

Summary
A large body of experimental evidence supports the hypothesis that T-helper 2 (Th2) cytokines orchestrate allergic airway inflammation in animal models. However, human asthma is heterogeneous with respect to clinical features, cellular sources of inflammation, and response to common therapies. This disease heterogeneity has been investigated using sputum cytology as well as unbiased clustering approaches using cellular and clinical data. Important differences in cytokine-driven inflammation may underlie this heterogeneity, and studies in human subjects with asthma have begun to elucidate these molecular differences. This molecular heterogeneity may be assessed by existing biomarkers (induced sputum evaluation or exhaled nitric oxide testing) or may require novel biomarkers. Effective testing and application of emerging therapies that target Th2 cytokines will depend on accurate and easily obtained biomarkers of this molecular heterogeneity in asthma. Furthermore, whether other non-Th2 cytokine pathways underlie airway inflammation in specific subsets of patients with asthma is an unresolved question and an important goal of future research using both mouse models and human studies.
Keywords: cytokine, biomarker, asthma, phenotype, clinical trials
Introduction
Although the discovery of many cytokines took place in the context of immune cells, a major subsequent focus has been to understand how their effects on non-immune cells manifest as clinical human disease. A wide range of non-immune cells have the ability to respond to the cytokine milieu, and many also have the ability to secrete cytokines (1). These two properties give many non-immune cells the ability to both effect and perpetuate an inflammatory environment. For example, epithelial cells can respond to tumor necrosis factor-α (TNF-α) by secreting interleukin-8 (IL-8), which recruits neutrophils to sites of injury (2). These cytokine responses and functions in non-immune cells provide mechanisms for disease, potential drug targets, and biomarkers to track disease progression and responses to targeted therapies.
Asthma is one disease in which we and many others have made substantial progress through an understanding of the roles of cytokines. Asthma is a chronic inflammatory disorder of the airways characterized by episodic and reversible airway obstruction and bronchial hyper-responsiveness. Asthma affects 7% of adults in America and is more prevalent in minority groups (Black and Puerto Rican) and in the impoverished (3). Because the prevalence of asthma is high and both its prevalence and burden have increased over the last several decades (4-6), reduction in asthma prevalence and improvement in treatments have been key components of public health programs such as Healthy People 2010 (7).
Allergic asthma, in which inflammation and airway obstruction are triggered by allergen exposure in atopic individuals, is the best studied form of the disease. Steps in the immunopathogenesis of allergic inflammation are complex, and the reader is referred to several recent reviews for detail (8, 9). Current models include the initiation of allergic responses in the airway by epithelial cells which release thymic stromal lymphopoietin (TSLP), IL-33, and IL-25 in response to allergens. TSLP mediates migration of dendritic cells (DCs), which present antigen to and promote differentiation of T-helper cells. T-helper cells mediate immunoglobulin E (IgE)-isotype switching in B cells. Antigen specific-IgE, in turn, binds to mast cells and basophils in the airway, augmenting local allergic responses. In addition, the IL-33 and IL-25 produced by epithelial cells promote IL-13 and IL-5 release from IL-25 receptor (IL-25R)+ natural helper cells in the airway (10-12), which then promote Th2 cell differentiation and further production of the Th2 cytokines IL-4, IL-5, and IL-13 (13-15) locally. In the process, memory T and B cells are generated that can facilitate more rapid responses to repeated stimulation and disease chronicity. The downstream consequences of IL-13 exposure on resident lung cells in asthma include goblet cell metaplasia and increased mucous production (16) and airway fibrosis (17). Other possible consequences include airway smooth muscle hyper-responsiveness (18-21) and remodeling (22).
Asthma heterogeneity and phenotypes
By definition, all asthmatics share common physiologic abnormalities of airflow limitation such as obstruction on spirometry, airway hyper-responsiveness to methacholine challenge, and symptoms that can include shortness of breath, chest tightness, wheeze, and cough. Despite these shared features, clinicians have long recognized the great heterogeneity in the severity of airway obstruction and symptoms, degree of reversibility, and the amount of improvement in response to medications. Up to 30-45% of asthmatics fail to have improvement in lung function with high doses of inhaled corticosteroids, the gold-standard anti-inflammatory therapy (23, 24). There is also variation in the triggers causing episodic airflow limitation such as exercise, allergens, aspirin or occupational irritants, the frequency and severity of exacerbations, and long-term outcomes such as irreversible loss of lung function due to airway remodeling.
In an attempt to understand the mechanisms for these differences and develop targeted therapeutics, many approaches have been taken to assign asthmatics to distinct phenotypes that can predict disease course and treatment response. Simple, univariate separations based on physiologic parameters such as the degree of obstruction on spirometry or frequency of symptoms and rescue medication use have been integral components of national guidelines for asthma classification and care (25), but these separations fall short of providing insight into molecular mechanisms. These guidelines have been very valuable in public health efforts to improve asthma education and care, specifically with respect to promotion of inhaled corticosteroid (ICS) use for asthma control. On balance, therefore, these coarse classification schemes have been very useful. But a better appreciation of disease heterogeneity and the treatment implications of that heterogeneity will be important in treating severe asthma and in the clinical application of emerging cytokine-based biologics, which are likely to be expensive. In addition, patients who do not respond to inhaled corticosteroids represent an unmet clinical need. Given the critical role of cytokines in asthma, our group has taken a molecular approach to distinguish subgroups based on differences in the composition of the cytokine milieu and its downstream effects. In this review, we summarize our findings and place them in the context of other attempts to find clinically and biologically meaningful phenotypes.
Cellular inflammatory phenotypes
An early approach to asthma classification rightfully focused on the type of cellular inflammation present upon microscopic observation of samples from diseased lungs. The goals with this approach as with others discussed below were to identify mechanisms, predictors, and drug targets, while recognizing heterogeneity in asthma. Not long after Paul Erlich described eosinophils in 1879, findings of eosinophils in airway tissues and sputum in asthma led to the general consensus that eosinophils were the sentinel inflammatory cell in that disease. Analyses of sputum, bronchoalveolar lavage, and endobronchial biopsy specimens from living asthmatics and post-mortem samples from fatal asthma has found that the majority of asthmatics have elevated eosinophils (26). However, not all asthmatics have eosinophils in their sputum, and this subset represents more than simply sampling error. Based on the cellular composition of induced sputum samples, non-eosinophilic asthma may be seen in up to 25% of asthmatics not on treatment and in up to 50% of those on treatment (27). A comparison of patients with and without eosinophilia revealed that non-eosinophilic asthma has a poor response to ICS, the gold-standard asthma treatment (28, 29). Furthermore, non-eosinophilic asthma is seen across a range of asthma severity, from mild, moderate, to severe, and in the severe group has been associated with a lower FEV1, fewer mast cells, and less sub-epithelial fibrosis (i.e. airway remodeling) (30).
Further refinement of this cellular classification has led to four categories based on induced sputum inflammatory cells: (i) eosinophilic, (ii) neutrophilic, (iii) mixed eosinophilic and neutrophilic, and (iv) paucigranulocytic, where there is no observable presence of inflammatory cells (27, 31, 32). Eosinophilia is most commonly seen in classic atopic asthma with allergen-mediated inflammation, and except for severe cases, generally has good response to inhaled corticosteroids in terms of reduced eosinophils, airway obstruction, and symptoms (27, 33-35). Neutrophilia has been noted in asthmatics with acute and chronic infection, obesity, smoking, and irritant exposure such as pollutants (27), in subsets of patients with severe asthma (30), and during acute asthma exacerbations (36). We have previously shown that neutrophilia is associated with reduced lung function, as measured by FEV1, independent of eosinophils (37, 38). Mixed neutrophilia and eosinophilia has been reported in refractory asthma. One study found that subjects with mixed neutrophilia and eosinophilia had lowest lung function, highest frequency of daily wheeze, and highest health care utilization (31). Most studies have concluded that asthmatics with low eosinophilia have poor response to steroid treatment (27).
Clinical phenotypes from cluster approaches
Two studies have made significant advances in our understanding of asthma heterogeneity through the use of multivariate approaches. These studies recognize the limitations of using only one variable, such as severity of airflow obstruction or type of cellular inflammation. Instead, they identify phenotypes by clustering asthmatics together based on similarity across multiple variables. Haldar et al. (39) used principal components analysis (PCA) and clinical experience to select a subset of variables that were measured in clinical practice—age of asthma onset, gender, atopic status, body mass index (BMI), peak flow variability, induced sputum eosinophil counts, and symptom scores. These variables were used in k-means clustering to identify 3 distinct clusters in mild to moderate asthmatics, one with early-onset atopic asthma and eosinophilia, another with a preponderance of obesity, females, and lack of eosinophilia, and a third with very mild disease and lack of airway eosinophilia. When applied to asthmatics with more severe, refractory disease [as defined by ATS criteria (40)], the same analysis revealed four clusters: two similar to the early-onset atopic, and obese non-eosinophilic clusters identified in milder asthmatics; the other two clusters had a dissociation between eosinophilia and asthma, one with early-onset and symptoms in the absence of eosinophilia, and the other with late-onset, minimal symptoms and marked sputum eosinophilia. These clusters were validated by their presence in an independent prospective cohort of severe asthmatics. The presence of an obese, non-eosinophilic phenotype combined with the known steroid-responsiveness of eosinophilic inflammation suggests an explanation for the lack of improvement with steroids seen in obese asthmatics (41, 42). That gender, age of onset, BMI, and atopic status were invariant across time and therapy suggests that the phenotypes identified by the multivariate clustering method might be stable characteristics of individuals, though this was not definitively shown in this study.
