

Background: PKD1 catalytic fragments accumulate during apoptosis; their cellular actions remain uncertain.
Results: A PKD1 truncation mutant lacking the N-terminal portion of the regulatory domain does not phosphorylate protein substrates or activate PKD1-dependent cellular responses.
Conclusion: The N-terminal portion of the regulatory domain (encompassing the C1 domain) is a positive regulator of PKD1 activity.
Significance: Proteolysis limits the cellular actions of PKD1.
Keywords: CREB, ERK, Protein Domains, Protein Kinase D (PKD), Protein Kinases, C1 Domain, PH Domain, Activation Loop, Autophosphorylation
Abstract
The canonical pathway for protein kinase D1 (PKD1) activation by growth factor receptors involves diacylglycerol binding to the C1 domain and protein kinase C-dependent phosphorylation at the activation loop. PKD1 then autophosphorylates at Ser916, a modification frequently used as a surrogate marker of PKD1 activity. PKD1 also is cleaved by caspase-3 at a site in the C1-PH interdomain during apoptosis; the functional consequences of this cleavage event remain uncertain. This study shows that PKD1-Δ1–321 (an N-terminal deletion mutant lacking the C1 domain and flanking sequence that models the catalytic fragment that accumulates during apoptosis) and PKD1-CD (the isolated catalytic domain) display high basal Ser916 autocatalytic activity and robust activity toward CREBtide (a peptide substrate) but little to no activation loop autophosphorylation and no associated activity toward protein substrates, such as cAMP-response element binding protein and cardiac troponin I. In contrast, PKD1-ΔPH (a PH domain deletion mutant) is recovered as a constitutively active enzyme, with high basal autocatalytic activity and high basal activity toward peptide and protein substrates. These results indicate that individual regions in the regulatory domain act in a distinct manner to control PKD1 activity. Finally, cell-based studies show that PKD1-Δ1–321 does not substitute for WT-PKD1 as an in vivo activator of cAMP-response element binding protein and ERK phosphorylation. Proteolytic events that remove the C1 domain (but not the autoinhibitory PH domain) limit maximal PKD1 activity toward physiologically relevant protein substrates and lead to a defect in PKD1-dependent cellular responses.
Introduction
Protein kinase D1 (PKD1)2 is the founding member of a family of structurally related serine/threonine kinases that have emerged as regulators of cardiac contraction and ventricular remodeling (1). PKD1 is structurally characterized by a C-terminal kinase domain that is distantly related to Ca2+/calmodulin-dependent protein kinases and an N-terminal regulatory domain consisting of a C1 motif that binds diacylglycerol-/phorbol ester-enriched membranes and a pleckstrin homology (PH) domain that participates in autoinhibitory intramolecular interactions. PKD1 activation is generally attributed to a phosphorylation-dependent mechanism involving PKC. Agonists that promote diacylglycerol accumulation co-localize PKD1 with novel PKC isoforms at membranes and promote novel PKC-dependent PKD1 phosphorylation at Ser744/Ser748 (a pair of highly conserved serine residues in the activation loop of the kinase domain; nomenclature based upon rodent sequence). Recent studies indicate that PKD1-Ser744/Ser748 phosphorylation also can be mediated by an autocatalytic reaction that assumes functional importance during chronic PKD activation by G protein-coupled receptor agonists (2, 3). Because the PKD1-Ser744/Ser748-phosphorylated enzyme typically autophosphorylates at Ser916, immunoblotting for PKD1-Ser916 phosphorylation is widely used as a surrogate measure of PKD1 activity (in place of more cumbersome direct measurements of enzyme activity). However, recent studies expose discrepancies between PKD1-Ser916 phosphorylation and PKD1 activity toward physiologically relevant protein substrates, indicating that conclusions based upon experiments that rely on Ser916 phosphorylation as a surrogate for PKD1 activity may be misleading (2).
