

Abstract
Pyrazinamidase of Mycobacterium tuberculosis catalyzes the conversion of pyrazinamide to the active molecule pyrazinoic acid. Reduction of pyrazinamidase activity results in a level of pyrazinamide resistance. Previous studies have suggested that pyrazinamidase has a metal-binding site and that a divalent metal cofactor is required for activity. To determine the effect of divalent metals on the pyrazinamidase, the recombinant wild-type pyrazinamidase corresponding to the H37Rv pyrazinamide-susceptible reference strain was expressed in Escherichia coli with and without a carboxy terminal. His-tagged pyrazinamidase was inactivated by metal depletion and reactivated by titration with divalent metals. Although Co2+, Mn2+, and Zn2+ restored pyrazinamidase activity, only Co2+ enhanced the enzymatic activity to levels higher than the wild-type pyrazinamidase. Cu2+, Fe2+, Fe3+, and Mg2+ did not restore the activity under the conditions tested. Various recombinant mutated pyrazinamidases with appropriate folding but different enzymatic activities showed a differential pattern of recovered activity. X-ray fluorescence and atomic absorbance spectroscopy showed that recombinant wild-type pyrazinamidase expressed in E. coli most likely contained Zn. In conclusion, this study suggests that M. tuberculosis pyrazinamidase is a metalloenzyme that is able to coordinate several ions, but in vivo, it is more likely to coordinate Zn2+. However, in vitro, the metal-depleted enzyme could be reactivated by several divalent metals with higher efficiency than Zn.
Introduction
Pyrazinamide (PZA) is one of the most important drugs against the latent stage of tuberculosis.1–3 PZA is a prodrug that is converted by bacterial pyrazinamidase (PZAse) to its active form, pyrazinoic acid. Because of its role in triggering the bactericidal activity of PZA, defective PZAse is often found in Mycobacterium tuberculosis PZA-resistant strains with mutations in the pncA gene that encodes PZAse.1,4–10
Important molecular characteristics of M. tuberculosis PZAse have been elucidated from crystallized homologous hydrolases such as N-Carbamoylsarcosine amidohydrolase (CHSase) from Arthrobacter (26% identical),11 PZAse from Pyrococcus horikoshii (37% identical),12 and PZAse from Acinetobacter baumanii (37% identical)13 as well as the recently crystallized structure of M. tuberculosis pyrazinamidase14 and a theoretical analysis of a modeled structure.15 According to these studies, the catalytic cavity comprises an active site (D8, A134, and C138) and a metal-binding site (D49, H51, and H71). The work by Du and others12 showed that Zn increased PZAse activity of P. horikoshii and that it was coordinated in crystals of this enzyme. Similarly, a PZAse of A. baumanii that cocrystallized with nicotinamide revealed a coordination with Zn and Fe in a 1:1 ratio,13 whereas the M. tuberculosis PZAse was successfully crystallized with Fe.14 Several studies with recombinant M. tuberculosis PZAses showed that mutations affecting the active or metal-binding site impaired the PZAse activity, whereas mutations occurring far from these sites had different levels of decreased enzyme activity.9,10,12,16–18 A recent study found that M. tuberculosis PZAse coordinated Mn2+ and Fe2+, and these ions restored activity after metal depletion.18
In this study, we tested the presence of metals in PZAse and measured the effect of divalent ions on enzyme activity and the kinetic parameters of (1) the metal-depleted recombinant M. tuberculosis wild-type PZAse expressed with and without a His-tag end and (2) various recombinant mutated PZAses with moderate and non-detectable enzyme activity.
Materials And Methods
Protein sample.
The wild-type PZAse corresponding to the H37Rv PZA-susceptible reference strain and nine mutated PZAses from PZA-resistant clinical isolates of M. tuberculosis were analyzed. These recombinant enzymes were cloned with a carboxy-terminal His-tag to be purified. These enzymes were previously studied and showed a single amino acid substitution with a variable PZAse activity.10,17 Mutations were of five types: mutation of the metal-binding residues (D49N and H51R), mutation of residues close to the active site (T135P), mutation of residues close to the metal-binding site (T67P and G78C), mutation of residue close to both active and metal-binding sites (D12G, D12A), and mutation of residues distant from both the active and metal-binding sites (G24D, F94L, and L116P) (Figure 1). To analyze the potential interference of the His-tag on the chelation or metal reactivation, the wild-type H37Rv PZAse without a His-tag was produced recombinantly and similarly evaluated.
Figure 1.
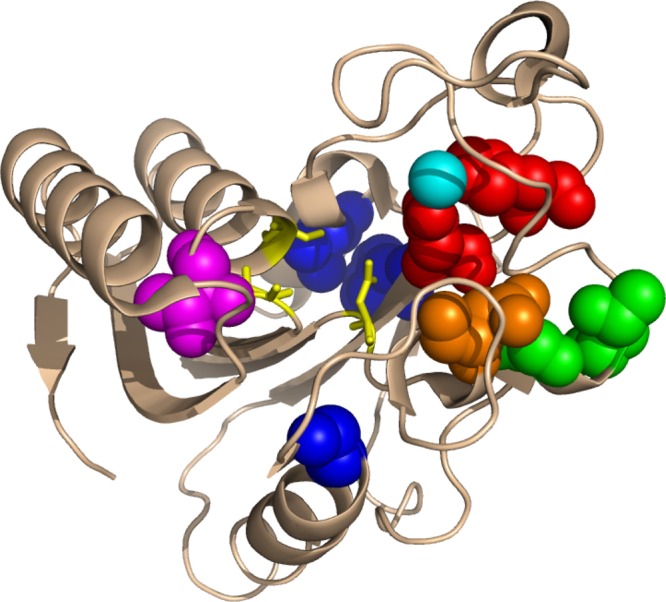
PZAse single amino acids mutations (Protein Data Bank 3GBC). The positions of the 10 PZAse single amino acid mutation studied in the present work are represented as spheres. PZAse active site residues and the bound metal ion are highlighted in yellow and cyan, respectively. Residues involved in the metal binding are depicted in red. Residues close to the active site, the metal-binding site, or both sites are in magenta, green, and orange, respectively. Residues distant from both the active and metal-binding sites are depicted in blue.
Protein purification.
The PZAses containing a carboxy-terminal His-tag were expressed in Escherichia coli and purified by affinity chromatography with Ni-nitrilotriacetic acid (NTA) column (GE Healthcare, Uppsala, Sweden) as described elsewhere.10,17
The wild-type PZAse without a His-tag was purified in a two-step ion exchange chromatography using DEAE Sephadex (Sigma, St. Louis, MO). Proteins were loaded in 75 mM Tris · HCl (pH 7.5). In the first step, the protein was eluted with 0.5 M NaCl. The semipure fraction was passed through a similar column and eluted with an NaCl gradient (0–1 M). The enzyme purity was determined by sodium dodecyl sulfate–polyacrylamide gel electrophoresis (SDS-PAGE).
