

Abstract
The cAMP response element-binding protein (CREB) is a nuclear transcription factor that is critical for normal and neoplastic hematopoiesis. Previous studies have demonstrated that CREB is a proto-oncogene whose overexpression promotes cellular proliferation in hematopoietic cells. Transgenic mice that overexpress CREB in myeloid cells develop a myeloproliferative disease with splenomegaly and aberrant myelopoiesis. However, CREB overexpressing mice do not spontaneously develop acute myeloid leukemia. In this study, we used retroviral insertional mutagenesis to identify genes that accelerate leukemia in CREB transgenic mice. Our mutagenesis screen identified several integration sites, including oncogenes Gfi1, Myb, and Ras. The Sox4 transcription factor was identified by our screen as a gene that cooperates with CREB in myeloid leukemogenesis. We show that the transduction of CREB transgenic mouse bone marrow cells with a Sox4 retrovirus increases survival and self-renewal of cells in vitro. Furthermore, leukemic blasts from the majority of acute myeloid leukemia patients have higher CREB, phosphorylated CREB, and Sox 4 protein expression. Sox4 transduction of mouse bone marrow cells results in increased expression of CREB target genes. We also demonstrate that CREB is a direct target of Sox4 by chromatin immunoprecipitation assays. These results indicate that Sox4 and CREB cooperate and contribute to increased proliferation of hematopoietic progenitor cells.
Introduction
Acute myeloid leukemia (AML) results from the accumulation of several genetic alterations that confer a proliferative or survival advantage to hematopoietic stem cells.1 More than 5000 total mutations with or without chromosomal aberrations in AML have been reported (http://cgap.nci.nih.gov/chromosomes/).2 Recent studies suggest that specific chromosomal translocations are found in less than 40% of AML patients.3 Furthermore, many of these genetic alterations are not sufficient to drive leukemogenesis. Therefore, identifying novel oncogenic alterations that cause leukemia and deciphering the signaling pathways that are affected is key to improving our understanding of this disease and identifying novel therapeutic targets.
The cAMP response element-binding protein (CREB) is a 43-kDa nuclear transcription factor that has been implicated in severalcancers, including leukemias.4,5 The transcription factor CREB is downstream of cell surface receptors and mitogens that are critical for normal and neoplastic hematopoiesis.6–9 We reported previously that CREB is a proto-oncogene whose overexpression promotes cellular transformation of hematopoietic cells. Transgenic mice overexpressing CREB in the myeloid lineage cells develop a myeloproliferative/myelodysplastic syndrome (MPD/MDS) after 1 year.6,9 In addition, a majority of patients with AML and acute lymphoid leukemia overexpress CREB in the bone marrow (BM).6,8
Previous reports demonstrated that 80% of patients with AML overexpressed CREB by Western blot analysis and ELISA.8 CREB also was phosphorylated and actively bound to the cyclic AMP response element consensus site at diagnosis but not in remission or in nonleukemia control samples.
Retroviral insertional mutagenesis (RIM) or retroviral tagging is a powerful tool that has been used for oncogene discovery.10–14 In this study, RIM was used to accelerate myeloid leukemia in CREB transgenic (TG) mice and to identify CREB-cooperating oncogenes. We inoculated CREB TG mice with the MOL4070LTR retrovirus. MOL4070LTR is a Moloney murine leukemia virus–based retrovirus capable of inducing myeloid leukemia at a higher frequency.15 In our mutagenesis screen, the sox4 transcription factor was identified as a potential cooperating oncogene. Retroviral transduction studies demonstrated that aberrant expression of sox4 synergizes with CREB to increase proliferation, self-renewal, and survival of bone marrow cells in culture. Furthermore, increased protein expression of Sox4, CREB, and phosphorylated CREB was observed in the majority of blast cells from AML patients. Taken together, our results suggest that sox4 and CREB cooperate to enhance myeloid cell proliferation.
Methods
Plasmid constructs
Retroviral vectors pMIG (MSCV-IRES-GFP) and pMIG-Sox4 were kindly provided by A. Perkins (University of Rochester). The pMIG-Sox4 vector was constructed by subcloning murine sox4 cDNA from pIRES-Sox4 into the multiple cloning site of pMIG.16
Cell culture
Human AML cell line HL60, ML-2, NB4, THP1, MV4;11, K562, and NOMO (ATCC) were cultured in media supplemented with 10% FBS.
Retroviral infection
The MOL4070LTR retrovirus was propagated and harvested as described previously.17 Newborn neonatal pups were inoculated with 1 × 105 infectious particles of MOL4070LTR retrovirus in 0.1 mL of medium intraperitoneally, 1 to 3 days after birth. C57BL/6 wild-type (WT) or CREB TG mice were housed and cared for in accordance with the recommendations of the Guide for the Care and Use of Laboratory Animals.18 The human mrp8 promoter up-regulates CREB in macrophage and monocytic lineage cells in CREB TG mice.6,9 Mice were under daily observation for signs and symptoms of leukemia, including lethargy, labored breathing, lordokyphosis, and abdominal distention. Mice were euthanized when these symptoms were observed. All murine studies were approved by the University of California, Los Angels (UCLA) Institutional Animal Care and Use Committee.
Pathologic evaluation and phenotype analysis
Moribund mice were euthanized, and liver, spleen, and bone marrow were harvested for analysis. Paraffin-embedded tissue sections were stained with H&E, and cytospin slides were prepared from bone marrow cell suspensions and stained with Wright-Giemsa. Flow cytometry was performed by staining single-cell suspensions from bone marrow with fluorescein isothiocyanate (FITC)–conjugated anti–mouse Gr-1, phycoerythrin-conjugated CD11b (Mac), allophycocyanin-conjugated CD90, and phycoerythrin-Cy5 CD19, FITC-conjugated anti–mouse CD3, phycoerythrin-conjugated B220, phycoerythrin-conjugated Ter119, FITC-conjugated Sca1, and allophycocyanin-conjugated CD117 (eBioscience).
Long-mediated PCR
Genomic DNA from affected mice was harvested from splenocytes, restricted, ligated to adapters, and amplified using adapter and viral specific primers. Retroviral integration sites were cloned using ligation-mediated (LM)–PCR. Amplicons were purified (QIAGEN), cloned into pGEM-T Easy (Promega), and transformed into DH5α Escherichia coli cells (Invitrogen).19 Plasmid DNA was isolated and sequenced. These sequences were mapped to the mouse genome to identify the chromosomal location using the National Center for Biotechnology Information mouse genome and Ensembl Genome Browser (http://www.ensembl.org/).20
Retroviral transduction
VSV-G pseudotyped retrovirus was produced by transient transfection of 293T cells. Vector supernatant was collected and concentrated by ultracentrifugation. Murine hematopoietic stem cells and progenitors were isolated from 6- to 12-week-old C57BL6 mice treated with 150 mg/kg 5-fluorouracil. Whole bone marrow cells were harvested 3 to 5 days after 5-fluorouracil treatment, and the red blood cells were lysed using ammonium chloride lysing solution. Whole bone marrow cells were maintained in culture for 48 hours in Iscove modified Dulbecco medium (IMDM) with 30% FCS, 10% BSA, 2.5 ng/mL murine stem cell factor (SCF), 10 ng/mL murine IL-3, and 25 ng/mL human IL-6 and 100 μL 2-mercaptoethanol. Cells were plated into a well coated with Retronectin (Clonetech Laboratories), incubated with viral supernatant in the presence of the same cytokines and harvested after 24 to 48 hours. Viral supernatant was subsequently removed, and fresh media were added to cultures. The transductants were then allowed to recover and expand for 24 hours before sorting for the green fluorescent protein (GFP)–selectable marker.