In addition to the canonical growth factor-dependent mechanism for PKD1 activation, PKD1 also is proteolytically cleaved by caspase-3 during induction of apoptosis. Caspase-3 cleaves PKD1 at a site in the C1-PH interdomain, generating a catalytic fragment that retains the PH, but not the C1, domain (4–6). Because the C1 and PH domains have been implicated in similar intramolecular interactions that limit basal catalytic activity, the functional consequences of cleavage at this site are not necessarily predictable, and they have been disputed (7, 8). Vántus et al. (5) concluded that this proteolytic event activates PKD1 based upon studies showing that the PKD1 cleavage product generated during apoptosis displays a modest increase in basal activity, compared with the basal activity of full-length PKD1. However, Häussermann et al. (9) performed a more comprehensive analysis and showed that the increased basal activity of the C-terminal cleavage product is inconsequential when compared with the high PS/PMA-dependent activity displayed by the full-length PKD1 enzyme; these results suggest that proteolysis limits maximal PKD1 activity. Of note, these previous studies measured PKD1 activity using assays that tracked autophosphorylation or phosphorylation of a peptide substrate; recent studies indicate that these approaches may be misleading because they do not necessarily provide valid surrogate readouts of PKD1 activity toward heterologous protein substrates (2). Therefore, this study uses cell-based and in vitro approaches (including assays that track phosphorylation of physiologically relevant substrates, such as cAMP-response element binding protein (CREB) and cardiac troponin I (cTnI)) to resolve uncertainties regarding the activity of PKD1 catalytic fragments.
EXPERIMENTAL PROCEDURES
Materials
Anti-PKD1-Ser(P)742 (numbering based upon human sequence, corresponding to rodent PKD1-Ser748) was from Abcam. Other antibodies were from Cell Signaling Technologies. CREB-MBP fusion protein was from Invitrogen. CREBtide was from Calbiochem.
PKD1 Mutants
Plasmids that drive expression of HA-tagged WT-PKD1, PKD1-SS/AA (harboring non-phosphorylatable alanines at Ser744/Ser748 in the activation loop), PKD1- ΔPH (PH domain deletion), and PKD1-CD (an isolated catalytic domain) generated by the Toker laboratory were obtained from Addgene. PKD1-Δ1–321 (an N-terminal deletion mutant lacking the C1 domain) was generously provided by Dr. Peter Storz. PKD1 expression plasmids were introduced into HEK293 cells by Effectene transfection reagent (Qiagen) according to the instruction manual. Cells were grown for 24 h and lysed in radioimmune precipitation buffer containing 1 mm sodium orthovanadate, 10 μg/ml aprotinin, 10 μg/ml leupeptin, 10 μg/ml benzamidine, 0.5 mm PMSF, 5 μm pepstatin A, and 0.1 μm calyculin. Cell extracts were used for immunoblotting experiments or subjected to immunoprecipitation with anti-HA tag-agarose (Roche Applied Science) for subsequent in vitro kinase assays.
In Vitro Kinase Assays
In vitro kinase assays were performed with PKD1 immunoprecipitated from 150 μg of starting cell extract. Incubations were performed in 110 μl of a reaction buffer containing 30 mm Tris-Cl, pH 7.5, 5.45 mm MgCl2, 0.65 mm EDTA, 0.65 mm EGTA, 0.1 mm DTT, 1.09 mm sodium orthovanadate, 0.1 μm calyculin, 0.55 μm PKI, 217 mm NaCl, 3.6% glycerol, and [γ-32P]ATP (10 μCi, 66 μm, unless indicated otherwise). The reaction buffers contained either 89 μg/ml phosphatidylserine plus 175 nm PMA or 30 μg/ml dextran sulfate, as indicated; control experiments establish that these concentrations result in maximal activation of WT-PKD1. Assays contained either 4 μg of troponin complex (consisting of equimolar concentrations of cardiac troponin I, cardiac troponin T, and cardiac troponin C, generously provided by Drs. John Solaro and Marius Sumandea) or recombinant human CREB-maltose-binding protein fusion construct (CREB-MBP, 1 μg/assay; BIOSOURCE). Incubations were for 30 min at 30 °C. PKD1 autophosphorylation was tracked with the CST anti-PKD1-Ser(P)744/Ser(P)748 (which is reported to track primarily Ser744 phosphorylation), the Cell Signaling Technology anti-PKD1-Ser(P)916 phosphorylation state specific antibodies, and the Abcam anti-PKD1-Ser(P)742 antibody (which was recently characterized as relatively selective for Ser742 in human PKD1, corresponding to Ser748 in rodent PKD1 (2)). Control experiments established that activity in these assays cannot be attributed to the co-precipitation of endogenous PKD1, PKD2, or other enzymes with Ser916 kinase activity; studies showing that endogenous PKD1 and PKD2 enzymes do not co-immunoprecipitate with PKD1-CD under our assay conditions and that kinase-inactive PKD1 pull-downs lack Ser916 kinase activity have been published (2, 10).