Enzymatic activity.
PZAse activity was measured by monitoring the appearance of pyrazinoic acid during the incubation time, which was described elsewhere,10,12 with the following modifications. The reaction mix included 3 μM PZAse, 50 mM phosphate buffer (pH 6.5), and 20 mM PZA. The reaction was incubated at 37°C for 3 minutes. These conditions resulted in less than 10% substrate conversion. A monotonic substrate consumption was observed until 15 minutes, where almost all substrate was consumed. To stop the reaction, 10 μL 20% ferrous ammonium sulphate and 890 μL 0.1 M glycine-HCl buffer (pH 3.4) were added. The precipitates were removed by centrifugation (12,000 × g for 10 minutes). The pyrazinoic acid in the supernatant was detected by measuring the absorbance at 450 nm.
Metal depletion.
The wild-type PZAse with His-tag and the one without the His-tag were inactivated by metal depletion. To inactivate PZAse, 34 μM purified enzyme was incubated with 80 mM ethylenediamine tetra acetic acid (EDTA) in 100 mM phosphate buffer (pH 6.5) for 6 hours at 25°C. Free and metal-bound EDTA was removed by ultrafiltration and completed by a washing with four volumes of 100 mM phosphate buffer (pH 6.5). Cellulose membranes with 10-kDa pore size were used in an Ultracel Amicon Ultrafiltration system (Millipore, Billerica, MA). Protein concentration was determined by the Bradford assay.19 As a control, a PZAse fraction received similar treatment by replacing EDTA with water. This control fraction was called non-chelated PZAse.
Effect of metal ions on enzyme activity.
The metal-depleted wild-type PZAse (10 μM) with and without the His-tag was reactivated by titration with different metals at concentrations of 0.001, 0.01, 0.1, 1, and 2 mM. Cu2+ was tested as a chloride salt, Zn2+ was tested as a nitrated salt, and Co2+, Fe2+, Fe3+, Mg2+, and Mn2+ were tested as sulphate salts. In addition, we tested Fe2+ and Fe3+ as chloride salts.
The metal-depleted wild-type PZAse was incubated with each metal ion at 37°C for 30 minutes followed by measurement of PZAse activity. The optimal metal concentration that reactivated the PZAse was selected as the minimum concentration that rendered the highest PZAse activity.
Each of the mutated metal-depleted PZAses was incubated with each metal at the respective optimal concentration. Incubation was performed at 37°C for 30 minutes followed by measurement of PZAse activity. To adjust for the residual activity after metal depletion, the percentage of recovered activity was estimated: (activity of reactivated metal-depleted PZAse – activity of metal-depleted PZAse)/(activity of non-chelated PZAse – activity of metal-depleted PZAse) × 100. All assays were performed in triplicate.
To determine the effect of metals on the activity of non-chelated wild-type PZAse, the enzymatic activity was measured after adding the metal ions to non-chelated PZAse in the same conditions used to reactivate the metal-depleted enzyme. The effect of the metals on the activity of wild-type non-chelated PZAse was expressed as a relative activity calculated as (activity of non-chelated PZAse with added metal)/(activity of non-chelated PZAse) × 100. The assays were run in triplicate.
Enzyme kinetic.
The kinetic parameters of the metal-depleted wild-type PZAse were measured after reactivation with the metal ions that most notably reactivated the enzyme activity (Co2+, Mn2+ and Zn2+) at their respective optimal concentrations using the procedure previously described.10 To measure the substrate affinity of PZAse, the Michaelis constant (Km) was estimated. To determine the number of times that each enzyme substrate complex converts substrate to its product per unit time, the catalytic constant (kcat) was estimated. To determine how efficiently PZA is converted into pyrazinoic acid (POA), the enzymatic efficiency20 (kcat/Km) was estimated using the protocol described in ref. 18.
Briefly, PZA was used from 0 to 4 mM and incubated with 1 μM PZAse in 50 mM sodium phosphate (pH 6.5). To prevent the hydrolysis of more than 10% of the initial PZA, 1 minute was used. The incubation period was increased to 2 hours for mutants with very low activity; 10 μL 20% FeNH4(SO4)2 were added and followed immediately by the addition of 890 μL 100 mM glycine-HCl (pH 3.4) to stop the reaction. Optical density was measured at 450 nm in a 96-well plate using 200 μL reaction.
The amount of POA produced was estimated by interpolation in a standard curve of known concentrations.
Data were fitted by a linear regression to the Lineweaver Burk plot (1/V versus 1/[S]21) as well as by a non-linear regression to the Michaelis–Menten equation.22 The assays were performed in triplicate. To compare the kinetic parameters between the reactivated PZAse and the non-chelated enzyme, a non-parametric statistical test was performed.
Circular dichroism analysis.
Circular dichroism (CD) spectra were recorded on a J-810 spectropolarimeter (Jasco, Japan) using a 0.2-mm path length quartz cuvette. Proteins and buffer solutions spectra were averaged from four scans in the range of 260–190 nm with a 20 nm · min−1 scan rate, setting a bandwidth of 1 nm. Measurements were performed with 156 μg/mL PZAses in 20 mM Na-phosphate (pH 7.0) and 1 mM β-mercaptoethanol.Buffer spectra were subtracted from the proteins spectra. “The results are expressed in molar ellipticity (degrees·cm2·decimol-1)
⌈Θ⌉=Θ/⌈10⋅c⋅d⌉
where Θ is the observed ellipticity in degrees, c is the molar concentration of the sample (mole/L), d is the optical path length in cm.”
Analysis of the CD data for secondary structure determination was carried out using the CDSSTR secondary structure fitting program (basis set 7)23,24 that is available online at the Dichroweb server (http://www.cryst.bbk.ac.uk/cdweb).25
Metal ion contents.
To detect the metals coordinated by the wild-type His-tag PZAse, elemental analysis in lyophilized enzyme and buffer control was done using X-ray fluorescence spectroscopy; 1.5 mL 0.5 mM recombinant wild-type PZAse in 20 mM Tris · HCl (pH 7.9) were frozen to –70°C for 24 hours and lyophilized. To adjust for the presence of contaminant metals, three control solutions were tested: (1) the buffer used to resuspend the PZAse (20 mM Tris [pH 7.9]), (2) the elution buffer used during protein purification (20 mM phosphate, 0.5 M NaCl, 60 mM imidazole [pH 7.4]), and (3) the soluble fraction from crude extract of non-transformed E. coli BL21 (DE3) pLys that was eluted with 60 mM imidazole in the His-Trap Ni-NTA column and concentrated in 20 mM Tris (pH 7.9). This strain was the same E. coli strain used to produce the recombinant PZAse.
A Cd109 source (25 mCi) was used to activate the Ag Kα radiation used to irradiate the sample. The characteristic X-ray emitted by the excited sample was detected by a Si (Li) detector (ORTEC) and interpreted with the Analysis of X-Ray Spectra by Iterative Least Squares Fitting (AXIL) software.26 An absolute method based on elemental sensitivity for semiquantitative analysis was used.27 This method can detect metals from K to Mo and from Cs to U. The elements between Mo and Cs cannot be detected, because the radioactive source excites Ag, resulting in a masking background signal close to Ag.