Hematopoietic progenitor assays
Methylcellulose-based culture (MC) of bone marrow cells (5-10 000 GFP-positive cells/mL MethoCult) was performed using MethoCult GF M3434 (StemCell Technologies) containing mouse SCF, mouse IL-3, human IL-6, and human erythropoietin. Colonies were counted after 7 to 14 days of culture. For serial replating assays, cells were harvested from the MC1 after 7 to 14 days of culture and replated in MC2 using GF M3434.
In vitro phenotype and survival analysis
For phenotype analysis, GFP-sorted cells (0.5 × 106) were expanded in IMDM supplemented with 30% FCS, 10% BSA, 2.5 ng/mL murine SCF, 10 ng/mL murine IL-3, and 25 ng/mL human IL-6 and 100μM 2-mercaptoethanol for 2 days. Cells were then harvested and phenotyped using CD11b and Gr-1 (Ly-6G) markers. For survival assays, 1 × 106 cells were plated in the absence of cytokines and limited serum (2% FBS). Cells were harvested at 16 to 18 hours and stained for markers of apoptosis (annexin V and 7-aminoactinomycin D [7-AAD]). For BrdU experiments, 0.5 × 106 cells were seeded at the proper concentrations and labeled with BrdU using the BrdU Flow cytometry kit (BD Biosciences). Cells were analyzed by flow cytometry and analyzed using the FlowJo Version 9.4.1 software (TreeStar).
Cell sorting
Total murine bone marrow cells were harvested, and RBCs were lysed with ammonium chloride lysing solution. Bone marrow cells were then preincubated with anti-CD16/32 (FcyRII-III; eBiosciences) or total mouse IgG (for common lymphoid progenitor [CMP] and granulocyte macrophage progenitor [GMP] populations only; Axell) to reduce nonspecific labeling. Stem cells and progenitors were isolated by staining with a lineage depletion cocktail that consisted of CD3ϵ, CD8α, B220, Mac-1, Gr-1, TER-119, TCRαβ, TCRγδ, IgM, and NKl.l FITC-labeled antibodies and immunostaining with fluorochrome-conjugated antibodies based on the following phenotypes: CMPs (Lin−CD127−Sca1−CD117hi CD16/32lo CD34+), GMPs (Lin−CD127−Sca1−CD117hiCD16/32hiCD34+), megakaryocyte erythroid progenitors (Lin−CD127−Sca1−CD117hiCD16/32loCD34), common lymphoid progenitors (Lin−CD127+Sca1LoCD117lo), and hematopoietic stem cells (LSK CD150 HSCs; Lin−CD150+Sca1HiCD117Hi, short-term (ST)–HSCs; LSK CD34+Flk2−CD150+, long-term (LT)-HSCs: LSK CD34−Flk2−CD150+, multipotent progenitors [MPPs]; LSK CD34+Flk2+CD150−). All the listed antibodies were purchased from eBioscience with the exception of CD150 (Biolegend). Antibody incubations were performed at 4°C for 30 minutes and sorted on a FACSAria flow cytometer (BD Biosciences).
Quantitative real-time analysis and RT-PCR
Total RNA was extracted from mouse bone marrow or patient sample bone marrow using TRIzol (Invitrogen), according to the manufacturer's instructions). The cDNA was synthesized using the SuperScript III First Strand Synthesis System (Invitrogen). To detect expression of Sox4 and CREB, reverse transcription-polymerase chain reaction (RT-PCR) was performed using oligo(dT) primers and PCR conditions according to the manufacturer's instructions. Quantitative real-time PCR analysis was performed in triplicate using the TaqMan probe system with human and/or mouse Sox4-, CREB-, bcl2-, cfos-, cnd1-, and glyceraldehyde-3-phosphate dehydrogenase (GAPDH)–specific probes and primers using the ABI StepOnePlus detection system (Applied Biosystems) according to manufacturer's instructions. Expression analysis was standardized to GAPDH expression.
Western blot analysis
Protein lysates were separated using SDS-PAGE. Immunoblotting was performed with anti-CREB (UBI), antiactin (Sigma-Aldrich), and anti-sox4 antibodies (Santa Cruz Biotechnology), as described previously.9
Chromatin immunoprecipitation
Chromatin immunoprecipitation (ChIP) was performed according to EZ-ChIP (Millipore) manual. Proteins in K562 or KG1 cells (2 × 107) lysates were cross-linked, harvested, and sonicated. Chromatin was precleared with protein A/G beads and then was immunoprecipitated with 5 μg of either anti-SOX4 antibody (Santa Cruz Biotechnology) or IgG control antibody. After centrifugation, beads were washed and the protein-DNA complexes were eluted; cross-links were reversed to free DNA. The immunoprecipitated DNA was then purified and PCR was performed. Input DNA was purified from the precleared chromatin. PCR was performed using normal PCR reaction program with primers against CREB promoter region: forward, 5′-TTACCAAGTCTGGATGGAAC-3′ and reverse, 5′-GGTGTGTCATTAACTGTTGAGAGC-3′.
Reverse phase protein arrays
Reverse phase protein array analysis was performed as described previously.21 In brief, patients and cell lines samples were lysed in an appropriate lysis buffer and printed in 4-point dilution curves in duplicate on nitrocellulose-coated slides (FAST slides; Whatman Schleicher & Schuell) with the 2470 Arrayer (Aushon BioSystems). Slides were stained for total protein content (Fast Green FCF; Sigma-Aldrich) and with 3 different previously validated antibodies using the Catalyzed Signal Amplification System kit (Dako North America). The images of stained slides were analyzed using the Microvigene software (VigeneTech) to generate normalized and background-subtracted single values for each protein in each sample: CREB (Millipore), CREB S133 (Cell Signaling Technology), and SOX4 (Abnova). Each antibody was previously subjected to extensive validation for single band specificity by Western blot analysis.
Patient selection
BM samples of 66 patients at diagnosis of AML collected at the Laboratory of Paediatric Hematology of the University Hospital in Padova, Italy, were used with institutional board approval. The enrollment criteria of the study followed the AIEOP-2002 AML pediatric protocol. Newly diagnosed AML, age range from 0 to 18 years, and written informed consent of the parents. Patients with granulocytic sarcoma, secondary AML, myelodysplastic syndrome, or Down syndrome, as well as patients with pretreatment duration longer than 14 days or patients with acute promyelocytic leukemia have been excluded from the present analysis. The initial diagnosis of AML was established according to the FAB-morphology classification and evaluated by multiparameter flow cytometry.22 Healthy BM cells from volunteers also were included as controls. Informed written consent was obtained from all subjects before bone marrow samples were taken, in compliance with the Declaration of Helsinki protocol.