Application of induced sputum analyses in clinical practice would require significant effort and standardization, and this approach has not penetrated health delivery systems in the US. Using only spirometric measurements and clinical characteristics, Moore et al. (43) provide a complementary view of asthma heterogeneity through multivariate clustering of subjects from the Severe Asthma Research Program, which includes the full range of asthma severity. In this study, hierarchical clustering of subjects with 34 variables representing demographics (sex, age, race), age of onset, duration, symptoms, medication use, health-care utilization rates, lung function, and atopy revealed 5 distinct clusters. Further analysis showed that the 11 most distinguishing variables included baseline and post-bronchodilator FEV1 and FVC, age of onset and asthma duration, sex, frequency of β2-agonist use, and dose of corticosteroids. Features consistent with allergic disease—early age of onset and atopy—were present in 3 clusters representing 76% of patients overall. Corresponding to the obese, non-eosinophilic cluster in the Haldar et al. analysis, the SARP study found an obese, female predominant, late-onset, non-atopic cluster accounting for 10% of the subjects studied, with moderate reduction in FEV1 and a need for frequent oral corticosteroid. Subjects with severe asthma were mostly divided between two clusters, one with early-onset, atopic disease and significant reversibility in FEV1 after administration of bronchodilator, and the other a female-predominant cluster with later-onset, less atopy, and poorer response to bronchodilator. Consistent with the theory that ongoing inflammation can lead to chronic airway remodeling and obstruction, longer disease duration was well-correlated with severe asthma and low lung function across all study subjects. An important contribution of this study was the creation of a decision tree from three variables readily available in the clinic: pre-bronchodilator FEV1, post-bronchodilator FEV1, and atopic status. This decision tree had 80% accuracy in classifying the study subjects into their assigned cluster, suggesting that with further validation and study, clinical clustering approaches have the potential to be used in the clinic to predict severity and treatment response.
Molecular phenotypes
Given advances in our understanding of the molecular pathways involved in asthma and in particular the key cytokines, there is a major advantage to using modern molecular techniques to characterize asthma heterogeneity. Creating subgroups of asthma based on the activity (or lack thereof) of specific cytokine pathways immediately identifies mechanisms underlying clinical phenotypes, new pharmaceutical targets, biomarkers for clinical trials of targeted pharmaceuticals, and has the potential to predict treatment response in the clinic.
Epithelial cells are anatomically well positioned to be major drivers of airway remodeling, including sub-epithelial fibrosis (44-47) and smooth muscle hyperplasia (48-50). Based on the knowledge that airway epithelial cells respond to the cytokine milieu to contribute to airway immune responses (51), our group has studied the gene expression profiles of epithelial brushings obtained through research bronchoscopy in a well-characterized cohort of asthmatics and healthy controls as a means of identifying distinct molecular phenotypes (52, 53). For characterization of molecular phenotypes based on native disease pathophysiology, we have focused first on mild to moderate asthmatics who are not using inhaled or systemic corticosteroids, as steroids likely alter epithelial gene expression. Among the most highly induced genes in mild asthmatics as compared to healthy controls were chloride channel, calcium-activated, family member 1 (CLCA1), periostin, and serine peptidase inhibitor, clade B (ovalbumin), member 2 (serpinB2, also known as plasminogen activator inhibitor-2). Induction of periostin, an integrin ligand and extracellular matrix protein with roles in cell adhesion, cell motility, matrix remodeling, and fibrosis (54, 55), and serpinB2 a member of the serpin class of proteases that promote fibrin formation and deposition and may be involved in the regulation of immune responses (56, 57), was confirmed at the protein level as well. All three of these genes were found to be directly regulated by IL-13 and IL-4 in isolated human airway epithelial cells in vitro (55, 58). These data established that the expression of three genes, CLCA1, periostin, and serpinB2, serves as a surrogate marker for the effects of IL-13/IL-4, and hence Th2 inflammation, in the airway.
Among the top 10 most differentially expressed genes in asthmatics were two mast cell proteases, tryptase and carboxypeptidase A3. Mast cells have been proposed to have a critical role in airway hyper-responsiveness (59, 60), though their presence in the epithelial compartment in asthmatics has varied across studies, perhaps due to small sample sizes, inclusion of subjects using inhaled corticosteroids, and imprecise methods of quantification. Using design-based stereology (61, 62), we showed that asthmatics have an accumulation of intraepithelial mast cells, and through immunohistochemistry, that these mast cells express tryptase and CPA3 (63) at the protein level. Stimulation of primary human bronchial epithelial cells at an air-liquid interface with IL-13 induced the expression of stem cell factor, a growth factor and attractant for mast cells. This induction of stem cell factor provides a mechanism for the increased numbers of intraepithelial mast cells in asthmatics. Additional studies of intraepithelial mast cells in asthma highlight their particular importance in severe asthma (64).
Further analysis of these study subjects showed that nearly half of those with asthma were indistinguishable from healthy controls based upon the epithelial brushing expression of the three surrogate markers of IL-13 exposure, periostin, CLCA1, and serpinB2 (53) (Fig. 1A). This finding suggested that our population of asthmatics was heterogeneous; some had Th2-high and others Th2-low inflammation. Increased expression of IL-13 and IL-5 in Th2-high subjects, as assessed by quantitative polymerase chain reaction (qPCR) in bronchial biopsy specimens, provided further confirmation for this molecular phenotype (Fig. 1B). Compared to healthy controls, both Th2-high and -low subjects had decreased FEV1, bronchodilator responsiveness, and positive allergen skin-prick tests. However, Th2-high subjects were enriched for airway hyper-responsiveness (as measured by the concentration of methacholine required to effect a 20% decline in FEV1 [PC20 methacholine]), serum IgE levels, and both blood and especially bronchoalveolar lavage eosinophilia. Th2-high subjects also had increased numbers of intra-epithelial mast cells (63). Through rigorous application of design-based stereology, we found that reticular basement membrane thickness, a measure of sub-epithelial fibrosis and hence airway remodeling, was significantly increased in Th2-high but not Th2-low subjects. Furthermore, there were dramatic differences in the types of epithelial mucin genes expressed in Th2-high and Th2-low subjects, with an increased MUC5AC/MUC5B ratio in Th2-high asthma. These data support the existence of a distinct asthma molecular phenotype as a result of increased Th2 activity, from which follow testable hypotheses about the molecular mechanisms of eosinophilic inflammation, airway hyper-responsiveness and airway remodeling.
Fig. 1. Expression levels of three IL-13 induced genes in the airway define two subgroups of asthmatics.

(A) Heatmap depicting unsupervised hierarchical clustering (Euclidean complete) of POSTN, CLCA1, and SERPINB2 expression levels in bronchial epithelial brushings across 42 subjects with asthma (denoted by ‘A’) and 28 healthy controls (‘H’). (B) Mean (+ SEM) expression levels of IL-4, IL-5, and IL-13 in bronchial biopsy homogenates obtained contemporaneously with bronchial brushings from a subset of subjects depicted in Figure 1A (cluster 1: 18 ‘Th2-high’ asthmatics, red bars; cluster 2: 14 healthy controls, grey bars, and 16 ‘Th2-low’ asthmatics, blue bars). Two-way correlations across all subjects between IL-4, IL-5, and IL-13 indicated at right (Spearman’s rank correlation).
We performed a randomized placebo-controlled trial of inhaled corticosteroids in this same study showing that the Th2-low phenotype did not respond with an increase in lung function to inhaled corticosteroids as did the Th2-high group (Fig. 2). After inhaled corticosteroids, the Th2-high group had a reduction in expression of periostin, CLCA1, serpinB2, and the mast cell proteases tryptase and carboxypeptidase (CPA3), the latter suggesting a mechanism by which inhaled corticosteroids improved lung function in the Th2-high subgroup. Further investigation of the differences between the Th2-high and -low groups may improve our understanding of corticosteroid insensitivity and resistance, which as mentioned earlier is a significant unmet clinical need.
Fig. 2. Responsiveness of ‘Th2-high’ asthma to inhaled steroids in a randomized placebo-controlled trial.
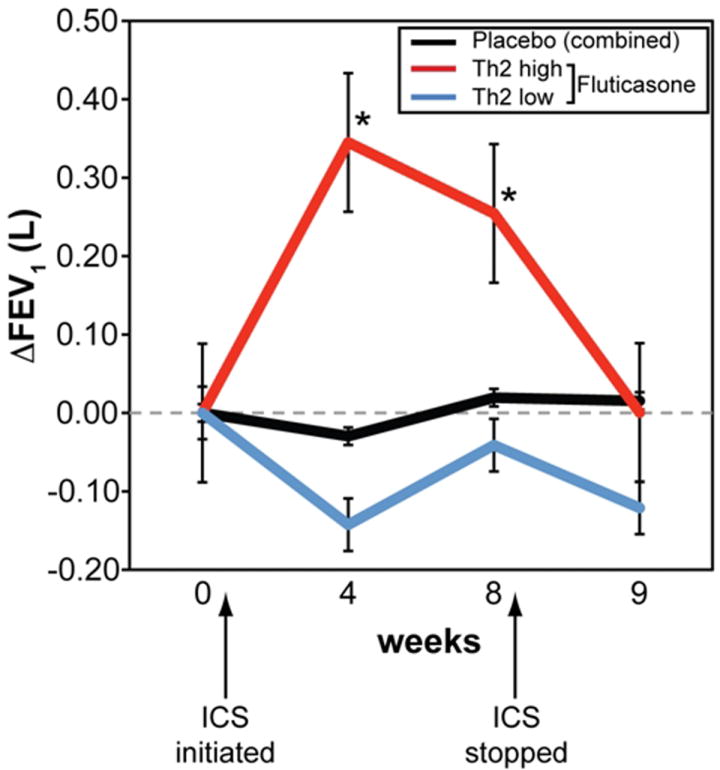
FEV1 measured at baseline (week 0), after 4 and 8 weeks on daily fluticasone (500μg BID), and one week after the cessation of fluticasone (week 9). N=6 Th2-low on fluticasone (blue line), 10 Th2-high on fluticasone (red) and 11 on placebo (black). * denotes p<0.05 for comparison with placebo.