Atomic absorbance.
The wild-type PZAse was tested for Zn concentration using atomic absorbance spectroscopy by the standard addition method28,29; 10 mL PZAse 0.05 mg/mL (2.48 μM) were prepared in 20 mM Tris · HCl (pH 7.9). Three standards of 10 mL were prepared containing 3, 4.5, and 6 μg Zn. The standard solutions were obtained from metallic Zn dissolved in HCl. The readings were done in the spectrometer Precisely Analyst 200 (Perkin Elmer, Waltham, MA) with a Zn lamp (wavelength = 213.9 nm, slit width = 1.8 nm). This assay was repeated three times.
Results
Optimal ion concentrations to reactivate the metal-depleted wild-type PZAse.
After 6 hours of incubation with 80 mM EDTA, the wild-type His-tagged PZAse activity was decreased by 28-fold compared with the non-chelated PZAse. The optimal concentrations of the metals to reactivate the wild-type metal-depleted PZAse were 1 mM for Mg2+, Co2+, and Mn2+ and 1.5 mM for Zn2+. These concentrations corresponded to the metal to enzyme molar ratios of 333:1 and 500:1. Fe2+, Fe3+, and Cu2+ did not notably recover the wild-type His-tagged PZAse activity. Because higher concentrations resulted in precipitation, these metals were tested at 0.1 mM. A similar behavior was observed in the wild-type PZAse without the His-tag end.
Effect of metal ions on the enzyme activity.
The non-chelated wild-type His-tagged PZAse showed an enzymatic activity of 15.4 (95% confidence interval [CI] = 10.8, 20.3) μmol POA · mg−1 PZAse · min−1 (Table 1). This activity did not change in presence of Co2+, Fe2+, Mg2+, and Mn2+, whereas Fe3+, Cu2+, and Zn2+ inhibited the PZAse activity (Figure 2A ). Cu2+ reduced the relative activity to less than 10%, Zn2+ reduced the relative activity to 50%, and Fe2+ reduced the relative activity to 75%. Sulphate and chloride salts of Fe2+ and Fe3+ showed a similar behavior, with the exception that Fe+2 chloride salt in contrast to the sulfate salt resulted in certain precipitation.
Table 1.
Effect of metal ions on the enzymatic activity of His-tagged metal-depleted mutated PZAses
| Mutated PZAse | Enzyme activity of non-chelated enzyme* | Co2+ | Cu2+ | Fe2+ | Fe3+ | Mg2+ | Mn2+ | Zn2+ |
|---|---|---|---|---|---|---|---|---|
| WT | 15.4 | 20.4 (136) | 0.53 (0) | 0.53 (0) | 0 (0) | 1.1 (3.3) | 13.7 (93.8) | 5 (37) |
| D12A | 11.2 | 31.9 (311.7) | 4.9 (34.4) | 2.1 (0) | 2 (0) | 2.3 (0.7) | 16.4 (150.3) | 4.2 (21.5) |
| D12G | 16.3 | 40.7 (261.3) | 3.4 (13.8) | 1.5 (0) | 1.4 (0.6) | 1.5 (1.1) | 16.9 (103.4) | 3.8 (14) |
| G24D | 11.8 | 8.2 (67) | 1.5 (4) | 1.2 (0) | 1.2 (0) | 1.1 (0) | 3.3 (20.1) | 1.3 (0.4) |
| Y34D | 10.4 | 2.6 (0) | 3.4 (6.7) | 3 (11) | 4.4 (14.8) | 4.5 (16.9) | 4.1 (0) | 3.8 (0) |
| G78C | 7.62 | 38.1 (809.1) | 2.1 (7.7) | 2.3 (0) | 2.4 (0) | 2.5 (0) | 28.8 (445.9) | 1.4 (0) |
| F94L | 25.2 | 6 (5.6) | 6.2 (7.6) | 5.8 (0.9) | 5.1 (0) | 5 (0.7) | 5.1 (1.6) | 4.7 (0.4) |
| T135P | 1.5 | 1.1 (65) | 0.2 (1.6) | 0.17 (0) | 0.2 (0) | 0.2 (0) | 0.5 (22.5) | 0.3 (3.3) |
| D136G | 11.5 | 5.7 (47.9) | 0.6 (0) | 0.32 (0) | 0.52 (0) | 0.53 (0) | 5.3 (45.6) | 1.3 (5.8) |
| D49N | ND | ND | ND | ND | ND | ND | ND | ND |
| H51R | ND | ND | ND | ND | ND | ND | ND | ND |
Enzymatic activity (mmol POA · mg−1 PZAse · min−1).
Enzymatic activity of the His-tagged mutated PZAses after reactivation with different metal ions. The percentage of recovered activity is in parenthesis. To adjust for the residual activity after metal depletion, the percentage of recovered activity was estimated as (activity of reactivated metal-depleted PZAse − activity of metal-depleted PZAse)/(activity of non-chelated PZAse – activity of metal-depleted PZAse) × 100. The data are presented as the medians of triplicate tests. ND = non-detectable activity; WT = wild-type H37Rv strain.
Figure 2.

Effect of metals on the relative activity of wild-type pyrazinamidase. (A) Non-chelated wild-type pyrazinamidase. (B) Metal-depleted wild-type His-tagged pyrazinamidase. (C) Metal-depleted wild-type pyrazinamidase without His-tag.
The metal-depleted wild-type His-tagged PZAse was highly reactivated, with Co2+ increasing about 40-fold of the metal-depleted PZAse activity (136% of recovered activity), Mn2+ increasing about 28-fold (94% of recovered activity), and Zn2+ increasing about 12-fold (37% of recovered activity). Fe2+ and Fe3+ (in sulphate and chloride salts), Cu2+, and Mg2+ recovered less than twofold of the metal-depleted wild-type His-tagged PZAse activity (Figure 2B).
No effect was observed in the presence of the His-tag in the PZAse. The non-chelated wild-type PZAse without His-tag showed a similar activity of 21.6 (95% CI = 15.9, 27.5) μmol POA · mg−1 PZAse · min−1) and a similar reactivation pattern to the corresponding wild-type His-tagged PZAse (Figure 2C).
In the mutated PZAses, a differential reactivation pattern was observed depending on the metal ion and the site of mutation (Table 1). Fe2+ and Fe3+ were tested as sulphate salts. Three of eight mutants with low to high activity (D12A, D12G, and G78C) showed a similar metal reactivation pattern as the wild-type PZAse in terms of the percentage of recovered activity, with the difference that, in these mutated enzymes, Cu2+ produced a slight reactivation (6–34%). G24D was not reactivated by any metal except for Co2+ and Mn2+. These two metals recovered activity in all the mutated enzymes except D49N, H51R, and Y34D. In contrast, the Y34D PZAse was slightly reactivated (6–16%) by Cu2+, Fe2+, Fe3+, and Mg2+. Two mutated enzymes that had undetectable activity bearing mutations in the metal-binding site (D49N and H51R) were not reactivated by any of the tested metals.