Results
CREB transgenic mice infected with retrovirus develop AML with shorter latency
We showed previously that human mrp8-CREB TG mice that overexpress CREB in macrophage and monocytic cells develop MPD and MDS but not AML after 1 year.6,9 This suggests that the transcription factor CREB is a relatively weak proto-oncogene and insufficient to cause leukemia. To identify genes that cooperate with CREB, we sought to investigate whether these mice would have an increased incidence of AML after MOL4070LTR infection. Inoculation of CREB TG or WT newborn mice with the MOL4070LTR retrovirus accelerated acute leukemias in both groups. However, the latency for disease development was significantly decreased in our CREB TG mice (Figure 1A-B). Infection of CREB TG mice with the recombinant MOL4070LTR retrovirus resulted in leukemia with a median latency of 9 months compared with 14 months for WT mice (P < .001 by the log-rank test). These results suggested that additional oncogenes accelerate the development of leukemia in CREB TG mice.
Figure 1.

Induction of leukemia in CREB transgenic mice by RIM. (A) Kaplan-Meier survival curve of WT and TG mice inoculated with MOL4070LTR retrovirus. Survival curve analysis showed significant difference between WT and TG MOL4070LTR-inoculated mice (P < .001 by log-rank test). (B) Characteristics of leukemia in mice infected with MOL4070LTR retrovirus. Transgenic mice have a 5-month decrease in median latency for disease compared with WT mice inoculated with the MOL4070LTR retrovirus. Both WT- (n = 21) and TG (n = 34)–infected mice that developed leukemia had greater hepatosplenomegaly and increased WBC compared with uninfected WT and TG mice (P < .002). (C) Enlargement of hematologic organs in leukemic mice infected with the MOL4070LTR retrovirus. Leukemic mice typically exhibit enlarged spleens and liver. (Left) Arrows point to enlarged liver (L) and spleen (S) of mouse with AML. (Right) Spleen from WT age-matched control (left side) and CREB TG mouse with AML (right side). (D) Wright-Giemsa–stained slide of peripheral blood and bone marrow from CREB TG mice with AML. This is a representative mouse with myeloid leukemia.
Infected WT and TG mice with leukemia developed hepatosplenomegaly with enlarged, pale spleens compared with uninfected WT mice (Figure 1B-C). Both infected WT and TG mice had greater hepatosplenomegaly and increased white blood cells (WBCs) compared with uninfected WT and TG mice (P < .002; Figure 1B). We observed a statistically significant difference between uninfected WT and CREB TG mouse WBCs and spleens (P < .001) but not livers (P = .5). Pathologic examination showed infiltration of leukemic cells with increased nuclear-to-cytoplasmic ratio and diffuse chromatin in the liver and spleen and myeloblasts in the peripheral blood and bone marrow (Figure 1D). Tissues were harvested for histologic and immunophenotypic analysis to classify the type of leukemia (Figure 2 and supplemental Figure 1, available on the Blood Web site; see the Supplemental Materials link at the top of the online article). The leukemia phenotype was determined using fluorescence-activated cell sorting and cell type–specific cell surface markers in bone marrow, spleen, and thymus tissues (Figure 2B-C and supplemental Figure 1). In total, bone marrow cells from 34 TG mice were immunophenotyped and the majority of leukemias were of myeloid origin (74% or 25/34 total mice analyzed; Figure 2C). Infected WT mice had a decreased penetrance for myeloid disease compared with TG (38% or 8/21 mice analyzed; P = .01). These results indicate that the MOL4070LTR retrovirus accelerates myeloid leukemia in CREB TG mice with higher penetrance (P < .05 between myeloid and lymphoid incidence in WT- and TG-infected mice), resulting in more rapid disease progression compared with infected WT mice.
Figure 2.

Histologic and phenotypic analysis of leukemic mice. (A) H&E-stained histologic sections of liver and spleens infiltrated by leukemic cells in diseased mice. Lower power magnifications, 40×. Higher power magnifications, ×100. Uninfected age-matched TG mouse tissues on the left, and TG MOL4070LTR infected mouse tissues on the right. (B) Immunophenotypic characterization of leukemias in MOL4070LTR-infected WT and TG mouse bone marrow by flow cytometry using the Gr-1 and Mac-1 myeloid markers, CD19 and B220 B-lymphocyte markers, CD90 and CD3 T-lymphocyte markers, Ter119 erythroid marker, and Sca1 and CD117 immature cell markers. (Top) Bone marrow from an uninfected TG mouse. Subsequent panels represent myeloid, lymphoid, and biphenotypic mouse leukemia cases. (C) Immunophenotypic percentages of leukemia/lymphoma in mice infected with the MOL4070LTR retrovirus as determined by flow cytometry. Graphs show percentage of leukemia type as a result of retroviral insertional mutagenesis (for WT n = 21, for TG n = 34). Biphenotypic denotes positive for both myeloid and lymphoid cell markers; and Other, denotesleukemia of immature cell type (only 1 of 21 cases).
Identification of integration sites
To identify possible cooperating oncogenes, genomic DNA was subjected to LM-PCR analysis to amplify genomic sequences upstream of the virus.23 Genomic DNA from spleens was harvested from mice with leukemia and subjected to Southern blot analysis to verify retroviral integration (data not shown). These sequences were mapped to the mouse genome to identify the chromosomal location. Blast analysis of retroviral integration sites (RISs) was performed on the publicly available Ensembl database and compared with known common integration sites on the Retroviral Tagged Cancer Gene Database (http://rtcgd.ncifcrf.gov/).24
We identified more than 100 RISs 100 kb from genes and several common integration sites (in > 2 mice) in MOL4070LTR-infected CREB TG mice that developed AML (Table 1). Sequencing of integration sites identified several previously known oncogenes, such as Evi1/Mds, Evi5/Gfi1, Myb/Ahi, and Ras. Common integration sites identified in multiple mice include Sox4, Evi5/Gfi1, Myb/Ahi, and Cbfa2t3. The highest incidence of integration in the mutagenesis screen was found to be in the sox4 gene (Table 1). Thirty-two percent of the TG mice that were analyzed by LM-PCR contained sox4 retroviral integration (8/25 CREB TG mice with myeloid leukemia analyzed). Of 4 myeloid leukemia cases screened in WT mice, none possessed retroviral integration in or nearby the sox4 gene. Integration of the sox4 gene was identified both upstream and downstream of the coding region, indicating that retroviral integration at this site possibly acts to insertionally activate sox4 expression. We confirmed overexpression of the sox4 gene by performing real-time PCR analysis on bone marrow from CREB TG mice that developed AML and possessed sox4 retroviral integration (Figure 3). These data suggest the possibility that sox4 deregulation cooperates with CREB to accelerate myeloid cell transformation.
Table 1.
CREB-associated CISs
| CIS gene | Protein family | Mouse chromosome | Human chromosome | No. hits | Accession | Mouse identification |
|---|---|---|---|---|---|---|
| Sox4 | HMG-box related | 13 | 6p22.3 | 8 | NP_033264.1 | 1,7,11,18,23,121,123,145 |
| Evi5/Gfi1 | zinc finger, Kruppel | 5 | 1p22 | 6 | NP_034408.1 | 2,3,32,121,123,140 |
| Myb/Ahi1 | myb related | 10 | 6q22-q23 | 3 | NP_034978.3 | 6,32,124 |
| Cbfa2t3h | ETO-related | 8 | 16q24 | 3 | NP_033954.1 | 3,18,143 |
CIS denotes common integration site.
Figure 3.