Another question of interest is whether the Th2-high and -low molecular phenotypes are stable characteristics of individual subjects or whether they represent the effect of a changing environment. In our data, the airway epithelial gene expression signature is stable for one week, confirming technical reproducibility. Our finding of increased airway remodeling in Th2-high subjects suggests that the phenotype has stability in an individual long enough to lead to structural changes (53). However, ongoing and future studies will be necessary to definitively test this hypothesis.
Treatment implications of asthma phenotypes
Clinical, cellular, and molecular phenotypes of asthma have already given clinicians a framework to better use available asthma treatments. In all three, groups with a greater degree of sputum eosinophilia tend to have improved response to inhaled and systemic corticosteroid treatment. This finding suggests that eosinophils are playing a causative role, an idea that has support from the known pro-apoptotic effect of corticosteroids on eosinophils (65). Eosinophilia has also guided the use of a novel, targeted asthma treatment—mepolizumab, a humanized monoclonal blocking antibody against IL-5, a key mediator in the differentiation, recruitment, and activation of eosinophils (66). Measurements in bronchial biopsy specimens and serum from subjects with asthma have shown increased levels of IL-5 (14), and increased serum IL-5 has been associated with a fall in FEV1 during the late asthmatic reaction (67). Early studies with mepolizumab were unfavorable. When infusion of a single dose was used in a double-blind randomized placebo-controlled trial in 24 mild asthmatics, blood and sputum eosinophils counts were reduced in the mepolizumab-treated patients following allergen challenge, yet there was no effect on airway hyper-responsiveness (68). Similarly, when infused monthly for 3 months in over 300 subjects with persistent asthma symptoms despite inhaled corticosteroid use, there was no change with mepolizumab treatment in a number of clinical endpoints including FEV1, despite a decrease in sputum and blood eosinophilia (69). These negative clinical studies with mepolizumab led to significant doubt with respect to the role of the eosinophil as an important cellular mediator in asthma (70). However, the results of clinical studies with mepolizumab improved with patient selection based on cellular phenotype of disease. Specifically, two trials demonstrated benefits of mepolizumab in patients with severe asthma who were selected based on persistent sputum eosinophilia (71, 72). The first of these studies was a randomized placebo-controlled trial, which found a reduced frequency of asthma exacerbations when the drug was given to subjects with refractory eosinophilic asthma, defined by sputum eosinophil percentage > 3% on at least one occasion in the previous 2 years, despite high-dose corticosteriod treatment (71). The second study was a randomized trial of mepolizumab in patients with sputum eosinophilia and airway symptoms despite continued treatment with prednisone. In that study, mepolizumab reduced the number of blood and sputum eosinophils and allowed prednisone sparing in those patients (72). These positive studies highlight the need to classify asthma heterogeneity to identify sub-populations that can benefit from targeted therapies.
Asthma phenotypes also have implications for future drug development. Neutrophilia has been seen most often in subjects with severe asthma, who are also likely to be on high doses of corticosteroids. This observation may be a result of the anti-apoptotic effect corticosteroids have on neutrophils (73) and may pave the development of neutrophil-targeted therapies for patients with symptoms despite treatment with high doses of inhaled or systemic corticosteroids.
Our discovery of Th2-high and low asthma phenotypes has the potential to inform the study and use of emerging therapies targeting Th2-mediated inflammation. Because effector cytokines of Th2-mediated inflammation such as IL-13, IL-4, and IL-5 have been assumed to play a role in the majority of allergic asthma, emerging therapies have targeted each of these cytokines using novel biologics (71, 72, 74, 75). However, as described above, we found that nearly half of our subjects had markedly low expression of IL-13 induced genes in epithelial brushing samples, and low transcript levels of IL-13 and IL-5 in bronchial biopsy specimens (53). The benefits of targeting anti-IL-13 therapy to the Th2-high group needs to be urgently studied given that there was minimal benefit in lung function in a recent clinical trial of IL-13 blocking antibody in mild, atopic asthmatics (75). One issue of central importance is whether improved strategies for patient selection will improve the outcomes of these trials (as was observed with mepolizumab). Another pressing question is whether the pharmacologic intervention should target IL-5 in addition to IL-4 and/or IL-13. In support of this notion, (i) we observed over-expression of both IL-13 and IL-5 in bronchial biopsies from Th2-high asthmatics, and (ii) IL-13 blockade alone was not associated with a reduction in sputum eosinophils in a recent clinical trial (75).
Other important issues include the identification of the role of other asthma therapies vis a vis these molecular phenotypes. For example, leukotriene antagonists are another popular option for the treatment of asthma, especially given that they can be orally administered. Whether their effects are similar across molecular sub-phenotypes of asthma is not known. However, since 15-lipoxygenase overexpression in alveolar macrophages is clearly confined to the Th2-high phenotype of asthma (53, 76), it is possible that sub-phenotype differences in leukotriene antagonist response will be observed in future studies. Furthermore, emerging strategies for the treatment of asthma include the development of inhibitors of prostaglandin D2 and its receptor (77-80). Whether patient selection for therapies targeting eicosanoids should be based on molecular phenotypes will be important in the clinical evaluation of these agents and requires further study as well.
Biomarkers of airway inflammation in asthma
The characterization of asthma phenotypes and the recognition that these phenotypes are associated with significant variability in responses to established and emerging therapies has fueled the search for biomarkers. Biomarkers have the potential to inform clinical trial investigators as to which subjects respond to therapies under study and why. As in many other areas of medicine, traditional clinical trials in asthma have made conclusions about the efficacy of drugs under study based on the average response compared to placebo or a standard treatment. The problem with this approach is that the presence of a difference in average response can be driven by a few outliers, leading to acceptance of a new treatment that only helps a subgroup. Conversely, the lack of a difference in average response can lead to rejection of a new treatment even though it may be beneficial for a subgroup. More widespread measurement, prospective validation and availability of biomarkers that are associated with treatment response in clinical trials could improve our ability to evaluate new treatments, and make them available to patients who will benefit the most.
Biomarkers can also provide guidance to treating physicians on when to escalate or de-escalate treatment. The practical utility of biomarkers such as exhaled nitric oxide (eNO) and induced sputum eosinophil counts have been studied in clinical trials, and blood eosinophilia and total serum IgE are likely being used less formally in the clinic to guide disease management. Ideal biomarkers would be easy to measure, inexpensive, and informative.
Induced sputum eosinophilia as a biomarker
Airway inflammation is generally thought to cause, and therefore precede, the development of symptoms and changes in lung function in asthma. Knowledge of the state of airway inflammation would allow for the adjustment of anti-inflammatory medication such as inhaled corticosteroids, with the possibility of preventing exacerbations. This approach would have an advantage over altering asthma treatments based on patient-reported symptoms or measurements of peak expiratory flow in the clinic, as these occur relatively later than the onset of inflammation. Inflammatory cells and soluble mediators in the airway lumen are readily accessed through the non-invasive technique of sputum induction, whereby after inhalation of a hypertonic or isotonic saline solution, expectorated sputum is collected (81, 82). Induced sputum neutrophil and eosinophil counts accurately reflect the corresponding values obtained during washes of the airway lumen through the more direct but invasive bronchoscopic technique (82, 83).
Eosinophilic inflammation is an abnormal finding present in a large fraction of asthmatic airway samples and in some cases has been shown to precede exacerbations by several weeks (84, 85). Dissociation between airway inflammation and airway hyper-responsiveness provides further rationale for the hypothesis that following airway inflammation can provide unique information not found from assessments of symptoms and lung function (86, 87). Three randomized controlled trials have studied the utility of following induced sputum eosinophil counts in moderate-to-severe asthmatics to tailor the dose of inhaled corticosteroids. All three trials found a reduction in the frequency and severity of asthma exacerbations when the inhaled corticosteroid dose was adjusted based on induced sputum eosinophil counts obtained at intervals of 1-3 months, compared to the standard-of-care method of dose adjustment by symptoms, lung function, or rescue medication use (88-90). The threshold of eosinophilia for dose adjustment ranged from 2-3%. However, despite this improvement in exacerbations, which were defined by an increase in symptoms or need for oral corticosteroids, there was no significant change in lung function assessed by peak expiratory flow or spirometry. Consistent with this discordant result was the lack of the ability of baseline induced sputum eosinophilia to predict improvement in FEV1 in response to inhaled corticosteroids in the randomized trial ‘Predicting Response to Inhaled Corticosteroid Efficacy (PRICE)’, which was performed by the NHLBI Asthma Clinical Research Network (24). These findings suggest a disconnect between symptoms and lung function in terms of their responses to increased doses of inhaled corticosteroids, which is difficult to explain given the known efficacy of steroids in improving lung function, and requires further investigation.
Exhaled nitric oxide as a biomarker
Despite potential utility as a biomarker, the induced sputum eosinophil count measurements have not been widely adopted in clinical care settings in the United States. The fraction of nitric oxide in exhaled breath (FeNO) has been proposed as an alternative non-invasive biomarker to follow airway inflammation that requires less expertise than sputum eosinophilia to measure accurately. FeNO is a highly reproducible measurement that is relatively easy to perform for both patients and providers (91). Nitric oxide is constitutively produced by a variety of cell types by the action of nitric oxide synthase (NOS) on L-arginine; for example, eNOS in endothelial cells and nNOS in neuronal cells. In the setting of cytokine-driven airway inflammation, many cells such as eosinophils and epithelial cells can increase NO production in part through increased transcription of inducible NOS (iNOS). FeNO levels are increased in asthmatics (92, 93), and higher levels of FeNO have been associated with eosinophilic airway inflammation (94-96).