Effect of metal ions on the kinetic parameters of metal-depleted wild-type PZAse.
The kinetic parameters of the wild-type His-tagged PZAse varied differentially when reactivated with Co2+, Mn2+, and Zn2+ (Table 2). Km of the non-chelated wild-type His-tagged PZAse was 2.27 mM, whereas its kcat was 681.16 min−1. Reactivation with Co2+ was associated to a higher kcat compared with Zn2+ or Mn2+, and it was slightly higher than kcat of the non-chelated enzyme. Metal-depleted PZAse reactivated with Co2+ or Mn2+ did not show a significantly different enzymatic efficiency (kcat/Km) than non-chelated PZAse (P = 0.070 and 0.056, respectively). However, enzymatic efficiency of the metal-depleted PZAse reactivated with Zn2+ was significantly lower than the non-chelated enzyme (P < 0.001). The reactivation with Mn2+ and Zn2+ was associated with a slightly lower Km than the non-chelated PZAse, whereas the reactivation with Co2+ resulted in a similar Km.
Table 2.
Effect of metal reactivation in the kinetic parameters of metal-depleted wild-type H37Rv PZAse
| Metals | Km (mM) | kcat (min−1) | Efficiency (mM−1 · min−1) | ||||
|---|---|---|---|---|---|---|---|
| Mean | 95% CI | Mean | 95% CI | Mean | 95% CI | P value* | |
| Non-chelated | 2.27 | ±0.20 | 681.16 | ±52.14 | 300.46 | ±6.22 | |
| Co2+ | 2.32 | ±0.24 | 772.89 | ±68.43 | 333.11 | ±8.81 | 0.070 |
| Mn2+ | 1.35 | ±0.07 | 448.69 | ±18.21 | 330.71 | ±6.09 | 0.056 |
| Zn2+ | 1.09 | ±0.06 | 168.28 | ±6.59 | 154.85 | ±3.93 | 0.001 |
P value (mean comparison test) corresponding to the comparison of the enzymatic efficiency between the reactivated metal-depleted PZAse and the non-chelated enzyme.
Enzymatic efficiency is defined as kcat/Km. The data are presented as the mean (CI) of triplicate tests. Non-chelated = wild-type PZAse non–metal-depleted.
CD.
An overlay of the CD spectra of the wild-type and the D12G, D12A, G24D, D49N, H51R, T76P, G78C, F94L, L116P, and T135P His-tagged PZAse mutants is shown in Figure 3. Visual inspection of the spectra indicates that the proteins have a mixed α-helix/β-sheet secondary structure content. Analysis of the CD data for secondary structure determination using the CDSSTR secondary structure fitting program23,24 showed that the point mutations in the analyzed PZAses do not virtually affect the secondary structure composition of each of these enzymes. Therefore, the lack of PZAse activity in D49N and H51R is not likely to be because of a problem of folding. Indeed, the D12G, D12A, and G24D mutants displayed the same content of α-helices and β-sheets as the wild type (20% and 30%, respectively), whereas the remaining mutants (T76P, F94L, and L116P) exhibited a 5–10% increase of α-helices at the expense of the β-sheets contents.
Figure 3.

CD spectra of the wild-type and mutant PZAses. The CD spectra of the wild-type and mutated PZAses show a similar pattern, confirming that a misfolding is not likely to be present.
X-ray fluorescence and atomic absorbance spectroscopy of wild-type PZAse.
The X-ray fluorescence radiation spectra of the excited wild-type His-tagged PZAse and the three controls (the buffer used to resuspend the PZAse, the elution buffer used during protein purification, and the soluble fraction from crude extract of non-transformed E. coli) showed the presence of several metal ions (Figure 4). Fe, Ni, Cu, and Pb were found in the wild-type PZAse, but they were also found in the same level in the controls. Zn was the only ion present in the PZAse fraction in a higher level than the controls. According to the X-ray fluorescence and the atomic absorbance spectroscopy, Zn was found at a concentration of 0.3 moles/mol PZAse (Zn:PZAse = 0.3:1).
Figure 4.

X-ray spectra of wild-type His-tagged pyrazinamidase. Blue, recombinant PZAse; green, 20 mM Tris·HCl (pH 7.9); red, 20 mM phosphate buffer, 0.5 M NaCl, and 60 mM imidazole (pH 7.4); pink, eluted and concentrated proteins of soluble proteins of E. coli BL21(DE3)pLys without plasmid. The red arrow is pointing to the peak corresponding to zinc in recombinant wild-type PZAse.
Discussion
The critical role of PZAse in determining M. tuberculosis resistance to PZA1,10,30,31 makes it important to understand the role of metal ions in the enzymatic activity and the consequences of the amino acid substitutions.
This study and the study in ref. 18 confirm that M. tuberculosis PZAse is a metalloenzyme that can be inactivated by removal of divalent ions, and it can be reactivated differentially by different metal ions. The level of enzymatic reactivation depends on the type of metal ion coordinated as well as the type of PZAse mutation. According to this study, Zn2+ seemed to be the most abundant metal to activate M. tuberculosis PZAse in vivo under our experimental conditions, although Co2+ and Mn2+ showed a higher in vitro reactivation capacity.
Co2+ was the metal that recovered the highest activity of the wild-type metal-depleted PZAse in vitro followed by Mn2+ and to a lesser extent, Zn2+. PZAse activity was recovered by Co2+ in almost all the mutants, except those mutations that were affected on the metal-binding site (D49N and H51R).
M. tuberculosis recombinant His-tagged PZAses expressed in E. coli have been used in several functional/kinetic studies.11,12,18,32 This study shows that the differential pattern of reactivation of the wild-type metal-depleted PZAse does not depend on the presence of the His-tag end. Moreover, the enzymatic activities of the His-tagged and the His-tag–free PZAses were found to be similar. This evidence confirms that the His-tag is not involved in the metal reactivation and does not affect the enzymatic activity of the wild-type M. tuberculosis PZAse. This result is shown for the first time in the M. tuberculosis PZAse, although several previous studies only analyzed His-tagged recombinant enzymes. Therefore, any effect of the His-tag in the metal-binding enzyme activity and metal reactivation is not significant; thus, any artifact is unlikely to be associated with it.
The enzymatic activity of the non-chelated recombinant PZAse was inhibited by Fe3+, Cu2+, and Zn2+. In the case of Fe3+ and Cu2+, incubation with these metals resulted in precipitation. The Fe2+ chloride salt in contrast to its sulfate salt resulted in certain precipitation being evidence of a level of instability associated to chlorine ions. This finding could explain the lack of reactivation in our hands exerted by the Fe+2 chloride salt previously detected.18 In the case of Zn2+, although it did not cause precipitation, it is possible that its addition reduced the activity of the recombinant purified PZAse because of competition with and substitution of a more efficient cofactor.
