

Abstract
Young (4 month) and aged (15–18 months) mice were given intranasal saline or γ--herpesvirus-68 infection. After 21 days, aged, but not young mice, showed significant increases in collagen content and fibrosis. There were no differences in viral clearance or inflammatory cells (including fibrocytes) between infected aged and young mice. Enzyme-linked immunosorbent assays showed increased transforming growth factor-β in whole lung homogenates of infected aged mice compared with young mice. When fibroblasts from aged and young mice were infected in vitro, aged, but not young, fibroblasts upregulate alpha-smooth muscle actin and collagen I protein. Infection with virus in vivo also demonstrates increased alpha-smooth muscle actin and collagen I protein and collagen I, collagen III, and fibronectin messenger RNA in aged fibroblasts. Furthermore, evaluation revealed that aged fibroblasts at baseline have increased transforming growth factor-β receptor 1 and 2 levels compared with young fibroblasts and are resistant to apoptosis. Increased responsiveness to transforming growth factor-β was verified by increased collagen III and fibronectin messenger RNA after treatment in vitro with transforming growth factor-β.
Keywords: Lung, Fibrosis, Herpesvirus, Aging
Background
Idiopathic pulmonary fibrosis (IPF) is a chronic parenchymal disorder of the lungs associated with a progressive loss of lung function (1). It is likely secondary to dysregulated repair in response to injury leading to destruction of normal lung architecture through fibroblast accumulation, myofibroblast differentiation, and the overproduction of collagen and other extracellular matrix proteins. IPF is known to be a disease of the elderly population, with incidence, prevalence, and mortality of the disease increasing with age. The highest prevalence occurs in patients older than the age of 75 years and age is an independent predictor of having usual interstitial pneumonia (the pathological correlate of IPF) on surgical lung biopsy in comparison with other interstitial pneumonias (2,3). Patients can present with stability over several years, with a steady decline in lung function, or with a sudden accelerated decline in clinical status termed an acute exacerbation (1,4). Unfortunately, mortality remains high, with median survival times of 3 years from the time of diagnosis, and there are currently no effective therapies (5).
Although studies in the past decade have advanced our understanding of the pathogenesis and clinical presentation of the disease, the exact cause of initial injury is still unknown. However, there is accumulating data that an occult viral infection may play a role, leading to chronic lung inflammation and an abnormal healing response in certain individuals. In particular, Epstein–Barr virus (EBV), a γ-herpesvirus, has been found to have the strongest association with pulmonary fibrosis (reviewed in (6)). The virus is known to infect most people, and the initial lytic phase is followed with a latent phase and the potential for reactivation (7). Human studies have found elevated levels of EBV-specific IgA and IgG antibodies in serum, elevated EBV viral capsid–specific IgA in bronchoalveolar lavage fluid, and increased presence of EBV DNA in the lungs of IPF patients when compared with controls (8–10). In addition, evidence of actively replicating EBV was found in type 2 alveolar epithelial cells (AECs) of IPF patients and infection of type 2 alveolar cells with EBV in vitro led to increased expression of transforming growth factor (TGF)-β, a potent fibrotic mediator (11,12). Another study evaluating four human herpesviruses, cytomegalovirus, EBV, human herpesvirus-7, and human herpesvirus-8, found evidence of infection in 96% of IPF patients versus 36% of controls (13). It should be noted, however, that not all studies have found an association between γ-herpesviral infection and IPF (14,15).
Investigators have used mouse models to gain further insight into the profibrotic effects of γ-herpesviruses on the lung. γHV-68, a murine γ-herpesvirus genetically similar to EBV and human herpesvirus-8, infects the respiratory tract with an initial lytic phase followed by latency in lung epithelial cells (16–18) and B cells (19). Infection of Balb/c mice with γHV-68 before intraperitoneal bleomycin caused increased lung collagen content, increased fibrosis, and higher inflammation scores (20). Preceding latent infection also promotes fibrosis to a subthreshold dose of fluorescein isothiocyanate or bleomycin (21), suggesting that virus may alter the lung environment and predispose individuals to develop fibrosis to subsequent stimuli. However, in these studies carried out in young wild-type mice, infection with γHV-68 alone did not cause fibrosis (21). In contrast, infection with γHV-68 can cause fibrosis in Th2-biased (interferon [IFN]-γ-receptor–deficient [IFNγ-R −/−]) mice. γHV-68 infection in these mice causes multiorgan fibrosis in the lungs, liver, spleen, and lymph nodes (22). Similar to patients with IPF, the viral-induced fibrosis in these mice was found to have areas of increased collagen deposition and severe peripheral fibrosis characterized by fibroblastic foci and TGF-β elevations (23). Later studies demonstrated that viral reactivation was key to fibrosis development in this Th2-biased model (24).
It is plausible that environmental differences in an aging lung are responsible for the increased susceptibility of elderly patients to lung fibrosis, possibly potentiated by viral infections. Senescence-accelerated prone mice have increased lung fibrosis, serum fibrocyte concentrations, and TGF-β production in response to bleomycin when compared with senescence-resistant mice (25). There are also aged mouse studies showing a Th2 shift in cytokine profile with increased T-cell production of interleukin (IL)-4 (26–28). In addition, increased endoplasmic reticular stress, decreased protein chaperones causing misfolded proteins, and abnormal handling of these proteins in the elderly patients drive apoptotic responses in AECs; an observation that has been implicated to promote pulmonary fibrosis (29–31). Finally, the aging lung is susceptible to changes in the immune system, predominantly in T cells important for control of viral replication (32–34).
We hypothesized that the environment of an aged lung may be sufficiently altered for γ-herpesvirus infection alone to cause fibrosis in wild-type mice. We compared collagen content and histology of aged (15–18 months) and young (4 month) mice 21 days after intranasal infection with γHV-68 and found increased fibrosis and collagen in aged mice in response to virus. This was not due to an inability to control viral replication in aged mice but rather due to an increased TGF-β production and a profibrotic fibroblast phenotype in aged mice. To our knowledge, this is the first demonstration of a circumstance (aging) in which viral infection alone is sufficient to cause fibrosis in wild-type mice.
MATERIALS AND METHODS
Mice
Male young C57Bl/6 mice (4 months old) and aged C57Bl/6 (15–18 months old) mice were purchased from the National Institute of Aging (Bethesda, MD). All procedures were approved by the University of Michigan Committee on the Use and Care of Animals.
γHV-68 Infection
After mice were anesthetized with a low dose of ketamine and xylazine, 5 × 104 to 1 × 105 plaque-forming units (pfu) of γHV-68 (American Type Culture Collection, Manassas, VA) diluted in 20 μL sterilized phosphate-buffered saline were administered intranasally.
Hydroxyproline Assay
Total lung collagen was quantified via the hydroxyproline assay as described previously (35). Briefly, lung lobes were removed from perfused mice, homogenized in phosphate-buffered saline, and hydrolyzed by the addition of hydrochloric acid (Sigma, St Louis, MO). Samples were then baked at 110°C for 12 hours and aliquots were assayed by adding chloramine T solution, followed by development with Erlich’s reagent at 65°C as previously described. Absorbance was measured at 550 nm, and the amount of hydroxyproline was determined against a standard curve generated using known concentrations of hydroxyproline standard (Sigma).
Histology
Hematoxylin and eosin staining was performed as described previously (36). Briefly, animals were euthanized and perfused via the heart with normal saline. Lungs were inflated with 10% neutral buffered formalin, removed, and fixed overnight in formalin before being dehydrated in 70% ethanol. Lungs were processed using standard procedures and embedded in paraffin. Sections (3–5 μm) were cut, mounted on slides, and stained with hematoxylin and eosin.