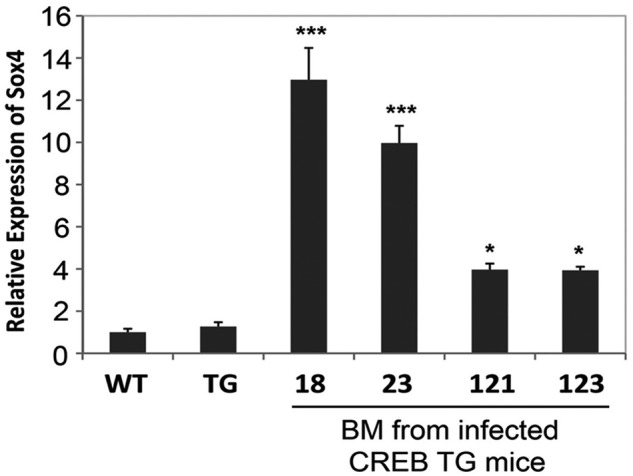
MOL4070LTR proviral integration. Real-time PCR analysis of Sox4 expression in the bone marrow of 4 representative CREB TG mice with leukemia. WT and TG control samples on graph represent Sox4 expression in bone marrow from normal WT and TG mice (ANOVA test P < .0001, Tukey *P < .05, **P < .01 ***P < .001). Real-time PCR experiments were performed in triplicate.
Sox4 and CREB increase proliferation and self-renewal in vitro
The goal of our RIM screen was to identify genes that potentially cooperate with CREB to accelerate myeloid leukemia. Because the CREB transgene is expressed under the control of the hMRP8 promoter in the monocytic and macrophage lineage, we analyzed only myeloid leukemias that developed in our mice. To study potential cooperation between CREB and Sox4, we analyzed the in vitro effects of Sox4 overexpression in mouse bone marrow cells from CREB TG or WT mice. Retroviral constructs that overexpressed Sox4 (kindly provided by Dr A. Perkins) were used to infect mouse bone marrow progenitor cells. The GFP-selectable marker cloned into these constructs allowed us to sort for positive cells by flow cytometry (20%-40% transduction efficiency; data not shown). In vitro colony assays demonstrated that Sox4 overexpression alone does not significantly increase colony formation in WT bone marrow progenitor cells (Figure 4A). However, Sox4 overexpression does enhance the self-renewal capabilities of WT bone marrow cells by ∼ 2-fold on serial replating. Both secondary and tertiary MethoCult cultures showed that Sox4-transduced cells have increased replating ability compared with vector control transduced bone marrow progenitors (Figure 4A). In addition, bone marrow cells from CREB TG mice transduced with Sox4 had a 2-fold increase in colony numbers compared with WT sox4 bone marrow transductants (P < .05). These data suggest that CREB and Sox4 enhance proliferation of progenitors in vitro. Serial replating assays in methylcellulose demonstrated that TG sox4-transduced cells also have increased self-renewal capacity compared with WT sox4-transduced cells.
Figure 4.

Sox4 overexpression enhances proliferation and self-renewal of myeloid cells. (A) WT and TG mouse bone marrow was transduced with the pMIG-Sox4 overexpressing vector or pMIG retroviral vector, sorted for GFP positivity, and seeded into semisolid MethoCult media. Colony scoring was performed 7 to 14 days after seeding or replating. Sox4 expression enhances self-renewal of cells in in vitro semisolid cultures. Sox4 expression in combination with overexpression of the proto-oncogene CREB enhances both proliferation and replating of mouse bone marrow transductants (ANOVA test P ≤ .001, Tukey analysis P values comparing groups/columns is represented above the data bars; *P < .05, **P < .01, ***P < .001) in 3 separate experiments. (Inset) Immunoblot detecting the expression of sox4, CREB, and actin-loading control of colonies grown on MethoCult. (B) Immunophenotypic characterization of WT and TG mouse bone marrow cells transduced with Sox4 retroviral construct or empty vector, sorted, and grown in liquid media supplemented with cytokines. CREB and sox4 synergize to enhance the proliferation or survival of Gr+Mac+ myeloid cells. (C) Graphical representation of panel B (1-way ANOVA test (P < .001). Tukey analysis shows a significant difference (P ≤ .05) in Gr-1+Mac-1+ percentages between WT vector vs TG vector, TG vector vs TG sox4, and WT sox4 vs TG sox4. Analysis was based on 3 independent experiments performed in triplicate.
To evaluate whether CREB and Sox4 overexpression have an effect on myeloid cell growth, we analyzed the immunophenotypic characteristics of sorted bone marrow cells grown in liquid culture in the presence of IL-3, IL-6, and SCF cytokines. The cytokine combination and growth media conditions that were used support short-term growth of all bone marrow progenitor types. After a 72-hour expansion period, bone marrow cells were stained with the Gr-1 and Mac-1 myeloid cell–specific markers. Phenotypic analysis showed that there was no significant difference between the frequency of myeloid cells in WT progenitors transduced with the empty vector and WT progenitors transduced with Sox4 (Figure 4B-C), suggesting that Sox4 alone does not affect or increase myeloid cell growth. As expected, we observed a significant increase in Gr-1+Mac-1+ (Tukey, after ANOVA P < .05) cells with TG mouse bone marrow progenitors transduced with vector compared with WT bone marrow. The frequency of Gr-1+Mac-1+ cells was higher with CREB TG bone marrow progenitor cells transduced with Sox4, compared with vector control–transduced cells (Tukey, after ANOVA P < .01). However, there were no clear lymphoid markers expressed. These results suggest that Sox4 alone does not affect myeloid cell proliferation and differentiation. However, CREB and Sox4 together enhance myeloid cell growth under these culture conditions.
Sox4 and CREB increase cell survival of bone marrow cells
To determine whether Sox4 enhances the survival of mouse bone marrow cells in vitro, we plated cells in the absence of cytokines and low serum (2% FBS) overnight and performed fluorescence-activated cell sorting analysis the next day using the annexin and 7-AAD (markers of apoptosis). Flow cytometric analysis showed that Sox4 overexpression conferred resistance to cell death in bone marrow cells by increasing the frequency of live cells and decreasing the percentage of annexin and 7-AAD double-positive cells (Figures 5A-B). Our results indicate that Sox4 overexpression provides a survival advantage in WT mouse bone marrow cells infected with the Sox4 retrovirus (ANOVA P < .0001, Tukey after ANOVA P ≤ .05, between WT vector and WT Sox4 live and annexin+7AAD+ populations; Figure 5A). Moreover, CREB and Sox4 overexpression cells had a greater increase in cell survival (and fewer apoptotic cells) compared with WT mouse Sox4-transduced cells (ANOVA P < .0001, Tukey after ANOVA P ≤ .05, between TG vector and TG Sox4 live and annexin+7AAD+ populations; Figure 5B). These results demonstrate that the CREB and Sox4 act to inhibit apoptosis in primary mouse bone marrow progenitor cells.
Figure 5.

Sox4 overexpression enhances survival and cell-cycle progression of mouse bone marrow cells in vitro. (A) Flow cytometry analysis of retrovirally transduced and sorted WT and TG bone marrow cells grown in the absence of cytokines and limited serum overnight. Sox4 transduction decreases apoptosis and enhances survival of bone marrow cells. (B) Graphical representation of panel A (ANOVA test, ***P < .0001). Tukey test shows a significant difference (P ≤ .05) between all the groups in live and annexin+7AAD+ double-positive populations. These data represent 3 separate experiments. (C) Cell-cycle analysis by BrdU incorporation. (Top) Percentage of cells in S phase. (Bottom right) Percentage of cells in G2/M. (Bottom left) Percentage of cells in G0 phase. (D) Graphical representation of 2 independent BrdU cell-cycle experiments (ANOVA test, ***P < .01). Tukey after ANOVA, P values are represented above the data bars (*P < .05, **P < .01). These results are representative of 2 independent experiments.
