

Abstract
Matrix metalloproteinases (MMPs) influence synaptic recovery following traumatic brain injury (TBI). Membrane type 5-matrix metalloproteinase (MT5-MMP) and a distintegrin and metalloproteinase-10 (ADAM-10) are membrane-bound MMPs that cleave N-cadherin, a protein critical to synapse stabilization. This study examined protein and mRNA expression of MT5-MMP, ADAM-10, and N-cadherin after TBI, contrasting adaptive and maladaptive synaptogenesis. The effect of MMP inhibition on MT5-MMP, ADAM-10, and N-cadherin was assessed during maladaptive plasticity and correlated with synaptic function. Rats were subjected to adaptive unilateral entorhinal cortical lesion (UEC) or maladaptive fluid percussion TBI+bilateral entorhinal cortical lesion (TBI+BEC). Hippocampal MT5-MMP and ADAM-10 protein was significantly elevated 2 and 7 days post-injury. At 15 days after UEC, each MMP returned to control level, while TBI+BEC ADAM-10 remained elevated. At 2 and 7 days, N-cadherin protein was below control. By the 15-day synapse stabilization phase, UEC N-cadherin rose above control, a shift not seen for TBI+BEC. At 7 days, increased TBI+BEC ADAM-10 transcript correlated with protein elevation. UEC ADAM-10 mRNA did not change, and no differences in MT5-MMP or N-cadherin mRNA were detected. Confocal imaging showed MT5-MMP, ADAM-10, and N-cadherin localization within reactive astrocytes. MMP inhibition attenuated ADAM-10 protein 15 days after TBI+BEC and increased N-cadherin. This inhibition partially restored long-term potentiation induction, but did not affect paired-pulse facilitation. Our results confirm time- and injury-dependent expression of MT5-MMP, ADAM-10, and N-cadherin during reactive synaptogenesis. Persistent ADAM-10 expression was correlated with attenuated N-cadherin level and reduced functional recovery. MMP inhibition shifted ADAM-10 and N-cadherin toward adaptive expression and improved synaptic function.
Key words: a distintegrin and metalloproteinase-10 (ADAM-10), brain injury, membrane type 5-matrix metalloproteinase (MT5-MMP), N-cadherin, synaptic plasticity
Introduction
Traumatic brain injury (TBI) induces multiple secondary pathologies, including diffuse axonal damage, deafferentation, and neuronal death (Hayes et al., 1992; Okonkwo and Povlishock, 2003; Povlishock and Christman, 1995). Deafferentation triggers reactive synaptogenesis to restore lost synaptic structure and function. However, successful synaptic recovery is dependent on a variety of factors, including injury complexity and severity. With TBI, this synaptic recovery is often attenuated or maladaptive. We have employed different experimental TBI models to contrast adaptive and maladaptive synaptic plasticity. Under adaptive conditions, synaptic reorganization occurs in serial post-injury phases: acute degradation of injured axons (1–5 days); subsequent regeneration of new synapses (6–15 days); and synaptic maturation (15–30 days+; Steward and Vinsant, 1983). The unilateral entorhinal cortex lesion (UEC) is a well-established model of hippocampal synaptic plasticity, resulting in adaptive reorganization (Steward et al., 1988). By contrast, the combination of moderate central fluid percussion TBI with bilateral entorhinal cortex lesion (TBI+BEC) leads to maladaptive synaptic plasticity, with persistent structural, functional, and behavioral deficits (Phillips and Reeves, 2001; Phillips et al., 1994). In the present study we use these models to explore how two membrane-bound matrix metalloproteinases and a synapse-associated substrate affect the success of reactive synaptogenesis induced by TBI.
Matrix metalloproteinases (MMPs) affect central nervous system (CNS) recovery in a variety of neuropathologies, including stroke, Alzheimer's disease, multiple sclerosis, and TBI (Cunningham et al., 2005; Falo et al., 2006; Truettner et al., 2005; Yong et al., 2001). During early post-injury phases, MMPs are thought to direct matrix proteolysis, preparing a malleable environment for synaptic reorganization. Release of MMPs from reactive astrocytes within the damaged region is an important factor in post-injury synaptic regeneration and stabilization (Falo et al., 2006; Muir et al., 2002; Wells et al., 1996). However, persistent glial reactivity, paired with elevated MMP expression, may produce excessive breakdown and poor synaptic recovery post-TBI (Falo et al., 2006; Kim et al., 2005; Rosenberg et al., 2001; Shigemori et al., 2006; Suehiro et al., 2004). In prior studies, we have shown that certain MMP/substrate pairs contribute to injury-dependent differences in the extent of synaptic plasticity generated (Falo et al., 2006,2008). For example, MMP-3/agrin expression was both spatially and temporally correlated with distinct phases of UEC adaptive synaptogenesis (Falo et al., 2008). However, under maladaptive conditions, secreted MMPs (MMP-2, −3, and −9) were persistently upregulated, suggesting the aberrant extension of lytic activity into periods when matrix stabilization is critical to synapse maturation (Falo et al., 2006; Kim et al. 2005). Importantly, this persistent MMP expression and activity was correlated with attenuated synaptogenesis and long-term functional deficits (Phillips et al., 1994). These findings support MMPs as integral mediators of successful synaptic plasticity following TBI.
The present study extends our understanding of how MMPs affect synaptogenesis by tracking the expression of two membrane-bound MMPs, membrane type 5-matrix metalloproteinase (MT5-MMP) and a distintegrin and metalloproteinase-10 (ADAM-10), during adaptive and maladaptive synaptic plasticity. Membrane-bound MMPs are anchored adjacent to their matrix substrates, permitting focal MMP/substrate interaction at specific sites targeted for synaptic formation. Here we examine the protein and mRNA profiles of hippocampal MT5-MMP and ADAM-10, along with their synaptic substrate N-cadherin, during injury-induced synaptogenesis. Each of these enzymes has been localized at hippocampal synapses or in surrounding astrocytes (Bandyopadhyay et al., 2006; Hayashita-Kinoh et al., 2001; Jaworski, 2000; Kieseier et al., 2003; Marcinkiewicz and Seidah, 2000; Sekine-Aizawa et al., 2001). Both enzymes are capable of cleaving the extracellular peptide sequences of N-cadherin in a process called shedding. Shedding is important for early stages of reactive synaptogenesis because N-cadherin normally functions as an adhesion molecule, binding synaptic junctions through its homotypic pre- and post-synaptic interaction. Release of this binding would be expected during the acute, plastic phases of injury-induced synaptic reorganization. During later stages of synaptogenesis, N-cadherin can also serve to guide developing synapses, as well as to stabilize and strengthen mature synaptic connections (Jaworski, 2000; Marcinkiewicz and Seidah, 2000; Monea et al., 2006; Reiss et al., 2005; Takeichi and Abe, 2005). Further synapse stabilization can be achieved through N-cadherin intracellular binding with catenins, molecules which are anchored to the pre- and post-synaptic cytoskeleton, preserving structural integrity (Takeichi and Abe, 2005). Several recent studies also support a regulatory role for both secreted MMPs and N-cadherin in the altered synaptic efficacy associated with long-term potentiation (Conant et al., 2010; Huntley et al., 2012; Wang et al., 2008). Thus, MT5-MMP- and ADAM-10-driven shedding of N-cadherin may modify synaptic structure and function through the course of injury-induced synaptogenesis.
To examine these potential MMP/substrate relationships, we investigated the protein expression of MT5-MMP, ADAM-10, and N-cadherin within the deafferented hippocampus over the first 2 weeks after UEC or TBI+BEC. We hypothesized that all three molecules would be significantly altered post-injury, and that inter-model differences in protein expression and distribution exist, particularly during the 7- to 15-day period of synapse regeneration and stabilization. We posited that changes in mRNA for these molecules would be observed at 7 days, the period of collateral sprouting and synapse formation. Finally, we predicted that MMP inhibition in the maladaptive model would alter the pattern of MT5-MMP, ADAM-10, and N-cadherin expression, supporting a functional plasticity more consistent with the adaptive condition.
Methods
Experimental animals
Adult male Sprague-Dawley rats (Hilltop Laboratory Animals Inc., Scottsdale, PA) weighing 300–400 g were used in this study. At 2, 7, and 15 days post-injury, three randomly divided groups were selected for biochemical or histological assessment: (1) unilateral entorhinal cortex lesion (UEC, n=41), (2) combined moderate central fluid percussion injury and bilateral entorhinal cortical lesion (TBI+BEC, n=88), and (3) sham fluid percussion injured (n=65). During surgical preparations, the animals were monitored for heart rate (beats per minute, bpm), arterial oxygen saturation (% O2), breath rate (breath per minute, brpm), pulse distention (in μm), and breath distention (in μm; MouseOx™; Starr Life Sciences, Oakmont, PA). Surgical procedures produced no significant changes in these physiological measurements. The contralateral hippocampus served as an internal paired-control for the UEC group, while a sham-injured control group (surgically prepared, not injured) was used for TBI+BEC. Two rats were housed per cage, with food and water ad libitum, and subjected to a 12-h dark-light cycle at 22°C. All protocols for injury and use of animals were approved by the Institutional Animal Care and Use Committee of Virginia Commonwealth University.
Surgical and injury procedures
Unilateral entorhinal cortical lesion
The entorhinal cortex lesion (UEC) protocol was a modification of the method of Loesche and Steward (1977). All rats were anesthetized with isoflurane (4% in carrier gas of 70% N2O and 30% O2) delivered via nose cone and placed in a stereotaxic frame. During all surgeries, body temperature was maintained at 37°C via thermostatic heating pad (Harvard Apparatus, Holliston, MA), and physiological measurements were monitored as described above. Briefly, a 3×5-mm portion of the skull was removed on the right side to expose the dura mater superior to the entorhinal cortex. The dura was cut and reflected to expose the cortical surface. Electrolytic lesions were made by passing a 1.5-mA current (40 sec duration) through a 0.2-mm Teflon-insulated wire electrode, positioned 10° to the perpendicular plane, and delivered at eight separate sites: 1.5 mm anterior to the transverse sinus, and 3, 4, and 5 mm lateral to midline; then 2, 4, and 6 mm ventral to the brain surface for the first two lateral measurements (3 and 4 mm), and 2 and 4 mm ventral to the brain surface for the 5-mm lateral measurement. After lesioning, the electrode was removed, the dura repositioned, the scalp sutured over the surgical site, and Bacitracin applied topically. The rats were closely monitored for recovery criteria, and then returned to their home cages.
Combined traumatic brain injury and bilateral entorhinal lesion
All animals were anesthetized as described above and placed in a stereotaxic frame. Body temperature and physiological measurements were also monitored with the MouseOx system. Briefly, rats received a 4.8-mm midline craniectomy midway between the bregma and the lambda, exposing but not breaching the underlying dura. Two steel screws were implanted 1 mm caudal to the coronal suture on the left, and 1 mm rostral to the lambdoid suture on the right. A modified Luer-Lock hub (2.6 mm inside diameter) was implanted in the craniectomy site and fixed with cyanoacrylate adhesive. Dental acrylic was placed around the hub and screws to secure the complex. The scalp was sutured closed to cover the hub and Bacitracin was applied. The rats were monitored closely during post-operative recovery and returned to their home cages.
Twenty-four hours after hub implantation, moderate central fluid percussion injury (2.0±0.1 atmospheres, atm) was induced as described by Dixon and associates (1987). The fluid percussion injury device consists of an acrylic glass cylinder (60 cm long, 4.5 cm diameter) filled with double distilled water. One end of the cylinder has a rubber-covered acrylic glass piston mounted on O-rings, and the opposite end is closed by a metal extra-cranial pressure transducer (model EPN-0300-100A; Entram Devices, Inc., Fairfield, NJ). Attached to the metal transducer is a 5-mm tube (2.6-mm inside diameter) that connects to the exposed female Luer-Lock hub at the time of injury. A metal pendulum strikes the rubber-covered piston, forcing a small injection of double distilled water into the closed cranial cavity, briefly displacing the brain tissue. Injury level is recorded by the extra-cranial transducer and reported (in atmospheres, atm) on an oscilloscope (model 5111; Tektronix, Beaverton, OR). Following injury the animals were ventilated with room air until spontaneous breathing resumed. The animals were monitored for timing of reflex recovery (paw, tail, corneal, righting, and pinna). The duration of righting reflex suppression was used as an index of traumatic unconsciousness. Following post-operative recovery, the animals were returned to their home cages for another 24-h period, after which they were re-anesthetized and subjected to bilateral entorhinal cortex lesions (Phillips et al., 1994). Lesions were performed using the UEC protocol described above. After lesioning, the scalp was sutured and Bacitracin was applied topically. The animals were monitored for post-operative recovery criteria and then returned to their home cages.
MMP-inhibitor administration
MT5-MMP and ADAM-10 activity was inhibited using a commercially available MMP inhibitor, GM6001 (Millipore, Billerica, MA). This hydroxamate compound chelates the active site Zn++, preventing substrate binding and rendering the enzyme inactive (Grobelny et al., 1992; Schultz et al., 1992). Although a broad-spectrum MMP inhibitor, GM6001 significantly blocks both MT5-MMP and ADAM-10 activity in vitro (Monea et al., 2006; Reiss et al., 2005). Animals subjected to maladaptive TBI+BEC received either GM6001 (10 mg/kg IP), or an equal volume of vehicle (4% carboxymethlycellulose, CMC sodium salt; Sigma-Aldrich, St. Louis, MO), once daily at 6 and 7 days post-injury. GM6001 dose and delivery matched in vivo studies showing effective MMP inhibition in other neurotrauma models (Sifringer et al., 2007; Wang and Tsirka, 2005). The post-injury timeframe for dosing was selected to coincide with maximal MT5-MMP and ADAM-10 protein elevation. No significant differences were detected between injured-untreated and injured-vehicle animals (data not shown).