Virus Plaque Assay
Lytic virus present in two lung lobes of infected mice was measured by plaque assay as described previously by plating dilutions of lung homogenates onto 3T12 cells and measuring pfu on day 5 (17).
Semiquantitative Real-Time Reverse Transcription–Polymerase Chain Reaction
Semiquantitative real-time reverse transcription–polymerase chain reaction (RT-PCR) was performed with ABI Prism 7000 thermocycler (Applied Biosystems, Foster City, CA) using a previously described protocol (37). Primers and probes, listed in Table 1, were made using Primer Express software (Applied Biosystems). All samples were run in triplicate.
Table 1.
Primers and Probes Used for Analysis
| Gene | Oligo | Sequences |
| M3 | Forward primer | AGTGGGCTCACGCTGTACTTGT |
| Reverse primer | TGTCTCTGCTCACTCCATTTGG | |
| Probe | CATGGGCAAGTGTTCATCTTAGCC | |
| gB | Forward primer | CGCTCATTACGGCCCAAA |
| Reverse primer | ACCACGCCCTGGACAACTC | |
| Probe | TTGCCTATGACAAGCTGACCACCA | |
| β-Actin | Forward primer | CCGTGAAAAGATGACCCAGATC |
| Reverse primer | CACAGCCTGGATGGCTACGT | |
| Probe | TTTGAGACCTTCAACACCCCCAGCCA | |
| Collagen I | Forward primer | TGACTGGAAGAGCGGAGAGTACT |
| Reverse primer | GGTCTGACCTGTCTCCATGTTG | |
| Probe | CTGCAACCTGGACGCCATCAAGG | |
| Collagen III | Forward primer | GGATCTGTCCTTTGCGATGAC |
| Reverse primer | GCTGTGGGCATATTGCACAA | |
| Probe | TGCCCCAACCCAGAGATCCCATTT | |
| Fibronectin | Forward primer | TCGAGCCCTGAGGATGGA |
| Reverse primer | GTGCAAGGCAACCACACTGA | |
| Probe | CTGCAGGGCCTCAGGCCGG | |
| TGF-βR1 | Forward primer | CTGCCATAACCGCACTGTCA |
| Reverse primer | CTTTTAAGGTGGTGCCCTCTGA | |
| Probe | CCGTGTGCCAAATGAAGAGGATCCA | |
| TGF-βR2 | Forward primer | GGCTTCGAACACCATGGAA |
| Reverse primer | CTAGAGGGCGGTGAACAACAG | |
| Probe | TGGATTGCCAGTGCTAACCCAG |
Analysis of Viral Genome Loads
DNA was prepared from cells isolated from mock-infected or γHV-68-infected mice using the Qiagen DNeasy Blood and Tissue Kit (Valencia, CA), and PCR was performed to detect the gB viral coding sequence as previously described (38). Values were compared with a standard curve consisting of gB plasmid DNA diluted at known copy numbers. Reported values were normalized to 100 ng of input DNA for each reaction and represent the copy number in mock-infected mice (background) subtracted from the virus-infected samples. For gB DNA analysis, the forward primer was 5′GGCCCAAATTCAATTTGCCT; the reverse primer, 5′CCCTGGACAACTCCTCAAGC; and the probe, 5′6-(FAM)-ACAAGCTGACCACCAGCGTCAACAAC-(TAMRA).
Enzyme-Linked Immunosorbent Assay
Cytokines and chemokines were measured in whole lung homogenate supernatants using DuoSet ELISA Development System kits (R&D Systems, Minneapolis, MN) following the manufacturer’s instructions. For analysis of TGF-β1, supernatants were first acidified to allow for measurements of total TGF-β1.
Collagenase Digests
Lungs were enzymatically digested using collagenase and DNase as described previously (36). Briefly, lungs were excised, minced, and enzymatically digested using digestion buffer (RPMI 1640, 5% fetal calf serum, antibiotics, 1 mg/mL collagenase [Roche, Chicago, IL], and 30 μg/mL DNase (Sigma), followed by passage through the bore of a 10-mL syringe. Contaminating erythrocytes were removed from the total cell suspension, which was then dispersed by passing through a 5-mL syringe before being filtered through a Nytex filter (Tetko, Kansas City, MO). An equal volume of 40% Percoll (Sigma–Aldrich) was added, and cells were centrifuged at 3,000 rpm (2095 × g) for 30 minutes without brake. The cell pellets were resuspended in complete medium, and leukocytes were counted on a hemocytometer in the presence of trypan blue. Recovered leukocytes were analyzed by flow cytometry.
Flow Cytometry
All antibodies were purchased from BD Biosciences (San Jose, CA) unless otherwise noted. Leukocytes from collagenase digests were incubated 15 minutes on ice with Fc Block before surface staining with anti-mouse CD45-PerCP-Cy5.5, followed by fixation–permeabilization with BD PharMingen Cytofix/Cytoperm kit according to the manufacturer's instructions. Cells were then stained with the following antibodies: rat anti-mouse CD4-PE, rat anti-mouse CD8-FITC, anti-mouse CD19-APC, mouse anti-mouse NK-1.1-APC, anti-mouse CD25-PE, anti-mouse GR-1-PE, anti-mouse TCR-β chain-FITC, and anti-mouse F4/80-APC (eBioscience, San Diego, CA). Cells were analyzed on a FACSCalibur (BD Biosciences) and FlowJo software (Tree Star Inc., Ashland, OR).
AEC Isolation
Type II AECswere isolated using dispase and DNase digestion of lungs as previously described (39). After the solution was passed successively through 100-, 40-, and 25-μm nylon mesh filters, bone marrow–derived cells were removed via anti-CD32 and anti-CD45 magnetic depletion. Mesenchymal cells were removed by overnight adherence in a Petri dish. The nonadherent cells, after this initial plating, were plated in 12-well plates coated with fibronectin. Resultant purity was approximately 94% as determined previously by intermediate filament staining. Cells were plated at 1 × 106 cells per well and cultured in serum-free media for 48 hours before collection of cell supernatants.
Alveolar Macrophage Isolation
Alveolar macrophages (AMs) were extracted from lungs by bronchoalveolar lavage as previously described (40). Briefly, these cells were collected in lavage fluid consisting of complete medium and 5 mM ethylenediaminetetraacetic acid. These cells were enumerated by counting on a hemocytometer before plating. Cells were allowed to adhere to tissue culture plates for 1 hour and then were washed to remove nonadherent cells, resulting in an approximately 95% AM culture. The cells were then cultured in serum-free media at 3 × 104 cells per well in a 96-well plate for 24 hours before collection of cell supernatants.
Mesenchymal Cell Isolation
Whole lung was minced and cultured for 14 days to enrich for mesenchymal cells in complete media. (See below for the recipe for Dulbecco’s modified eagle medium.). If mice were infected in vivo prior to cell isolation, complete media were supplemented with 10 μM cidofovir (Gilead Sciences Inc., Foster City, CA) to prevent viral reactivation and cell lysis. Uninfected mesenchymal cells were cultured in serum-free media on six-well plates at 5 × 105 cells per well for 48 hours with or without additional treatment (2 ng/mL TGF-β or 0.1 pfu per cell γHV-68) before isolating RNA or protein lysates.
Western Blots
Western blots were performed on fibroblasts isolated from uninfected young and aged mice as previously described (36). Antibodies used to detect collagen I were rabbit polyclonal sera from Rockland (Gilbertsville, PA), mouse monoclonal antibodies used to detect alpha-smooth muscle actin were from Sigma, glyceraldehyde 3-phosphate dehydrogenase was identified with rabbit antibodies from Cell Signaling Technology (Danvers, MA), and β-actin was identified with mouse monoclonal antibodies from Sigma. Secondary reagents were goat anti-rabbit IgG-HRP and goat anti-mouse IgG-HRP from Pierce (Rockford, IL). Blots were scanned and densitometry was performed using the Image J program downloadable from http://imagej.nih.gov/ij/download.html.