Despite the strong rationale for the use of FeNO as an asthma biomarker, the results of clinical trials which apply FeNO to guide treatment have been mixed. A randomized trial of 118 subjects with a primary care diagnosis of asthma did not show a difference in exacerbation frequency or total inhaled corticosteroids used over a 1 year period when comparing an asthma treatment strategy based on measurement of FeNO versus following standard guidelines (97). Two other studies also showed no change in exacerbation frequency, although in one inhaled corticosteroid dose was reduced significantly (98), and in the other bronchial hyper-responsiveness improved in children (99). Possible reasons for the poor performance of FeNO include high sensitivity but poor specificity for eosinophilic inflammation based on the cut-off selected in specific studies (97), the existence of non-eosinophilic asthma, and confounding by comorbidities that influence FeNO such as nasal polyps (100).
The Th2-high signature as a biomarker
The association between the Th2-high molecular asthma phenotype and a favorable response to inhaled corticosteroids suggests a potential role for this signature as a biomarker to guide the decision to start, continue, or change the dose of inhaled corticosteroids. The likely pathophysiologic connection between the Th2-high phenotype and Th2 cytokines such as IL-13, IL-4, and IL-5 indicates that such a biomarker would also aid in the development and clinical testing of cytokine-targeted therapies. We identified this phenotype by comparing genome-wide expression profiling of epithelial brushing samples from asthmatics to those obtained from healthy controls. Th2-low asthmatics have expression levels of three Th2-cytokine-induced genes, CLCA1, periostin, and serpinB2, that overlap with expression levels in healthy controls, and Th2-high asthmatics have higher expression (53). By this definition, assigning Th2-high or -low status to an unknown sample would require comparison to a set of known asthmatics and healthy controls. The current monetary cost, human resources, and invasiveness involved with obtaining a bronchoscopic sample and analyzing multiple expression arrays to type one unknown are prohibitive. Fortunately, given that the phenotype can be characterized using the airway epithelial expression of just three genes, CLCA1, periostin, and serpinB2, a lower-cost, faster, and less variable PCR-based assay can be designed. We have found that when the distribution of airway epithelial brush expression in healthy controls is obtained from a simple arithmetic mean of the quantitative RT-PCR copy numbers of these three genes, a three-gene-mean value greater than 2 standard deviations above the mean of this distribution identifies a distinct Th2-high group. Th2-high or -low status can be reproducibly determined for an unknown sample by comparing a qPCR-based three-gene-mean (of CLCA1, periostin, and serpinB2) to a previously run reference distribution of healthy controls using a recently described method for inter-run qPCR calibration (101). Further studies are required to validate the use of this epithelial signature as a ‘gold-standard’ biomarker in research protocols that employ bronchoscopy.
While airway epithelial gene expression may be a valuable biomarker of Th2-driven inflammation in clinical trials, invasive bronchoscopic specimens would not be available in clinical practice. High-throughput techniques have provided an extremely valuable, publicly available knowledge base of over a thousand proteins detectable in human serum (102, 103). This information should allow screening of gene expression markers identified using genomic approaches that have serum or plasma protein counterparts. Clinical labs are well equipped to use immunoassays to make inexpensive, fast, and accurate measurements of serum and plasma proteins.
Published studies also support the idea that organ inflammation is reflected in gene expression changes in peripheral blood mononuclear cells (PBMCs) and whole blood cell populations. Kidney and heart transplant patients have altered expression of many genes during organ rejection (104, 105). Whole blood gene expression signatures have also been studied as biomarkers in autoimmune disease and in tuberculosis (106-109). In the lung, whole genome expression profiling of cultured blood neutrophils showed 317 differentially expressed genes between asthmatic patients with and without sputum eosinophilia (110). Further evidence that human asthma is a systemic disease comes from a comparison of gene expression from purified peripheral blood CD4+ T cells, which showed a number of differentially regulated genes including TGF-β (111). Finally, Hankonarson et al. (112) were able to discriminate between corticosteroid responders and non-responders through genes that were differentially induced or downregulated in PBMCs stimulated with IL-1β/TNF-α.
Success in using PBMCs or whole blood gene expression profiles for phenotyping in other diseases provides a strong rationale for investigating the utility of this approach in making a clinically relevant biomarker of Th2 inflammation in asthma. PCR and array-based technologies yield rapid measurements of multiple genes in PBMCs, making possible the use of multivariate approaches that consider quantitative interactions between genes. These approaches, also known as machine learning algorithms, leverage information from multiple genes, an approach shown to have the power to discriminate between disease phenotypes in cancer (113), and that has even led to the development of gene expression-based tests that are FDA-approved for clinical use (105, 114-116).
Beyond Th2-driven asthma phenotypes
Whether non-Th2 pathways of inflammation underlie Th2-low phenotypes of human asthma is as yet unknown and requires further investigation. The predominant view on the role of the classic Th1-Th2 dichotomy has been that the higher the ratio of Th2 to Th1 cytokines, the more likely the development of asthma (117). This prevailing paradigm of the Th2 to Th1 balance, however, has met with some conflicting evidence from mouse models of asthma. The adoptive transfer of antigen-specific Th1 cells into ovalbumin-challenged mice led to the development of airway hyper-responsiveness and airway inflammation that was independent of IL-13 and IL-4 in one study (118) and dependent on IL-13 and IL-18 in another (119). Interestingly, adoptive transfer of antigen-specific Th1 cells into antigen-challenged mice led to the development of IFN-γ-dependent airway hyper-responsiveness that failed to improve with dexamethasone treatment (120), suggesting a role for Th1-dependent inflammation in corticosteroid resistance.
Recently, Th17 cells were added as a third distinct T-helper cell subset, and the presence of inflammation driven by these cells may underlie a clinically relevant disease phenotypes. Th17 cells are characterized by the secretion of IL-17A, IL-17F, IL-21, and IL-22, but not IFN-γ or IL-4 (121, 122). The differentiation of naive human T cells into the Th17 class can be induced by TGF-β and IL-6, requires IL-23 for maintenance of the phenotype, and is independent of lineage specific transcription factors T-bet (for Th1) and GATA-3 and c-Maf (for Th2) (123). Most parenchymal cells, including airway epithelial cells, express receptors for Th17 cytokines including IL-17A and IL-17F, and signaling through these receptors leads to the production of pro-inflammatory factors such as IL-6, IL-1, TNF-α, and IL-8 (CXCL8, a neutrophil chemoattractant).
Studies in the literature support a role for IL-17 in human asthma. Bronchoalveolar lavage samples from patients with asthma have increased numbers of IL-17A-producing cells compared to healthy controls (124), and similar findings in induced sputum also point to a positive correlation with the severity of airway hyper-responsiveness (125-127). Furthermore, evidence of Th17 activity through identification of specific Th17 cells and IL-17A/F has also been found in human airway tissue (128-130) and peripheral blood with a positive correlation with asthma severity (131, 132). IL-17A stimulation of epithelial cells in vitro leads to the production of the secreted mucins MUC5AC and MUC5B, which contribute to the asthma phenotype (133, 134). The role of Th17 cytokines in neutrophil recruitment is unclear. Although IL-8, a potent neutrophil chemoattractant induced by IL-17, has been associated with neutrophil influx in Th17 mouse models of asthma, human studies are conflicting depending on the lung compartment studied. There is one report of a correlation between IL-17 and neutrophils in induced sputum (125), another with a weak correlation in sputum, but no association with neutrophils found in the bronchial submucosa across a range of asthma severity (130). Unresolved questions in human asthma include whether this pathway contributes to corticosteroid resistance as was found in a mouse model of Th17-mediated allergic airway inflammation and airway hyper-responsiveness (135), and whether Th17 cells mediate Th2 independent neutrophilic inflammation as shown in IL-17F transgenic and knockout mice (136). Th17-mediated inflammation may also co-exist with the Th2-high phenotype and augment Th2 responses (136, 137). If there is evidence for Th1 or Th17-mediated inflammation in a subgroup of asthmatics, biomarkers and targeted therapeutics can be developed.
Alternatively, is it possible that airway inflammation does not play a dominant role in the clinical manifestations of a subset of patients with Th2-low asthma? Airway hyper-responsiveness, an excessive airway narrowing in response to a variety of stimuli, is a cardinal feature of asthma, and is the result of an abnormal airway smooth muscle response to mediators of contraction such as histamine and acetylcholine. Although a pro-inflammatory cytokine milieu and infiltration of smooth muscle with inflammatory cells is likely a major influence on abnormal airway smooth muscle contraction, intrinsic abnormalities in the signal transduction or contractile functions of smooth muscle cells in human asthma are largely unstudied. The possibility of a larger degree of airway smooth muscle hyperplasia or hypertrophy also needs investigation as a mechanism in Th2-low asthma.
Conclusion
A large body of evidence supports the hypothesis that Th2 cytokines orchestrate the airway inflammation associated with allergic asthma, and we are collectively poised to specifically target these pathways in human disease with emerging cytokine based therapies. Effective application of these new therapies, however, will depend on the development of biomarkers of molecular heterogeneity in asthma. Biomarkers for patient selection will be important in clinical trials (to maximize the chances of success) and in implementation in the clinic (where there will be pressure from payers to target these relatively expensive biologics to the patients who will truly benefit rather than simply add them on to existing regimens in patients with refractory disease). It is also possible that other cytokine pathways underlie airway inflammation in Th2-low asthma, and genomic/molecular approaches may elucidate the contribution of the pathways in specific subsets of patients with asthma. Other patients with Th2-low asthma may have the disease on the basis of non-inflammatory factors such as intrinsic abnormalities of airway smooth muscle contractility or remodeling. Distinguishing these other pathways which may contribute to asthma will be an important goal of future research using both mouse models (9) and human studies.
Acknowledgments
This work is supported in part by the NIH grants HL007185 (NRB), HL095372, HL097591, and N01-08-08 (PGW) and a Research grant from Genentech (PGW). PGW is a co-inventor with Genentech on a patent related to asthma diagnostics.