CREBtide kinase assays were performed in a similar manner, with 0.36 mg/ml CREBtide (or the indicated concentration of CREBtide in Fig. 3) included as the PKD1 substrate. Assays were terminated by adding 30 μl of 350 mm phosphoric acid followed by centrifugation at 14,000 × g for 10 min. 50 μl of each supernatant was spotted onto phosphocellulose filter papers (P-81), dropped into 75 mm phosphoric acid, washed (three times for 5 min), dried, and counted for radioactivity.
FIGURE 3.

PKD1-ΔPH is a constitutively phosphorylated enzyme that displays a high level of CREBtide and CREB kinase activity. WT-PKD1 and PKD1-ΔPH immunoprecipitated from HEK293 cells were used in IVKAs without or with PMA or DS (with CREB as substrate) as described under “Experimental Procedures.” Left, autoradiography and immunoblotting for PKD1 activation loop and Ser916 phosphorylation. Right, 32P incorporation into PKD1 (top) or CREB (bottom) was quantified by PhosphorImager. The data are from a single experiment, with identical results obtained in two separate experiments.
RESULTS
Initial studies compared the in vitro catalytic activities of WT-PKD1, PKD1-Δ1–321 (a PKD1 N-terminal truncation mutant lacking the extreme N terminus and the C1 domain), and PKD1-CD (a PKD1 catalytic domain fragment lacking the entire regulatory domain; Fig. 1). Assays were performed in parallel with either full-length CREB or cTnI as substrate, with the following end points used to track PKD1 activity. 1) PKD1 autophosphorylation was tracked as 32P incorporation into PKD1 by PhosphorImager analysis. 2) PKD1 autophosphorylation at Ser916 was detected by immunoblot analysis. 3) PKD1 activation loop autophosphorylation was tracked with anti-PKD-Ser(P)744/Ser(P)748, which primarily recognizes PKD1-Ser744 phosphorylation, and anti-PKD1-Ser(P)748, which preferentially recognizes PKD1-Ser748 phosphorylation (nomenclature based upon rodent sequence (2, 3)). 4) PKD1 phosphorylation of full-length CREB or cTnI was tracked by immunoblot analysis with phosphorylation state specific antibodies that recognize CREB-Ser133 or cTnI-Ser23/Ser24 phosphorylation and by PhosphorImager analysis to track 32P incorporation into these proteins. Finally, a separate series of kinase assays were performed in parallel to track PKD1 phosphorylation of CREBtide, a 15-residue peptide based upon the PKD phosphorylation site at Ser133 in CREB (CKRREILSRRPS*YRK). We used CREBtide (rather than syntide-2, the peptide used in most previous studies) to avoid sequence differences that might influence substrate recognition/phosphorylation; this permits comparisons of PKD1 activity toward peptide (CREBtide) versus protein (CREB) substrates.
FIGURE 1.
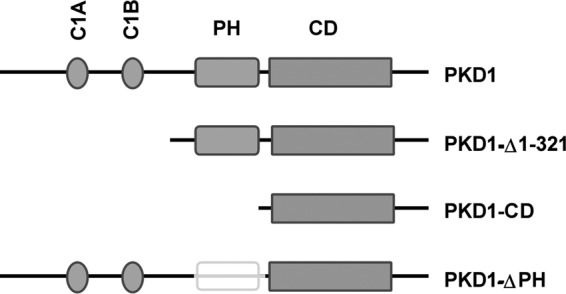
Schematic of PKD1 structure and mutants used in this study. PKD1 contains tandem C1 domains, a PH domain, and a CD. PKD1-Δ1–321, deletion of the first 321 residues at the N-terminal domain; PKD1-CD, catalytic domain fragment; PKD1-ΔPH, deletion of the PH domain.