Apoptosis Assay
Primary lung fibroblasts isolated from naive mice as above were used for experiments between passage 4 and 6. For assessment of apoptosis, cells were seeded into 35-mm dishes in complete media. When the cells reached 80% confluence, the media were changed to serum-free media. Apoptosis was assessed 40 hours later using the Caspase-3 Colorimetric Assay Kit (Assay Designs, Ann Arbor, MI) according to the manufacturer’s protocol.
Reagents Used
Complete media were Dulbecco’s modified eagle medium (Lonza, Walkersville, MD) with 10% fetal bovine serum (Fisher, Pittsburgh, PA), 1% penicillin–streptomycin (Gibco/Invitrogen, Carlsbad, CA), 1% L-glutamine (Fisher), and 0.1% amphotericin B (Lonza). Serum-free media were Dulbecco’s modified eagle medium with 1% bovine serum albumin (Sigma), 1% penicillin–streptomycin, 1% L-glutamine, and 0.1% amphotericin B.
Statistical Analyses
Statistical significance was measured by analysis of variance (three or more groups) or unpaired Student’s t-test (two groups) using GraphPad Prism 5 software (San Diego, CA); data represent mean ± standard error of the mean; p < .05 was considered significant.
RESULTS
Latent γ-Herpesvirus Infection in Aged Mice Is Associated With Increased Fibrosis After 21 days
Previously, we have shown that γ-herpesvirus achieves latency in mice after 14 days of infection and that γHV-68 infection alone in young mice does not cause fibrosis after 21 days (21). To determine whether γ-herpesvirus infection induced pulmonary fibrosis in aged mice, we infected five young mice and five aged mice with 1 × 105 pfu γHV-68 intranasally. For controls, the same amount of saline was introduced intranasally into three young mice and three aged mice. Lungs were harvested 21 days after infection or saline administration. Hydroxyproline assay was performed on whole lung homogenates to assess for collagen content. There was a statistically significant increase in hydroxyproline content between aged mice infected with virus compared with aged saline controls and young mice infected with virus (Figure 1A). There was no difference between young mice infected with virus and young saline controls. This was confirmed by hematoxylin and eosin staining of lung sections showing increased destruction of normal lung architecture in aged mice infected with virus (Figure 1B). This demonstrated that virus alone could induce fibrosis and increase collagen production in aged, but not in young, mice.
Figure 1.
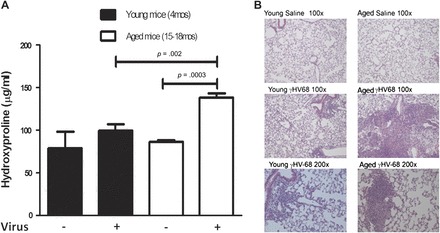
Increased fibrosis in aged mice 21 days following γHV-68 infection. Aged and young mice were intranasally infected with 1 × 105 plaque-forming units of γHV-68 (n = 5 per group) or saline (n = 3 per group). After 21 days, lungs were harvested and hydroxyproline assay was performed on right lungs while left lungs were used for histology. (A) Hydroxyproline assay results showing significantly increased hydroxyproline in aged mice infected with γHV-68 when compared with aged controls given saline (p = .0003) and young mice infected with γHV-68 (p = .002). There also was no difference in hydroxyproline between infected young mice and young controls given saline. (B) Hematoxylin and eosin staining showing increased disruption of lung architecture, inflammation, and fibrosis in aged mice infected with γHV-68 compared with infected young mice and aged and young saline controls. Results are representative of two similar experiments.
Aged Mice Effectively Control Lytic γ HV-68 Infection
Given the possibility of age-associated decline in viral immunity, we next evaluated whether the increased fibrotic response in infected aged mice was due to decreased lytic viral clearance. We performed a viral plaque assay at 7 days following infection with lungs of infected aged and young mice and found significantly more lytic virus present in young infected mice (Figure 2A). This was confirmed by increased levels in young mice of messenger RNA encoding viral gene products, gB and M3, 7 days following infection as evaluated by real-time RT-PCR (Figure 2B). Thus, it appears that aged mice are able to control initial γHV-68 viral replication and appear to do so more effectively than younger mice.
Figure 2.
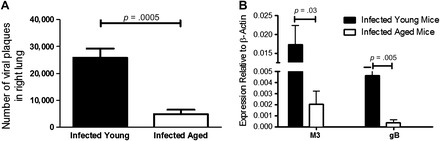
Aged mice are able to effectively clear lytic virus 7 days after infection. Aged mice and young mice (n = 5 per group) were intranasally infected with 5 × 104 plaque-forming units of γHV-68. After 7 days, lungs were harvested. (A) Viral plaque assay on right lungs demonstrates that there is less preformed virus at day 7 following infection in infected aged mice versus infected young mice (p = .0005). (B) Real-time reverse transcription–polymerase chain reaction performed on left lungs demonstrates decreased lytic viral gene expression, both M3 (p = .03) and gB (p = .005), in aged mice vs young mice.
Aged Mice Do Not Harbor Increased Latent γ HV-68
We also measured total viral gB DNA in whole lung homogenates at 21 days following infection by real-time PCR as a measure of latent viral load and found a trend toward increased viral DNA in young mice, although this was not statistically significant (Figure 3). This confirmed that the increased fibrosis found in aged mice in response to γHV-68 infection was not due to increased viral lytic replication or an inability of aged mice to effectively control latent viral infection.
Figure 3.
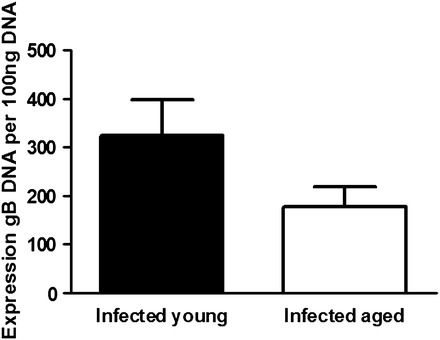
Latent viral load is not different in infected young and aged mice 21 days after infection. Aged and young mice (n = 3 per group) were infected with 1 × 105 plaque-forming units of γHV-68 intranasally. After 21 days, lungs were harvested and viral gB DNA content was measured by real-time polymerase chain reaction (p,nonsignificant).
Increased Production of TGF-β but Not Type 2 Cytokines in Infected Aged Lungs
We next wanted to determine if inflammatory and profibrotic cytokines were elevated in aged mice infected with γHV-68. We performed enzyme-linked immunosorbent assays on whole lung homogenates taken from aged and young mice 21 days after infection. In initial studies, we found no significant differences in cytokine production between young and aged lungs at baseline (data not shown). Yet, after infection with γHV-68, we found increased amounts of TGF-β production at day 21 in aged mice (Figure 4A). However, there was no difference in type 2 cytokines (IL-4, IL-13; Figure 4B and C), type 1 inflammatory cytokines (IFNγ, IL-12), or tumor necrosis factor-α (data not shown). Thus, although aged mice infected with γHV-68 do secrete more of the profibrotic cytokine, TGF-β, they do not show evidence of a fibrosis-prone Th2 environment.
Figure 4.