To assess the cell-cycle status of sorted bone marrow cells, we also pulsed bone marrow cells with BrdU and stained them with anti-BrdU and 7-AAD markers. Flow cytometric analysis showed that Sox4 alone does not significantly increase the percentage of cells in S or G2/M (Figure 5C-D). In contrast, CREB overexpression increases the percentage of both S and G2/M phase cells compared with vector control–transduced WT bone marrow cells. Furthermore, CREB TG bone marrow cells transduced with Sox4 retrovirus had increased percentage of cells in S phase compared with vector control–transduced cells (Figure 5C-D). We performed transplantation experiments with CREB TG bone marrow transduced with Sox4 retrovirus into lethally irradiated recipient mice; however, these cells did not engraft (data not shown).
Expression of CREB and its target genes
To investigate the underlying molecular mechanism by which the Sox4 and CREB work cooperatively to enhance the proliferation of myeloid cells, we extracted RNA from WT and TG bone marrow transductants and performed quantitative real-time PCR analysis to examine of CREB and its known target genes. Our results demonstrated that retroviral transduction of Sox4 into WT bone marrow cells resulted in increased CREB expression by 2.3-fold (Figure 6A). This difference in CREB expression between WT vector and WT sox4 was significant (by t test, P < .001). When both CREB and Sox4 were overexpressed in normal mouse bone marrow progenitors (CREB transgenic + vector vs CREB transgenic + sox4), we observed an 11-fold increase in CREB expression (Figure 6A). Sox4 expression was 6- to 7-fold higher in Sox4-transduced cells compared with vector control–transduced cells (Figure 6B). These data suggest that overexpression of Sox4 in CREB TG mouse bone marrow progenitor cells further increases CREB expression.
Figure 6.

Real-time PCR analysis of sox4 expression in mouse bone marrow transductants and adult hematopoiesis. Real-time PCR analysis of creb (A), sox4 (B), cyclin d1 (C), bcl2 (D), and c-fos (E) genes in mouse bone marrow cells, transduced with pMIG-R1 vector or the pMIGR1-sox4 construct. WV = WT + vector, WS = WT + sox4, TV = CREB transgenic + vector, TS = CREB transgenic + sox4 (Tukey *P < .05, **P < .01 ***P < .001). (F) QRT-PCR expression analysis of SOX4 in LSK HSCs, LSK CD150 HSCs, CMPs, GMPs, megakaryocyte erythroid progenitors (MEPs), and differentiated cells. Relative fold is in comparison to Gr-1Mac-1 double-positive myeloid cells. These data are representative of 3 experiments. (G) Sox4 expression in LT-HSCs, ST-HSCs, and MPPs and LSK HSCs and LSK CD150 HSCs for comparison. Relative fold shown is in relation to LSKCD150 HSC. Tukey after ANOVA, P values are represented above the data bars (*P < .05, **P < .01, ***P < .001). These results are from 2 independent experiments. (H) Sox4 binds to the CREB promoter. K562 cells (left) and KG1 cells (right) were cross-linked, harvested, and sonicated, and then chromatin was immunoprecipitated with either anti-Sox4 antibody or IgG control antibody. PCR was performed with primers against CREB promoter region. Input DNA purified from precleared chromatin was used as PCR positive control.
To determine whether Sox4 increases expression of CREB target genes, we performed quantitative real-time PCR analysis using primers against known CREB target genes cyclin D1, bcl-2, and c-fos. We found that the expression of cyclin D1, a regulator of G1 to S transition, followed a similar pattern as CREB expression and coincided with our results demonstrating increased percentage of cells in S phase.6,17 Cyclin D1 expression is slightly increased in WT sox4 transductants (4.6-fold), further increased in the TG mock–transduced bone marrow cells (15.5-fold) and was highest in CREB and Sox4-overexpressing cells (45.5-fold; Figure 6C). We also observed a similar increase in the expression of bcl2 in Sox4-transduced CREB TG bone marrow cells compared with WT or vector contro–transduced bone marrow cells (Figure 6D). Similarly, we observed an increase in the expression of c-fos in sox4-transduced CREB TG bone marrow cells compared with vector-transduced cells, although not as dramatic (Tukey after ANOVA P < .05; Figure 6E). Increase in c-fos expression in WT bone marrow progenitors transduced with Sox4 was minimal and not significant. Therefore, increased proliferation in the presence of sox4 and CREB overexpression may be because of the effects of known CREB downstream target genes.
To examine the contribution of known Sox4 target genes, we performed real-time quantitative PCR for PU.1, β-catenin, TGF-β, p53, and notch. Gli-1 also was examined but was not expressed in progenitor cells transduced with vector or Sox4 retrovirus (data not shown). The results demonstrated a lesser yet statistically significant increase in Sox4 target gene expression but not as dramatic as the increase in CREB expression (supplemental Figure 4). Therefore, although known Sox4 target genes are elevated, a greater effect was observed on CREB expression and known CREB target genes, suggesting that pathways downstream of CREB, more than other Sox4 target genes, play an important role in myeloid cell proliferation, cell-cycle progression, and survival.
CREB is a direct target of Sox4
To test the possibility that Sox4 increases CREB expression and hematopoietic progenitor cells proliferation through a direct interaction, we investigated whether CREB could be a downstream target of Sox4. We identified 5 putative Sox4 binding sites (AACAA A/T G) in the CREB 5′ upstream regulatory region and performed ChIP assays with K562 and KG-1 human myeloid leukemia cell lines. ChIP experiments demonstrated that Sox4 directly binds to the CREB promoter region ∼ 9 kb upstream of the start site but not other sites in the promoter containing a Sox4 binding site AACAATG (Figure 6H and supplemental Figure 5). Therefore, our results suggest that through direct binding to the CREB promoter, Sox4 induces CREB expression and up-regulation of CREB target genes.
Characterization of sox4 expression in hematopoiesis
Our functional studies demonstrated that overexpression of Sox4 canenhance the self-renewal and survival properties of mouse bone marrow cells in culture. We hypothesized that Sox4 could perhaps play a role in progenitor or stem cell maintenance and function. Quantitative real-time PCR analysis was performed on distinct bone marrow cell populations (Figure 6F-G and supplemental Figure 3). Gene expression analysis showed that Sox4 was expressed at low levels in the majority of mature lineage-positive cells, including Gr-1, Mac-1, Gr-1Mac-1, and Ter-119–positive cells (Figure 6F). B220-positive cells, however, express very high levels of Sox4, as do the common lymphoid progenitors. This was expected, because previous reports have shown that sox4 plays a role in lymphopoiesis.25,26 Sox4 gene expression is increased in CMPs compared with Gr-1 (4-fold), Mac-1 (1.8-fold), and Gr-1Mac-1 (160-fold) populations. It is also increased in the committed myeloid progenitor population, GMPs (P < .05) compared with mature myeloid cells, Gr-1, Mac-1, Gr-1Mac-1 (t test between groups).