Western blotting
At 2, 7, and 15 days following UEC or TBI+BEC, a random subset of animals from each group (UEC n=24; TBI+BEC n=39; sham-injured n=35) was selected for time course analysis of hippocampal MT-5 MMP, ADAM-10, and N-cadherin, using Western blot (WB) methods. In parallel studies with MMP inhibition, subsets of TBI+BEC animals received GM6001 (n=6), or vehicle (n=7), on days 6 and 7 after BEC, and were sacrificed at 15 days post-injury for hippocampal protein analysis. The rats were anesthetized (4% isoflurane in carrier gas of 70% N2O and 30% O2), decapitated, and the whole hippocampus was dissected free. Tissue was homogenized in ice-cold T-PER (Pierce Protein Research Products, Rockford, IL), and centrifuged (8000×g) for 5 min at 4°C. Supernatants were removed and assayed for protein concentration (Shimadzu UV-160, Shimadzu Scientific, Columbia, MD; FLUOstar Optima, BMG Lab Technologies, Inc., Durham, NC). For blot preparation, 5 μg of each sample was mixed with XT Sample Buffer/Reducing Agent (Bio-Rad Laboratories, Hercules, CA), and heated to 95°C. Proteins were resolved on a 4–12% Bio-Tris gel (Bio-Rad) and transferred to PVDF membranes. Post-blotted gels were stained and inspected for even protein load and transfer. The membranes were blocked in 5% milk TBS-Tween (TBS-T), and then probed with primary antibody (MT5-MMP rabbit polyclonal N-terminus, 1:1000, Abcam, Cambridge, MA; ADAM-10 rabbit polyclonal C-terminus, 1:1000, Sigma-Aldrich; N-cadherin mouse monoclonal C-terminus, 1:1000, BD Transductions Laboratories, San Jose, CA) in 5% milk TBS-T. After overnight incubation at 4°C, the blots were washed with milk TBS-T, incubated in secondary antibody (IgG goat anti-mouse, 1:20,000, Rockland, Gilbertsville, PA; IgG bovine anti-rabbit, 1:20,000, Santa Cruz Biotechnologies, Santa Cruz, CA), and then washed with TBS-T. Antibody binding was visualized with enhanced chemiluminescence substrate, SuperSignal (Thermo Scientific, Rockford, IL). Positive signals were imaged using the Syngene G: BOX system, and data were collected in relative optical density (ROD) and analyzed with GeneTools programs (Syngene, Frederick, MD). Results are expressed as percentages of paired control samples run on the same membrane. Load control experiments were performed with β-actin (mouse monoclonal, 1:3000, Sigma-Aldrich) signal detection.
Immunohistochemistry
At 2, 7, and 15 days following UEC or TBI+BEC a random subset of animals (UEC, n=7; TBI+BEC, n=13; sham-injured, n=13) was selected for qualitative immunohistochemical (IHC) analysis of protein distribution. The rats were anesthetized with a lethal dose of sodium pentobarbital (90 mg/kg IP), and perfused first with 0.9% NaCl, followed by aldehyde fixative (4% paraformaldehyde in 0.1 M NaHPO4 buffer or PBS, pH 7.4). The brains were removed and stored in buffer overnight at 4°C. Coronal vibratome sections (30–40 μm) of the hippocampus were prepared for immunofluorescence visualization. Briefly, the sections were pre-incubated in blocking buffer (fish gelatin in PBS+0.05% Triton X-100), washed in PBS, and then incubated overnight at 4°C in primary antibody (MT5-MMP rabbit polyclonal N-terminus, 1:250, Abcam; N-cadherin goat polyclonal N-terminus, 1:100, Santa Cruz Biotechnologies; ADAM-10 goat polyclonal C-terminus, 1:500, Santa Cruz Biotechnologies; glial fibrillary acidic protein [GFAP] mouse monoclonal, 1:20,000, Millipore; CD-11 mouse monoclonal, 1:500, BD Biosciences, San Jose, CA; Iba-1 rabbit polyclonal, 1:250, Wako, Osaka, Japan). After overnight incubation in paired primary antibody combinations, the sections were washed in PBS and then blocking buffer. In a dark room, the secondary fluorescent antibody (Alexa-Fluor 488 donkey anti-goat or donkey anti-rabbit, 1:1000 and Alexa-Fluor 594 donkey anti-mouse, 1:1000, Invitrogen, Carlsbad, CA) was applied for 1–2 h, followed by PBS washes. The sections were then float-mounted in PB onto Probe On Plus glass slides (Fisher Scientific, Pittsburgh, PA), and cover-slipped (1.5 μm thickness) using Vectashield (Vector Laboratories, Burlingame, CA). Tissue was qualitatively analyzed for MT5-MMP, N-cadherin, and ADAM-10 co-localization with GFAP (astrocytes) and CD-11/Iba-1 (microglia) using the Leica TCS-SP2 AOBS confocal laser scanning microscope.
Quantitative reverse transcriptase polymerase chain reaction
At 7 days after either UEC (n=5), TBI+BEC (n=4), or sham injury (n=4), a random subset of animals was anesthetized (4% isoflurane in carrier gas of 70% N2O and 30% O2), decapitated, and the brains removed. The 7-day time point was selected based on WB and IHC results, which showed the greatest elevation in MT5-MMP and ADAM-10 protein within the deafferented zone at this time. Total RNA was extracted from enriched dentate molecular layer (ML) dissections with Trizol, the extracts were subjected to Qc analysis, and quantitative reverse transcriptase polymerase chain reaction (qRT-PCR) performed with Taqman® Assay Reagents. Specific primer pairs for MT5-MMP (Rn00582114 m1), ADAM-10 (Rn01530753 m1), and N-cadherin (Rn00580099 m1) were obtained from Inventoried Assays (Applied Biosystems, Foster City, CA). Each primer was 5′ labeled with FAM (6-carboxyfluorescein), and 3′ labeled with a dark quencher. Efficiency was determined with 10× serial dilutions of template, and cyclophilin A was used as endogenous control. PCR was performed in the ABI Prism 7500 Sequence Detection System (Applied Biosystems), using the TaqMan® One-Step PCR Master Mix Reagents Kit. Samples were run triplicate under specific cycling conditions (48°C/30 min, 95°C/10 min; 40 cycles of 95°C/15 sec, and 60°C/1 min), and fold change in expression was calculated by the 2−ΔΔCt method (Dumur et al., 2009).
Electrophysiological recording
Random subsets of rats from the TBI+BEC+GM6001 (n=9), TBI+BEC+vehicle (n=8), and sham-injured (n=12) groups were prepared for in vitro electrophysiological recording. Each rat was anesthetized with 4% isoflurane for 4 min, and the brain rapidly removed. Coronal 450-μm slices were cut into artificial cerebrospinal fluid (aCSF; 2–4°C) containing (in mM): NaCl 124, KCl 3, MgSO4 2, CaCl2 2, NaH2PO4 1.3, NaHCO3 26, glucose 10 (pH 7.4), saturated with 95% O2/5% CO2. All slices containing the mid-dorsal hippocampus were saved, which yielded 4 to 6 slices per brain. Slices were gradually equilibrated to 34°C in oxygenated aCSF for at least 1 h, and then transferred to a submersion-type recording chamber perfused with aCSF at a rate of 2–3 mL/min.
A bipolar stimulating electrode (Teflon-insulated tungsten, with inter-tip distance of 0.2–0.3 mm) was lowered into the stratum radiatum of CA1 to stimulate Schaffer collateral fibers. A recording electrode (glass micropipette filled with aCSF; resistance 6–8 MegOhm) was also placed in the stratum radiatum, approximately 0.5–0.7 mm medial to the stimulating electrode, to measure evoked population excitatory postsynaptic potentials (field EPSPs, or fEPSPs). The depth of the electrodes in the slice was adjusted to achieve maximum fEPSP amplitude. Stimulation was constant current stimulus-isolated square wave pulses (0.20-msec duration), delivered at one pulse every 30 sec (0.033 Hz). Signals were amplified (bandpass=d.c. to 5 kHz), digitized at 20 kHz, and digitally stored for off-line analysis. fEPSP slopes were measured as rate of descent over a 0.5-msec interval in the initial negative-going phase of the fEPSP (ClampFit v.8.2 software; Axon Instruments, Inc., Union City, CA).
An input-output function was generated for each slice by varying stimulus intensity in 10 equal steps from sub-threshold level to the stimulus intensity that evoked a maximum fEPSP. To assure measurement stability, all evoked potentials were repeated 4 times, at each separate level of stimulation intensity, and statistical analyses were performed only on the averaged signals. Following the determination of the input-output function, two paradigms of neural functional plasticity were implemented: long-term potentiation (LTP), and paired-pulse facilitation (PPF).
Stimulus amplitude was adjusted to evoke a fEPSP response that was 50% of maximum, based on the prior input-output analysis, and a 30-min period of baseline recording was obtained at this stimulus level at a rate of 0.033 Hz. LTP was then induced by theta-burst high-frequency stimulation (HFS), consisting of a total of 10 bursts of four pulses at 100 Hz, with 200 msec separating the onset of each burst. The magnitude of LTP was then evaluated for a period of 60 min, using the same stimulation parameters (0.033 Hz) as during the pre-HFS baseline sample.
A subset of rats (n=6) from each group was further assessed for PPF. In the PPF procedure, two identical stimulus pulses, sub-threshold for target cell discharges (i.e., not evoking population spikes), and separated by either 50, 100, or 150 msec, usually evoke non-identical responses. The second augmented response has been interpreted as being due, in large part, to residual Ca2+ loaded into pre-synaptic terminals during the first response, which has not been buffered or cleared prior to the second response (Leung and Fu, 1994). Paired-pulse plasticity was calculated at each interpulse interval (50, 100, and 150 msec), using the ratio of the second fEPSP slope to the first (fEPSP2/fEPSP1).
Statistical analysis
All results were expressed as percent of control samples and reported as mean±standard error of the mean (SEM). The significance of the difference in densitometric measures (ROD) of WB immunobinding was analyzed using analysis of variance (ANOVA), followed by post-hoc analyses using the Dunnett multiple comparison test (SPSS v.11.5). Differences in fold changes of qRT-PCR mRNA measures were tested for significance using the single F statistic against a 100% control level. Electrophysiological results were evaluated with mixed-model ANOVAs, with between-subjects factor of analytic group (uninjured control, TBI+BEC [untreated], and TBI+BEC [GM6001- treated]). Repeated measures factor was time relative to application of theta-burst stimulation (for LTP analyses), or inter-pulse interval (for PPF analyses), with planned pair-wise comparisons computed based on simple effects (Keppel, 1991; Levine, 1991). For LTP baseline and post-HFS data, mean fEPSP slopes were aggregated into 2-min epochs used for graphical and statistical analyses. Outcome assessments were performed with the experimenter blinded to injury or treatment condition. A probability of less than 0.05 was considered statistically significant for all experiments.
Results
MT5-MMP, ADAM-10, and N-cadherin protein expression during synaptogenesis
Overall, WB results revealed a single band with predominant change for each MMP and N-cadherin following UEC and TBI+BEC (Fig. 1). For ADAM-10, signal was observed at 70 kDa, which is consistent with the reported active form of the enzyme (Maretzky et al., 2008; Reiss et al., 2005). In the case of MT5-MMP, the principal immunopositive band was detected at 80 kDa, larger than the predicted 58kDa active form. When the MT5-MMP membrane was re-probed with antibody against its endogenous inhibitor tissue inhibitor of metalloproteinase-2 (TIMP-2) (24 kDa), the TIMP-2 signal co-migrated with MT5-MMP (data not shown), suggesting that the 80-kDa band represents either a modified form of pro-MT5-MMP (Llano et al., 1999), or the active enzyme bound to TIMP-2 (Wang et al., 1999). N-cadherin signal was detected at approximately 118 kDa, consistent with the reported size for its full-length form (Maarouf et al., 2008; Reiss et al., 2005; Tanaka et al., 2000).
FIG. 1.

Time course of hippocampal membrane type 5-matrix metalloproteinase (MT5-MMP), a distintegrin and metalloproteinase-10 (ADAM-10), and N-cadherin protein expression following either unilateral entorhinal cortical lesion (UEC) or traumatic brain injury+bilateral entorhinal cortical lesion (TBI+BEC). Western blot (WB) probe identified major 80-kDa MT5-MMP, 70-kDa ADAM-10, and 118-kDa N-cadherin forms expressed after UEC (A). A significant increase in each MMP relative to contralateral control was observed at 2 and 7 days, with paired reduction in the expression of substrate N-cadherin at 2 and 7 days. By 15 days post-UEC, each MMP was normalized to contralateral expression, while N-cadherin protein was significantly elevated over control values. In the TBI+BEC model (B), WB probe revealed the same major 80-kDa MT5-MMP, 70-kDa ADAM-10, and 118-kDa N-cadherin signals as with UEC. A significant MMP increase relative to sham-injured controls was observed at 2 and 7 days as in UEC, and reduction of substrate N-cadherin was observed at both 2 and 7 days. In contrast to UEC, only MT5-MMP was normalized to control at 15 days, with ADAM-10 remaining significantly elevated, and N-cadherin expression no different from sham-injured cases. Representative gel images are shown below for each protein/time point and β-actin (42-kDa) load control (in A: I, ipsilateral; C, contralateral; *p<0.05, **p<0.01; in B: I, combined injured; S, sham-injured control; *p<0.05, **p<0.01; ROD, relative optical density).