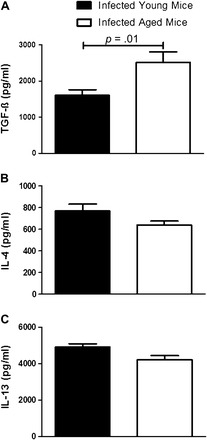
Infection with γHV-68 induces more transforming growth factor (TGF)-β but not interleukin (IL)-4 or IL-13 production in aged mice 21 days following infection. Aged and young mice were infected with 1 × 105 plaque-forming units of γHV-68 and lungs were harvested 21 days later. Enzyme-linked immunosorbent assays were run on whole lung homogenates for profibrotic and inflammatory cytokines. Significant increases were found in infected aged lungs compared with young lungs in (A) TGF-β production (p = .01, n = at least 11 per group pooled from three experiments). However, there was no difference between groups in (B) IL-4 production (p =.14, n = at least 5 per group) and (C) IL-13 production (p =.06, n = at least 5 per group).
Increased Production of TGF-β Is Mainly Due to Differences in Production by Infected Aged Fibroblasts
In order to understand which cells were responsible for the increased TGF-β production seen in infected aged lungs, we infected aged and young mice with γHV-68. After 21 days, we isolated AMs, fibroblasts, and AECs. Equal numbers of each cell type were plated from each group. After 24 or 48 hours (macrophages or fibroblasts, respectively), the cell supernatants were collected, and TGF-β enzyme-linked immunosorbent assay was performed. We found that infected aged fibroblasts produce twice the amount of TGF-β produced by infected young fibroblasts (Figure 5A). In addition, a threefold increase in TGF-β production was seen by infected aged AMs compared with infected young AMs, but note that the overall levels were significantly smaller amounts than produced by fibroblasts (Figure 5B). No significant differences were seen between infected aged and young AECs (data not shown). Therefore, fibroblasts appear to be the main cell type contributing to the increased TGF-β seen in infected aged mice, with some contribution also coming from macrophages.
Figure 5.
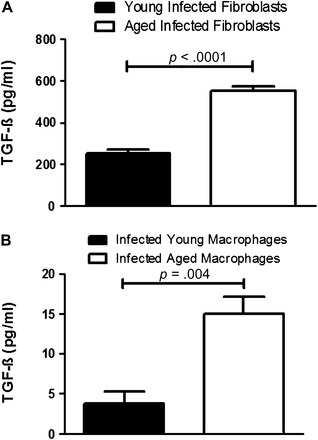
Increased transforming growth factor (TGF)-β production in aged fibroblasts and macrophages infected with γHV-68 in vivo compared with young infected fibroblasts at 21 days following infection. Aged and young mice were infected with 1 × 105 plaque-forming units of γHV-68 and euthanized 21 days later. Fibroblasts were isolated and 5 × 105 cells were cultured in six-well plates for 48 hours in serum-free media. TGF-β enzyme-linked immunosorbent assay (ELISA) was performed on cell supernatants. (A) Fibroblast TGF-β production (n = 4 in each group, p < .0001). Alveolar macrophages were isolated by performing bronchoalveolar lavages on mice and allowing the cells to adhere to plastic after 1 hour. A total of 3 × 104 cells were cultured in 96-well plates for 24 hours in serum-free media. TGF-β ELISA was performed on cell supernatants. (B) Macrophage TGF-β production (n = at least four per group, p = .004).
Equal Numbers of Inflammatory Cells and Fibrocytes Are Recruited to Lungs of Aged and Young Mice After γHV-68 Infection
Since aging is sometimes associated with increased numbers of inflammatory cells (26), we hypothesized that the viral-induced fibrosis in aged mice was due to increased inflammation. We performed a collagenase digest on the lungs of infected mice at days 12 and 21 following γHV-68 infection and counted the total number of inflammatory cells. There was no difference in total cell count of inflammatory cells between infected aged and young mice at either time point, although the total inflammatory cell count did drop over time as expected (Figure 6A). We next performed flow cytometry on the isolated lung leukocytes in order to determine if the composition of inflammatory cells was different between young and aged mice at day 12. The population of cell types was similar between aged and young mice (Figure 6B). Similar profiles of inflammatory cells were also noted between young and old mice at day 21 following infection (data not shown). These data suggest that differences in inflammatory cells were not responsible for the increased fibrosis found in aged mice infected with γHV-68.
Figure 6.
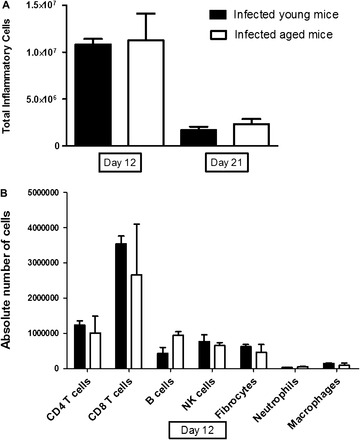
Total inflammatory cell and fibrocyte populations are similar between aged and young mice 12 and 21 days after infection. Aged and young mice were infected with 1 × 105 plaque-forming units of γHV-68 and lungs were harvested at either 12 days or 21 days following infection (n = 3 per group). Collagenases were performed on the lungs. (A) Total number of inflammatory cells from both groups at each time point was evaluated. There was a decline in total inflammatory cells as expected over time in both aged and young mice but no difference was found between groups at either time point. (B) Flow cytometry was performed on the collagenase cell digests at day 12 but no significant differences were found. Additionally, no differences were noted at day 21 between either groups (not shown).
Given the work by Xu and colleagues (25) implicating increased fibrocytes in the serum of senescence-accelerated prone mice in a bleomycin model of pulmonary fibrosis, we also measured fibrocyte numbers by flow cytometry in the lung leukocytes isolated as above. We found no difference in the number of fibrocytes between infected aged and young mice at day 12 (Figure 6B) or day 21 following infection (data not shown).
Fibroblasts From Aged Mice Display a Profibrotic Phenotype in Response to Infection With γHV-68
Because there did not appear to be differences in recruitment of cells to the lungs of our aged mice after infection and because we were able to determine that aged fibroblasts were the predominant source of TGF-β after infection, we suspected that functional differences in resident fibroblasts were playing a role in increased fibrosis. We isolated fibroblasts from the lungs of uninfected aged and young mice and plated equal numbers of fibroblasts in six-well plates. Half the wells were treated with 0.1 pfu per cell of γHV-68, whereas the rest were mock infected as controls. After 48 hours, protein lysates were extracted from the cells and analyzed by Western blot. We found that in vitro infection with virus caused aged fibroblasts to upregulate their collagen and alpha-smooth muscle actin protein production. This increase over baseline expression was not seen in young infected fibroblasts (Figure 7).
Figure 7.
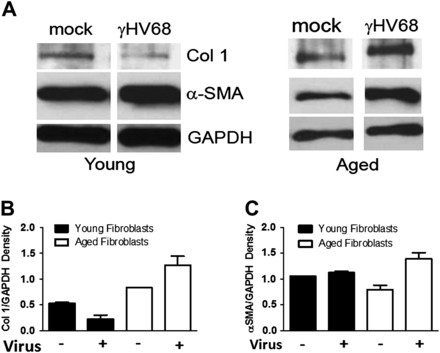
γHV-68 infection in vitro induces alpha-smooth muscle actin (α-SMA) and collagen I in aged fibroblasts. Fibroblasts from the lungs of aged and young mice were isolated and 5 × 105 cells were cultured in six-well plates for 48 hours after treatment with 0.1 plaque-forming units per cell γHV-68 or mock infection. Protein lysates were made and protein concentration was evaluated by Western blot. (A) Western blot showing an increase in both α-SMA and collagen I production in infected aged fibroblasts compared with aged fibroblasts at baseline. This increase in fibrotic protein production was not seen in young infected fibroblasts compared with baseline. Note these Western blots were from separate experiments and thus the amount of collagen I, α-SMA, and glyceraldehyde 3-phosphate dehydrogenase (GAPDH) cannot be directly compared between them, only relative increases can be calculated within each group. (B) Density of collagen I band in the Western blot when normalized to GAPDH confirming the increase in collagen seen in infected aged, but not young, fibroblasts (n = 2). (C) Density of α-SMA in the Western blot when normalized to GAPDH confirming the increase in α-SMA seen in infected aged, but not young, fibroblasts (n = 2).