The kinetic parameters of the reactivated wild-type metal-depleted PZAse varied differentially depending on the coordinated metal in vitro. Although small, a change was not only observed in kcat but also in Km, indicating that the affinity for the substrate depends on the type of metal coordinated. The reactivation with Co2+ was associated with the highest kcat and the highest enzymatic efficiency, surpassing the wild type.
These results suggest that there is no unique metal cofactor that activates PZAse, and several metals can play this role. Previous studies have described metalloproteins that can use different metals as cofactors with equivalent efficiency.18,33–36 In previous reports, two independent groups have conducted very similar analyses on M. tuberculosis PZAse and the effects of divalent metals. The results produced by these two laboratories are largely divergent from those results reported here. The work by Zhang and others18 found that PZAse activity was efficiently recovered by the addition of Mn and Fe from apoenzyme but not the addition of Zn. In a more recent study, Seiner and others35 performed electron paramagnetic resonance (EPR) spectroscopy on M. tuberculosis PZAse and observed the presence of Mn2+ bound to the enzyme. In addition, the metal seemed to be tightly bound, and the addition of EDTA resulted in a decrease in activity. These different results suggest that experimental conditions, including expression of the recombinant protein, are critical for the metal binding to PZAse.
Several studies have shown that Co2+ is a universal enhancer of metalloenzymes37 and may be having the same effect in PZAse. The fact that PZAse is reactivated with Co2+, Mn2+, and Zn2+ could be attributed to their characteristic of being transition metals able to coordinate six bonds in their divalent form with similar covalent radii (1.26, 1.39, and 1.31 Å, respectively).38 Interestingly, the majority of enzymes that coordinate Zn2+ in vivo are highly activated by Co2+ in vitro.39 Although Co2+ strongly stimulated the PZAse activity, it is unlikely to be the naturally most used metal being coordinated by the PZAse in vivo. Co2+ is found in very low concentrations in living organisms,40 although cobalt is essential to all animals, including humans, because it is a key constituent of cobalamin, also known as vitamin B12.41
Mn2+ also showed an important reactivation of PZAse activity, which is similar to what has been proven in other hydrolases.42 Our results alternatively suggest that, under in vitro conditions, PZAse also prefers Mn, which is, apart from Co, the only metal capable of restoring nearly 100% of the enzymatic activity of the wild-type PZAse and several mutants; also, it showed no inhibitory activity when added to the holoenzyme in kinetic assays. By contrast, Zn restored only 37% of the activity from metal-depleted PZAse, failed to restore the activity of several mutants, and behaved as inhibitor in the kinetic measurements with the holoenzyme.
The X-ray fluorescence assay, after controlling with the three selected blanks, showed that Zn2+ was found in the recombinant M. tuberculosis PZAse and was not found in any of the controls. However, Fe2+, Ni2+, Cu2+, and Pb2+ were found in the recombinant PZAse, but these metals were also found in the controls in similar concentrations. Therefore, under the conditions used in this study, Zn2+ was the most abundant metal coordinating PZAse. The presence of Zn2+ in the wild-type PZAse was confirmed by the atomic absorption spectroscopy. The low Zn2+:PZAse ratio detected in this study (0.3:1) has also been observed in other amidohydrolases.20 This low ratio is possibly because of a low protein concentration used or what we believe is more likely, the high production of protein in a heterologous recombinant expression system with a limited source of intracellular Zn2+, which might be affected similarly as other mutants.
This differential affinity could be explained by electrostatic interactions with the ion or subtle alterations of the active site cavity structure in response to the presence of the coordinated ion. The latter is observed in the P. horikoshii pyrazinamidase. The atomic coordinates of the homologue enzyme crystallized with and without Zn (Protein Data Bank 1IM5 and 1ILW, respectively) showed differences in the orientation of some of the amino acid side chains close to the active site (data not shown).12 It could also be possible that a significant amount of metal-depleted PZAse began to unfold, because it was unable to easily rebind metals other than Co2+ and Mn2+. This mechanism of metal selectivity should be further investigated.
Our study was conducted independently of another study with a similar experimental approach.18 In contrast to our results, that study found that Fe2+ and Mn2+ were able to reactivate the enzymatic activity of the metal-depleted wild-type PZAse, whereas Zn2+ did not have any detectable effect. Although both studies used similar methodologies (i.e., His-tagged recombinant proteins expressed in E. coli BL21 and purification on Ni-NTA columns), these discrepancies could result from slight differences in the experimental conditions, like a difference of metal ions present in the buffers.
In our previous study,10 the enzymatic activity of a His-tagged PZAse was estimated as 38.40 (interquartile range = 18.36) μmol POA · mg−1 PZAse · min−1, and its kinetic parameters were Km = 1.24 ± 0.44 mM and kcat = 1005.41 ± 199.91 min−1. In a recent study,18 the enzymatic activity of a recombinant His-tagged H37Rv PZAse was estimated as 89.6 U · mg−1 protein (1 U = amount of PZAse required to produce 1 μmol POA per minute), which is about sixfold the value that we estimated. In another study,12 the 37% identical P. horikoshii His-tagged PZAse showed an enzymatic activity of 17.3 μmol POA · mg−1 protein · min−1, which is similar to what we found in M. tuberculosis H37Rv. Another recent study working with a His-tagged H37Rv PZAse35 estimated a Km value of 300 μM (0.3 mM) and kcat value of 3.8 s−1 (228 min−1) for pyrazinamide. Although not identical, all estimates are similar or at least are in the same order of magnitude as the ones measured in this study. Discrepancies maybe caused by different facts such as the level of purification of the enzymes to the different enzymatic incubation periods. We verified that the linearity of PZA hydrolysis is satisfied within the first 3 minutes of reaction when using 3 μM pure enzyme; however, this result may change if the level of purity is different.
The reactivation pattern of the wild-type H37Rv PZAse showed that the percentage of recovered activity was highest with Co (136%) followed by Mn (93.8%) and Zn (37%). If recovered activities below 17% are neglected because of biological non-significance, most of mutated PZAses showed a similar pattern as the wild type (D12G, G24D, G78C, T135P, and D136G). The high recovered activity in mutation close to the metal-binding site G78C and in a lesser extent, mutations D12A and D12G requires additional structural analysis to understand this observation. We can speculate that this mutation enhances the activity by an increased affinity to the metals or alteration of the efficiency of the catalysis at some point. PZAse mutants Y34D and F94L showed a low reactivation (in the range considered as biologically non-significant) with all the metals tested, although they were enzymatically active before the metal depletion. Without a structural analysis, we can speculate that these mutations, although distant from the metal-binding site, could distantly affect the metal coordination site structure and reduce the affinity to metals under in vitro but not in vivo conditions. PZAse mutants D49N and H51R did not show a detectable activity either before or after the metal depletion. D49 and H51 constitute, together with H71, the metal-binding site. The change of the sign of charge (negative to positive) prevents the coordination with a metal cation. Cu slightly reactivated the enzymatic activity of D12A (34.4%). Additional structural analysis is required to understand this observation.