To further characterize Sox4 expression in hematopoiesis, we fractionated the HSCs into LSK HSCs, LSK CD150+ HSCs, MPPs, and LT- and ST-HSCs (Figure 6G and supplemental Figure 3). Quantitative real-time PCR analysis of Sox4 expression in these stem cell populations showed that LSK CD150+ HSCs, ST-HSCs, and LT-HSCs do not express high levels of the sox4 gene compared with LSKs. However, the LSK HSC (Tukey after ANOVA, P < .05 compared with mature myeloid cells) and MPP (Tukey after ANOVA, P < .01 compared with LSK HSCs) progenitor populations highly express sox4. Although these data do not establish the Sox4 as a stem cell gene, they do suggest that it is a gene expressed in immature progenitors and may play a role in early hematopoiesis.
Elevated CREB and Sox4 expression in AML patients
To examine Sox4 expression in primary AML patient bone marrow samples at diagnosis and AML cell lines, we performed Western blot analysis with CREB, phospho-CREB, and Sox4 antisera. Almost all AML cell lines and patient samples expressed CREB, phospho-CREB, and Sox4 protein (Figure 7A-C). Quantitative real-time PCR expression analysis also was performed with RNA extracted from newly diagnosed AML patient bone marrow samples. There was a weak correlation among 39 AML patients between relative expression of CREB and Sox4 mRNA (Spearman correlation 0.30, P = .07; data not shown). To determine whether Sox4, CREB, and phospho-CREB proteins were overexpressed in human AML, the reverse phase protein array was performed to measure the relative protein expression of CREB, phospho-CREB, and Sox4 in bone marrow samples from 66 pediatric AML patients (ages 0-18 years; Figure 7D). Expression for each protein is represented as fold change relative to normal bone marrow and displayed on a logarithmic scale. The difference in the variation of CREB and Sox4 expression by Western blot analysis is most likely because of different sample groups; 1 group from Italy (reverse phase protein assay) and the other group from the United States (Western blot analysis), and also may reflect differences in processing of bone marrow samples between the 2 groups. Our results demonstrate a strong correlation between protein expression of Sox4, CREB, and phospho-CREB in pediatric AML patients (P < .0001).
Figure 7.

Sox4 and CREB are overexpressed in human AML. (A-C) Western blots with lysates from AML cell lines and pediatric AML bone marrow samples at diagnosis probed with CREB, phospho-CREB, and Sox4 antisera. Sample PZ2 is normal BM control. (D) Sox4 and CREB protein expressions are highly correlated in human AML. Protein expression of Sox4, CREB, and phospho-CREB was quantitated using reverse phase protein array using bone marrow samples from 66 patients with newly diagnosed childhood AML (age range, 0-18 years). Expression is represented as fold change relative to normal bone marrow and displayed on a logarithmic scale. Pearson correlations (r) for log protein expression are significantly different from zero (P < .0001). The first principal component (straight line) accounts for 89% of the total variation.
Discussion
AML results from the accumulation of several oncogenic hits.1,2 Many of the reported TG mouse models of leukemia with single genetic lesions do not develop leukemia but instead, myeloproliferative disease.27–30 In our experiments, neonatal CREB TG or WT mice were inoculated with a slow-transforming retrovirus that accelerated the development of leukemia over a period of months. Previous work from our laboratory demonstrated that CREB TG mice that overexpress CREB in myeloid cells develop a myeloproliferative disorder (MPD/MDS) but not AML. Despite several lines of evidence suggesting that CREB overexpression contributes to transformation, it is not sufficient to induce myeloid leukemia. Similar observations have been described with other mouse models of leukemia, including AML1-ETO, PML-RARα, and FLT3-internal tandem duplication transgenic mice, resulting in myeloproliferative disorders but not full transformation to AML.27–30 These findings are consistent with the fact that myeloid leukemias result from the accumulation of multiple genetic alterations.
In this study, we sought to use RIM to accelerate leukemogenesis in our CREB TG mice and to identify known or novel oncogenes that enhance transformation with CREB. Our RIM screen successfully accelerated leukemogenesis in our CREB TG mice with decreased latency compared with WT mice. We identified greater than 100 RISs, some of which potentially cooperate with CREB in the development of human AML. Many of the integration sites identified in this study are well-characterized oncogenes in myeloid leukemia, including Gfi1, Myb Cbfa2t3h, and Ras, that encode members of signaling pathways that play a role in proliferation, survival, differentiation, and cell-cycle regulation.31–36
The leukemias generated by this study contained more than 1 retroviral integration site (3-8 RISs per tumor). Our mutagenesis screen also identified genes whose functions are not fully known in myeloid cells. We found recurrent integration of virus in proximity to the sox4-coding region in our CREB TG mice that developed myeloid disease. The role of this gene in myelopoiesis and myeloid leukemia is not well understood. Sox4-deficient mice are embryonic lethal and die because of cardiac failure and impaired lymphocyte development.37–39 In adult mice, the sox4 gene is expressed in the gonads, thymus, lymphocytes, and heart, and it is thought to regulate cell fate and development. In humans, Sox4 overexpression or amplification has been associated with various solid tumors, including prostate, lung, breast cancer, and bladder carcinoma.25,26,40–43 Sox4 inhibition by small interfering RNA induces apoptosis of prostate cancer cells.26 Sox4 also has been shown to be overexpressed in breast cancer cell lines and is thought to potentiate transformation by regulating the expression of breast cancer oncogene ErbB2.44 Given that Sox4 plays a key role in development and cell fate, one can hypothesize that re-expression of this gene in differentiated cells could result in the dedifferentiation of mature cells with increased self-renewal and antiapoptotic properties.
In other RIM studies, Sox4 has been identified as a site of retroviral integration.24,45,47 Boyd et al demonstrated that the Sox4 cooperates with Evi1 in the AKXD-23 mouse model of myeloid leukemia.16 Recently, Bies et al identified Sox4 in their RIM screen as a potential oncogene that collaborates with the p15Ink4b tumor suppressor to accelerate myeloid leukemogenesis.23 They reported that Sox4 overexpression synergizes with p15Ink4b inactivation to increase bone marrow colony formation. More recently, Sox4 was demonstrated to cooperate with decreased sfpi1(PU.1) expression in transduction-transplantation experiments resulting in AML.48 Therefore, deregulation of Sox4 has been show to be an important step in myeloid transformation. We are the first to demonstrate that Sox4 and CREB are cooperating oncogenes.
Despite studies that demonstrated that Sox4 overexpression is associated with tumorigenesis, the role of Sox4 in transformation is poorly understood. Very few studies have examined the biologic effects of Sox4 overexpression in tumors. Our studies suggest that Sox4 and CREB cooperate to increase myeloid proliferation and cell-cycle progression. Future studies will focus on cooperation between CREB and the other oncogenes, that is, Gfi1, Myb, and Cbfa2t3h, which were identified in this study. In conclusion, our results provide evidence that Sox4 is a potential oncogene that cooperates with CREB to enhance transformation of hematopoietic progenitor cells thereby contributing to AML.
Supplementary Material
Acknowledgments
The authors thank Rachel Ochoa (Stanford University) for technical assistance.
This work was supported by the National Institutes of Health grants R01 HL75826 and R01 HL83077 (K.M.S.). K.M.S. is a scholar of the Leukemia & Lymphoma Society. M.C. is supported by a supplement through American Recovery and Reinvestment Act of 2009 funding through R01 HL83077.