We next infected aged and young mice in vivo with γHV-68 or introduced saline as a control and isolated fibroblasts 21 days after infection. Forty-eight hours after plating equal numbers of fibroblasts from each group in six-well plates, we collected protein lysates and performed a Western blot for protein analysis. Again, we found that viral infection in vivo caused aged fibroblasts to upregulate their collagen and alpha-smooth muscle actin protein production far more than seen in young fibroblasts (Figure 8). We also extracted RNA from cultures of fibroblasts from infected mice and analyzed them by real-time RT-PCR. Of note, initial studies comparing uninfected aged and young fibroblast RNA production showed no significant increases in collagen I, collagen III, or fibronectin expression in aged mice (Figure 9A). After 21 days of infection, we found that collagen I, collagen III, and fibronectin messenger RNA were significantly increased in aged fibroblasts compared with young fibroblasts (Figure 9B). Thus, there are clear functional differences between aged and young fibroblasts following viral infection, with aged infected fibroblasts showing a predominantly profibrotic, myofibroblast phenotype.
Figure 8.
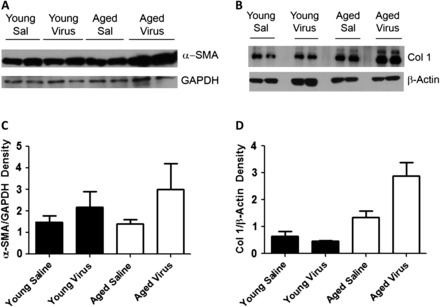
γHV-68 infection in vivo also upregulates alpha-smooth muscle actin (α-SMA) and collagen I protein production in aged fibroblasts. Aged and young mice were infected with 1 × 105 plaque-forming units of γHV-68 or equal amount of saline for controls. Twenty-one days after infection, lungs were harvested and fibroblasts were isolated (n = 2 per group). Fibroblasts were plated at 5 × 105 cells in six-well plates and were cultured for 48 hours. Protein lysates were made and protein concentration was evaluated by Western blot. (A) Western blot showing increased α-SMA in aged infected fibroblasts compared with aged fibroblasts with saline controls, young infected fibroblasts and young fibroblasts with saline controls. (B) Western blot showing increased collagen I production by infected aged fibroblasts compared with other groups. (C) Densitometry showing differences in α-SMA between the four groups. (D) Densitometry showing differences in collagen I between the four groups.
Figure 9.
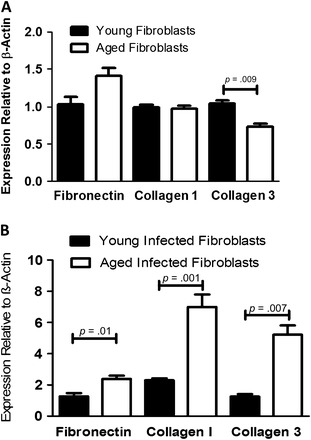
Aged fibroblasts do not produce increased collagen I, collagen III, or fibronectin messenger RNA expression at baseline compared with young fibroblasts, but do following infection. (A) Lungs from aged and young mice were harvested and fibroblasts were isolated. A total of 5 × 105 cells were cultured in six-well plates for 48 hours before RNA from each well was isolated and real-time reverse transcription–polymerase chain reaction (RT-PCR) was performed (n =3 per group). (B) Aged and young mice were infected with 1 × 105 plaque-forming units of γHV-68. After 21 days, fibroblasts were isolated from lungs of these mice. A total of 5 × 105 cells were cultured in six-well plates for 48 hours before RNA from each well was isolated and real-time RT-PCR was performed (n = 4 per group).
Fibroblasts From Aged Mice Have Increased Sensitivity to TGF-β With Increased Expression of TGF-β Receptor 1 and Receptor 2
TGF-β is a known promoter of fibrosis and is a potent mediator of fibroblast differentiation to myofibroblasts (41). It has also been shown that fibroblasts from the lungs of IPF patients have increased TGF-β receptor (TGF-βR) density on their surface (42). Given the increases in whole lung TGF-β found in infected aged mice, we hypothesized that the difference in fibrotic potential between aged and young fibroblasts was TGF-β mediated. We isolated fibroblasts from untreated aged and young mice and extracted their total RNA. After analysis by real-time RT-PCR, we found that aged fibroblasts have a significant three- to fourfold increase in TGF-βR1 and TGF-βR2 messenger RNA expression compared with young fibroblasts (Figure 10A). This suggested that aged fibroblasts at baseline were more responsive to TGF-β than young fibroblasts.
Figure 10.
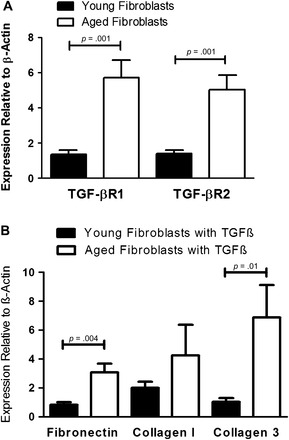
Increased production of transforming growth factor (TGF)-β receptor 1 (TGF-βR1) and receptor 2 (TGF-βR2) messenger RNA in aged fibroblasts compared with young fibroblasts and increased fibrotic response of aged fibroblasts treated with TGF-β. (A) Fibroblasts were isolated from aged and young mice and plated in six-well plates at 5 × 105cells per well. After 48 hours, RNA was isolated from cells and real-time reverse transcription–polymerase chain reaction (RT-PCR) was performed (n = at least 5 per group). (B) Fibroblasts were isolated from uninfected aged and young mice and plated in six-well plates at 5 × 105 cells per well with or without 2 ng/mL TGF-β. After 48 hours, RNA was isolated from cells and real-time RT-PCR was performed (n = at least 5 per group).
To confirm increased sensitivity to TGF-β, we added 2 ng/mL of TGF-β to aged and young fibroblasts in vitro. Similar to results seen after viral infection in vivo, we found that aged fibroblasts had significantly increased messenger RNA levels of fibronectin and collagen III (Figure 10B). In addition, there was a trend toward increased collagen I production by aged fibroblasts (for baseline values between young and aged fibroblasts, see Figure 9A). Thus, aged fibroblasts are more likely to be prone to fibrosis due to their increased sensitivity to TGF-β. It is likely a combination of their increased responsiveness to TGF-β and the increased production of TGF-β found in aged lungs after viral infection that causes γHV-68-induced pulmonary fibrosis in aged but not in young mice.
Fibroblasts From Aged Mice Are Resistant to Apoptosis
To determine whether lung fibroblasts from aged mice showed altered susceptibility to apoptosis, fibroblasts from aged and young mice were seeded and grown to 80% confluence in complete media (which provides an endogenous source of TGF-β). Next fibroblasts were serum starved to induce apoptosis for 40 hours and caspase 3 activity was measured. Figure 11 demonstrates that young fibroblasts are more susceptible to apoptosis (264 ± 18.4 units/mL caspase 3) than similarly cultured aged fibroblasts (193 ± 21 units/mL caspase 3, n = 4, p .04). Thus, in total, aged fibroblasts are both more responsive to TGF-β production induced by viral infection and more resistant to apoptosis.