The CD measurements did not reveal significant differences in terms of percentages of α-helices, β-sheets, turns, and random coils content between the wild-type PZAse and the investigated mutants. It is worth note that the disruption of the metal-binding site as a result of the D49N and H51R mutations, although provoking the inactivation of the enzyme, leaves the secondary structure content of the protein virtually unaltered. It is tempting to hypothesize that metal binding may stabilize the tertiary structure of the enzyme without affecting significantly the overall secondary structure composition or the local structural determinants, thus explaining why the enzymatic activity can be readily restored after metal depletion by chelating agents.
The study by Du and others12 working in an homology model based on the 37% identical P. horikoshii PZAse suggested that M. tuberculosis PZAse has a metal-binding site with an octahedral ligand structure coordinated by two histidines (H51 and H71), one aspartic acid (D49), and three water molecules.12 This coordination is characteristic of a Zn2+-binding site in metalloproteins.39,43 Recently, the PZAse crystal structure of M. tuberculosis was determined (Protein Data Bank 3GBC). The crystal structure reveals a coordination of Fe2+ and Mn2+ in the metal-binding site; however, it was noted that Mn2+ and Zn2+ were in alternate conformation. It is worthy of note that both Fe2+ and Mn2+ have been included in the crystallographic refinement with the very same positional coordinates but an occupancy factor of 0.5. Another recent study reported that the crystal structures of the PZAse from A. baumanii cocrystallized with nicotinamide and pyrazinoic acid. The analysis of PZAse from A. baumanii revealed the presence of Fe and Zn in a 1:1 ratio, with trace of Mn.13
Furthermore, the Metalloprotein Data Base, which compiles the metalloproteins reported in the Protein Data Bank,44 shows that, among the 219 proteins reported with a metal coordination site comprised by two histidines and one aspartic acid, 101 proteins bind Zn2+, 56 proteins bind Fe2+, 24 proteins bind Ni2+, 18 proteins bind Cd2+, 14 proteins bind Mn2+, 5 proteins bind Cu2+, and 1 protein binds Ca2+. These proteins are classified within 10 classes of proteins, including esterases, transferases, immunoglobulins, oxidoreductases, lyases, dehydrogenases, toxins, blood clotting proteins, apoptosis proteins, and hydrolases. Similarly, among the 332 proteins reported with a metal coordination site comprised by three histidines and one aspartic acid (i.e., identical to the one of the PZAse), 192 proteins bind Zn2+, 74 proteins bind Fe2+, 48 proteins bind Mn2+, 14 proteins bind Cu2+, 2 proteins bind Cd2+, and 2 proteins bind Co2+. Therefore, Zn2+ is very likely to be coordinated by a His-His-Asp site such as the one present in the M. tuberculosis PZAse.
The concentration of proteins and metals in a typical cell are about 10−6 M in a 1:1 ratio.45 Our results showed almost 300-fold more metal than enzyme was required to reactivate the chelated PZAse in vitro under the experimental conditions that we tested. This fact was also observed in other studies with hydrolases46,47 in contrast to other metalloenzymes that require lower concentrations of ions for their reactivation.46 It is possible that, in vivo, the activation of a metal-depleted wild-type PZAse may not be spontaneously favorable. The fact that enzymatic activity was reconstituted by titration with metal ions at unphysiologically high concentrations suggests that it is possible that assisted mechanisms to favor metal coordination by the M. tuberculosis PZAse are present in vivo. Thus, it is possible that metal ions coordination with PZAse may be mediated by a metallochaperone.48
Multidrug-resistant tuberculosis (MDR-TB) strains associated to the mutated PZAses D49N and H51R are highly prevalent in Peru and are probably linked to epidemic clones that were shown in a previous study.6,9,16 Consequently, the reactivation of these strains would have meant a therapeutic benefit, and its testing was of public health interest. Mutations directly affecting the metal-binding site (D49N and H51R) completely impaired the PZAse activity. The lack of activity on these mutated enzymes confirms that D49 and H51 are critical residues in the PZA hydrolysis. Unfortunately, none of the tested metal ions were able to reactivate the enzymatic activity in these mutated PZAses.
Conclusion
We found that the M. tuberculosis PZAse is a metalloenzyme. The level of enzyme reactivation depends on the type of metal ion coordinated as well as the type of PZAse mutation. Under the experimental conditions tested in this study, the evidence shows that, in vivo, Zn2+ is likely to be the most abundant metal coordinated by the His-tagged wild-type PZAse. However, in vitro, PZAse could bind and could be reactivated also by Co2+ and Mn2+. Therefore, several metals could play the role of cofactors depending on their availability in the local environment. An experimental clarification will pose an interesting challenge for future structural and functional studies.
ACKNOWLEDGMENTS
We are grateful to Jon Lopez and César Lopez for their input in the exploratory assays at the beginning of this study. We are also grateful to Christian Jacinto for his advice with the atomic absorbance and Jesus Castagnetto, Claudia Machicado, and Daniel Guerra for their general advice and comments during the conducting of this study.
Footnotes
Financial support: This research was funded by National Institute of Allergy and Infectious Diseases, National Institutes of Health United States Award 1R01TW008669-01, Third World Academy of Science (TWAS) Grant 08-070RG/BIO/LA-UNESCO FR:3240204464, Programme for Research and Training in Tropical Diseases (TDR)-World Health Organization Reference 2009/53662-0, and Fundación Instituto Hipólito Unanue. P.S. and M.Z. were supported by Tropical Medicine Research Centers (TMRC) New Tools to Understand and Control Endemic Parasites Grant 1 P01 AI51976 and Global Research Training Grant 3 D43 TW006581.
Authors' addresses: Patricia Sheen, Patricia Ferrer, Gina Christiansen, Paola Moreno-Román, Andrés H. Gutiérrez, Jun Sotelo, Wilfredo Evangelista, Patricia Fuentes, Daniel Rueda, Myra Flores, and Mirko Zimic, Laboratorios de Investigación y Desarrollo, Facultad de Ciencias, Universidad Peruana Cayetano Heredia, Lima, Perú, E-mails: patricia.sheen@upch.pe, ferrerp@gmail.com, ginachristiansen1@yahoo.com, paola.moreno@upch.pe, ahgn5@hotmail.com, jihusan@gmail.com, willyef@gmail.com, pfuentesbonilla@gmail.com, ldrueda.raez@gmail.com, meff_uni@yahoo.com, and mzimic@jhsph.edu. Robert H. Gilman, Department of International Health, Johns Hopkins Bloomberg School of Public Health, Baltimore, MD, E-mail: rgilman@jhsph.edu. Paula Olivera and José Solis, Instituto Peruano de Energía Nuclear, Lima, Perú, E-mails: polivera@IPEN.GOB.PE and jsolis@uni.edu.pe. Alessandro Pesaresi and Doriano Lamba, Istituto di Cristallografia, Consiglio Nazionale delle Ricerche, Area Science Park—Basovizza, Trieste, Italy, E-mails: alessandro.pesaresi@ts.ic.cnr.it and doriano.lamba@ts.ic.cnr.it.