References
- 1.Saenz SA, Taylor BC, Artis D. Welcome to the neighborhood: epithelial cell-derived cytokines license innate and adaptive immune responses at mucosal sites. Immunol Rev. 2008;226:172–190. doi: 10.1111/j.1600-065X.2008.00713.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Newcomb DC, Sajjan US, Nagarkar DR, Goldsmith AM, Bentley JK, Hershenson MB. Cooperative effects of rhinovirus and TNF-{alpha} on airway epithelial cell chemokine expression. Am J Physiol Lung Cell Mol Physiol. 2007;293:L1021–1028. doi: 10.1152/ajplung.00060.2007. [DOI] [PubMed] [Google Scholar]
- 3.Jeanne EM, Rudd RA, Johnson CA, King M, Minor RA, Bailey C, Scalia MR, Akinbami LJ. MMWR: Surveillance Summaries: National Surveillance for Asthma --- United States, 1980--2004. CDC; 2007. [PubMed] [Google Scholar]
- 4.Evans R, 3rd, Mullally DI, Wilson RW, Gergen PJ, Rosenberg HM, Grauman JS, Chevarley FM, Feinleib M. National trends in the morbidity and mortality of asthma in the US. Prevalence, hospitalization and death from asthma over two decades: 1965-1984. Chest. 1987;91:65S–74S. [PubMed] [Google Scholar]
- 5.Mannino DM, Homa DM, Pertowski CA. MMWR: Surveillance Summaries: National Surveillance for Asthma --- United States, 1960--1995. CDC; 1998. [PubMed] [Google Scholar]
- 6.Mannino DM, Homa DM, Akinbami LJ, Moorman JE, Gwynn C, Redd SC. MMWR: Surveillance Summaries: National Surveillance for Asthma --- United States, 1980--1999. CDC; 2002. [PubMed] [Google Scholar]
- 7.US Department of Health and Human Services. Healthy People 2010: Respiratory Diseases [Goal 24] Washington, DC: US Government Printing Office; 2000. [Google Scholar]
- 8.Locksley RM. Asthma and allergic inflammation. Cell. 2010;140:777–783. doi: 10.1016/j.cell.2010.03.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kim HY, DeKruyff RH, Umetsu DT. The many paths to asthma: phenotype shaped by innate and adaptive immunity. Nat Immunol. 2010;11:577–584. doi: 10.1038/ni.1892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Fort MM, et al. IL-25 induces IL-4, IL-5, and IL-13 and Th2-associated pathologies in vivo. Immunity. 2001;15:985–995. doi: 10.1016/s1074-7613(01)00243-6. [DOI] [PubMed] [Google Scholar]
- 11.Moro K, et al. Innate production of T(H)2 cytokines by adipose tissue-associated c-Kit(+)Sca-1(+) lymphoid cells. Nature. 2009;463:540–544. doi: 10.1038/nature08636. [DOI] [PubMed] [Google Scholar]
- 12.Price AE, Liang HE, Sullivan BM, Reinhardt RL, Eisley CJ, Erle DJ, Locksley RM. Systemically dispersed innate IL-13-expressing cells in type 2 immunity. Proc Natl Acad Sci USA. 2010;107:11489–11494. doi: 10.1073/pnas.1003988107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Grunig G, et al. Requirement for IL-13 independently of IL-4 in experimental asthma. Science. 1998;282:2261–2263. doi: 10.1126/science.282.5397.2261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Humbert M, et al. Elevated expression of messenger ribonucleic acid encoding IL-13 in the bronchial mucosa of atopic and nonatopic subjects with asthma. J Allergy Clin Immunol. 1997;99:657–665. doi: 10.1016/s0091-6749(97)70028-9. [DOI] [PubMed] [Google Scholar]
- 15.Wills-Karp M, Luyimbazi J, Xu X, Schofield B, Neben TY, Karp CL, Donaldson DD. Interleukin-13: central mediator of allergic asthma. Science. 1998;282:2258–2261. doi: 10.1126/science.282.5397.2258. [DOI] [PubMed] [Google Scholar]
- 16.Shim JJ, Dabbagh K, Ueki IF, Dao-Pick T, Burgel PR, Takeyama K, Tam DC, Nadel JA. IL-13 induces mucin production by stimulating epidermal growth factor receptors and by activating neutrophils. Am J Physiol Lung Cell Mol Physiol. 2001;280:L134–140. doi: 10.1152/ajplung.2001.280.1.L134. [DOI] [PubMed] [Google Scholar]
- 17.Zhu Z, Homer RJ, Wang Z, Chen Q, Geba GP, Wang J, Zhang Y, Elias JA. Pulmonary expression of interleukin-13 causes inflammation, mucus hypersecretion, subepithelial fibrosis, physiologic abnormalities, and eotaxin production. J Clin Invest. 1999;103:779–788. doi: 10.1172/JCI5909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Akiho H, Blennerhassett P, Deng Y, Collins SM. Role of IL-4, IL-13, and STAT6 in inflammation-induced hypercontractility of murine smooth muscle cells. Am J Physiol Gastrointest Liver Physiol. 2002;282:G226–232. doi: 10.1152/ajpgi.2002.282.2.G226. [DOI] [PubMed] [Google Scholar]
- 19.Eum SY, Maghni K, Tolloczko B, Eidelman DH, Martin JG. IL-13 may mediate allergen-induced hyperresponsiveness independently of IL-5 or eotaxin by effects on airway smooth muscle. Am J Physiol Lung Cell Mol Physiol. 2005;288:L576–584. doi: 10.1152/ajplung.00380.2003. [DOI] [PubMed] [Google Scholar]
- 20.Grunstein MM, Hakonarson H, Leiter J, Chen M, Whelan R, Grunstein JS, Chuang S. IL-13-dependent autocrine signaling mediates altered responsiveness of IgE-sensitized airway smooth muscle. Am J Physiol Lung Cell Mol Physiol. 2002;282:L520–528. doi: 10.1152/ajplung.00343.2001. [DOI] [PubMed] [Google Scholar]
- 21.Tliba O, Deshpande D, Chen H, Van Besien C, Kannan M, Panettieri RA, Jr, Amrani Y. IL-13 enhances agonist-evoked calcium signals and contractile responses in airway smooth muscle. Br J Pharmacol. 2003;140:1159–1162. doi: 10.1038/sj.bjp.0705558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Espinosa K, Bosse Y, Stankova J, Rola-Pleszczynski M. CysLT1 receptor upregulation by TGF-beta and IL-13 is associated with bronchial smooth muscle cell proliferation in response to LTD4. J Allergy Clin Immunol. 2003;111:1032–1040. doi: 10.1067/mai.2003.1451. [DOI] [PubMed] [Google Scholar]
- 23.Szefler SJ, Martin RJ, King TS, Boushey HA, Cherniack RM, Chinchilli VM, Craig TJ, Dolovich M, Drazen JM, Fagan JK, et al. Significant variability in response to inhaled corticosteroids for persistent asthma. J Allergy Clin Immunol. 2002;109:410–418. doi: 10.1067/mai.2002.122635. [DOI] [PubMed] [Google Scholar]
- 24.Martin RJ, et al. The Predicting Response to Inhaled Corticosteroid Efficacy (PRICE) trial. J Allergy Clin Immunol. 2007;119:73–80. doi: 10.1016/j.jaci.2006.10.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.National Heart Lung and Blood Institute. Expert Panel report 3: Guidelines for the Diagnosis and Management of Asthma: US Department of Health and Human Services and the NIH. 2007 [Google Scholar]
- 26.Wenzel SE. Eosinophils in asthma--closing the loop or opening the door? N Engl J Med. 2009;360:1026–1028. doi: 10.1056/NEJMe0900334. [DOI] [PubMed] [Google Scholar]
- 27.Haldar P, Pavord ID. Noneosinophilic asthma: a distinct clinical and pathologic phenotype. J Allergy Clin Immunol. 2007;119:1043–1052. doi: 10.1016/j.jaci.2007.02.042. quiz 1053-1044. [DOI] [PubMed] [Google Scholar]
- 28.Pavord ID, Brightling CE, Woltmann G, Wardlaw AJ. Non-eosinophilic corticosteroid unresponsive asthma. Lancet. 1999;353:2213–2214. doi: 10.1016/S0140-6736(99)01813-9. [DOI] [PubMed] [Google Scholar]
- 29.Green RH, Brightling CE, Woltmann G, Parker D, Wardlaw AJ, Pavord ID. Analysis of induced sputum in adults with asthma: identification of subgroup with isolated sputum neutrophilia and poor response to inhaled corticosteroids. Thorax. 2002;57:875–879. doi: 10.1136/thorax.57.10.875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Wenzel SE, et al. Evidence that severe asthma can be divided pathologically into two inflammatory subtypes with distinct physiologic and clinical characteristics. Am J Respir Crit Care Med. 1999;160:1001–1008. doi: 10.1164/ajrccm.160.3.9812110. [DOI] [PubMed] [Google Scholar]
- 31.Hastie AT, Moore WC, Meyers DA, Vestal PL, Li H, Peters SP, Bleecker ER. Analyses of asthma severity phenotypes and inflammatory proteins in subjects stratified by sputum granulocytes. J Allergy Clin Immunol. 2010;125:1028–1036. e1013. doi: 10.1016/j.jaci.2010.02.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Porsbjerg C, Lund TK, Pedersen L, Backer V. Inflammatory subtypes in asthma are related to airway hyperresponsiveness to mannitol and exhaled NO. J Asthma. 2009;46:606–612. doi: 10.1080/02770900903015654. [DOI] [PubMed] [Google Scholar]
- 33.Berry M, Morgan A, Shaw DE, Parker D, Green R, Brightling C, Bradding P, Wardlaw AJ, Pavord ID. Pathological features and inhaled corticosteroid response of eosinophilic and non-eosinophilic asthma. Thorax. 2007;62:1043–1049. doi: 10.1136/thx.2006.073429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Brightling CE, Green RH, Pavord ID. Biomarkers predicting response to corticosteroid therapy in asthma. Treat Respir Med. 2005;4:309–316. doi: 10.2165/00151829-200504050-00002. [DOI] [PubMed] [Google Scholar]