WB analysis of hippocampal extracts ipsilateral to the UEC lesion revealed significant injury-induced changes in MT5-MMP, ADAM-10, and N-cadherin protein between 2 and 15 days post-lesion, when expressed as a percent of contralateral controls (Fig. 1A). The 80-kDa MT5-MMP signal was significantly elevated at 2 and 7 days post-injury (177.23±20.77%, p<0.01; 202.06±21.90%, p<0.01), as was the active 70-kDa form of ADAM-10 (213.15±8.26%, p<0.01; 200.89±34.62%, p<0.05). Full length N-cadherin expression was significantly decreased at 2 and 7 days (68.39±6.94%, p<0.01; 74.23±11.77%, p<0.05), and showed an increase over controls at 15 days (128.27±9.00%, p<0.05). At all post-injury intervals, the N-cadherin protein level was inversely related to that of each enzyme, suggesting specific enzyme/substrate interactions during UEC-induced synaptogenesis.
Hippocampal extracts were also examined for MT5-MMP, ADAM-10, and N-cadherin protein expression following maladaptive TBI+BEC insult (Fig. 1B). Overall, the pattern of elevated MMP and reduced substrate expression at 2 and 7 days post-injury was similar to UEC. The 80-kDa MT5-MMP was significantly elevated above sham levels at 2 and 7 days (126.73±9.85%, p<0.05; 195.72±11.01%, p<0.01). By 15 days, MT5-MMP expression was no longer different from control. The 70-kDa ADAM-10 was also significantly increased at 2 and 7 days following TBI+BEC (156.34±18.18%, p<0.01; 267.17±23.54%, p<0.01); however, in contrast to UEC, ADAM-10 remained elevated over controls at 15 days (230.78±30.41, p<0.01). Under conditions of maladaptive plasticity, full-length N-cadherin showed an acute reduction similar to UEC, and was significantly decreased at both 2 and 7 days (75.11±6.91%, p<0.05; 81.03±4.80%, p<0.05), but failed to show a difference from control values at 15 days. Thus, two important differences were observed when adaptive plasticity fails: (1) an upregulation of active ADAM-10 across all time intervals, and (2) the attenuation of the N-cadherin protein increase at 15 days post-injury. These results suggest that a persistent activation of ADAM-10 reduces the capacity for N-cadherin reorganization during the period of synapse maturation and stabilization.
Distribution of MT5-MMP, ADAM-10, and N-cadherin at sites of synaptic plasticity
Immunofluorescence labeling was used to determine MT5-MMP, ADAM-10, and N-cadherin distribution within the deafferented dentate gyrus, as illustrated by the representative sections shown in Figures 2–4. Overall, we observed a qualitative change in the distribution of each protein over time within the deafferented ML, which was predominantly localized to stellate cells of the neuropil. These cells were confirmed as astrocytes (GFAP-positive).
FIG. 2.

Time course of a distintegrin and metalloproteinase-10 (ADAM-10) protein distribution in the deafferented dentate molecular layer. Confocal microscopy shows representative single-channel (green) images of ADAM-10 molecular layer staining at 2, 7, and 15 days after traumatic brain injury+bilateral entorhinal cortical lesion (TBI+BEC) (A–C). The enzyme was predominantly localized within stellate cell bodies and processes (arrows), and this signal was increased over sham-injured controls (D). ADAM-10 cellular localization changed over time post-injury, showing broad molecular layer distribution at 2–7 days, increased intensity at 7 days, and focal molecular layer distribution over the region of synapse reformation at 15 days (GCL, granule cell layer; ML, molecular layer; scale bar=20 μm).
FIG. 4.

Glial localization of membrane type 5-matrix metalloproteinase (MT5-MMP), a distintegrin and metalloproteinase-10 (ADAM-10), and N-cadherin within the deafferented dentate molecular layer. Confocal microscopy shows co-localization of each MMP and N-cadherin (green), with the astrocyte marker glial fibrillary acidic protein (GFAP, red) in representative single channel and overlay images. MT5-MMP (A–C) and N-cadherin (G–I) appeared as a punctate signal, both diffuse within the neuropil and focally within reactive astrocytes (arrows). ADAM-10 signal was predominant in cell bodies and processes of reactive astrocytes (arrows), uniformly filling these structures (D–F; scale bar=20 μm in A–F, 10 μm in G–I).
Based on WB results showing that ADAM-10 expression was both higher and persistently elevated during maladaptive plasticity, we profiled tissue localization of this membrane-bound MMP over time for TBI+BEC. While hippocampal ADAM-10 was detected in low-level punctate staining and in processes surrounding neuronal cell bodies, the predominant sites of signal change were seen in cells within the deafferented ML. Relative to sham animals, ML ADAM-10 was elevated across the entire post-injury time course, and a clear shift in neuropil distribution occurred at 15 days post-injury (Fig. 2). At 2 and 7 d, ADAM-10 was localized primarily to stellate-shaped cells and their processes across the full extent of the ML. However, at the 15-day synaptic stabilization phase, the ADAM-10 signal was concentrated in cells of the heavily deafferented outer ML. We also observed the most intense ADAM-10 signal at 7 days, which is the post-injury interval with maximal enzyme expression in tissue extracts. Further, since WB analysis shows an aberrant elevation of active ADAM-10 at 15 days after TBI+BEC, these IHC results suggest that persistent enzyme activity occurs within the zone of synaptogenesis, and plays a role in the altered N-cadherin processing that occurs with maladaptive recovery.
Confocal imaging of MT5-MMP, ADAM-10, and N-cadherin also revealed model differences in pattern and intensity of ML labeling. We focused on contrasting adaptive and maladaptive profiles at the 7- and 15-day time intervals, given that these periods match peak WB enzyme expression and mark the onset of synaptic stabilization (Fig. 3). Post-injury signal for each MMP was elevated when compared to controls, a result consistent with changes observed in tissue extracts. As for ADAM-10, MT5-MMP was primarily visualized in stellate-shaped ML cells, and the combined TBI+BEC model showed more intense labeling than UEC. This result is consistent with the fact that the maladaptive model is a more complex injury. Both MMPs were distributed throughout the ML, but tended to be more concentrated in the outer deafferented zone. We also noted differences in intracellular localization of ADAM-10 and MT5-MMP. Both were high in stellate-cell bodies; however, ADAM-10 labeling was more uniform in appearance and extended throughout the multiple processes of these cells. The MT5-MMP signal was more punctate over the entire neuropil, did not entirely fill the cell somas, and showed less process labeling. Diffuse punctate MT5-MMP signal would be predicted from published studies linking this enzyme to synaptic junctions (Monea et al., 2006). Confocal imaging of N-cadherin at 15 days revealed the most pronounced model differences. After UEC, N-cadherin labeling was strong within both stellate-shaped cells and focal punctate sites, and was concentrated over the outer ML, the principal zone of reactive synaptogenesis. By contrast, with maladaptive TBI+BEC, N-cadherin signal was low, similar to sham controls in intensity, and appeared in punctate clusters throughout the ML. Diffuse staining of the neuropil was also observed, possibly representing reactive synaptic sites. Overall, these results show that deafferentation induces distinct ADAM-10, MT5-MMP, and N-cadherin distribution, and that this response differs during adaptive and maladaptive synaptogenesis. These data are also consistent with enhanced MMP cleavage of N-cadherin under maladaptive conditions, and suggest the possibility that N-cadherin may be subject to proteolysis within reactive ML glia.
FIG. 3.
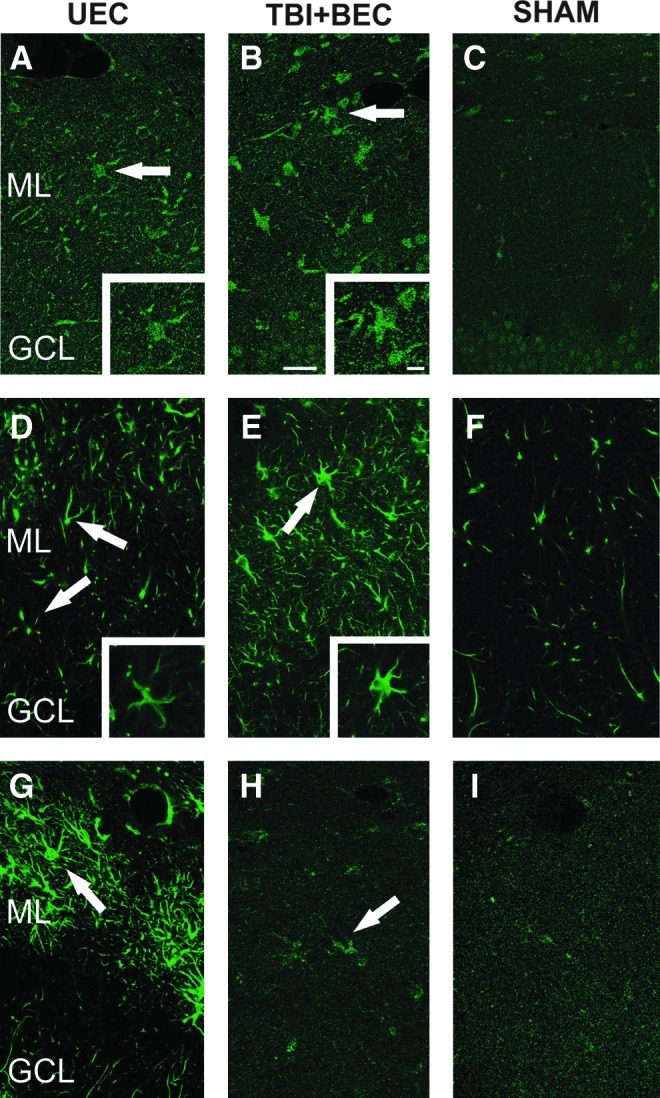
Differences in cellular distribution of membrane type 5-matrix metalloproteinase (MT5-MMP), a distintegrin and metalloproteinase-10 (ADAM-10), and N-cadherin after unilateral entorhinal cortical lesion (UEC) and traumatic brain injury+bilateral entorhinal cortical lesion (TBI+BEC). Confocal microscopy shows representative single-channel (green) images of MT5-MMP (A–C), and ADAM-10 (D–F) staining in the dentate molecular layer at 7 days post-injury. Overall intensity of enzyme labeling was greater in the TBI+BEC cases (B and E) than in UEC (A and D), and increased over paired controls (C and F). Each protein was principally found in stellate cells of the deafferented zone (arrows). MT5-MMP (A and B) showed both diffuse and cellular punctate labeling in each model, while ADAM-10 (D and E) more extensively filled cell bodies and processes (see high-magnification insets). At 15 days after UEC (G), N-cadherin showed strong stellate cell localization (arrow) in the outer molecular layer, while the TBI+BEC signal (H) was diffuse, with modest punctate cell labeling (arrow; GCL, granule cell layer; ML, molecular layer; scale bar=20 μm in panels A–I, 10 μm in the insets for A, B, D, and E).
In order to identify which cell types within the deafferented ML contain membrane-bound MMPs and N-cadherin, we performed double-label confocal imaging of ADAM-10, MT5-MMP, and N-cadherin with astrocyte- (GFAP) and microglial-specific (CD-11/Iba-1) cell markers. These two cells are the principal reactive glia induced by hippocampal deafferentation (Dong et al., 2004; Poirier et al., 1991; Steward et al., 1993; Wang et al., 2005), and can each produce MMPs following CNS insult (Gottschall et al., 1996; Rosenberg et al., 2001; Sbai et al., 2010). In the present experiments, we compared both UEC adaptive and TBI+BEC maladaptive conditions 7 days after injury, matching the survival interval when initial IHC model comparisons were made. The results were similar for both injury models, showing that the stellate-shaped cells each containing MMP and N-cadherin were reactive astrocytes. This co-localization is illustrated by the images from TBI+BEC animals in Figure 4. ADAM-10 signal filled astrocyte cell bodies and processes throughout the ML. MT5-MMP was also localized within GFAP-positive astrocytes; however, this signal was much less intense, diffuse, and failed to fill the cells or their processes as did ADAM-10. Based on these observations, it appears likely that reactive astrocytes produce or process ADAM-10 and MT5-MMP during reactive synaptogenesis. Interestingly, N-cadherin was also localized within GFAP-positive astrocytes, principally in large somatic clusters, with scattered punctate signal along astrocytic processes. Overall, this pattern of enzyme/substrate distribution provides further evidence that ADAM-10 and MT5-MMP may locally target molecules like N-cadherin during synapse reconstruction. While each molecule showed localization within astrocytes, co-localization within microglia was absent (data not shown), suggesting that reactive microglia are not major mediators of the ADAM-10 and MT5-MMP response during synaptic plasticity.
MT5-MMP, ADAM-10, and N-cadherin mRNA expression during synaptogenesis
WB and IHC experiments revealed significant shifts in MT5-MMP, ADAM-10, and N-cadherin protein within the deafferented hippocampus, pointing to elevated localization in the reactive astrocytes of the ML. Nevertheless, it remained unclear whether these changes were associated with altered gene transcription. To test this possibility we conducted qRT-PCR experiments on ML RNA extracts using specific primers to MT5-MMP, ADAM-10, and N-cadherin mRNA (Fig. 5). The 7-day time point was selected because it represented maximal MMP increase and significant N-cadherin reduction, as well as the interval prior to persistent ADAM-10 elevation in the maladaptive model. In addition, MT5-MMP protein expression was normalized between 7 and 15 days for each injury, suggesting a possible shift in transcriptional regulation at this time. Overall, qRT-PCR results showed no significant transcript change at 7 days in the UEC model. A change in transcript was detected only for ADAM-10 in the TBI+BEC cases.
FIG. 5.

Membrane type 5-matrix metalloproteinase (MT5-MMP), a distintegrin and metalloproteinase-10 (ADAM-10), and N-cadherin mRNA expression in the deafferented dentate molecular layer at 7 days after unilateral entorhinal cortical lesion (UEC) and traumatic brain injury+bilateral entorhinal cortical lesion (TBI+BEC). Quantitative reverse transcriptase polymerase chain reaction (qRT-PCR) showed significant elevation of ADAM-10 transcript relative to sham-injured controls in the TBI+BEC model. No change was detected for ADAM-10 mRNA after UEC. Similarly, MT5-MMP and N-cadherin (N-Cad) failed to exhibit significant transcript differences relative to controls in both models (**p<0.01).