References
- 1.Konno K, Feldmann FM, McDermott W. Pyrazinamide susceptibility and amidase activity of tubercle bacilli. Am Rev Respir Dis. 1967;95:461–469. doi: 10.1164/arrd.1967.95.3.461. [DOI] [PubMed] [Google Scholar]
- 2.Mitchison DA. The action of antituberculosis drugs in short-course chemotherapy. Tubercle. 1985;66:219–225. doi: 10.1016/0041-3879(85)90040-6. [DOI] [PubMed] [Google Scholar]
- 3.Steele MA, Des Prez RM. The role of pyrazinamide in tuberculosis chemotherapy. Chest. 1988;94:845–850. doi: 10.1378/chest.94.4.845. [DOI] [PubMed] [Google Scholar]
- 4.Butler WR, Kilburn JO. Susceptibility of Mycobacterium tuberculosis to pyrazinamide and its relationship to pyrazinamidase activity. Antimicrob Agents Chemother. 1983;24:600–601. doi: 10.1128/aac.24.4.600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Miller MA, Thibert L, Desjardins F, Siddiqi SH, Dascal A. Testing of susceptibility of Mycobacterium tuberculosis to pyrazinamide: comparison of Bactec method with pyrazinamidase assay. J Clin Microbiol. 1995;33:2468–2470. doi: 10.1128/jcm.33.9.2468-2470.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Scorpio A, Lindholm-Levy P, Heifets L, Gilman R, Siddiqi S, Cynamon M, Zhang Y. Characterization of pncA mutations in pyrazinamide-resistant Mycobacterium tuberculosis. Antimicrob Agents Chemother. 1997;41:540–543. doi: 10.1128/aac.41.3.540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Hirano K, Takahashi M, Kazumi Y, Fukasawa Y, Abe C. Mutation in pncA is a major mechanism of pyrazinamide resistance in Mycobacterium tuberculosis. Tuber Lung Dis. 1997;78:117–122. doi: 10.1016/s0962-8479(98)80004-x. [DOI] [PubMed] [Google Scholar]
- 8.Cheng SJ, Thibert L, Sanchez T, Heifets L, Zhang Y. pncA mutations as a major mechanism of pyrazinamide resistance in Mycobacterium tuberculosis: spread of a monoresistant strain in Quebec, Canada. Antimicrob Agents Chemother. 2000;44:528–532. doi: 10.1128/aac.44.3.528-532.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Zimic M, Sheen P, Quiliano M, Gutierrez A, Gilman RH. Peruvian and globally reported amino acid substitutions on the Mycobacterium tuberculosis pyrazinamidase suggest a conserved pattern of mutations associated to pyrazinamide resistance. Infect Genet Evol. 2010;10:346–349. doi: 10.1016/j.meegid.2009.11.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Sheen P, Ferrer P, Gilman RH, Lopez-Llano J, Fuentes P, Valencia E, Zimic MJ. Effect of pyrazinamidase activity on pyrazinamide resistance in Mycobacterium tuberculosis. Tuberculosis (Edinb) 2009;89:109–113. doi: 10.1016/j.tube.2009.01.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Lemaitre N, Callebaut I, Frenois F, Jarlier V, Sougakoff W. Study of the structure-activity relationships for the pyrazinamidase (PncA) from Mycobacterium tuberculosis. Biochem J. 2001;353:453–458. doi: 10.1042/0264-6021:3530453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Du X, Wang W, Kim R, Yakota H, Nguyen H, Kim SH. Crystal structure and mechanism of catalysis of a pyrazinamidase from Pyrococcus horikoshii. Biochemistry. 2001;40:14166–14172. doi: 10.1021/bi0115479. [DOI] [PubMed] [Google Scholar]
- 13.Fyfe PK, Rao VA, Zemla A, Cameron S, Hunter WN. Specificity and mechanism of Acinetobacter baumanii nicotinamidase: implications for activation of the front-line tuberculosis drug pyrazinamide. Angew Chem Int Ed Engl. 2009;48:9176–9179. doi: 10.1002/anie.200903407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Petrella S, Gelus-Ziental N, Maudry A, Laurans C, Boudjelloul R, Sougakoff W. Crystal structure of the pyrazinamidase of Mycobacterium tuberculosis: insights into natural and acquired resistance to pyrazinamide. PLoS One. 2011;6:e15785. doi: 10.1371/journal.pone.0015785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Quiliano M, Gutierrez AH, Gilman RH, Lopez C, Evangelista W, Sotelo J, Sheen P, Zimic M. Structure-activity relationship in mutated pyrazinamidases from Mycobacterium tuberculosis. Bioinformation. 2011;6:335–339. doi: 10.6026/97320630006335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Lemaitre N, Sougakoff W, Truffot-Pernot C, Jarlier V. Characterization of new mutations in pyrazinamide-resistant strains of Mycobacterium tuberculosis and identification of conserved regions important for the catalytic activity of the pyrazinamidase PncA. Antimicrob Agents Chemother. 1999;43:1761–1763. doi: 10.1128/aac.43.7.1761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Sheen P. Molecular Diagnosis of Pyrazinamide Resistance and Molecular Understanding of the Pyrazinamidase Functionality in Mycobacterium tuberculosis. Baltimore, MD: Johns Hopkins University; 2008. [Google Scholar]
- 18.Zhang H, Deng JY, Bi LJ, Zhou YF, Zhang ZP, Zhang CG, Zhang Y, Zhang XE. Characterization of Mycobacterium tuberculosis nicotinamidase/pyrazinamidase. FEBS J. 2008;275:753–762. doi: 10.1111/j.1742-4658.2007.06241.x. [DOI] [PubMed] [Google Scholar]
- 19.Simonian M, Smith J. Short Protocols in Molecular Biology. New York, NY: John Wiley & Sons; 1999. [Google Scholar]
- 20.Shapir N, Pedersen C, Gil O, Strong L, Seffernick J, Sadowsky MJ, Wackett LP. TrzN from Arthrobacter aurescens TC1 is a zinc amidohydrolase. J Bacteriol. 2006;188:5859–5864. doi: 10.1128/JB.00517-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Fersht A. Structure and Mechanism in Protein Science: A Guide to Enzyme Catalysis and Protein Folding. New York, NY: W.H. Freeman and Company; 1999. The basic equations of enzyme kinetics; pp. 103–131. [Google Scholar]
- 22.Michaelis L, Menten M. Kinetics of invertase action. Biochem Z. 1913;49:333–369. [Google Scholar]