- 35.Gibson PG. Inflammatory phenotypes in adult asthma: clinical applications. Clin Respir J. 2009;3:198–206. doi: 10.1111/j.1752-699X.2009.00162.x. [DOI] [PubMed] [Google Scholar]
- 36.Fahy JV, Kim KW, Liu J, Boushey HA. Prominent neutrophilic inflammation in sputum from subjects with asthma exacerbation. J Allergy Clin Immunol. 1995;95:843–852. doi: 10.1016/s0091-6749(95)70128-1. [DOI] [PubMed] [Google Scholar]
- 37.Woodruff PG, et al. Relationship between airway inflammation, hyperresponsiveness, and obstruction in asthma. J Allergy Clin Immunol. 2001;108:753–758. doi: 10.1067/mai.2001.119411. [DOI] [PubMed] [Google Scholar]
- 38.Woodruff PG, Fahy JV. A role for neutrophils in asthma? Am J Med. 2002;112:498–500. doi: 10.1016/s0002-9343(02)01105-1. [DOI] [PubMed] [Google Scholar]
- 39.Haldar P, et al. Cluster analysis and clinical asthma phenotypes. Am J Respir Crit Care Med. 2008;178:218–224. doi: 10.1164/rccm.200711-1754OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.American Thoracic Society. Proceedings of the ATS workshop on refractory asthma: current understanding, recommendations, and unanswered questions. Am J Respir Crit Care Med. 2000;162:2341–2351. doi: 10.1164/ajrccm.162.6.ats9-00. [DOI] [PubMed] [Google Scholar]
- 41.Sutherland ER, Lehman EB, Teodorescu M, Wechsler ME. Body mass index and phenotype in subjects with mild-to-moderate persistent asthma. J Allergy Clin Immunol. 2009;123:1328–1334. e1321. doi: 10.1016/j.jaci.2009.04.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Sutherland TJ, et al. The association between obesity and asthma: interactions between systemic and airway inflammation. Am J Respir Crit Care Med. 2008;178:469–475. doi: 10.1164/rccm.200802-301OC. [DOI] [PubMed] [Google Scholar]
- 43.Moore WC, et al. Identification of asthma phenotypes using cluster analysis in the Severe Asthma Research Program. Am J Respir Crit Care Med. 2010;181:315–323. doi: 10.1164/rccm.200906-0896OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Sullivan P, Stephens D, Ansari T, Costello J, Jeffery P. Variation in the measurements of basement membrane thickness and inflammatory cell number in bronchial biopsies. Eur Respir J. 1998;12:811–815. doi: 10.1183/09031936.98.12040811. [DOI] [PubMed] [Google Scholar]
- 45.Brewster CE, Howarth PH, Djukanovic R, Wilson J, Holgate ST, Roche WR. Myofibroblasts and subepithelial fibrosis in bronchial asthma. Am J Respir Cell Mol Biol. 1990;3:507–511. doi: 10.1165/ajrcmb/3.5.507. [DOI] [PubMed] [Google Scholar]
- 46.Roche WR, Beasley R, Williams JH, Holgate ST. Subepithelial fibrosis in the bronchi of asthmatics. Lancet. 1989;1:520–524. doi: 10.1016/s0140-6736(89)90067-6. [DOI] [PubMed] [Google Scholar]
- 47.Ferrando RE, Nyengaard JR, Hays SR, Fahy JV, Woodruff PG. Applying stereology to measure thickness of the basement membrane zone in bronchial biopsy specimens. J Allergy Clin Immunol. 2003;112:1243–1245. doi: 10.1016/j.jaci.2003.09.038. [DOI] [PubMed] [Google Scholar]
- 48.Benayoun L, Druilhe A, Dombret MC, Aubier M, Pretolani M. Airway structural alterations selectively associated with severe asthma. Am J Respir Crit Care Med. 2003;167:1360–1368. doi: 10.1164/rccm.200209-1030OC. [DOI] [PubMed] [Google Scholar]
- 49.Woodruff PG, et al. Hyperplasia of smooth muscle in mild to moderate asthma without changes in cell size or gene expression. Am J Respir Crit Care Med. 2004;169:1001–1006. doi: 10.1164/rccm.200311-1529OC. [DOI] [PubMed] [Google Scholar]
- 50.Bara I, Ozier A, Tunon de Lara JM, Marthan R, Berger P. Pathophysiology of bronchial smooth muscle remodelling in asthma. Eur Respir J. 2010;36:1174–1184. doi: 10.1183/09031936.00019810. [DOI] [PubMed] [Google Scholar]
- 51.Kuperman DA, et al. Dissecting asthma using focused transgenic modeling and functional genomics. J Allergy Clin Immunol. 2005;116:305–311. doi: 10.1016/j.jaci.2005.03.024. [DOI] [PubMed] [Google Scholar]
- 52.Woodruff PG, et al. Genome-wide profiling identifies epithelial cell genes associated with asthma and with treatment response to corticosteroids. Proc Natl Acad Sci USA. 2007;104:15858–15863. doi: 10.1073/pnas.0707413104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Woodruff PG, et al. T-helper type 2-driven inflammation defines major subphenotypes of asthma. Am J Respir Crit Care Med. 2009;180:388–395. doi: 10.1164/rccm.200903-0392OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Sidhu SS, Yuan S, Innes AL, Kerr S, Woodruff PG, Hou L, Muller SJ, Fahy JV. Roles of epithelial cell-derived periostin in TGF-beta activation, collagen production, and collagen gel elasticity in asthma. Proc Natl Acad Sci USA. 2010;107:14170–14175. doi: 10.1073/pnas.1009426107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Takayama G, et al. Periostin: a novel component of subepithelial fibrosis of bronchial asthma downstream of IL-4 and IL-13 signals. J Allergy Clin Immunol. 2006;118:98–104. doi: 10.1016/j.jaci.2006.02.046. [DOI] [PubMed] [Google Scholar]
- 56.Schroder WA, et al. SerpinB2 deficiency modulates Th1Th2 responses after schistosome infection. Parasite Immunol. 2010;32:764–768. doi: 10.1111/j.1365-3024.2010.01241.x. [DOI] [PubMed] [Google Scholar]
- 57.Schroder WA, et al. A physiological function of inflammation-associated SerpinB2 is regulation of adaptive immunity. J Immunol. 2010;184:2663–2670. doi: 10.4049/jimmunol.0902187. [DOI] [PubMed] [Google Scholar]
- 58.Zhou Y, et al. Characterization of a calcium-activated chloride channel as a shared target of Th2 cytokine pathways and its potential involvement in asthma. Am J Respir Cell Mol Biol. 2001;25:486–491. doi: 10.1165/ajrcmb.25.4.4578. [DOI] [PubMed] [Google Scholar]
- 59.Williams CM, Galli SJ. Mast cells can amplify airway reactivity and features of chronic inflammation in an asthma model in mice. J Exp Med. 2000;192:455–462. doi: 10.1084/jem.192.3.455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Yu M, Tsai M, Tam SY, Jones C, Zehnder J, Galli SJ. Mast cells can promote the development of multiple features of chronic asthma in mice. J Clin Invest. 2006;116:1633–1641. doi: 10.1172/JCI25702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Ochs M. A brief update on lung stereology. J Microsc. 2006;222:188–200. doi: 10.1111/j.1365-2818.2006.01587.x. [DOI] [PubMed] [Google Scholar]
- 62.Weibel ER, Hsia CC, Ochs M. How much is there really? Why stereology is essential in lung morphometry. J Appl Physiol. 2007;102:459–467. doi: 10.1152/japplphysiol.00808.2006. [DOI] [PubMed] [Google Scholar]
- 63.Dougherty RH, et al. Accumulation of intraepithelial mast cells with a unique protease phenotype in T(H)2-high asthma. J Allergy Clin Immunol. 2010;125:1046–1053. e1048. doi: 10.1016/j.jaci.2010.03.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Balzar S, et al. Mast cell phenotype, location and activation in severe asthma. Data from The Severe Asthma Research Program. Am J Respir Crit Care Med. 2010 doi: 10.1164/rccm.201002-0295OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Meagher LC, Cousin JM, Seckl JR, Haslett C. Opposing effects of glucocorticoids on the rate of apoptosis in neutrophilic and eosinophilic granulocytes. J Immunol. 1996;156:4422–4428. [PubMed] [Google Scholar]
- 66.Hart TK, et al. Preclinical efficacy and safety of mepolizumab (SB-240563), a humanized monoclonal antibody to IL-5, in cynomolgus monkeys. J Allergy Clin Immunol. 2001;108:250–257. doi: 10.1067/mai.2001.116576. [DOI] [PubMed] [Google Scholar]
- 67.van der Veen MJ, Van Neerven RJ, De Jong EC, Aalberse RC, Jansen HM, van der Zee JS. The late asthmatic response is associated with baseline allergen-specific proliferative responsiveness of peripheral T lymphocytes in vitro and serum interleukin-5. Clin Exp Allergy. 1999;29:217–227. doi: 10.1046/j.1365-2222.1999.00466.x. [DOI] [PubMed] [Google Scholar]
- 68.Leckie MJ, et al. Effects of an interleukin-5 blocking monoclonal antibody on eosinophils, airway hyper-responsiveness, and the late asthmatic response. Lancet. 2000;356:2144–2148. doi: 10.1016/s0140-6736(00)03496-6. [DOI] [PubMed] [Google Scholar]