Quantitative RT-PCR analysis of ML samples at 7 days post-UEC showed that the ipsilateral ADAM-10 transcript was slightly higher than in contralateral controls, but this difference was not significant. Similarly, MT5-MMP and N-cadherin each had average transcript levels lower than controls, but these shifts were not significant. Thus, changes in MT5-MMP, ADAM-10, and N-cadherin protein seen in WB analysis at 7 days were not temporally correlated with altered mRNA. This suggests that UEC-induced effects on enzyme gene transcription occur at earlier post-injury intervals. Further, if the N-cadherin increase at 15 days after UEC depends on gene transcription, such regulation is likely to occur after the 7-day time point.
By contrast, TBI+BEC samples showed significant elevation of ML ADAM-10 mRNA relative to sham-injured controls (166.27±9.56%; p<0.01), providing evidence for time-matched transcriptional regulation of ADAM-10 expression 7 days after injury. These results are also consistent with the continued elevation of ADAM-10 protein seen at 15 days after TBI+BEC. Notably, average MT5-MMP mRNA expression was slightly higher than in controls, but not significantly different. Again, if transcript elevation was responsible for the 7-day MT5-MMP increase, it likely occurred prior to that time. N-cadherin transcript also did not significantly change after TBI+BEC, suggesting that the reduction in N-cadherin protein seen at 7 days is due to either increased lysis or a reduced translation. Taken together, the present WB and qRT-PCR results indicate that under conditions of maladaptive plasticity, aberrant ADAM-10 expression can be transcriptionally regulated, affecting phases of synapse formation and stabilization.
MMP inhibition after TBI+BEC: Effects on N-cadherin and ADAM-10 protein expression
In order to determine whether membrane-bound MMP activity affects N-cadherin expression during synaptic plasticity, we tested the effect of MMP inhibition on hippocampal N-cadherin levels at 15 days following TBI+BEC. Since no specific pharmacological inhibitor for MT5-MMP and ADAM-10 is available, we chose to apply the broad-spectrum MMP inhibitor GM6001, which blocks both enzymes. GM6001 was applied 6–7 days post-injury, targeting the time point with high enzyme and low N-cadherin expression. N-cadherin, MT5-MMP, and ADAM-10 expression were assayed by WB at 15 days post-injury, the interval when N-cadherin response differed between models. Overall, MMP inhibition significantly increased N-cadherin protein compared with injured-untreated cases. The active form of ADAM-10 was significantly reduced relative to injured-untreated animals, and there was no effect on MT5-MMP expression.
Following GM6001 treatment (Fig. 6), full-length N-cadherin was significantly elevated over sham-injured controls (122.29±12.87; p<0.05), whereas its level in injured-untreated cases was no different from sham-injured cases. When injured-treated and injured-untreated cases were compared, N-cadherin protein was significantly increased, by 44% (p<0.05). For active ADAM-10, injured-treated cases were no longer different from sham-injured controls, in contrast to the injured-untreated group (230.78±30.41%, p<0.01). Further, between the two injured groups there was a 48% reduction in ADAM-10 following GM6001 (p<0.01). By contrast, MT5-MMP in the injured-treated group remained similar to sham-injured controls, and did not significantly change from injured-untreated cases. These results show that MMP inhibition during maladaptive plasticity attenuates active ADAM-10 expression and increases N-cadherin levels, generating an enzyme/substrate profile more consistent with the adaptive UEC model. They also suggest that ADAM-10, rather than MT5-MMP, plays a more significant role in N-cadherin proteolysis during TBI-induced synapse reorganization.
FIG. 6.

Hippocampal membrane type 5-matrix metalloproteinase (MT5-MMP), a distintegrin and metalloproteinase-10 (ADAM-10), and N-cadherin (N-Cad) protein expression in the traumatic brain injury+bilateral entorhinal cortical lesion (TBI+BEC) model following MMP inhibition with GM6001. Western blot (WB) probe shows that MMP inhibition at 6–7 days after TBI+BEC produced a significant reduction in 15-day post-injury ADAM-10 relative to untreated injured cases. In treated cases ADAM-10 protein expression was no longer different from sham-injured controls. This was paired with a significant treatment-induced increase in 15-day N-cadherin protein over untreated-injured cases, and a significant elevation relative to control values. MT5-MMP expression remained no different from sham-injured controls, and failed to show any significant change after GM6001 treatment. Representative gel images are shown below for each protein/treatment condition (I, combined injured; S, sham-injured control; *p<0.05, **p<0.01 relative to untreated-injured animals; §p<0.05, §§p<0.01 relative to sham-injured controls; ROD, relative optical density).
MMP inhibition after TBI+BEC: Effects on synaptic functional plasticity
In order to test whether MMP inhibition at 6–7 days post-injury altered subsequent functional indices of synaptic plasticity, we assessed the effects of this prior inhibition on LTP and PPF in the Schaffer collateral-CA1 pathway at 15 days after TBI+BEC. Application of the theta-burst HFS generally elevated fEPSP slopes during the 60-min post-HFS period. Example evoked waveforms from each group are shown in Figure 7A, illustrating potentials acquired before and after HFS. At the standardized current intensity (50% of maximum, based on input-output functions) fEPSP waveforms had only small amplitude population spikes, or lacked them altogether, but fEPSPs evoked after HFS often developed large population spikes (see example labeled “ps” in Fig. 7A).
FIG. 7.

Effects of traumatic brain injury+bilateral entorhinal cortical lesion (TBI+BEC) injury and GM6001 treatment on long-term potentiation (LTP). (A) Representative traces from a sham control, a TBI+BEC (untreated), and from a TBI+BEC (GM6001-treated) rat, during pre-high-frequency stimulation (HFS) baseline recording, and at 10 and 60 min after HFS. Baseline traces show field excitatory postsynaptic potential (fEPSP) slope measurement (“e”). Population spikes (“ps”) were observed following HFS. (B) Time course of mean fEPSP slopes spanning 10 min pre-HFS to 60 min post-HFS (arrow shows time of theta-burst HFS). Separate analyses were conducted of the period from 0–15 min post-HFS (P1), and 16–60 min post-HFS (P2). (C) The mean level of LTP was significantly suppressed in injured rats from 0–15 min post-HFS (P1; *p<0.05). LTP in GM6001-treated rats was no different from control levels. No significant injury or drug effects were observed from 16–60 min post-HFS (P2).
In uninjured control rats, the HFS trains induced a significant increase in fEPSP slope (as percent of control: 158.0±8.6%; p<0.001) when averaged across the 60-min post-HFS monitoring period. However, as measured at 15 days after the TBI+BEC injury, LTP was weak and quite variable (Fig. 7B). Again averaging across 0–60 min post-HFS, the TBI+BEC (untreated) group exhibited a 36.1±15.5% elevation of fEPSP slope, but this narrowly missed significance. In contrast, HFS induced a significant 45.8±10.8% average increase in TBI+BEC+GM6001 rats (p<0.01). These results indicated that the HFS was effective in inducing a functional plasticity: a significant 58% and 46% enhancement for controls and injured-treated rats, respectively, but did not significantly elevate responses in injured-untreated rats. A separate issue was whether these magnitudes of LTP were statistically different among the three groups. As a first approach to this question, we analyzed whether the mean fEPSP, averaged over entire 60 min of post-HFS monitoring, was significantly different among the three groups. This analysis indicated that there were no statistically significant differences among any of the three groups, probably due to the highly variable responses in rats subjected to TBI+BEC.
Inspection of Figure 7B suggested a possible systematic difference among groups during the first 15 min post-HFS. Specifically, the rate of post-HFS response increases in the injured-untreated group tended to be delayed and variable in comparison to the control and injured-treated groups. Accordingly, analyses were also conducted separately for this early period (P1=0–15 min post-HFS), and for P2=16–60 min post-HFS. While this division is arbitrary, for theoretical reasons it is useful to explore indications that injury or drug treatments may preferentially impact activity-dependent plasticity at early stages. For example, molecular cascades and structural modifications initiated by HFS may be quite labile during the early phases of plasticity, and especially susceptible to neuropathological conditions during this time. Analyses targeted at period P1 revealed that the average LTP induced in TBI+BEC (untreated) rats was significantly below that of uninjured control rats (122.65±18.08% versus 155.82±4.01%, p<0.05; Fig. 7C). In contrast, during this same interval (0–15 min post-HFS) the level of LTP in TBI+BEC+GM6001 rats (142.26±9.49%) was no different from uninjured controls (155.82±4.01%), or from TBI+BEC (untreated) rats (122.65±18.08%; Fig. 7C). To the extent that a significant effect of injury was only observed in the TBI+BEC (untreated) group, and not in the TBI+BEC+GM6001 group, these results are consistent with a neuroprotective effect of GM6001 on LTP induction under these injury conditions. However, any neuroprotective benefit was limited to this early interval, because no statistically significant differences were noted among the groups during 16–60 min post-HFS (Fig. 7C).
While studies of LTP have documented the involvement of multiple postsynaptic mechanisms, less evidence has accumulated for a presynaptic role in LTP induction (Malenka and Bear, 2004). Because N-cadherin participates in the linkage and stabilization of presynaptic terminals with postsynaptic structures, it is useful to consider if TBI+BEC injury, and GM6001 treatment, alter presynaptic functional plasticity in a manner comparable to their effects on LTP. To approach this issue we also conducted the paired-pulse facilitation (PPF) procedure. While a postsynaptic role in this form of short-term plasticity cannot be entirely ruled out, our results suggest that using low levels of stimulus current, PPF reflects predominantly presynaptic functioning. Multiple authors have interpreted the augmentation of the second response to reflect residual Ca2+ loaded into presynaptic terminals during the first response (Creager et al., 1980; Leung and Fu, 1994; Lomo, 1971; Zucker, 1989).
In the present study, all groups showed a similar pattern of responses to the paired stimulus presentations. An example set of paired fEPSPs, obtained from a control rat, are shown in Figure 8A. Amplitude and waveform of the second field potential in each pair were contingent on the interpulse interval, with the greatest augmentation of fEPSP2 seen at an interval of 50 msec. Stimulus currents were held constant during this procedure, set individually for each slice to ensure that fEPSP1 was stimulated below the threshold for a population spike. However, it was commonly observed that a small population spike was visible in fEPSP2, especially at 50- and 100-msec intervals (see example “ps” in Fig. 8A). At interpulse intervals of 50 mec, fEPSP2 was elevated over fEPSP1 by an average of 69.55±7.30%, for all slices tested. The mean magnitude of PPF at the 50-msec interpulse interval ranged from 81.92±13.82% (injured untreated) to 59.47±14.52% (injured-treated), with controls showing an intermediate magnitude 67.24±9.46%. However, these differences did not reach statistical significance. All groups showed the predicted pattern of decreased PPF as the interpulse interval was increased to 100 msec, and then to 150 msec (Fig. 8B). Nevertheless, the same lack of statistically significant group differences were noted at these longer intervals, with overall PPF magnitudes of 51.69% and 33.16% observed at 100 and 150 msec, respectively. Taken together, analysis of the PPF data indicated that neither the TBI+BEC injury nor the GM6001 compound significantly altered this form of short-term plasticity.
FIG. 8.

Effects of traumatic brain injury+bilateral entorhinal cortical lesion (TBI+BEC) injury and GM6001 treatment on paired-pulse facilitation (PPF). (A) Representative paired-pulse waveforms for interpulse intervals of 50, 100, and 150 msec, from a sham-injured control rat. The dotted portion of the traces abbreviates the period between evoked responses. Stimulus current is sub-threshold for population spikes in the first response of each pair (field excitatory postsynaptic potential 1 [fEPSP1]). Second responses are augmented, and often include small population spikes (“ps”). (B) Mean paired-pulse ratios (fEPSP2/fEPSP1×100) for the experimental groups exhibited similar decreases as interpulse interval was increased from 50 to 150 msec.
Discussion
The current study examined the spatio-temporal expression of the membrane-bound MMPs MT5-MMP and ADAM-10, along with their substrate N-cadherin, under conditions of adaptive and maladaptive synaptic plasticity. Overall, both MT5-MMP and ADAM-10 displayed time-dependent increases during reactive synaptogenesis, a pattern associated with high glial expression. Elevated enzyme was paired with reduction of the synaptic adhesion protein N-cadherin. These data suggest a selective post-injury interaction between enzyme and substrate affecting synapse remodeling. We also showed, for the first time, differences in the expression and distribution of these proteins when injury promotes maladaptive synaptic plasticity. Elevation of ADAM-10 protein and mRNA in the maladaptive cases extended into post-injury periods when under adaptive conditions, expression would be normalized. Aberrant ADAM-10 expression was correlated with attenuated N-cadherin during nascent synapse stabilization. MMP inhibition in the maladaptive model reduced ADAM-10 and increased N-cadherin expression, approaching the profile seen in the adaptive condition. Effects were detected in both the amount of protein produced and the stability of LTP in the recovering circuitry. Together, these results suggest that membrane-bound MMPs target N-cadherin following TBI, affecting the reorganization of synaptic structure. Importantly, this response is influenced by the type and complexity of neuropathology induced.