Footnotes
The online version of the article contains a data supplement.
The publication costs of this article were defrayed in part by page charge payment. Therefore, and solely to indicate this fact, this article is hereby marked “advertisement” in accordance with 18 USC section 1734.
Authorship
Contribution: S.S. designed and performed the research, analyzed data, and wrote the paper; K.M.S. designed the research, wrote the paper, and supervised the project; C.K. and M.C. performed the research and analyzed data; J.B. and L.W. aided in research design and contributed vital reagents; E.-C.C. performed the ChIP experiments; E.M.L. assisted with the statistical analysis; E.M. performed the AML and patient sample Western blot analysis; B.A. performed the reverse phase protein arrays; and M.P. provided the research design, provided patient samples, and supervised the experiments with K.M.S.
Conflict-of-interest disclosure: The authors declare no competing financial interests.
Correspondence: Kathleen M. Sakamoto, Division of Pediatric Hematology/Oncology/Stem Cell Transplantation and Cancer Biology, Lucile Packard Children's Hospital, Stanford University School of Medicine, CCSR 1215C, 269 Campus Dr, Stanford, CA 94305-5162; e-mail: kmsakamo@stanford.edu.
References
- 1.Fialkow PJ, Janssen JW, Bartram CR. Clonal remissions in acute nonlymphocytic leukemia: evidence for a multistep pathogenesis of the malignancy. Blood. 1991;77(7):1415–1417. [PubMed] [Google Scholar]
- 2.Mitelman F, Johansson B, Mertens F, editors. Mitelman Database of Chromosome Aberrations and Gene Fusions in Cancer. 2010. [Accessed June 1, 2012]. http://cgap.nci.nih.gov/Chromosomes/Mitelman.
- 3.Look AT. Oncogenic transcription factors in the human acute leukemias. Science. 1997;278(5340):1059–1064. doi: 10.1126/science.278.5340.1059. [DOI] [PubMed] [Google Scholar]
- 4.Mayr B, Montminy M. Transcriptional regulation by the phosphorylation-dependent factor CREB. Nat Rev Mol Cell Biol. 2001;2(8):599–609. doi: 10.1038/35085068. [DOI] [PubMed] [Google Scholar]
- 5.Siu YT, Jin DY. CREB—a real culprit in oncogenesis. FEBS J. 2007;274(13):3224–3232. doi: 10.1111/j.1742-4658.2007.05884.x. [DOI] [PubMed] [Google Scholar]
- 6.Cheng JC, Kinjo K, Judelson DR, et al. CREB is a critical regulator of normal hematopoiesis and leukemogenesis. Blood. 2008;111(3):1182–1192. doi: 10.1182/blood-2007-04-083600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Crans-Vargas HN, Landaw EM, Bhatia S, Sandusky G, Moore TB, Sakamoto KM. Expression of cyclic adenosine monophosphate response-element binding protein in acute leukemia. Blood. 2002;99(7):2617–2619. doi: 10.1182/blood.v99.7.2617. [DOI] [PubMed] [Google Scholar]
- 8.Pigazzi M, Ricotti E, Germano G, Faggian D, Arico M, Basso G. cAMP response element binding protein (CREB) overexpression CREB has been described as critical for leukemia progression. Haematologica. 2007;92(10):1435–1437. doi: 10.3324/haematol.11122. [DOI] [PubMed] [Google Scholar]
- 9.Shankar DB, Cheng JC, Kinjo K, et al. The role of CREB as a proto-oncogene in hematopoiesis and in acute myeloid leukemia. Cancer Cell. 2005;7(4):351–362. doi: 10.1016/j.ccr.2005.02.018. [DOI] [PubMed] [Google Scholar]
- 10.Dudley JP. Tag, you're hit: retroviral insertions identify genes involved in cancer. Trends Mol Med. 2003;9(2):43–45. doi: 10.1016/s1471-4914(03)00003-0. [DOI] [PubMed] [Google Scholar]
- 11.Mikkers H, Allen J, Knipscheer P, et al. High-throughput retroviral tagging to identify components of specific signaling pathways in cancer. Nat Genet. 2002;32(1):153–159. doi: 10.1038/ng950. [DOI] [PubMed] [Google Scholar]
- 12.Mikkers H, Berns A. Retroviral insertional mutagenesis: tagging cancer pathways. Adv Cancer Res. 2003;88:53–99. doi: 10.1016/s0065-230x(03)88304-5. [DOI] [PubMed] [Google Scholar]
- 13.Touw IP, Erkeland SJ. Retroviral insertion mutagenesis in mice as a comparative oncogenomics tool to identify disease genes in human leukemia. Mol Ther. 2007;15(1):13–19. doi: 10.1038/sj.mt.6300040. [DOI] [PubMed] [Google Scholar]
- 14.Uren AG, Kool J, Berns A, van Lohuizen M. Retroviral insertional mutagenesis: past, present and future. Oncogene. 2005;24(52):7656–7672. doi: 10.1038/sj.onc.1209043. [DOI] [PubMed] [Google Scholar]
- 15.Wolff L, Koller R, Hu X, Anver MR. A Moloney murine leukemia virus-based retrovirus with 4070A long terminal repeat sequences induces a high incidence of myeloid as well as lymphoid neoplasms. J Virol. 2003;77(8):4965–4971. doi: 10.1128/JVI.77.8.4965-4971.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Boyd KE, Xiao YY, Fan K, et al. Sox4 cooperates with Evi1 in AKXD-23 myeloid tumors via transactivation of proviral LTR. Blood. 2006;107(2):733–741. doi: 10.1182/blood-2003-05-1626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Zhang X, Odom DT, Koo SH, et al. Genome-wide analysis of cAMP-response element binding protein occupancy, phosphorylation, and target gene activation in human tissues. Proc Natl Acad Sci U S A. 2005;102(12):4459–4464. doi: 10.1073/pnas.0501076102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.National Research Council. Guide for the Care and Use of Laboratory Animals. 8th Ed. Washington, DC: The National Academics Press; 2011. [Google Scholar]
- 19.Slape C, Hartung H, Lin YW, Bies J, Wolff L, Aplan PD. Retroviral insertional mutagenesis identifies genes that collaborate with NUP98-HOXD13 during leukemic transformation. Cancer Res. 2007;67(11):5148–5155. doi: 10.1158/0008-5472.CAN-07-0075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Spudich GM, Fernandez-Suarez XM. Touring Ensembl: a practical guide to genome browsing. BMC Genomics. 2010;11:295. doi: 10.1186/1471-2164-11-295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Accordi B, Espina V, Giordan M, et al. Functional protein network activation mapping reveals new potential molecular drug targets for poor prognosis pediatric BCP-ALL. PloS One. 2010;5(10):e13552. doi: 10.1371/journal.pone.0013552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Pession A, Rondelli R, Basso G, et al. Treatment and long-term results in children with acute myeloid leukaemia treated according to the AIEOP AML protocols. Leukemia. 2005;19(12):2043–2053. doi: 10.1038/sj.leu.2403869. [DOI] [PubMed] [Google Scholar]