- 69.Flood-Page P, Swenson C, Faiferman I, Matthews J, Williams M, Brannick L, Robinson D, Wenzel S, Busse W, Hansel TT, et al. A study to evaluate safety and efficacy of mepolizumab in patients with moderate persistent asthma. Am J Respir Crit Care Med. 2007;176:1062–1071. doi: 10.1164/rccm.200701-085OC. [DOI] [PubMed] [Google Scholar]
- 70.Flood-Page PT, Menzies-Gow AN, Kay AB, Robinson DS. Eosinophil’s role remains uncertain as anti-interleukin-5 only partially depletes numbers in asthmatic airway. Am J Respir Crit Care Med. 2003;167:199–204. doi: 10.1164/rccm.200208-789OC. [DOI] [PubMed] [Google Scholar]
- 71.Haldar P, et al. Mepolizumab and exacerbations of refractory eosinophilic asthma. N Engl J Med. 2009;360:973–984. doi: 10.1056/NEJMoa0808991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Nair P, et al. Mepolizumab for prednisone-dependent asthma with sputum eosinophilia. N Engl J Med. 2009;360:985–993. doi: 10.1056/NEJMoa0805435. [DOI] [PubMed] [Google Scholar]
- 73.Liles WC, Dale DC, Klebanoff SJ. Glucocorticoids inhibit apoptosis of human neutrophils. Blood. 1995;86:3181–3188. [PubMed] [Google Scholar]
- 74.Wenzel S, Wilbraham D, Fuller R, Getz EB, Longphre M. Effect of an interleukin-4 variant on late phase asthmatic response to allergen challenge in asthmatic patients: results of two phase 2a studies. Lancet. 2007;370:1422–1431. doi: 10.1016/S0140-6736(07)61600-6. [DOI] [PubMed] [Google Scholar]
- 75.Gauvreau GM, et al. The effects of IL-13 blockade on allergen-induced airway responses in mild atopic asthma. Am J Respir Crit Care Med. 2010 doi: 10.1164/rccm.201008-1210OC. [DOI] [PubMed] [Google Scholar]
- 76.Chu HW, Balzar S, Westcott JY, Trudeau JB, Sun Y, Conrad DJ, Wenzel SE. Expression and activation of 15-lipoxygenase pathway in severe asthma: relationship to eosinophilic phenotype and collagen deposition. Clin Exp Allergy. 2002;32:1558–1565. doi: 10.1046/j.1365-2222.2002.01477.x. [DOI] [PubMed] [Google Scholar]
- 77.Norman P. DP(2) receptor antagonists in development. Expert Opin Investig Drugs. 2010;19:947–961. doi: 10.1517/13543784.2010.500019. [DOI] [PubMed] [Google Scholar]
- 78.Stebbins KJ, et al. Therapeutic efficacy of AM156, a novel prostanoid DP2 receptor antagonist, in murine models of allergic rhinitis and house dust mite-induced pulmonary inflammation. Eur J Pharmacol. 2010;638:142–149. doi: 10.1016/j.ejphar.2010.04.031. [DOI] [PubMed] [Google Scholar]
- 79.Weber JE, et al. Identification and characterisation of new inhibitors for the human hematopoietic prostaglandin D2 synthase. Eur J Med Chem. 2009;45:447–454. doi: 10.1016/j.ejmech.2009.10.025. [DOI] [PubMed] [Google Scholar]
- 80.Yoshimura T, Yoshikawa M, Otori N, Haruna S, Moriyama H. Correlation between the prostaglandin D(2)/E(2) ratio in nasal polyps and the recalcitrant pathophysiology of chronic rhinosinusitis associated with bronchial asthma. Allergol Int. 2008;57:429–436. doi: 10.2332/allergolint.o-08-545. [DOI] [PubMed] [Google Scholar]
- 81.Fahy JV, Liu J, Wong H, Boushey HA. Cellular and biochemical analysis of induced sputum from asthmatic and from healthy subjects. Am Rev Respir Dis. 1993;147:1126–1131. doi: 10.1164/ajrccm/147.5.1126. [DOI] [PubMed] [Google Scholar]
- 82.Fahy JV, Wong H, Liu J, Boushey HA. Comparison of samples collected by sputum induction and bronchoscopy from asthmatic and healthy subjects. Am J Respir Crit Care Med. 1995;152:53–58. doi: 10.1164/ajrccm.152.1.7599862. [DOI] [PubMed] [Google Scholar]
- 83.Keatings VM, Evans DJ, O’Connor BJ, Barnes PJ. Cellular profiles in asthmatic airways: a comparison of induced sputum, bronchial washings, and bronchoalveolar lavage fluid. Thorax. 1997;52:372–374. doi: 10.1136/thx.52.4.372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Pizzichini MM, Pizzichini E, Clelland L, Efthimiadis A, Pavord I, Dolovich J, Hargreave FE. Prednisone-dependent asthma: inflammatory indices in induced sputum. Eur Respir J. 1999;13:15–21. doi: 10.1183/09031936.99.13101599. [DOI] [PubMed] [Google Scholar]
- 85.Jatakanon A, Lim S, Barnes PJ. Changes in sputum eosinophils predict loss of asthma control. Am J Respir Crit Care Med. 2000;161:64–72. doi: 10.1164/ajrccm.161.1.9809100. [DOI] [PubMed] [Google Scholar]
- 86.Crimi E, Spanevello A, Neri M, Ind PW, Rossi GA, Brusasco V. Dissociation between airway inflammation and airway hyperresponsiveness in allergic asthma. Am J Respir Crit Care Med. 1998;157:4–9. doi: 10.1164/ajrccm.157.1.9703002. [DOI] [PubMed] [Google Scholar]
- 87.Rosi E, Ronchi MC, Grazzini M, Duranti R, Scano G. Sputum analysis, bronchial hyperresponsiveness, and airway function in asthma: results of a factor analysis. J Allergy Clin Immunol. 1999;103:232–237. doi: 10.1016/s0091-6749(99)70496-3. [DOI] [PubMed] [Google Scholar]
- 88.Green RH, et al. Asthma exacerbations and sputum eosinophil counts: a randomised controlled trial. Lancet. 2002;360:1715–1721. doi: 10.1016/S0140-6736(02)11679-5. [DOI] [PubMed] [Google Scholar]
- 89.Chlumsky J, Striz I, Terl M, Vondracek J. Strategy aimed at reduction of sputum eosinophils decreases exacerbation rate in patients with asthma. J Int Med Res. 2006;34:129–139. doi: 10.1177/147323000603400202. [DOI] [PubMed] [Google Scholar]
- 90.Jayaram L, et al. Determining asthma treatment by monitoring sputum cell counts: effect on exacerbations. Eur Respir J. 2006;27:483–494. doi: 10.1183/09031936.06.00137704. [DOI] [PubMed] [Google Scholar]
- 91.Kharitonov SA, Gonio F, Kelly C, Meah S, Barnes PJ. Reproducibility of exhaled nitric oxide measurements in healthy and asthmatic adults and children. Eur Respir J. 2003;21:433–438. doi: 10.1183/09031936.03.00066903a. [DOI] [PubMed] [Google Scholar]
- 92.Kharitonov SA, Yates D, Robbins RA, Logan-Sinclair R, Shinebourne EA, Barnes PJ. Increased nitric oxide in exhaled air of asthmatic patients. Lancet. 1994;343:133–135. doi: 10.1016/s0140-6736(94)90931-8. [DOI] [PubMed] [Google Scholar]
- 93.Alving K, Weitzberg E, Lundberg JM. Increased amount of nitric oxide in exhaled air of asthmatics. Eur Respir J. 1993;6:1368–1370. [PubMed] [Google Scholar]
- 94.Jatakanon A, Lim S, Kharitonov SA, Chung KF, Barnes PJ. Correlation between exhaled nitric oxide, sputum eosinophils, and methacholine responsiveness in patients with mild asthma. Thorax. 1998;53:91–95. doi: 10.1136/thx.53.2.91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Payne DN, Adcock IM, Wilson NM, Oates T, Scallan M, Bush A. Relationship between exhaled nitric oxide and mucosal eosinophilic inflammation in children with difficult asthma, after treatment with oral prednisolone. Am J Respir Crit Care Med. 2001;164:1376–1381. doi: 10.1164/ajrccm.164.8.2101145. [DOI] [PubMed] [Google Scholar]
- 96.Berry MA, Shaw DE, Green RH, Brightling CE, Wardlaw AJ, Pavord ID. The use of exhaled nitric oxide concentration to identify eosinophilic airway inflammation: an observational study in adults with asthma. Clin Exp Allergy. 2005;35:1175–1179. doi: 10.1111/j.1365-2222.2005.02314.x. [DOI] [PubMed] [Google Scholar]
- 97.Shaw DE, Berry MA, Thomas M, Green RH, Brightling CE, Wardlaw AJ, Pavord ID. The use of exhaled nitric oxide to guide asthma management: a randomized controlled trial. Am J Respir Crit Care Med. 2007;176:231–237. doi: 10.1164/rccm.200610-1427OC. [DOI] [PubMed] [Google Scholar]
- 98.Smith AD, Cowan JO, Brassett KP, Herbison GP, Taylor DR. Use of exhaled nitric oxide measurements to guide treatment in chronic asthma. N Engl J Med. 2005;352:2163–2173. doi: 10.1056/NEJMoa043596. [DOI] [PubMed] [Google Scholar]
- 99.Pijnenburg MW, Bakker EM, Hop WC, De Jongste JC. Titrating steroids on exhaled nitric oxide in children with asthma: a randomized controlled trial. Am J Respir Crit Care Med. 2005;172:831–836. doi: 10.1164/rccm.200503-458OC. [DOI] [PubMed] [Google Scholar]
- 100.Guida G, et al. Determinants of exhaled nitric oxide in chronic rhinosinusitis. Chest. 2010;137:658–664. doi: 10.1378/chest.09-0667. [DOI] [PubMed] [Google Scholar]
- 101.Vermeulen J, et al. External oligonucleotide standards enable cross laboratory comparison and exchange of real-time quantitative PCR data. Nucleic Acids Res. 2009;37:e138. doi: 10.1093/nar/gkp721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Omenn GS, et al. Overview of the HUPO Plasma Proteome Project: results from the pilot phase with 35 collaborating laboratories and multiple analytical groups, generating a core dataset of 3020 proteins and a publicly-available database. Proteomics. 2005;5:3226–3245. doi: 10.1002/pmic.200500358. [DOI] [PubMed] [Google Scholar]