Figure 11.
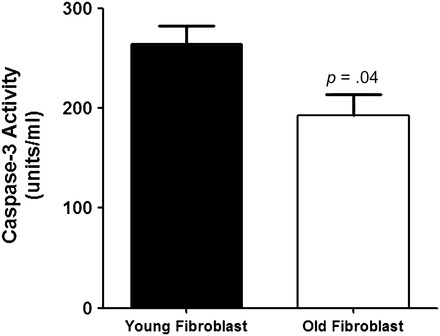
Aged fibroblasts are resistant to apoptosis. Fibroblasts from young and aged mice were cultured to 80% confluence in complete media before being switched to serum-free media for an additional 40 hour. Caspase 3 activity was measured in cellular lysates using a caspase 3 colorimetric assay according to the manufacturer’s instructions. Data shown represent the mean ± standard error of the mean of four independent experiments (p = .04).
DISCUSSION
To our knowledge, this is the first report of γHV-68 inducing pulmonary fibrosis in the absence of additional fibrotic stimuli and without genetic alterations in the lung environment. Contrary to our initial expectations, increased susceptibility to viral-induced fibrosis in the aged lung is not due to alterations in Th1 or Th2 cytokines, recruitment of inflammatory cells, or recruitment of fibrocytes. Furthermore, susceptibility in aged mice is not associated with an inability to control γHV-68 replication or latency. Rather, it appears that aged resident fibroblasts are skewed toward a profibrotic phenotype with increased responsiveness to TGF-β. This is associated with increased alpha-smooth muscle actin production over baseline levels and increased production of extracellular matrix secondary to the increased levels of TGF-β produced by infected aged fibroblasts. Additionally, aged fibroblasts are resistant to apoptosis.
Previous work by Mora and colleagues (24) in IFNγ receptor knockout mice with a Th2 cytokine bias showed that viral replication was key in that mouse model for γHV-68 to produce pulmonary fibrosis. Our results demonstrate that aged mice develop a more robust fibrotic response despite improved lytic control of virus replication when compared with young mice. We have seen a similar disconnect between viral replication and fibrotic response in mice without TLR9 signaling that have been infected with γHV-68 (43). TLR9 knockout and wild-type mice were infected with γHV-68 14 days after treatment with bleomycin and harvested 7 days later. Lungs from TLR9 knockout mice showed increased collagen expression compared with wild-type mice but no significant difference in viral replication. Thus, the degree of viral replication does not necessarily correlate with subsequent pulmonary fibrosis severity. It is also interesting that infection of fibroblasts from young mice actually decreases collagen synthesis, whereas infection of fibroblasts from aged mice does not (Figure 7). It may be the case that increased viral replication in young mice may in fact divert cellular machinery away from host proteins or may result in lysis of infected cells.
We have also seen that viral reactivation is not necessary for latent γHV-68 to augment fibrosis in response to subsequent stimuli such as bleomycin or fluorescein isothiocyanate (21). Although our experiments did not specifically address reactivation, the fact that the viral DNA load is not increased in aged mice suggests that significant viral reactivation is likely not a feature of γHV-68-induced lung fibrosis in aged mice. Our data corroborate studies recently published by Yager and colleagues (44) showing no evidence of decreased viral immunity, increased viral reactivation, or increased viral latent load in mice infected with γHV-68 as they age. Finally, unlike the IFNγ receptor knockout mouse model, aged mice demonstrate a fibrotic response to viral infection in the absence of a Th2 cytokine bias. Because type 1 cytokines are largely responsible for control of viral replication, it is presumed that the Th2 cytokine bias allows viral reactivation to occur and this repetitive injury is necessary to cause virus-induced fibrosis in young mice (24). However, this Th2 bias does not appear necessary in aged mice for fibrosis to occur.
We were surprised that our aged mice did not show evidence of increased fibrocyte accumulation in their lungs in response to γHV-68 infection. Xu and colleagues (25) showed that senescence-accelerated prone mice, phenotypic equivalents to aged mice, developed worse bleomycin-induced fibrosis than did senescence-accelerated resistant mice. They also found increased TGF-β production and increased serum fibrocytes in their bleomycin-treated senescence-accelerated prone mice (25). Additionally, we have previously shown that increased numbers of fibrocytes accumulate when viral infection occurs prior to or after fibrotic stimulus (17,21). Given these prior studies, we anticipated that increased fibrosis in aged mice would be associated with increased fibrocyte accumulation. However, this does not appear to be the case in our model; both aged and young mice accumulated similar numbers of fibrocytes in response to viral infection. Given our studies in the mesenchymal cells, however, it is likely that the fibrocytes that accumulate in aged mice are more responsive to TGF-β and thus potentially skewed to a profibrotic phenotype.
Although the inflammatory cell populations appeared to be similar between the infected young and aged mice, we did not evaluate for specific differences such as the presence of alternatively activated macrophages, which have been shown to be recruited to sites of active viral-induced fibrosis in Th2-biased mice and to express fibrotic mediators (45). Because our aged mice showed no increase in IL-4 or IL-13 following infection, we think it unlikely that alternatively activated macrophages are preferentially accumulated. It remains a formal possibility however that the increased levels of TGF-β that accumulate following infection in aged mice may serve to promote alternatively activated macrophage differentiation.
Previous work by our laboratory has shown that multiple cell types can be latently infected with γHV-68 and produce TGF-β in response to infection, but mesenchymal cells appear to produce far more TGF-β than any other cell type (21,46). Our results in aged mice display a similar trend demonstrating that infected fibroblasts in aged mice are the predominant source of TGF-β. Although AECs are well known to be a reservoir for latent γHV-68 infection (21,47), we did not find evidence of increased TGF-β release by infected aged AECs compared with infected young AECs. One possibility is that the activation of TGF-β by its main integrin, αvβ6, requires direct cell-to-cell contact, and thus, active TGF-β may be rapidly bound by epithelial cells and unavailable to be measured in the supernatants (48). If integrin expression is altered in aged mice, this could result in increased cell-associated activation of TGF-β in aged AECs that may have been missed in our studies of supernatants. Additionally, AMs are a potential reservoir for γHV-68 (46), and although we did find increases in secretion of TGF-β by infected macrophages from aged mice, the total levels were much lower than those noted in fibroblasts.
Our results demonstrate that aged fibroblasts have increased levels of TGF-βR1 and TGF-βR2 making them functionally more susceptible to TGF-β-induced fibrotic responses. It is interesting that this phenotype recapitulates the observation that TGF-βR density is increased on the surface of fibroblasts taken from lung tissue of patients with IPF (42). Like our aged mice, IPF patients have an increased sensitivity to TGF-β (42). We do not yet know whether the differences in TGF-βR1 and TGF-βR2 that characterize aged fibroblasts are also present on aged epithelial cells. Recent work by Degryse and colleagues (49) showed that mice with a selective deficiency in TGF-βR2 on AECs had improved epithelial cell survival and decreased epithelial mesenchymal transition, resulting in markedly attenuated bleomycin-induced fibrosis. Therefore, if our aged mice have increased TGF-βR2 on their epithelial cells as they do on fibroblasts, this alteration would be predicted to decrease epithelial cell survival and promote epithelial mesenchymal transition. This may be especially relevant because epithelial cells are a known reservoir for γHV-68 (21,47) and EBV infection is associated with epithelial mesenchymal transition in human epithelial cells (12,50). Further study is needed to determine if alterations in cell types other than fibroblasts are also responsible for the viral-induced fibrosis seen in aged mice.