- 23.Bies J, Sramko M, Fares J, et al. Myeloid-specific inactivation of p15Ink4b results in monocytosis and predisposition to myeloid leukemia. Blood. 2010;116(6):979–987. doi: 10.1182/blood-2009-08-238360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Akagi K, Suzuki T, Stephens RM, Jenkins NA, Copeland NG. RTCGD: retroviral tagged cancer gene database. Nucleic Acids Res. 2004;32:D523–D527. doi: 10.1093/nar/gkh013. Database issue. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Scharer CD, McCabe CD, Ali-Seyed M, Berger MF, Bulyk ML, Moreno CS. Genome-wide promoter analysis of the SOX4 transcriptional network in prostate cancer cells. Cancer Res. 2009;69(2):709–717. doi: 10.1158/0008-5472.CAN-08-3415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Liu P, Ramachandran S, Ali Seyed M, et al. Sex-determining region Y box 4 is a transforming oncogene in human prostate cancer cells. Cancer Res. 2006;66(8):4011–4019. doi: 10.1158/0008-5472.CAN-05-3055. [DOI] [PubMed] [Google Scholar]
- 27.Early E, Moore MA, Kakizuka A, et al. Transgenic expression of PML/RARalpha impairs myelopoiesis. Proc Natl Acad Sci U S A. 1996;93(15):7900–7904. doi: 10.1073/pnas.93.15.7900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Grisolano JL, Wesselschmidt RL, Pelicci PG, Ley TJ. Altered myeloid development and acute leukemia in transgenic mice expressing PML-RAR alpha under control of cathepsin G regulatory sequences. Blood. 1997;89(2):376–387. [PubMed] [Google Scholar]
- 29.He LZ, Tribioli C, Rivi R, et al. Acute leukemia with promyelocytic features in PML/RARalpha transgenic mice. Proc Natl Acad Sci U S A. 1997;94(10):5302–5307. doi: 10.1073/pnas.94.10.5302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Li L, Piloto O, Nguyen HB, et al. Knock-in of an internal tandem duplication mutation into murine FLT3 confers myeloproliferative disease in a mouse model. Blood. 2008;111(7):3849–3858. doi: 10.1182/blood-2007-08-109942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Brown AL, Wilkinson CR, Waterman SR, et al. Genetic regulators of myelopoiesis and leukemic signaling identified by gene profiling and linear modeling. J Leukoc Biol. 2006;80(2):433–447. doi: 10.1189/jlb.0206112. [DOI] [PubMed] [Google Scholar]
- 32.Farr C, Gill R, Katz F, Gibbons B, Marshall CJ. Analysis of ras gene mutations in childhood myeloid leukaemia. Br J Haematol. 1991;77(3):323–327. doi: 10.1111/j.1365-2141.1991.tb08578.x. [DOI] [PubMed] [Google Scholar]
- 33.Farr CJ, Saiki RK, Erlich HA, McCormick F, Marshall CJ. Analysis of RAS gene mutations in acute myeloid leukemia by polymerase chain reaction and oligonucleotide probes. Proc Natl Acad Sci U S A. 1988;85(5):1629–1633. doi: 10.1073/pnas.85.5.1629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Khandanpour C, Thiede C, Valk PJ, et al. A variant allele of Growth Factor Independence 1 (GFI1) is associated with acute myeloid leukemia. Blood. 2010;115(12):2462–2472. doi: 10.1182/blood-2009-08-239822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Schmidt M, Koller R, Haviernik P, Bies J, Maciag K, Wolff L. Deregulated c-Myb expression in murine myeloid leukemias prevents the up-regulation of p15(INK4b) normally associated with differentiation. Oncogene. 2001;20(43):6205–6214. doi: 10.1038/sj.onc.1204821. [DOI] [PubMed] [Google Scholar]
- 36.Wolff L, Schmidt M, Koller R, et al. Three genes with different functions in transformation are regulated by c-Myb in myeloid cells. Blood Cells Mol Dis. 2001;27(2):483–488. doi: 10.1006/bcmd.2001.0409. [DOI] [PubMed] [Google Scholar]
- 37.Ya J, Schilham MW, de Boer PA, Moorman AF, Clevers H, Lamers WH. Sox4-deficiency syndrome in mice is an animal model for common trunk. Circ Res. 1998;83(10):986–994. doi: 10.1161/01.res.83.10.986. [DOI] [PubMed] [Google Scholar]
- 38.Schilham MW, Moerer P, Cumano A, Clevers HC. Sox-4 facilitates thymocyte differentiation. Eur J Immunol. 1997;27(5):1292–1295. doi: 10.1002/eji.1830270534. [DOI] [PubMed] [Google Scholar]
- 39.Schilham MW, Oosterwegel MA, Moerer P, et al. Defects in cardiac outflow tract formation and pro-B-lymphocyte expansion in mice lacking Sox-4. Nature. 1996;380(6576):711–714. doi: 10.1038/380711a0. [DOI] [PubMed] [Google Scholar]
- 40.Graham JD, Hunt SM, Tran N, Clarke CL. Regulation of the expression and activity by progestins of a member of the SOX gene family of transcriptional modulators. J Mol Endocrinol. 1999;22(3):295–304. doi: 10.1677/jme.0.0220295. [DOI] [PubMed] [Google Scholar]
- 41.Chang CH, Scott GK, Kuo WL, et al. ESX: a structurally unique Ets overexpressed early during human breast tumorigenesis. Oncogene. 1997;14(13):1617–1622. doi: 10.1038/sj.onc.1200978. [DOI] [PubMed] [Google Scholar]
- 42.Aaboe M, Birkenkamp-Demtroder K, Wiuf C, et al. SOX4 expression in bladder carcinoma: clinical aspects and in vitro functional characterization. Cancer Res. 2006;66(7):3434–3442. doi: 10.1158/0008-5472.CAN-05-3456. [DOI] [PubMed] [Google Scholar]
- 43.Medina PP, Castillo SD, Blanco S, et al. The SRY-HMG box gene, SOX4, is a target of gene amplification at chromosome 6p in lung cancer. Hum Mol Genet. 2009;18(7):1343–1352. doi: 10.1093/hmg/ddp034. [DOI] [PubMed] [Google Scholar]
- 44.Dong C, Wilhelm D, Koopman P. Sox genes and cancer. Cytogenet Genome Res. 2004;105(2-4):442–447. doi: 10.1159/000078217. [DOI] [PubMed] [Google Scholar]
- 45.Shin MS, Fredrickson TN, Hartley JW, et al. High-throughput retroviral tagging for identification of genes involved in initiation and progression of mouse splenic marginal zone lymphomas. Cancer Res. 2004;64(13):4419–4427. doi: 10.1158/0008-5472.CAN-03-3885. [DOI] [PubMed] [Google Scholar]
- 46.Suzuki T, Shen H, Akagi K, et al. New genes involved in cancer identified by retroviral tagging. Nat Genet. 2002;32(1):166–174. doi: 10.1038/ng949. [DOI] [PubMed] [Google Scholar]
- 47.Lund AH, Turner G, Trubetskoy A, et al. Genome-wide retroviral insertional tagging of genes involved in cancer in Cdkn2a-deficient mice. Nat Genet. 2002;32(1):160–165. doi: 10.1038/ng956. [DOI] [PubMed] [Google Scholar]
- 48.Aue G, Du Y, Cleveland SM, et al. Sox4 cooperates with PU. 1 haploinsufficiency in murine myeloid leukemia. Blood. 2011;118(17):4674–4681. doi: 10.1182/blood-2011-04-351528. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.