyl (0.30 equiv), 1,4-dinitrobenzene (0.30 equiv), hydroquinone (0.30 equiv), 4-methoxyphenol (1.0 equiv), and butylatedhydroxytoluene (1.0 equiv). The conversion of 1 and yield of 2a varied considerably depending on the identity of the scavenger employed.
- [23].a) Beringer FM, Geering EJ, Kuntz I, Mausner M. J. Phys. Chem. 1956;60:141. [Google Scholar]; b) Caserio MC, Glusker DL, Roberts JD. J. Am. Chem. Soc. 1959;81:336. [Google Scholar]; c) Lockhart TP. J. Am. Chem. Soc. 1983;105:1940. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.