Finally, our results indicate that fibroblasts in aged mice are more resistant to induction of apoptosis caused by serum deprivation. This indicates that fibroblasts within dense fibrotic areas (sites that may represent areas of serum deprivation) may live longer and thus have prolonged potential to secrete extracellular matrix. Taken together, our results demonstrate that aged mice produce increased levels of TGF-β in response to γHV-68 infection largely due to increased production by infected aged fibroblasts. These same aged fibroblasts have increased sensitivity to TGF-β signaling and reduced sensitivity to apoptosis. Based on these findings, we suggest that these alterations may predispose aged individuals who experience acute or reactivated herpesvirus infections to develop a fibrotic lung response. These observations provide mechanistic insight into why fibrosis is associated with aging (3,42).
FUNDING
This work was supported by the National Institutes of Health grants HL087846 (G.B.T.) and AI065543 (B.B.M.), and funding from the Atlantic Philanthropies (USA) Inc., American College of Chest Physicians/ the Chest Foundation, John A. Hartford Foundation, and Association of Specialty Professors(J.C.H.).
References
- 1.American Thoracic Society. Idiopathic pulmonary fibrosis: diagnosis and treatment. International consensus statement. Am J Respir Crit Care Med. 2000;161:646–664. doi: 10.1164/ajrccm.161.2.ats3-00. [DOI] [PubMed] [Google Scholar]
- 2.Raghu G, Weycker D, Edelsberg J, et al. Incidence and prevalence of idiopathic pulmonary fibrosis. Am J Respir Crit Care Med. 2006;174(7):810–816. doi: 10.1164/rccm.200602-163OC. [DOI] [PubMed] [Google Scholar]
- 3.Fell CD, Martinez FJ, Liu LX, et al. Clinical predictors of a diagnosis of idiopathic pulmonary fibrosis. Am J Respir Crit Care Med. 2010;181(8):832–837. doi: 10.1164/rccm.200906-0959OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Collard HR, Moore BB, Flaherty KR, et al. Acute exacerbations of idiopathic pulmonary fibrosis. Am J Respir Crit Care Med. 2007;176(7):636–643. doi: 10.1164/rccm.200703-463PP. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Akira M, Hamada H, Sakatani M, et al. CT findings during phase of accelerated deterioration in patients with idiopathic pulmonary fibrosis. AJR Am J Roentgenol. 1997;168(1):79–83. doi: 10.2214/ajr.168.1.8976924. [DOI] [PubMed] [Google Scholar]
- 6.Naik PK, Moore BB. Viral infection and aging as cofactors for the development of pulmonary fibrosis. Expert. 2010;4(6):759–771. doi: 10.1586/ers.10.73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Kutok JL, Wang F. Spectrum of Epstein-Barr virus-associated diseases. Annu Rev Pathol. 2006;1:375–404. doi: 10.1146/annurev.pathol.1.110304.100209. [DOI] [PubMed] [Google Scholar]
- 8.Manika K, Alexiou-Daniel S, Papakosta D, et al. Epstein-Barr virus DNA in bronchoalveolar lavage fluid from patients with idiopathic pulmonary fibrosis. Sarcoidosis Vasc Diffuse Lung Dis. 2007;24(2):134–140. [PubMed] [Google Scholar]
- 9.Stewart JP, Egan JJ, Ross AJ, et al. The detection of Epstein-Barr virus DNA in lung tissue from patients with idiopathic pulmonary fibrosis. Am J Respir Crit Care Med. 1999;159(4 Pt 1):1336–1341. doi: 10.1164/ajrccm.159.4.9807077. [DOI] [PubMed] [Google Scholar]
- 10.Vergnon JM, Vincent M, de Thé G, et al. Cryptogenic fibrosing alveolitis and Epstein-Barr virus: an association? Lancet. 1984;2(8406):768–771. doi: 10.1016/s0140-6736(84)90702-5. [DOI] [PubMed] [Google Scholar]
- 11.Egan JJ, Stewart JP, Haselton PS, et al. Epstein-Barr virus replication within pulmonary epithelial cells in cryptogenic fibrosing alveolitis. Thorax. 1995;50(12):1234–1239. doi: 10.1136/thx.50.12.1234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Malizia AP, Keating DT, Smith SM, et al. Alveolar epithelial cell injury with Epstein-Barr virus upregulates TGFbeta1 expression. Am J Physiol Lung Cell Mol Physiol. 2008;295(3):L451–L460. doi: 10.1152/ajplung.00376.2007. [DOI] [PubMed] [Google Scholar]
- 13.Tang YW, Johnson JE, Browning PJ, et al. Herpesvirus DNA is consistently detected in lungs of patients with idiopathic pulmonary fibrosis. J Clin Microbiol. 2003;41(6):2633–2640. doi: 10.1128/JCM.41.6.2633-2640.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Wangoo A, Shaw RJ, Diss TC, et al. Cryptogenic fibrosing alveolitis: lack of association with Epstein-Barr virus infection. Thorax. 1997;52(10):888–891. doi: 10.1136/thx.52.10.888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Zamo A, Poletti V, Reghellin D, et al. HHV-8 and EBV are not commonly found in idiopathic pulmonary fibrosis. Sarcoidosis Vasc Diffuse Lung Dis. 2005;22(2):123–128. [PubMed] [Google Scholar]
- 16.Efstathiou S, Ho YM, Hall S, et al. Murine herpesvirus 68 is genetically related to the gammaherpesviruses Epstein-Barr virus and herpesvirus saimiri. J Gen Virol. 1990;71(Pt 6):1365–1372. doi: 10.1099/0022-1317-71-6-1365. [DOI] [PubMed] [Google Scholar]
- 17.McMillan TR, Moore BB, Weinberg JB, et al. Exacerbation of established pulmonary fibrosis in a murine model by gammaherpesvirus. Am J Respir Crit Care Med. 2008;177(7):771–180. doi: 10.1164/rccm.200708-1184OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Virgin HWIV, Latreille P, Wamsley P, et al. Complete sequence and genomic analysis of murine gammaherpesvirus 68. J Virol. 1997;71(8):5894–5904. doi: 10.1128/jvi.71.8.5894-5904.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Flano E, Husain SM, Sample JT, et al. Latent murine gamma-herpesvirus infection is established in activated B cells, dendritic cells, and macrophages. J Immunol. 2000;165(2):1074–1081. doi: 10.4049/jimmunol.165.2.1074. [DOI] [PubMed] [Google Scholar]
- 20.Lok SS, Haider Y, Howell D, et al. Murine gammaherpes virus as a cofactor in the development of pulmonary fibrosis in bleomycin resistant mice. Eur Respir J. 2002;20(5):1228–1232. doi: 10.1183/09031936.02.00272902. [DOI] [PubMed] [Google Scholar]
- 21.Vannella KM, Luckhardt TR, Wilke CA, et al. Latent herpesvirus infection augments experimental pulmonary fibrosis. Am J Respir Crit Care Med. 2010;181:465–477. doi: 10.1164/rccm.200905-0798OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Ebrahimi B, Dutia BM, Brownstein DG, et al. Murine gammaherpesvirus-68 infection causes multi-organ fibrosis and alters leukocyte trafficking in interferon-gamma receptor knockout mice. Am J Pathol. 2001;158(6):2117–2125. doi: 10.1016/s0002-9440(10)64683-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Mora AL, Woods CR, Garcia A, et al. Lung infection with gamma-herpesvirus induces progressive pulmonary fibrosis in Th2-biased mice. Am J Physiol Lung Cell Mol Physiol. 2005;289(5):L711–L721. doi: 10.1152/ajplung.00007.2005. [DOI] [PubMed] [Google Scholar]