Membrane-bound MMPs and the time course of synaptogenesis
Given their transmembrane anchoring, membrane-bound MMPs are well positioned to interact with extracellular substrates at specific loci along the cell surface. In the context of neuroplasticity, membrane-bound MMPs are found at sites critical to this process, within axons and the synaptic junction (Jangouk et al., 2009; Monea et al., 2006). Our study investigated post-injury tissue expression and distribution of MT5-MMP and ADAM-10 in the deafferented hippocampus, a structure known for its ability to exhibit synaptic plasticity. We found that these two membrane-bound MMPs were upregulated during the early degenerative/regenerative phases of reactive synaptogenesis, and localized to the zone of synaptic reorganization. Such upregulation of membrane-bound MMPs is consistent with their posited role during CNS synapse development. MT5-MMP and ADAM-10 are each found in axonal growth cones and developing dendritic spines, as well as within surrounding neuroglia (Hayashita-Kinoh et al., 2001; Kieseier et al., 2003). Transcript for MT-5 MMP is highly regulated during brain development, showing persistent mRNA elevation in the parenchyma of the adult hippocampus, olfactory bulb, and cerebellum, regions exhibiting continued synaptic plasticity (Jaworski, 2000). Based on these patterns, the two enzymes have been proposed to facilitate outgrowth of neurites and formation of new synapses (Aizawa et al., 2001; Bernstein et al., 2003; Hayashita-Kinoh et al., 2001; Sekine-Marcinkiewicz and Seidah, 2000), major components of the plasticity processes explored in the present study. In vitro studies suggest that MT5-MMP and ADAM-10 mediate this outgrowth through proteoglycan cleavage, and can guide growing axons by an ephrin signaling mechanism (Hattori et al., 2000; Hayashita-Kinho et al., 2001). ADAM-10 has also been associated with proteolytic activation of neuregulin and Notch, driving downstream signaling pathways (Lathia et al., 2008; Tousseyn et al., 2009). This interaction can regulate a variety of growth-related processes, including neurogenesis, axonal/dendritic extension, and synapse formation during both development and disease states (Lathia et al., 2008; Yang et al., 2006). While less is known about the role of MT5-MMP in CNS injury, enzyme upregulation has been linked to the reorganization of Aβ fibers within the spinal cord following in vivo sciatic nerve damage (Komori et al., 2004). Interestingly, it exhibits reduced expression under conditions of severe experimental autoimmune encephalomyelitis in the rodent (Toft-Hansen et al., 2007). Evidence for the ADAM-10 response to CNS insult supports its involvement with both excitotoxic and inflammatory challenge, each prominent in the UEC and TBI+BEC models. For example, in the hippocampal model of seizure induction by kainic acid injection, ADAM-10 protein and transcript was elevated in the dentate gyrus, a site where mossy fibers exhibited extensive sprouting and synaptogenesis (Ortiz et al., 2005). A growing body of literature also supports extensive ADAM involvement in the physiological processing of amyloid precursor protein (APP) and its secretion, particularly that of ADAM-10, which has α-secretase properties (Asai et al., 2003). This relationship is important in the context of TBI since APP expression and its contribution to amyloid-β generation, are reported markers of acute axonal pathology and cell death after injury (Ciallella et al., 2002; Loane et al., 2009; Olsson et al., 2004; Smith et al., 2003). In models of multiple sclerosis and experimental autoimmune encephalomyelitis, ADAM-10 is upregulated in reactive glia located at the vascular interface, suggesting a role in proteolytic activation of inflammatory cytokines (Kieseier et al., 2003; Plumb et al., 2005; Toft-Hansen et al., 2007), molecules marking acute TBI pathology. Together these studies support the present findings that both MT5-MMP and ADAM-10 are critical to the success of injury-induced synaptic plasticity.
Overall, the time course of MT5-MMP and ADAM-10 expression during reactive synaptogenesis was similar to that we and others have reported for secreted MMPs, with one exception. Both membrane-bound MMPs were increased at 2 and 7 days post-injury, but while adaptive plasticity showed a return to control levels at 15 days, the maladaptive model revealed a persistent increase in ADAM-10. In previous studies we have shown similar 2- to 7-day increases in protein and mRNA expression of gelatinases A and B (MMPs 2 and 9), and stromelysin-1 (MMP-3) for both models; however, in each case, the secreted MMPs returned to control levels of expression by 15 days (Falo et al., 2006; Kim et al., 2005; Phillips and Reeves, 2001). This time-dependent shift in expression suggests that MMPs are more critical to the local matrix and cell-cell modifications required for early degenerative and initial synaptogenic periods of recovery. During the subsequent synapse stabilization phase, MMP attenuation would support the re-emergence of substrate proteins which anchor new synaptic junctions. Early elevation of secreted MMPs is well documented during neuroplasticity in models of neurotrauma, including controlled cortical impact, and lateral fluid percussion injury, as well as after olfactory bulb lesion, and cerebral ischemia (Costanzo and Perrino, 2008; Costanzo et al., 2006; Fujimoto et al., 2008; Romanic et al., 1998; Truettner et al., 2005; Wang et al., 2000). For example, diffuse percussive TBI elevates MMP-9 within 4 h post-injury, with peak expression at 1–3 days for MMPs 2 and 9 (Truettner et al., 2005). Similarly, cerebral focal ischemia generates MMP-9 increases at 12 h after arterial occlusion, and a delayed MMP-2 response with a maximum level at 5 days (Romanic et al., 1998). This temporal pattern of secreted MMP activity is also replicated in the olfactory bulb after olfactory nerve transection (Costanzo and Perrino, 2008; Costanzo et al., 2006). While a longer elevation of MMP-2 has been reported following spinal cord injury (Hsu et al., 2006), the effect is correlated with management of the extensive glial scar formation in that model. In fact, most studies document a profile consistent with that observed for membrane-bound MMPs in the present experiments, showing an acute post-injury elevation of enzyme and peak activity ranging from 1–7 days after injury. The novel finding here is the discovery that a model of aberrant plasticity shows selective elevation of active ADAM-10 continuing into the second week post-injury, a period when recovering models have attenuated MMP expression.
During deafferentation-induced synaptic plasticity, we also observed a pronounced increase of membrane-bound MT5-MMP and ADAM-10 within reactive astrocytes. In prior studies with the UEC and TBI+BEC models, the secreted MMPs 2, 3, and 9 were similarly upregulated in astrocytes of the deafferented dentate gyrus (Falo et al., 2006; Kim et al., 2005; Phillips and Reeves, 2001). While soluble MMPs released from astrocytes affect diffuse areas of matrix, our current results suggest that MT5-MMP and ADAM-10 in glial processes contribute to synapse stability through targeting ligand molecules like N-cadherin, or the proteoglycans phosphacan (Harris et al., 2011) and brevican (Mayer et al., 2005; Yuan et al., 2002). MMP localization and production within astrocytes each are supported by several published studies modeling axonal/synaptic injury. For example, induction of the immune response with lipopolysaccharide (LPS) results in astrocyte reactivity and the synthesis of active MMP-2 and proMMP-9 protein (Gottschall and Yu, 1995; Rosenberg et al., 2001). Similarly, peri-lesion reactive astrocytes express MMPs 1 and 2 following spinal cord injury (Buss et al., 2007; Hsu et al., 2006), and middle cerebral artery occlusion induces upregulation of MMPs 2, 3, and 9 in reactive astrocytes (Rosenberg et al., 2001). An increase in glial transcript for secreted MMPs, as well as membrane-type MT1-MMP, has been shown with LPS treatment (Wells et al., 1996). More recently, in vitro studies support MT3-MMP as a critical mediator of Nogo-66 receptor shedding, a process which releases soluble peptide fragments that promote axon outgrowth (Ferraro et al., 2011). ADAM family members are also constitutively expressed within reactive astrocytes. ADAM-10 was found to be elevated in astrocytes and macrophages around blood vessels of multiple sclerosis patients (Kieseier et al., 2003), and ADAM-8 was induced in astrocytes at sites of CNS degeneration in the Wobbler mouse (Schlomann et al., 2000). While our IHC results identified reactive astrocytes as sites of MT5-MMP and ADAM-10 expression, they failed to show enzyme localization within microglia, which is consistent with other CNS inflammation and injury studies of these MMPs (Kieseier et al., 2003; Toft-Hansen et al., 2007). By contrast, secreted MMPs can be produced and released by microglia after in vitro activation or under in vivo ischemic injury conditions (Gottschall et al., 1995; Rosenberg et al., 2001). Thus, while secreted MMPs can be sourced by either astrocytes or microglia in response to CNS insults, the reactive astrocyte appears to the principal site for synthesis and/or processing of MT5-MMP and ADAM-10 during injury-induced synaptic plasticity.
N-cadherin: A critical membrane-bound MMP substrate for synaptic plasticity
An important finding from the present study was the temporal correlation between membrane-bound MMP elevation and N-cadherin expression. At the earlier 2- to 7-day post-injury interval, increased enzyme was matched with significant reductions in N-cadherin protein. Differences in the expression of the two membrane-bound MMPs, however, suggest that they may serve distinct roles over the time course of reactive synaptogenesis, with active ADAM-10 preferentially targeting N-cadherin. While synaptic adhesion molecules are linked to lysis by secreted MMPs (Nagy et al., 2006; Wang et al., 2008), in vitro studies also show that N-cadherin can be processed by either MT5-MMP or ADAM-10 (Monea et al., 2006; Reiss et al., 2005). Moreover, in vivo hippocampal deafferentation activates ADAM family member ADAMTS (a disintegrin and metalloprotease with thrombospondin motifs), promoting proteolysis of the matrix molecule brevican to facilitate synaptic remodeling (Mayer et al., 2005). In the present study ADAM-10 was observed in its active form, while MT5-MMP detection was consistent with TIMP-2-bound enzyme. Although more specific immunoprecipitation studies will be required to verify the latter interaction, we have observed WB co-localization for MT5-MMP and TIMP-2 (unpublished data), supporting their association in our tissue extracts. In a TIMP-2-bound form, MT5-MMP can activate proMMP-2 at the cell membrane (Pei et al., 1999; Llano et al., 1999; Nagase et al., 2006; Sternlicht and Werb, 2001; Wang et al., 1999), generating a ternary complex which anchors proenzyme in a position accessible for cleavage and activation by a second, unbound MT5-MMP (Maskos and Bode, 2003; Strongin et al., 1995). Given that proMMP-2 is elevated in the deafferented dentate of both UEC and TBI+BEC at 2–7 days after injury (Phillips and Reeves, 2001), it is reasonable to predict that the role of MT5-MMP involves matrix destabilization of the regenerative environment through gelatinase activation. In fact, each component of the MT5-MMP/TIMP-2/proMMP-2 complex is expressed in reactive astrocytes (Conant and Gottschall, 2005), consistent with our results in the UEC and TBI+BEC models.
During adaptive plasticity, the subsequent 15-day period of synapse stabilization showed reduced MT5-MMP and ADAM-10 expression, coupled with elevated N-cadherin. By contrast, in TBI+BEC maladaptive plasticity ADAM-10 failed to normalize at 15 days and N-cadherin did not increase. Notably, the MT5-MMP protein level was no different from control in either model at 15 days. These results suggest that N-cadherin is a molecular target for membrane-bound MMPs during lesion-induced synaptogenesis, and that change in its expression during critical periods of synapse stabilization contributes to poor synaptic recovery. Such an interpretation is supported by published reports that N-cadherin mediates morphological dynamics in normal dendritic spines (Mysore et al., 2007), as well as activity-driven spine changes seen with seizure-induced mossy fiber sprouting (Shan et al., 2002), or after high-frequency stimulation to induce LTP (Bozdagi et al., 2000; Tang et al., 1998). We tested this potential relationship between membrane-bound MMPs and N-cadherin during maladaptive synaptogenesis by applying the MMP inhibitor GM6001, and now provide evidence for ADAM-10 lysis of N-cadherin as a determinant of regenerative efficacy. GM6001 selectively reduced the expression of active ADAM-10 protein in this model, which was concurrent with an elevation of N-cadherin during synaptic stabilization. This effect of enzyme inhibition on protein expression is likely due to its block of injury-induced ECM change, which promotes transcriptional upregulation of MMPs (Conant and Gottschall, 2005). For example, post-ischemic alteration of hippocampal ECM/cellular contact can trigger intracellular signaling by protein focal adhesion kinase, leading to upregulation of MMPs 2 and 9 (Zalewska et al., 2003). Alternatively, MMP inhibition may influence N-cadherin expression by affecting its synaptic binding to β-catenin, which, upon release from N-cadherin, serves as a transcription factor, potentially reducing N-cadherin mRNA production (McCusker and Alfandari, 2009). In unpublished experiments, we have observed a three- to sixfold increase in β-catenin fragment generation at 7 days after TBI+BEC, supportive of the latter possibility. Overall, our results suggest that ADAM-10 inhibition contributes to elevated N-cadherin expression and improved synaptic stability under maladaptive conditions. Sustained N-cadherin expression is reported to underlie both structural and physiological plasticity at the synapse (Bozdagi et al., 2000; Mysore et al., 2007; Tang et al., 1998).
In the context of activity-induced synaptic plasticity, GM6001 treatment also generated a modest attenuation of LTP deficits following maladaptive synaptogenesis. The re-emergence of LTP is considered a correlate of synaptic recovery and improved cognitive function (Reeves and Steward, 1986). In earlier studies we used cognitive recovery as a metric to distinguish adaptive versus maladaptive recovery after TBI (Phillips and Reeves, 2001). The UEC model exhibits this recovery within 2 weeks post-injury, while cognitive deficits persist up to 60 days after TBI+BEC. Using the MMP inhibitor FN-439, we have reported that post-injury MMP inhibition alters the structural synaptic reorganization underlying UEC functional recovery (Reeves et al., 2003) and attenuates cognitive deficits induced by TBI+BEC insult (Falo et al., 2006). Prior in vivo experiments also showed inhibition of the capacity to generate LTP within the Schaffer collateral-CA1 pathway after TBI+BEC (Reeves et al., 1997b), an effect which correlates with the persistent cognitive deficits produced in this model, and is reproduced here using in vitro slices. The present experiments are the first to examine the effect of MMP inhibition on LTP after TBI+BEC. However, it is important to recognize that our design assessed the capacity to induce LTP 8 days after drug treatment. Thus we were probing time-dependent drug effects on the post-injury processes of fiber-remodeling and synaptogenesis, not the effect of MMP inhibition at the time of LTP trials. Earlier studies from our laboratory have used this approach to show that acute post-lesion MMP inhibition with FN-439 suppressed the re-emergence of LTP at 7 days post-lesion in the adaptive UEC model (Reeves et al., 2003). The present results reveal that early phases of LTP may be weak or unstable under pathological conditions of elevated ADAM-10 and reduced N-cadherin. They also indicate that MMP inhibition improves the stability of LTP in tandem with re-emergence of adaptive patterns of ADAM-10 and N-cadherin expression; however, the effect on LTP was modest and restricted to the initial 15 min following HFS.