- 23.Sreerama N, Venyaminov SY, Woody RW. Estimation of protein secondary structure from circular dichroism spectra: inclusion of denatured proteins with native proteins in the analysis. Anal Biochem. 2000;287:243–251. doi: 10.1006/abio.2000.4879. [DOI] [PubMed] [Google Scholar]
- 24.Sreerama N, Woody RW. Estimation of protein secondary structure from circular dichroism spectra: comparison of CONTIN, SELCON, and CDSSTR methods with an expanded reference set. Anal Biochem. 2000;287:252–260. doi: 10.1006/abio.2000.4880. [DOI] [PubMed] [Google Scholar]
- 25.Whitmore L, Wallace BA. DICHROWEB, an online server for protein secondary structure analyses from circular dichroism spectroscopic data. Nucleic Acids Res. 2004;32:W668–W673. doi: 10.1093/nar/gkh371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Van Espen P, Nullens H, Adams F. A computer analysis of X-ray fluorescence spectra. Nuclear Instruments and Methods. 1977;142:243–250. [Google Scholar]
- 27.Van Espen P, Nullens H, Adams F. Calibration of tube excited energy-dispersive X-ray spectrometers with thin film standards and with fundamental constants. X-ray Spectrometry. 1981;10:64–68. [Google Scholar]
- 28.Bader M. A systematic approach to standard addition methods in instrumental analysis. J Chem Educ. 1980;57:703–706. [Google Scholar]
- 29.Lewin AC, Doughty PA, Flegg L, Moore GR, Spiro S. The ferric uptake regulator of Pseudomonas aeruginosa has no essential cysteine residues and does not contain a structural zinc ion. Microbiology. 2002;148:2449–2456. doi: 10.1099/00221287-148-8-2449. [DOI] [PubMed] [Google Scholar]
- 30.Trivedi SS, Desai SG. Pyrazinamidase activity of Mycobacterium tuberculosis a test of sensitivity to pyrazinamide. Tubercle. 1987;68:221–224. doi: 10.1016/0041-3879(87)90058-4. [DOI] [PubMed] [Google Scholar]
- 31.Wade MM, Zhang Y. Mechanisms of drug resistance in Mycobacterium tuberculosis. Front Biosci. 2004;9:975–994. doi: 10.2741/1289. [DOI] [PubMed] [Google Scholar]
- 32.Boshoff HI, Mizrahi V. Purification, gene cloning, targeted knockout, overexpression, and biochemical characterization of the major pyrazinamidase from Mycobacterium smegmatis. J Bacteriol. 1998;180:5809–5814. doi: 10.1128/jb.180.22.5809-5814.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Henderson JN, Zhang J, Evans BW, Redding K. Disassembly and degradation of photosystem I in an in vitro system are multievent, metal-dependent processes. J Biol Chem. 2003;278:39978–39986. doi: 10.1074/jbc.M304299200. [DOI] [PubMed] [Google Scholar]
- 34.Pozo-Dengra J, Martinez-Gomez AI, Martinez-Rodriguez S, Clemente-Jimenez JM, Rodriguez-Vico F, Las Heras-Vazquez FJ. Evaluation of substrate promiscuity of an L-carbamoyl amino acid amidohydrolase from Geobacillus stearothermophilus CECT43. Biotechnol Prog. 2010;26:954–959. doi: 10.1002/btpr.410. [DOI] [PubMed] [Google Scholar]
- 35.Seiner DR, Hegde SS, Blanchard JS. Kinetics and inhibition of nicotinamidase from Mycobacterium tuberculosis. Biochemistry. 2010;49:9613–9619. doi: 10.1021/bi1011157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Zhang FL, Fu HW, Casey PJ, Bishop WR. Substitution of cadmium for zinc in farnesyl: protein transferase alters its substrate specificity. Biochemistry. 1996;35:8166–8171. doi: 10.1021/bi960574+. [DOI] [PubMed] [Google Scholar]
- 37.Kgayama T, Ohe T. Purification and properties of an aromatic amidase from Pseudomonas sp. GDI 211. Agric Biol Chem. 1990;53:2565–2571. [Google Scholar]
- 38.Barbalace K. Periodic Table of Elements 1995 – 2012. 1995. www.EnvironmentalChemistry.com Available at: Accessed July 2010.
- 39.Vallet M, Faus J, Garcia E, Moratal J. Introducción a la Química Bioinorgánica: Editorial Síntesis. 2003. pp. 332–334. [Google Scholar]
- 40.Wackett L, Orme-Johnson W, Walsh C. Transition metal enzymes in bacterial metabolism. In: Beveridge T, Doyle R, editors. Metal Ions and Bacteria. New York, NY: John Wiley & Sons; 1989. pp. 165–206. [Google Scholar]
- 41.Smith I, Carlson B. Trace Metals in the Environment. Volume 6. Ann Arbor, MI: Ann Arbor Science Publising Inc; 1981. [Google Scholar]
- 42.Kanyo ZF, Scolnick LR, Ash DE, Christianson DW. Structure of a unique binuclear manganese cluster in arginase. Nature. 1996;383:554–557. doi: 10.1038/383554a0. [DOI] [PubMed] [Google Scholar]
- 43.Seibert CM, Raushel FM. Structural and catalytic diversity within the amidohydrolase superfamily. Biochemistry. 2005;44:6383–6391. doi: 10.1021/bi047326v. [DOI] [PubMed] [Google Scholar]
- 44.Castagnetto JM, Hennessy SW, Roberts VA, Getzoff ED, Tainer JA, Pique ME. MDB: the Metalloprotein Database and Browser at The Scripps Research Institute. Nucleic Acids Res. 2002;30:379–382. doi: 10.1093/nar/30.1.379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Sols A, Marco R. Concentrations of metabolites and binding sites. Implications in metabolic regulation. Curr Top Cell Regul. 1970;2:227–273. [Google Scholar]
- 46.Guerra DG, Vertommen D, Fothergill-Gilmore LA, Opperdoes FR, Michels PA. Characterization of the cofactor-independent phosphoglycerate mutase from Leishmania mexicana mexicana. Histidines that coordinate the two metal ions in the active site show different susceptibilities to irreversible chemical modification. Eur J Biochem. 2004;271:1798–1810. doi: 10.1111/j.1432-1033.2004.04097.x. [DOI] [PubMed] [Google Scholar]
- 47.de Carvalho LP, Blanchard JS. Kinetic analysis of the effects of monovalent cations and divalent metals on the activity of Mycobacterium tuberculosis alpha-isopropylmalate synthase. Arch Biochem Biophys. 2006;451:141–148. doi: 10.1016/j.abb.2006.03.030. [DOI] [PubMed] [Google Scholar]
- 48.Patzer SI, Hantke K. The zinc-responsive regulator Zur and its control of the znu gene cluster encoding the ZnuABC zinc uptake system in Escherichia coli. J Biol Chem. 2000;275:24321–24332. doi: 10.1074/jbc.M001775200. [DOI] [PubMed] [Google Scholar]