- 103.Anderson NL, et al. The human plasma proteome: a nonredundant list developed by combination of four separate sources. Mol Cell Proteomics. 2004;3:311–326. doi: 10.1074/mcp.M300127-MCP200. [DOI] [PubMed] [Google Scholar]
- 104.Shen-Orr SS, et al. Cell type-specific gene expression differences in complex tissues. Nat Methods. 2010;7:287–289. doi: 10.1038/nmeth.1439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Pham MX, et al. Gene-expression profiling for rejection surveillance after cardiac transplantation. N Engl J Med. 2010;362:1890–1900. doi: 10.1056/NEJMoa0912965. [DOI] [PubMed] [Google Scholar]
- 106.Assassi S, et al. Systemic sclerosis and lupus: points in an interferon-mediated continuum. Arthritis Rheum. 2010;62:589–598. doi: 10.1002/art.27224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Berry MP, et al. An interferon-inducible neutrophil-driven blood transcriptional signature in human tuberculosis. Nature. 2010;466:973–977. doi: 10.1038/nature09247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Chaussabel D, et al. A modular analysis framework for blood genomics studies: application to systemic lupus erythematosus. Immunity. 2008;29:150–164. doi: 10.1016/j.immuni.2008.05.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Pascual V, Chaussabel D, Banchereau J. A genomic approach to human autoimmune diseases. Annu Rev Immunol. 2010;28:535–571. doi: 10.1146/annurev-immunol-030409-101221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Barnes PJ. The cytokine network in asthma and chronic obstructive pulmonary disease. J Clin Invest. 2008;118:3546–3556. doi: 10.1172/JCI36130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Hansel NN, et al. Oligonucleotide-microarray analysis of peripheral-blood lymphocytes in severe asthma. J Lab Clin Med. 2005;145:263–274. doi: 10.1016/j.lab.2005.02.010. [DOI] [PubMed] [Google Scholar]
- 112.Hakonarson H, et al. Profiling of genes expressed in peripheral blood mononuclear cells predicts glucocorticoid sensitivity in asthma patients. Proc Natl Acad Sci USA. 2005;102:14789–14794. doi: 10.1073/pnas.0409904102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Golub TR, et al. Molecular classification of cancer: class discovery and class prediction by gene expression monitoring. Science. 1999;286:531–537. doi: 10.1126/science.286.5439.531. [DOI] [PubMed] [Google Scholar]
- 114.Buyse M, et al. Validation and clinical utility of a 70-gene prognostic signature for women with node-negative breast cancer. J Natl Cancer Inst. 2006;98:1183–1192. doi: 10.1093/jnci/djj329. [DOI] [PubMed] [Google Scholar]
- 115.Deng MC, et al. Noninvasive discrimination of rejection in cardiac allograft recipients using gene expression profiling. Am J Transplant. 2006;6:150–160. doi: 10.1111/j.1600-6143.2005.01175.x. [DOI] [PubMed] [Google Scholar]
- 116.Dumur CI, et al. Interlaboratory performance of a microarray-based gene expression test to determine tissue of origin in poorly differentiated and undifferentiated cancers. J Mol Diagn. 2008;10:67–77. doi: 10.2353/jmoldx.2008.070099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Finotto S, et al. Development of spontaneous airway changes consistent with human asthma in mice lacking T-bet. Science. 2002;295:336–338. doi: 10.1126/science.1065544. [DOI] [PubMed] [Google Scholar]
- 118.Cui J, et al. TH1-mediated airway hyperresponsiveness independent of neutrophilic inflammation. J Allergy Clin Immunol. 2005;115:309–315. doi: 10.1016/j.jaci.2004.10.046. [DOI] [PubMed] [Google Scholar]
- 119.Hayashi N, Yoshimoto T, Izuhara K, Matsui K, Tanaka T, Nakanishi K. T helper 1 cells stimulated with ovalbumin and IL-18 induce airway hyperresponsiveness and lung fibrosis by IFN-gamma and IL-13 production. Proc Natl Acad Sci USA. 2007;104:14765–14770. doi: 10.1073/pnas.0706378104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Yang M, Kumar RK, Foster PS. Pathogenesis of steroid-resistant airway hyperresponsiveness: interaction between IFN-gamma and TLR4/MyD88 pathways. J Immunol. 2009;182:5107–5115. doi: 10.4049/jimmunol.0803468. [DOI] [PubMed] [Google Scholar]
- 121.Park H, et al. A distinct lineage of CD4 T cells regulates tissue inflammation by producing interleukin 17. Nat Immunol. 2005;6:1133–1141. doi: 10.1038/ni1261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Harrington LE, et al. Interleukin 17-producing CD4+ effector T cells develop via a lineage distinct from the T helper type 1 and 2 lineages. Nat Immunol. 2005;6:1123–1132. doi: 10.1038/ni1254. [DOI] [PubMed] [Google Scholar]
- 123.Miossec P, Korn T, Kuchroo VK. Interleukin-17 and type 17 helper T cells. N Engl J Med. 2009;361:888–898. doi: 10.1056/NEJMra0707449. [DOI] [PubMed] [Google Scholar]
- 124.Molet S, et al. IL-17 is increased in asthmatic airways and induces human bronchial fibroblasts to produce cytokines. J Allergy Clin Immunol. 2001;108:430–438. doi: 10.1067/mai.2001.117929. [DOI] [PubMed] [Google Scholar]
- 125.Bullens DM, Truyen E, Coteur L, Dilissen E, Hellings PW, Dupont LJ, Ceuppens JL. IL-17 mRNA in sputum of asthmatic patients: linking T cell driven inflammation and granulocytic influx? Respir Res. 2006;7:135. doi: 10.1186/1465-9921-7-135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Barczyk A, Pierzchala W, Sozanska E. Interleukin-17 in sputum correlates with airway hyperresponsiveness to methacholine. Respir Med. 2003;97:726–733. doi: 10.1053/rmed.2003.1507. [DOI] [PubMed] [Google Scholar]
- 127.Sun YC, Zhou QT, Yao WZ. Sputum interleukin-17 is increased and associated with airway neutrophilia in patients with severe asthma. Chin Med J (Engl) 2005;118:953–956. [PubMed] [Google Scholar]
- 128.Al-Ramli W, et al. T(H)17-associated cytokines (IL-17A and IL-17F) in severe asthma. J Allergy Clin Immunol. 2009;123:1185–1187. doi: 10.1016/j.jaci.2009.02.024. [DOI] [PubMed] [Google Scholar]
- 129.Pene J, et al. Chronically inflamed human tissues are infiltrated by highly differentiated Th17 lymphocytes. J Immunol. 2008;180:7423–7430. doi: 10.4049/jimmunol.180.11.7423. [DOI] [PubMed] [Google Scholar]
- 130.Doe C, et al. Expression of the T helper 17-associated cytokines IL-17A and IL-17F in asthma and COPD. Chest. 2010;138:1140–1147. doi: 10.1378/chest.09-3058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Agache I, Ciobanu C, Agache C, Anghel M. Increased serum IL-17 is an independent risk factor for severe asthma. Respir Med. 2010;104:1131–1137. doi: 10.1016/j.rmed.2010.02.018. [DOI] [PubMed] [Google Scholar]
- 132.Zhao Y, Yang J, Gao YD, Guo W. Th17 immunity in patients with allergic asthma. Int Arch Allergy Immunol. 2009;151:297–307. doi: 10.1159/000250438. [DOI] [PubMed] [Google Scholar]
- 133.Chen Y, Thai P, Zhao YH, Ho YS, DeSouza MM, Wu R. Stimulation of airway mucin gene expression by interleukin (IL)-17 through IL-6 paracrine/autocrine loop. J Biol Chem. 2003;278:17036–17043. doi: 10.1074/jbc.M210429200. [DOI] [PubMed] [Google Scholar]
- 134.Fujisawa T, Velichko S, Thai P, Hung LY, Huang F, Wu R. Regulation of airway MUC5AC expression by IL-1beta and IL-17A; the NF-kappaB paradigm. J Immunol. 2009;183:6236–6243. doi: 10.4049/jimmunol.0900614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.McKinley L, Alcorn JF, Peterson A, Dupont RB, Kapadia S, Logar A, Henry A, Irvin CG, Piganelli JD, Ray A, et al. TH17 cells mediate steroid-resistant airway inflammation and airway hyperresponsiveness in mice. J Immunol. 2008;181:4089–4097. doi: 10.4049/jimmunol.181.6.4089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Yang XO, Chang SH, Park H, Nurieva R, Shah B, Acero L, Wang YH, Schluns KS, Broaddus RR, Zhu Z, et al. Regulation of inflammatory responses by IL-17F. J Exp Med. 2008;205:1063–1075. doi: 10.1084/jem.20071978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Wang YH, et al. A novel subset of CD4(+) T(H)2 memory/effector cells that produce inflammatory IL-17 cytokine and promote the exacerbation of chronic allergic asthma. J Exp Med. 2010;207:2479–2491. doi: 10.1084/jem.20101376. [DOI] [PMC free article] [PubMed] [Google Scholar]