There are several potential mechanisms that could underlie this limited LTP response. The effect might simply be due to the fact that our 6- to 7-day dosing only partially normalized ADAM-10 expression at 15 days. Since extracellular N-cadherin can bind pre- and post-synaptic elements to stabilize synapse growth and function (Bozdagi et al., 2004; Takeichi and Abe, 2005), maximizing its protection against proteolysis at 15 days would certainly impact the potential for LTP generation (Tang et al., 1998). It is also possible that collateral inhibition of the secreted MMPs 3 and 9 contributes to the attenuated LTP response reported here. While each of these MMPs has been shown to support LTP maintenance and synaptic stability (Conant et al., 2010; Meighan et al., 2007; Nagy et al., 2006; Wang et al., 2000), such effects were demonstrated by the application of MMP inhibitors at the time of LTP trials, conditions that do not directly address the time-dependent processes of the present experimental paradigm. A more likely explanation of the effect of GM6001 on LTP would be that it attenuates ADAM-10 lytic shedding of N-cadherin, thereby increasing stabilization of the post-synaptic density (Isaac et al., 1995; Liao et al., 1995), and facilitating the trafficking of synaptic AMPA receptors (Bredt and Nicoll, 2003; Malenka and Nicoll, 1999; Nuriya and Huganir, 2006). During LTP induction, AMPA receptors are inserted into the post-synaptic membrane, strengthening the synaptic response (Malenka and Bear, 2004). Following ADAM-10 inhibition, our observation that N-cadherin increases in tandem with reduced LTP deficit suggests that restoration of intact N-cadherin could act by facilitating AMPA receptor placement to improve synaptic efficacy. Such N-cadherin facilitation of synaptic efficacy would also be supported by its intracellular binding to p120 catenin, a molecule critical to cytoskeletal stabilization in the postsynaptic spine, as well as the control of Ca2+ influx through voltage-activated Ca2+ channels (Marrs et al., 2009). Future studies tracking the expression and distribution of molecules like the AMPA receptor and p120 catenin under conditions of MMP inhibition are needed to test these possibilities. Nevertheless, it is clear that MMPs influence synaptic activity in multiple ways, and that the application of MMP inhibitors to affect outcome must be considered in the context of both time post-injury and injury condition.
Finally, given that paired pulse facilitation (PPF) deficits have also been reported after central (Reeves et al., 2000) and lateral (Cao et al., 2006) fluid percussion insults, we tested the effect of MMP inhibition on PPF following TBI+BEC. GM6001 failed to alter PPF following TBI+BEC, suggesting that PPF-associated presynaptic activity (Leung and Fu, 1994; Zucker et al., 1989) is less likely to be related to ADAM-10 and N-cadherin interactions under conditions of maladaptive plasticity. Prior studies have used paired-pulses evoked at stimulus intensities suprathreshold for eliciting recurrent inhibitory activity, and documented disturbances to excitatory/inhibitory balance after ischemia (Chang et al., 1989), kindling (Kamphuis et al., 1992), and TBI (Reeves et al., 1997a). In contrast, PPF (based on stimulus levels subthreshold for postsynaptic discharge, as used in the present study) has been assessed in fewer injury paradigms. It is notable that recent measurements in the intact hippocampal mossy fiber-CA3 projection, showed that MMP inhibition did not alter PPF, but did reduce LTP in the first minutes following HFS (Wojtowicz and Mozrzymas, 2010). While the importance of MMP mediation in Schaffer collateral PPF is controversial (Meighan et al., 2007; Nagy et al., 2006), our results suggest that in the complex TBI+BEC insult, MMPs have less influence on PPF short-term plasticity. Collectively, our electrophysiological data are consistent with the hypothesis that postsynaptic activity may be more vulnerable to changes in ADAM-10/N-cadherin expression, preferentially contributing to greater deficits in synaptic efficacy under maladaptive conditions.
Conclusions
The goal of this study was to assess the role of membrane-bound MMPs in successful reactive synaptogenesis. A comparison of adaptive and maladaptive synaptic plasticity revealed both similarities and differences in expression of MT5-MMP, ADAM-10, and their synaptic substrate N-cadherin. MT5-MMP and ADAM-10 proteins were elevated during degenerative and regenerative phases of synaptogenesis, supporting a role in synaptic reorganization following TBI. However, only ADAM-10 showed a transcript increase, commensurate with the onset of sprouting and synapse formation in the maladaptive model. With poor synaptic recovery, ADAM-10 protein was persistently elevated during synapse stabilization, matched by aberrantly low N-cadherin. Importantly, MMP inhibition in the maladaptive model attenuated ADAM-10 protein expression, elevated N-cadherin, and altered the stability of the early phase of LTP induction. We conclude that, in addition to secreted MMPs, membrane-bound MMPs also contribute to synaptic plasticity and influence the extent of successful outcome. They provide an additional approach for future studies exploring how maladaptive plasticity might be altered to an adaptive form, increasing the efficacy of both structural and functional reorganization after TBI.
Acknowledgments
The authors wish to thank Lesley K. Harris, Raiford T. Black, and Nancy N. Lee for their excellent technical assistance. Quantitative RT-PCR was conducted through the VCU Molecular Diagnostics Laboratory, Department of Pathology, in collaboration with Dr. Cathy I. Dumur and Tana L. Blevins. Microscopy was performed at the VCU Department of Anatomy and Neurobiology Microscopy Facility, which is funded in part by NIH/NINDS Center Core Grant NS047463. These studies were supported by NIH/NINDS grants NS057758, NS056247, and NS044372.
Author Disclosure Statement
No competing financial interests exist.
References
- Asai M. Hattori C. Szabo B. Sasagawa N. Maruyama K. Tanuma S. Ishiura S. Putative function of ADAM9, ADAM10, and ADAM17 as APP alpha-secretase. Biochem. Biophys. Res. Commun. 2003;301:231–235. doi: 10.1016/s0006-291x(02)02999-6. [DOI] [PubMed] [Google Scholar]
- Bandyopadhyay S. Hartley D.M. Cahill C.M. Lahiri D.K. Chattopadhyay N. Rogers J.T. Interleukin-1alpha stimulates non-amyloidogenic pathway by alpha-secretase (ADAM-10 and ADAM-17) cleavage of APP in human astrocytic cells involving p38 MAP kinase. J. Neurosci. Res. 2006;84:106–118. doi: 10.1002/jnr.20864. [DOI] [PubMed] [Google Scholar]
- Bernstein H.G. Bukowska A. Krell D. Bogerts B. Ansorge S. Lendeckel U. Comparative localization of ADAMs 10 and 15 in human cerebral cortex normal aging, Alzheimer's disease and Down syndrome. J. Neurocytol. 2003;32:152–160. doi: 10.1023/b:neur.0000005600.61844.a6. [DOI] [PubMed] [Google Scholar]
- Bozdagi O. Shan W. Tanaka H. Benson D.L. Huntley G.W. Increasing numbers of synaptic puncta during late-phase LTP: N-cadherin is synthesized, recruited to synaptic sites, and required for potentiation. Neuron. 2000;28:245–259. doi: 10.1016/s0896-6273(00)00100-8. [DOI] [PubMed] [Google Scholar]
- Bozdagi O. Valcin M. Poskanzer K. Tanaka H. Benson D.L. Temporally distinct demands for classic cadherins in synapse formation and maturation. Mol. Cell. Neurosci. 2004;27:509–521. doi: 10.1016/j.mcn.2004.08.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bredt D.S. Nicoll R.A. AMPA receptor trafficking at excitatory synapses. Neuron. 2003;40:361–379. doi: 10.1016/s0896-6273(03)00640-8. [DOI] [PubMed] [Google Scholar]
- Buss A. Pech K. Kakulas B.A. Martin D. Schoenen J. Noth J. Brook G.A. Matrix metalloproteinases and their inhibitors in human traumatic spinal cord injury. BMC Neurol. 2007;7:17. doi: 10.1186/1471-2377-7-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cao R. Hasuo H. Ooba S. Akasu T. Zhang X. Facilitation of glutamatergic synaptic transmission in hippocampal CA1 area of rats with traumatic brain injury. Neurosci. Lett. 2006;401:136–141. doi: 10.1016/j.neulet.2006.03.005. [DOI] [PubMed] [Google Scholar]
- Chang H.S. Steward O. Kassell N.F. Decreases in excitatory synaptic transmission and increases in recurrent inhibition in the rat dentate gyrus after transient cerebral ischemia. Brain Res. 1989;505:220–224. doi: 10.1016/0006-8993(89)91446-7. [DOI] [PubMed] [Google Scholar]
- Ciallella J.R. Ikonomovic M.D. Paljug W.R. Wilbur Y.I. Dixon C.E. Kochanek P.M. Marion D.W. DeKosky S.T. Changes in expression of amyloid precursor protein and interleukin-1beta after experimental traumatic brain injury in rats. J. Neurotrauma. 2002;19:1555–1567. doi: 10.1089/089771502762300229. [DOI] [PubMed] [Google Scholar]
- Conant K., editor; Gottschall P., editor. Matrix Metalloproteinases in the Central Nervous System. Imperial College Press; London: 2005. [Google Scholar]
- Conant K. Wang Y. Szklarczyk A. Dudak A. Mattson M.P. Lim S.T. Matrix metalloproteinase-dependent shedding of intercellular adhesion molecule-5 occurs with long-term potentiation. Neuroscience. 2010;166:508–521. doi: 10.1016/j.neuroscience.2009.12.061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Costanzo R.M. Perrino L.A. Peak in matrix metaloproteinases-2 levels observed during recovery from olfactory nerve injury. Neuroreport. 2008;19:327–331. doi: 10.1097/WNR.0b013e3282f50c7b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Costanzo R.M. Perrino L.A. Kobayashi M. Response of matrix metalloproteinase-9 to olfactory nerve injury. Neuroreport. 2006;17:1787–1791. doi: 10.1097/WNR.0b013e32800fef87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Creager R. Dunwiddie T. Lynch G. Paired-pulse and frequency facilitation in the CA1 region of the in vitro rat hippocampus. J. Physiol. 1980;299:409–424. doi: 10.1113/jphysiol.1980.sp013133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cunningham L.A. Wetzel M. Rosenberg G.A. Multiple roles for MMPs and TIMPs in cerebral ischemia. Glia. 2005;50:329–339. doi: 10.1002/glia.20169. [DOI] [PubMed] [Google Scholar]
- Dixon C.E. Lyeth B.G. Povlishock J.T. Findling R.L. Hamm R.J. Marmarou A. Young H.F. Hayes R.L. A fluid percussion model of experimental brain injury in the rat. J. Neurosurg. 1987;67:110–119. doi: 10.3171/jns.1987.67.1.0110. [DOI] [PubMed] [Google Scholar]
- Dong J.H. Ying G.X. Zhou C.F. Entorhinal deafferentation induces the expression of profilin mRNA in the reactive microglial cells in the hippocampus. Glia. 2004;47:102–108. doi: 10.1002/glia.10355. [DOI] [PubMed] [Google Scholar]
- Dumur C.I. Ladd A.C. Wright H.V. Penberthy L.T. Wilkinson D.S. Powers C.N. Garrett C.T. DiNardo L.J. Genes involved in radiation therapy response in head and neck cancers. Laryngoscope. 2009;119:91–101. doi: 10.1002/lary.20005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Falo M.C. Fillmore H.L. Reeves T.M. Phillips L.L. Matrix metalloproteinase-3 expression profile differentiates adaptive and maladaptive synaptic plasticity induced by traumatic brain injury. J. Neurosci. Res. 2006;84:768–781. doi: 10.1002/jnr.20986. [DOI] [PubMed] [Google Scholar]
- Falo M.C. Reeves T.M. Phillips L.L. Agrin expression during synaptogenesis induced by traumatic brain injury. J. Neurotrauma. 2008;25:769–783. doi: 10.1089/neu.2008.0511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferraro G.B. Morrison C.J. Overall C.M. Strittmatter S.M. Fournier A.E. Membrane-type matrix metalloproteinase-3 regulates neuronal responsiveness to myelin through Nogo-66 receptor 1 cleavage. J. Biol. Chem. 2011;286:31418–31424. doi: 10.1074/jbc.M111.249169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fujimoto M. Takagi Y. Aoki T. Hayase M. Marumo T. Gomi M. Nishimura M. Kataoka H. Hashimoto N. Nozaki K. Tissue inhibitor of metalloproteinases protect blood-brain barrier disruption in focal cerebral ischemia. J. Cereb. Blood Flow Metab. 2008;28:1674–1685. doi: 10.1038/jcbfm.2008.59. [DOI] [PubMed] [Google Scholar]