Fig. 2 shows that WT-PKD1 is activated in vitro by PMA and by dextran sulfate, another potent PKD1 agonist (11). PMA and dextran sulfate increase WT-PKD1 autophosphorylation, detected as an increase in PKD1 autophosphorylation at Ser916 (Fig. 2A), a decrease in the electrophoretic mobility of the protein in SDS-PAGE (Fig. 2A), and an increase in 32P incorporation into the enzyme (Fig. 2B). PMA and dextran sulfate induce similar high levels of 32P incorporation into WT-PKD1 and WT-PKD1-Ser916 autophosphorylation. However, WT-PKD1 activation loop autophosphorylation is prominent only in assays with PMA; the dextran sulfate-activated WT-PKD1 enzyme displays only a very modest increase in activation loop autophosphorylation. Nevertheless, PMA- and dextran sulfate-activated WT-PKD1 enzymes display similar high levels of trans phosphorylation of CREBtide (Fig. 2E). PMA and dextran sulfate also increase PKD1 activity toward full-length CREB or cTnI, measured by immunoblot analysis as CREB-Ser133 or cTnI-Ser23/Ser24 phosphorylation (Fig. 2A) or by PhosphorImager analysis as 32P incorporation (Fig. 2, C and D). In fact, the level of CREB and cTnI phosphorylation in assays with dextran sulfate (which elicits only a minor increase in activation loop phosphorylation) is considerably higher than in assays with PMA; these agonist-dependent differences in CREB and cTnI phosphorylation were detected both in immunoblotting studies that track CREB-Ser133 and cTnI-Ser23/Ser24 phosphorylation (Fig. 2A) and by PhosphorImager analyses quantifying 32P incorporation (Fig. 2, C and D). Although the dextran sulfate-dependent increase in phosphorylation might in theory be due to a change in the conformation of substrates used in the kinase assays, an effect on the enzyme is considerably more likely because 1) dextran sulfate exerts an obvious effect on the enzyme (it slows the electrophoretic mobility of PKD1 and enhances PKD1-Ser916 autophosphorylation); 2) dextran sulfate increases PKD1 phosphorylation of CREB and cTnI, two structurally different substrates; and 3) dextran sulfate does not enhance CREB-Ser133 phosphorylation by protein kinase A (data not shown). An effect of dextran sulfate to alter the conformation of two different target substrates (CREB and cTnI) in a manner that specifically enhances their phosphorylation by PKD1 but not by PKA is highly unlikely.
FIGURE 2.

PKD1-Δ1–321 and PKD1-CD are CREBtide kinases that autophosphorylate at Ser916 but do not effectively autophosphorylate their activation loop or trans phosphorylate CREB or cTnI. WT-PKD1, PKD1-Δ1–321, and PKD1-CD were immunoprecipitated from HEK293 cells and used in IVKAs in buffers without or with PMA or dextran sulfate (with CREB, cTnI, or CREBtide as substrate) as described under “Experimental Procedures.” A, immunoblotting for PKD1 activation loop and Ser916 autophosphorylation, PKD1 protein (to show similar recovery and loading of each enzyme), CREB-Ser133 phosphorylation, and cTnI-Ser23/Ser24 phosphorylation. B, C, and D, 32P incorporation into PKD1, cTnI, and CREB quantified by PhosphorImager. All results are from a single experiment, with similar results obtained in two separate experiments. E, PKD1-dependent CREBtide phosphorylation was examined in triplicate as described under “Experimental Procedures”; data are from a single experiment, with identical results obtained in two separate experiments. F, WT-PKD1 and PKD1-S744A/S748A (PKD1-SS/AA) were immunoprecipitated from HEK293 cells, and IVKAs were performed in buffers without or with PMA or DS with CREB as substrate. Immunoblotting was for PKD1-Ser916 and CREB-Ser133 phosphorylation or PKD1 protein (to show equal recovery of WT-PKD1 and PKD1-SS/AA), with 32P incorporation into CREB quantified by PhosphorImager. Data are from a single experiment that was replicated in two separate experiments.
PKD1-Δ1–321 is predicted to display characteristics of a constitutively active enzyme because it harbors an N-terminal deletion that encompasses the C1 domain. However, Fig. 2 shows that deletion of the N-terminal half of the regulatory domain leads to an increase in only certain basal catalytic activities. PKD1-Δ1–321 displays high basal autocatalytic activity (measured as 32P incorporation by PhosphorImager and Ser916 autophosphorylation by immunoblot analysis) and high basal CREBtide activity (Fig. 2, A, B, and E). These results replicate the phenotype originally described for PKD1-ΔC1 (which harbors a more restricted deletion of the C1 domain, residues 145–353 (7)). However, additional measurements (not included in previous studies of the PKD1-ΔC1 mutant) show that PKD1-Δ1–321 does not autophosphorylate at Ser744 or Ser748, and it displays no basal activity toward full-length CREB or cTnI (Fig. 2, A, C, and D). A PKD1-CD mutant lacking the entire regulatory domain shows a similar phenotype; PKD1-CD retains basal Ser916 (and to a lesser extent Ser748) autocatalytic activity and CREBtide kinase activity, but PKD1-CD does not undergo an electrophoretic mobility shift during the IVKA, it incorporates only trace amounts of 32P (by PhosphorImager analysis), it does not autophosphorylate at Ser744, and it does not phosphorylate full-length CREB and cTnI. Collectively, these results indicate that PKD1 truncation mutants harboring a deletion of the entire regulatory domain (PKD1-CD) or the N-terminal half of the regulatory domain (PKD1-Δ1–321) display constitutive activity toward peptide but not protein substrates.