- 24.Mora AL, Torres-Gonzalez E, Rojas M, et al. Control of virus reactivation arrests pulmonary herpesvirus-induced fibrosis in IFN-gamma receptor-deficient mice. Am J Respir Crit Care Med. 2007;175(11):1139–1150. doi: 10.1164/rccm.200610-1426OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Xu J, Gonzalez ET, Iyer SS, et al. Use of senescence-accelerated mouse model in bleomycin-induced lung injury suggests that bone marrow-derived cells can alter the outcome of lung injury in aged mice. J Gerontol A Biol Sci Med Sci. 2009;64(7):731–739. doi: 10.1093/gerona/glp040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Aw D, Silva AB, Palmer DB. Immunosenescence: emerging challenges for an ageing population. Immunology. 2007;120(4):435–446. doi: 10.1111/j.1365-2567.2007.02555.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Gardner EM, Murasko DM. Age-related changes in Type 1 and Type 2 cytokine production in humans. Biogerontology. 2002;3(5):271–290. doi: 10.1023/a:1020151401826. [DOI] [PubMed] [Google Scholar]
- 28.Mu XY, Thoman ML. The age-dependent cytokine production by murine CD8+ T cells as determined by four-color flow cytometry analysis. J Gerontol A Biol Sci Med Sci. 1999;54(3):B116–B123. doi: 10.1093/gerona/54.3.b116. [DOI] [PubMed] [Google Scholar]
- 29.Isler JA, Skalet AH, Alwine JC. Human cytomegalovirus infection activates and regulates the unfolded protein response. J Virol. 2005;79(11):6890–6899. doi: 10.1128/JVI.79.11.6890-6899.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Castriotta RJ, Eldadah BA, Foster WM, et al. Workshop on idiopathic pulmonary fibrosis in older adults. Chest. 2010;138(3):693–703. doi: 10.1378/chest.09-3006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Erickson RR, Dunning LM, Holtzman JL. The effect of aging on the chaperone concentrations in the hepatic, endoplasmic reticulum of male rats: the possible role of protein misfolding due to the loss of chaperones in the decline in physiological function seen with age. J Gerontol A Biol Sci Med Sci. 2006;61(5):435–443. doi: 10.1093/gerona/61.5.435. [DOI] [PubMed] [Google Scholar]
- 32.Goronzy JJ, Lee WW, Weyand CM. Aging and T-cell diversity. Exp Gerontol. 2007;42(5):400–406. doi: 10.1016/j.exger.2006.11.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Khan N, Hislop A, Gudgeon N, et al. Herpesvirus-specific CD8 T cell immunity in old age: cytomegalovirus impairs the response to a coresident EBV infection. J Immunol. 2004;173(12):7481–7489. doi: 10.4049/jimmunol.173.12.7481. [DOI] [PubMed] [Google Scholar]
- 34.Stowe RP, Kozlova EV, Yetman DL, et al. Chronic herpesvirus reactivation occurs in aging. Exp Gerontol. 2007;42(6):563–570. doi: 10.1016/j.exger.2007.01.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Moore B, Paine RIII, Christensen P, et al. Protection from pulmonary fibrosis in the absence of CCR2 signaling. J Immunol. 2001;167:4368–4377. doi: 10.4049/jimmunol.167.8.4368. [DOI] [PubMed] [Google Scholar]
- 36.Kolodsick JE, Toews GB, Jakubzick C, et al. Protection from fluorescein isothiocyanate-induced fibrosis in IL-13-deficient, but not IL-4-deficient, mice results from impaired collagen synthesis by fibroblasts. J Immunol. 2004;172(7):4068–4076. doi: 10.4049/jimmunol.172.7.4068. [DOI] [PubMed] [Google Scholar]
- 37.Ballinger MN, Paine RIII, Serezani CH, et al. Role of granulocyte macrophage colony-stimulating factor during gram-negative lung infection with Pseudomonas aeruginosa. Am J Respir Cell Mol Biol. 2006;34(6):766–774. doi: 10.1165/rcmb.2005-0246OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Nguyen Y, McGuffie BA, Anderson VE, et al. Gammaherpesvirus modulation of mouse adenovirus type 1 pathogenesis. Virology. 2008;380(2):182–190. doi: 10.1016/j.virol.2008.07.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Charbeneau RP, Christensen PJ, Chrisman CJ, et al. Impaired synthesis of prostaglandin E2 by lung fibroblasts and alveolar epithelial cells from GM-CSF-/- mice: implications for fibroproliferation. Am J Physiol Lung Cell Mol Physiol. 2003;284(6):L1103–L1111. doi: 10.1152/ajplung.00350.2002. [DOI] [PubMed] [Google Scholar]
- 40.Ballinger MN, Aronoff DM, McMillan TR, et al. Critical role of prostaglandin E2 overproduction in impaired pulmonary host response following bone marrow transplantation. J Immunol. 2006;177(8):5499–5508. doi: 10.4049/jimmunol.177.8.5499. [DOI] [PubMed] [Google Scholar]
- 41.Hu B, Wu Z, Phan SH. Smad3 mediates transforming growth factor-beta-induced alpha-smooth muscle actin expression. Am J Respir Cell Mol Biol. 2003;29(3 Pt 1):397–404. doi: 10.1165/rcmb.2003-0063OC. [DOI] [PubMed] [Google Scholar]
- 42.Raghu G, Chen YY, Rusch V, et al. Differential proliferation of fibroblasts cultured from normal and fibrotic human lungs. Am Rev Respir Dis. 1988;138(3):703–708. doi: 10.1164/ajrccm/138.3.703. [DOI] [PubMed] [Google Scholar]
- 43.Luckhardt TR, Coomes SM, Trujillo G, et al. TLR9-induced interferon beta is associated with protection from gammaherpesvirus-induced exacerbation of lung fibrosis. Fibrogenesis Tissue Repair. 2011;4:18. doi: 10.1186/1755-1536-4-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Yager EJ, Kim IJ, Freeman ML, et al. Differential impact of ageing on cellular and humoral immunity to a persistent murine gamma-herpesvirus. Immun Ageing. 2010;7:3. doi: 10.1186/1742-4933-7-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Mora AL, Torres-Gonzalez E, Rojas M, et al. Activation of alveolar macrophages via the alternative pathway in herpesvirus-induced lung fibrosis. Am J Respir Cell Mol Biol. 2006;35(4):466–473. doi: 10.1165/rcmb.2006-0121OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Stoolman JS, Vannella KM, Coomes SM, et al. Latent infection by gammaherpesvirus stimulates profibrotic mediator release from multiple cell types. Am J Physiol Lung Cell Mol Physiol. 2011;300(2):L274–L285. doi: 10.1152/ajplung.00028.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Stewart JP, Usherwood EJ, Ross A, et al. Lung epithelial cells are a major site of murine gammaherpesvirus persistence. J Exp Med. 1998;187(12):1941–1951. doi: 10.1084/jem.187.12.1941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Sheppard D. Integrin-mediated activation of latent transforming growth factor beta. Cancer Metastasis Rev. 2005;24(3):395–402. doi: 10.1007/s10555-005-5131-6. [DOI] [PubMed] [Google Scholar]
- 49.Degryse AL, Tanjore H, Xu XC, et al. TGFβ signaling in lung epithelium regulates bleomycin induced alveolar injury and fibroblast recruitment. Am J Physiol Lung Cell Mol Physiol. 2011;300(6):L887–L897. doi: 10.1152/ajplung.00397.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Malizia AP, Lacey N, Walls D, et al. CUX1/Wnt signaling regulates epithelial mesenchymal transition in EBV infected epithelial cells. Exp Cell Res. 2009;315(11):1819–1831. doi: 10.1016/j.yexcr.2009.04.001. [DOI] [PubMed] [Google Scholar]