- Gottschall P.E. Deb S. Regulation of matrix metalloproteinase expression in astrocytes, microglia and neurons. Neuroimmunomodulation. 1996;3:69–75. doi: 10.1159/000097229. [DOI] [PubMed] [Google Scholar]
- Gottschall P.E. Yu X. Cytokines regulate gelatinase A and B (matrix metalloproteinase 2 and 9) activity in cultured rat astrocytes. J. Neurochem. 1995;64:1513–1520. doi: 10.1046/j.1471-4159.1995.64041513.x. [DOI] [PubMed] [Google Scholar]
- Gottschall P.E. Yu X. Bing B. Increased production of gelatinase B (matrix metalloproteinase-9) and interleukin-6 by activated rat microglia in culture. J. Neurosci. Res. 1995;42:335–342. doi: 10.1002/jnr.490420307. [DOI] [PubMed] [Google Scholar]
- Grobelny D. Poncz L. Galardy R.E. Inhibition of human skin fibroblast collagenase, thermolysin, and Pseudomonas aeruginosa elastase by peptide hydroxamic acids. Biochemistry. 1992;31:7152–7154. doi: 10.1021/bi00146a017. [DOI] [PubMed] [Google Scholar]
- Harris J.L. Reeves T.M. Phillips L.L. Phosphacan and receptor protein tyrosine phosphatase b expression mediates deafferentation induced synaptogenesis. Hippocampus. 2011;21:81–92. doi: 10.1002/hipo.20725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hattori M. Osterfield M. Flanagan J.G. Regulated cleavage of a contact-mediated axon repellant. Science (NY) 2000;289:1360–1365. doi: 10.1126/science.289.5483.1360. [DOI] [PubMed] [Google Scholar]
- Hayashita-Kinoh H. Kinoh H. Okada A. Komori K. Itoh Y. Chiba T. Kajita M. Yana I. Seiki M. Membrane-type 5 matrix metalloproteinase is expressed in differentiated neurons and regulates axonal growth. Cell Growth Differ. 2001;12:573–580. [PubMed] [Google Scholar]
- Hayes R.L. Jenkins L.W. Lyeth B.G. Neurotransmitter-mediated mechanisms of traumatic brain injury: acetylcholine and excitatory amino acids. J. Neurotrauma. 1992;9(Suppl. 1):S173–S187. [PubMed] [Google Scholar]
- Hsu J.Y. McKeon R. Goussev S. Werb Z. Lee J.U. Trivedi A. Noble-Haeusslein L.J. Matrix metalloproteinase-2 facilitates wound healing events that promote functional recovery after spinal cord injury. J. Neurosci. 2006;26:9841–9850. doi: 10.1523/JNEUROSCI.1993-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huntley G.W. Elste A.M. Patil S.B. Bozdagi O. Benson D.L. Steward O. Synaptic loss and retention of different classic cadherins with LTP-associated synaptic structural remodeling in vivo. Hippocampus. 2012;22:17–28. doi: 10.1002/hipo.20859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Isaac J.T. Nicoll R.A. Malenka R.C. Evidence for silent synapses: implications for the expression of LTP. Neuron. 1995;15:427–434. doi: 10.1016/0896-6273(95)90046-2. [DOI] [PubMed] [Google Scholar]
- Jangouk P. Dehmel T. Meyer Zu Horste G. Ludwig A. Lehmann H.C. Kieseier B.C. Involvement of ADAM10 in axonal outgrowth and myelination of the peripheral nerve. Glia. 2009;57:1765–1774. doi: 10.1002/glia.20889. [DOI] [PubMed] [Google Scholar]
- Jaworski D.M. Developmental regulation of membrane type-5 matrix metalloproteinase (MT5-MMP) expression in the rat nervous system. Brain Res. 2000;860:174–177. doi: 10.1016/s0006-8993(00)02035-7. [DOI] [PubMed] [Google Scholar]
- Kamphuis W. Gorter J.A. Wadman W.J. Lopes da Silva F.H. Hippocampal kindling leads to different changes in paired-pulse depression of focal evoked field potentials in area CA1 and in the fascia dentata. Neurosci. Lett. 1992;141:101–105. doi: 10.1016/0304-3940(92)90344-7. [DOI] [PubMed] [Google Scholar]
- Keppel G., editor. Design and Analysis: A Researcher's Handbook. 3rd. Prentice Hall; Englewood Cliffs, NJ: 1991. [Google Scholar]
- Kieseier B.C. Pischel H. Neuen-Jacob E. Tourtellotte W.W. Hartung H.P. ADAM-10 and ADAM-17 in the inflamed human CNS. Glia. 2003;42:398–405. doi: 10.1002/glia.10226. [DOI] [PubMed] [Google Scholar]
- Kim H.J. Fillmore H.L. Reeves T.M. Phillips L.L. Elevation of hippocampal MMP-3 expression and activity during trauma-induced synaptogenesis. Exp. Neurol. 2005;192:60–72. doi: 10.1016/j.expneurol.2004.10.014. [DOI] [PubMed] [Google Scholar]
- Komori K. Nonaka T. Okada A. Kinoh H. Hayashita-Kinoh H. Yoshida N. Yana I. Seiki M. Absence of mechanical allodynia and Abeta-fiber sprouting after sciatic nerve injury in mice lacking membrane-type 5 matrix metalloproteinase. FEBS Lett. 2004;557:125–128. doi: 10.1016/s0014-5793(03)01458-3. [DOI] [PubMed] [Google Scholar]
- Lathia J.D. Mattson M.P. Cheng A. Notch: from neural development to neurological disorders. J. Neurochem. 2008;107:1471–1481. doi: 10.1111/j.1471-4159.2008.05715.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leung L.S. Fu X.W. Factors affecting paired-pulse facilitation in hippocampal CA1 neurons in vitro. Brain Res. 1994;650:75–84. doi: 10.1016/0006-8993(94)90209-7. [DOI] [PubMed] [Google Scholar]
- Levine G. A Guide to SPSS for Analysis of Variance. Lawrence Erlbaum Associates; Hillsdale, NJ: 1991. [Google Scholar]
- Liao D. Hessler N.A. Malinow R. Activation of postsynaptically silent synapses during pairing-induced LTP in CA1 region of hippocampal slice. Nature. 1995;375:400–404. doi: 10.1038/375400a0. [DOI] [PubMed] [Google Scholar]
- Llano E. Pendas A.M. Freije J.P. Nakano A. Knauper V. Murphy G. Lopez-Otin C. Identification and characterization of human MT5-MMP, a new membrane-bound activator of progelatinase a over expressed in brain tumors. Cancer Res. 1999;59:2570–2576. [PubMed] [Google Scholar]
- Loane D.J. Pocivavsek A. Moussa C.E. Thompson R. Matsuoka Y. Faden A.I. Rebeck G.W. Burns M.P. Amyloid precursor protein secretases as therapeutic targets for traumatic brain injury. Nat. Med. 2009;15:377–379. doi: 10.1038/nm.1940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loesche J. Steward O. Behavioral correlates of denervation and reinnervation of the hippocampal formation of the rat: recovery of alternation performance following unilateral entorhinal cortex lesions. Brain Res. Bull. 1977;2:31–39. doi: 10.1016/0361-9230(77)90022-3. [DOI] [PubMed] [Google Scholar]
- Lomo T. Potentiation of monosynaptic EPSPs in the perforant path-dentate granule cell synapse. Exp. Brain Res. 1971;12:46–63. [PubMed] [Google Scholar]
- Maarouf C.L. Daugs I.D. Spina S. Vidal R. Kokjohn T.A. Patton R.L. Kalback W.M. Luehrs D.C. Walker D.G. Castano E.M. Beach T.G. Ghetti B. Roher A.E. Histopathological and molecular heterogeneity among individuals with dementia associated with Presenilin mutations. Mol. Neurodegen. 2008;3:20. doi: 10.1186/1750-1326-3-20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malenka R.C. Bear M.F. LTP and LTD: an embarrassment of riches. Neuron. 2004;44:5–21. doi: 10.1016/j.neuron.2004.09.012. [DOI] [PubMed] [Google Scholar]
- Malenka R.C. Nicoll R.A. Long-term potentiation–a decade of progress? Science. 1999;285:1870–1874. doi: 10.1126/science.285.5435.1870. [DOI] [PubMed] [Google Scholar]
- Marcinkiewicz M. Seidah N.G. Coordinated expression of beta-amyloid precursor protein and the putative beta-secretase BACE and alpha-secretase ADAM10 in mouse and human brain. J. Neurochem. 2000;75:2133–2143. doi: 10.1046/j.1471-4159.2000.0752133.x. [DOI] [PubMed] [Google Scholar]
- Maretzky T. Scholz F. Koten B. Proksch E. Saftig P. Reiss K. ADAM10-mediated E-cadherin release is regulated by proinflammatory cytokines and modulates keratinocyte cohesion in eczematous dermatitis. J. Invest. Dermatol. 2008;128:1737–1746. doi: 10.1038/sj.jid.5701242. [DOI] [PubMed] [Google Scholar]
- Marrs G.S. Theisen C.S. Bruses J.L. N-cadherin modulates voltage activated calcium influx via RhoA, p120-catenin, and myosin-actin interaction. Mol. Cell. Neurosci. 2009;40:390–400. doi: 10.1016/j.mcn.2008.12.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maskos K. Bode W. Structural basis of matrix metalloproteinases and tissue inhibitors of metalloproteinases. Mol. Biotechnol. 2003;25:241–266. doi: 10.1385/MB:25:3:241. [DOI] [PubMed] [Google Scholar]
- Mayer J. Hamel M.G. Gottschall P.E. Evidence for proteolytic cleavage of brevican by the ADAMTSs in the dentate gyrus after excitotoxic lesion of the mouse entorhinal cortex. BMC Neurosci. 2005;6:52. doi: 10.1186/1471-2202-6-52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCusker C.D. Alfandari D. Life after proteolysis: Exploring the signaling capabilities of classical cadherin cleavage fragments. Commun. Integr. Biol. 2009;2:155–157. doi: 10.4161/cib.7700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meighan P.C. Meighan S.E. Davis C.J. Wright J.W. Harding J.W. Effects of matrix metalloproteinase inhibition on short- and long-term plasticity of Schaffer collateral/CA1 synapses. J. Neurochem. 2007;102:2085–2096. doi: 10.1111/j.1471-4159.2007.04682.x. [DOI] [PubMed] [Google Scholar]
- Monea S. Jordan B.A. Srivastava S. DeSouza S. Ziff E.B. Membrane localization of membrane type 5 matrix metalloproteinase by AMPA receptor binding protein and cleavage of cadherins. J. Neurosci. 2006;26:2300–2312. doi: 10.1523/JNEUROSCI.3521-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muir E.M. Adcock K.H. Morgenstern D.A. Clayton R. von Stillfried N. Rhodes K. Ellis C. Fawcett J.W. Rogers J.H. Matrix metalloproteases and their inhibitors are produced by overlapping populations of activated astrocytes. Brain Res. Mol. Brain Res. 2002;100:103–117. doi: 10.1016/s0169-328x(02)00132-8. [DOI] [PubMed] [Google Scholar]
- Mysore S.P. Tai C.Y. Schuman E.M. Effects of N-cadherin disruption on spine morphological dynamics. Front. Cell. Neurosci. 2007;1:1–14. doi: 10.3389/neuro.03.001.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nagase H. Visse R. Murphy G. Structure and function of matrix metalloproteinases and TIMPs. Cardiovasc. Res. 2006;69:562–573. doi: 10.1016/j.cardiores.2005.12.002. [DOI] [PubMed] [Google Scholar]
- Nagy V. Bozdagi O. Matynia A. Balcerzyk M. Okulski P. Dzwonek J. Costa R.M. Silva A.J. Kaczmarek L. Huntley G.W. Matrix metalloproteinase-9 is required for hippocampal late-phase long-term potentiation and memory. J. Neurosci. 2006;26:1923–1934. doi: 10.1523/JNEUROSCI.4359-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nuriya M. Huganir R.L. Regulation of AMPA receptor trafficking by N-cadherin. J. Neurochem. 2006;97:652–661. doi: 10.1111/j.1471-4159.2006.03740.x. [DOI] [PubMed] [Google Scholar]
- Okonkwo D. Povlishock J. Immunosuppressants in traumatic brain injury. In: Povlishock J.T., editor. Immunosuppressant Analogs in Neuroprotection. Humana Press; Totowa, NJ: 2003. p. 263. [Google Scholar]
- Olsson A. Csajbok L. Ost M. Hoglund K. Nylen K. Rosengren L. Nellgard B. Blennow K. Marked increase of beta-amyloid(1–42) and amyloid precursor protein in ventricular cerebrospinal fluid after severe traumatic brain injury. J. Neurol. 2004;251:870–876. doi: 10.1007/s00415-004-0451-y. [DOI] [PubMed] [Google Scholar]
- Ortiz R.M. Karkkainen I. Huovila A.P. Honkaniemi J. ADAM9, ADAM10, and ADAM15 mRNA levels in the rat brain after kainic acid-induced status epilepticus. Brain Res. Mol. Brain Res. 2005;137:272–275. doi: 10.1016/j.molbrainres.2005.03.008. [DOI] [PubMed] [Google Scholar]
- Pei D. Identification and characterization of the fifth membrane-type matrix metalloproteinase MT5-MMP. J. Biol. Chem. 1999;274:8925–8932. doi: 10.1074/jbc.274.13.8925. [DOI] [PubMed] [Google Scholar]
- Phillips L.L. Reeves T.M. Interactive pathology following traumatic brain injury modifies hippocampal plasticity. Restor. Neurol. Neurosci. 2001;19:213–235. [PubMed] [Google Scholar]
- Phillips L.L. Lyeth B.G. Hamm R.J. Povlishock J.T. Combined fluid percussion brain injury and entorhinal cortical lesion: a model for assessing the interaction between neuroexcitation and deafferentation. J. Neurotrauma. 1994;11:641–656. doi: 10.1089/neu.1994.11.641. [DOI] [PubMed] [Google Scholar]
- Plumb J. Cross A.K. Surr J. Haddock G. Smith T. Bunning R.A. Woodroofe M.N. ADAM-17 and TIMP3 protein and mRNA expression in spinal cord white matter of rats with acute experimental autoimmune encephalomyelitis. J. Neuroimmunol. 2005;164:1–9. doi: 10.1016/j.jneuroim.2005.02.021. [DOI] [PubMed] [Google Scholar]
- Poirier J. Hess M. May P.C. Finch C.E. Astrocytic apolipoprotein E mRNA and GFAP mRNA in hippocampus after entorhinal cortex lesioning. Brain Res. Mol. Brain Res. 1991;11:97–106. doi: 10.1016/0169-328x(91)90111-a. [DOI] [PubMed] [Google Scholar]
- Povlishock J.T. Christman C.W. The pathobiology of traumatically induced axonal injury in animals and humans: a review of current thoughts. J. Neurotrauma. 1995;12:555–564. doi: 10.1089/neu.1995.12.555. [DOI] [PubMed] [Google Scholar]
- Reeves T.M. Steward O. Emergence of the capacity for LTP during reinnervation of the dentate gyrus: evidence that abnormally shaped spines can mediate LTP. Exp. Brain Res. 1986;65:167–175. doi: 10.1007/BF00243839. [DOI] [PubMed] [Google Scholar]
- Reeves T.M. Zhu J. Povlishock J.T. Phillips L.L. The effect of combined fluid percussion and entorhinal cortical lesions on long-term potentiation. Neuroscience. 1997b;77:431–444. doi: 10.1016/s0306-4522(96)00486-1. [DOI] [PubMed] [Google Scholar]
- Reeves T.M. Kao C.Q. Phillips L.L. Bullock M.R. Povlishock J.T. Presynaptic excitability changes following traumatic brain injury in the rat. J. Neurosci. Res. 2000;60:370–379. doi: 10.1002/(SICI)1097-4547(20000501)60:3<370::AID-JNR12>3.0.CO;2-B. [DOI] [PubMed] [Google Scholar]
- Reeves T.M. Lyeth B.G. Phillips L.L. Hamm R.J. Povlishock J.T. The effects of traumatic brain injury on inhibition in the hippocampus and dentate gyrus. Brain Res. 1997a;757:119–132. doi: 10.1016/s0006-8993(97)00170-4. [DOI] [PubMed] [Google Scholar]
- Reeves T.M. Prins M.L. Zhu J. Povlishock J.T. Phillips L.L. Matrix metalloproteinase inhibition alters functional and structural correlates of deafferentation-induced sprouting in the dentate gyrus. J. Neurosci. 2003;23:10182–10189. doi: 10.1523/JNEUROSCI.23-32-10182.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reiss K. Maretzky T. Ludwig A. Tousseyn T. de Strooper B. Hartmann D. Saftig P. ADAM10 cleavage of N-cadherin and regulation of cell-cell adhesion and beta-catenin nuclear signaling. EMBO J. 2005;24:742–752. doi: 10.1038/sj.emboj.7600548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Romanic A.M. White R.F. Arleth A.J. Ohlstein E.H. Barone F.C. Matrix metalloproteinase expression increases after cerebral focal ischemia in rats: inhibition of matrix metalloproteinase-9 reduces infarct size. Stroke. 1998;29:1020–1030. doi: 10.1161/01.str.29.5.1020. [DOI] [PubMed] [Google Scholar]
- Rosenberg G.A. Sullivan N. Esiri M.M. White matter damage is associated with matrix metalloproteinases in vascular dementia. Stroke. 2001;32:1162–1168. doi: 10.1161/01.str.32.5.1162. [DOI] [PubMed] [Google Scholar]
- Sbai O. Ould-Yahoui A. Ferhat L. Gueye Y. Bernard A. Charrat E. Mehanna A. Risso J.J. Chauvin J.P. Fenouillet E. Rivera S. Khrestchatisky M. Differential vesicular distribution and trafficking of MMP-2, MMP-9, and their inhibitors in astrocytes. Glia. 2010;58:344–366. doi: 10.1002/glia.20927. [DOI] [PubMed] [Google Scholar]
- Schlomann U. Rathke-Hartlieb S. Yamamoto S. Jockusch H. Bartsch J.W. Tumor necrosis factor alpha induces a metalloprotease-disintegrin, ADAM8 (CD 156): implications for neuron-glia interactions during neurodegeneration. J. Neurosci. 2000;20:7964–7971. doi: 10.1523/JNEUROSCI.20-21-07964.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schultz G.S. Strelow S. Stern G.A. Chegini N. Grant M.B. Galardy R.E. Grobelny D. Rowsey J.J. Stonecipher K. Parmley V. Treatment of alkali-injured rabbit corneas with a synthetic inhibitor of matrix metalloproteinases. Invest. Ophthalmol. Vis. Sci. 1992;33:3325–3331. [PubMed] [Google Scholar]
- Sekine-Aizawa Y. Hama E. Watanabe K. Tsubuki S. Kanai-Azuma M. Kanai Y. Arai H. Aizawa H. Iwata N. Saido T.C. Matrix metalloproteinase (MMP) system in brain: identification and characterization of brain-specific MMP highly expressed in cerebellum. Eur. J. Neurosci. 2001;13:935–948. doi: 10.1046/j.0953-816x.2001.01462.x. [DOI] [PubMed] [Google Scholar]
- Shan W. Yoshida M. Wu X.R. Huntley G.W. Colman D.R. Neural (N-) cadherin, a synaptic adhesion molecule, is induced in hippocampal mossy fiber axonal sprouts by seizure. J. Neurosci. Res. 2002;69:292–304. doi: 10.1002/jnr.10305. [DOI] [PubMed] [Google Scholar]
- Shigemori Y. Katayama Y. Mori T. Maeda T. Kawamata T. Matrix metalloproteinase-9 is associated with blood-brain barrier opening and brain edema formation after cortical contusion in rats. Acta Neurochir. Suppl. 2006;96:130–133. doi: 10.1007/3-211-30714-1_29. [DOI] [PubMed] [Google Scholar]
- Sifringer M. Stefovska V. Zentner I. Hansen B. Stepulak A. Knaute C. Marzahn J. Ikonomidou C. The role of matrix metalloproteinases in infant traumatic brain injury. Neurobiol. Dis. 2007;25:526–535. doi: 10.1016/j.nbd.2006.10.019. [DOI] [PubMed] [Google Scholar]
- Smith D.H. Chen X.H. Iwata A. Graham D.I. Amyloid beta accumulation in axons after traumatic brain injury in humans. J. Neurosurg. 2003;98:1072–1077. doi: 10.3171/jns.2003.98.5.1072. [DOI] [PubMed] [Google Scholar]
- Sternlicht M.D. Werb Z. How matrix metalloproteinases regulate cell behavior. Annu. Rev. Cell. Dev. Biol. 2001;17:463–516. doi: 10.1146/annurev.cellbio.17.1.463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steward O. Vinsant S.L. The process of reinnervation in the dentate gyrus of the adult rat: A quantitative electron microscope analysis of terminal proliferation and reactive synaptogenesis. J. Comp. Neurol. 1983;214:370–386. [Google Scholar]
- Steward O. Kelley M.S. Torre E.R. The process of reinnervation in the dentate gyrus of adult rats: temporal relationship between changes in the levels of glial fibrillary acidic protein (GFAP) and GFAP mRNA in reactive astrocytes. Exp. Neurol. 1993;124:167–183. doi: 10.1006/exnr.1993.1187. [DOI] [PubMed] [Google Scholar]
- Steward O. Vinsant S. Davis L. The process of reinnervation in the dentate gyrus of adult rats: An ultrastructural study of changes in presynaptic terminals as a result of sprouting. J. Comp. Neurol. 1988;267:203. doi: 10.1002/cne.902670205. [DOI] [PubMed] [Google Scholar]
- Strongin A.Y. Collier I. Bannikov G. Marmer B.L. Grant G.A. Goldberg G.I. Mechanism of cell surface activation of 72-kDa type IV collagenase. Isolation of the activated form of the membrane metalloprotease. J. Biol. Chem. 1995;270:5331–5338. doi: 10.1074/jbc.270.10.5331. [DOI] [PubMed] [Google Scholar]
- Suehiro E. Fujisawa H. Akimura T. Ishihara H. Kajiwara K. Kato S. Fujii M. Yamashita S. Maekawa T. Suzuki M. Increased matrix metalloproteinase-9 in blood in association with activation of interleukin-6 after traumatic brain injury: influence of hypothermic therapy. J. Neurotrauma. 2004;21:1706–1711. doi: 10.1089/neu.2004.21.1706. [DOI] [PubMed] [Google Scholar]
- Takeichi M. Abe K. Synaptic contact dynamics controlled by cadherin and catenins. Trends Cell. Biol. 2005;15:216–221. doi: 10.1016/j.tcb.2005.02.002. [DOI] [PubMed] [Google Scholar]
- Tanaka H. Shan W. Phillips G.R. Arndt K. Bozdagi O. Shapiro L. Huntley G.W. Benson D.L. Colman D.R. Molecular modification of N-cadherin in response to synaptic activity. Neuron. 2000;25:93–107. doi: 10.1016/s0896-6273(00)80874-0. [DOI] [PubMed] [Google Scholar]
- Tang L. Hung C.P. Schuman E.M. A role for the cadherin family of cell adhesion molecules in hippocampal long-term potentiation. Neuron. 1998;20:1165–1175. doi: 10.1016/s0896-6273(00)80497-3. [DOI] [PubMed] [Google Scholar]
- Toft-Hansen H. Babcock A.A. Millward J.M. Owens T. Down regulation of membrane type-matrix metalloproteinases in the inflamed or injured central nervous system. J. Neuroinflammation. 2007;4:24. doi: 10.1186/1742-2094-4-24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tousseyn T. Thathiah A. Jorissen E. Raemaekers T. Konietzko U. Reiss K. Maes E. Snellinx A. Serneels L. Nyabi O. Annaert W. Saftig P. Hartmann D. De Strooper B. ADAM10, the rate-limiting protease of regulated intramembrane proteolysis of Notch and other proteins, is processed by ADAMS-9, ADAMS-15, and the gamma-secretase. J. Biol. Chem. 2009;284:11738–11747. doi: 10.1074/jbc.M805894200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Truettner J.S. Alonso O.F. Dalton Dietrich W. Influence of therapeutic hypothermia on matrix metalloproteinase activity after traumatic brain injury in rats. J. Cereb. Blood Flow Metab. 2005;25:1505–1516. doi: 10.1038/sj.jcbfm.9600150. [DOI] [PubMed] [Google Scholar]
- Wang J. Tsirka S.E. Neuroprotection by inhibition of matrix metalloproteinases in a mouse model of intracerebral haemorrhage. Brain. 2005;128:1622–1633. doi: 10.1093/brain/awh489. [DOI] [PubMed] [Google Scholar]
- Wang W.Y. Dong J.H. Liu X. Wang Y. Ying G.X. Ni Z.M. Zhou C.F. Vascular endothelial growth factor and its receptor Flk-1 are expressed in the hippocampus following entorhinal deafferentation. Neuroscience. 2005;134:1167–1178. doi: 10.1016/j.neuroscience.2005.04.064. [DOI] [PubMed] [Google Scholar]
- Wang X.B. Bozdagi O. Nikitczuk J.S. Zhai Z.W. Zhou Q. Huntley G.W. Extracellular proteolysis by matrix metalloproteinase-9 drives dendritic spine enlargement and long-term potentiation coordinately. Proc. Natl. Acad. Sci. USA. 2008;105:19520–19525. doi: 10.1073/pnas.0807248105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang X. Jung J. Asahi M. Chwang W. Russo L. Moskowitz M.A. Dixon C.E. Fini M.E. Lo E.H. Effects of matrix metalloproteinase-9 gene knock-out on morphological and motor outcomes after traumatic brain injury. J. Neurosci. 2000;20:7037–7042. doi: 10.1523/JNEUROSCI.20-18-07037.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang X. Yi J. Lei J. Pei D. Expression, purification and characterization of recombinant mouse MT5-MMP protein products. FEBS Lett. 1999;462:261–266. doi: 10.1016/s0014-5793(99)01534-3. [DOI] [PubMed] [Google Scholar]
- Wells G.M. Catlin G. Cossins J.A. Mangan M. Ward G.A. Miller K.M. Clements J.M. Quantitation of matrix metalloproteinases in cultured rat astrocytes using the polymerase chain reaction with a multi-competitor cDNA standard. Glia. 1996;18:332–340. [PubMed] [Google Scholar]
- Wojtowicz T. Mozrzymas J.W. Late phase of long-term potentiation in the mossy fiber-CA3 hippocampal pathway is critically dependent on metalloproteinase activity. Hippocampus. 2010;20:917–921. doi: 10.1002/hipo.20787. [DOI] [PubMed] [Google Scholar]
- Yuan W. Matthews R.T. Sandy J.D. Gottschall P.E. Association between protease-specific proteolytic cleavage of brevican and synaptic loss in the dentate gyrus of kainate-treated rats. Neuroscience. 2002;114:1091–1101. doi: 10.1016/s0306-4522(02)00347-0. [DOI] [PubMed] [Google Scholar]
- Yang P. Baker K.A. Hagg T. The ADAMs family: coordinators of nervous system development, plasticity and repair. Prog. Neurobiol. 2006;79:73–94. doi: 10.1016/j.pneurobio.2006.05.001. [DOI] [PubMed] [Google Scholar]
- Yong V.W. Power C. Forsyth P. Edwards D.R. Metalloproteinases in biology and pathology of the nervous system. Nat. Rev. Neurosci. 2001;2:502–511. doi: 10.1038/35081571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zalewska T. Ziemka-Nalecz M. Sarnowska A. Domanska-Janik K. Transient forebrain ischemia modulates signal transduction from extracellular matrix in gerbil hippocampus. Brain Res. 2003;977:62–69. doi: 10.1016/s0006-8993(03)02742-2. [DOI] [PubMed] [Google Scholar]
- Zucker R.S. Short-term synaptic plasticity. Annu. Rev. Neurosci. 1989;12:13–31. doi: 10.1146/annurev.ne.12.030189.000305. [DOI] [PubMed] [Google Scholar]