We previously demonstrated that a PKD1 mutant harboring an S744A/S748A substitution retains in vitro lipid-dependent Ser916 autocatalytic activity, but no lipid cofactor-dependent CREB kinase activity, and that PKD1-S744A/S748A phosphorylates CREB when assays are performed with dextran sulfate. This result (which is replicated in Fig. 2F) argues that dextran sulfate activates PKD1 via a mechanism that bypasses the requirement for activation loop phosphorylation. Because PKD1-Δ1–321 and PKD1-CD display similar activation loop autophosphorylation defects, we examined whether dextran sulfate rescues the CREB and cTnI kinase activity of these mutants. Fig. 2, A–E, shows that PKD1-Δ1–321 and PKD1-CD acquire CREB and cTnI kinase activity when assays are performed with dextran sulfate.
The observation that the PKD1-Δ1–321 mutant (lacking the C1 domain) displays a defect in trans phosphorylation of protein substrates runs counter to the prevailing notion that the C1 domain is an inhibitory regulator of PKD1 activity. However, it is worth noting that 1) PKD1-ΔC1 was originally characterized as a constitutively active enzyme based on studies that tracked in vitro autophosphorylation or phosphorylation of syntide-2 (7) (activity toward physiologically relevant protein substrates was not examined and might differ), and 2) this conclusion has been disputed; Hausser et al. (12) identified similar basal (lipid-independent) autocatalytic activities for WT-PKD1 and PKD1-ΔC1.
We performed a similar analysis of the in vitro kinase activity of PKD-ΔPH, a PH domain truncation mutant that was also previously characterized as a constitutively active enzyme (8). Fig. 3 shows that PKD1-ΔPH is recovered from resting HEK293 cells as a constitutively phosphorylated/active enzyme. PKD1-ΔPH displays a high level of basal Ser744, Ser748, and Ser916 phosphorylation. PKD1-ΔPH autophosphorylates further (incorporates 32P and undergoes an electrophoretic mobility shift) during in vitro kinase assays without or with agonists (PMA or dextran sulfate). PKD1-ΔPH also displays a high level of basal CREB kinase activity that is not further increased by PMA or dextran sulfate. A similar high level of basal PKD1-ΔPH phosphorylation of cTnI also was detected (data not shown). Quantification of a series of experiments by PhosphorImager analysis showed that basal 32P incorporation into CREB is 18.2 ± 1.5% higher in assays with PKD1-ΔPH than in assays with WT-PKD1 (n = 4, p < 0.05; representative experiment in Fig. 3). Collectively, these results indicate that the basal phosphorylation and activity profiles of PKD1-ΔPH and PKD1-Δ1–321 are vastly different; only PKD1-ΔPH is recovered with a high level of constitutive Ser744/Ser748 phosphorylation and CREB kinase activity. These results challenge the notion that an N-terminal deletion encompassing the C1 domain generates a constitutively active enzyme. Rather, these results indicate that the C1 and PH domains exert distinct effects on PKD1 activity; the PH domain engages in autoinhibitory interactions that restrict PKD1 activity, whereas the extreme N-terminal region of the regulatory domain (containing the C1 domain) is required for optimal PKD1 activity.
An N-terminal deletion that removes the C1 domain prevents in vitro PMA-dependent PKD1 activation (Fig. 2) and in vivo PKD1 co-localization with activated forms of PKC at lipid membranes (13), but Fig. 4A shows that the C1 domain deletion does not completely abrogate in vivo PMA responsiveness. WT-PKD1 and PKD1-Δ1–321 are recovered from resting cells with similar low levels of constitutive Ser916 and no detectable phosphorylation at Ser744/Ser748. PMA treatment results in a robust increase in WT-PKD1 phosphorylation at Ser744/Ser748 and Ser916 and a more modest increase in PKD1-Δ1–321 phosphorylation at Ser744/Ser748 and Ser916. The observation that PKD1-Δ1–321 retains some in vivo PMA responsiveness was surprising and provided the rationale to examine whether PKD1-Δ1–321 might acquire CREB kinase activity in vivo in PMA-treated cells.
FIGURE 4.
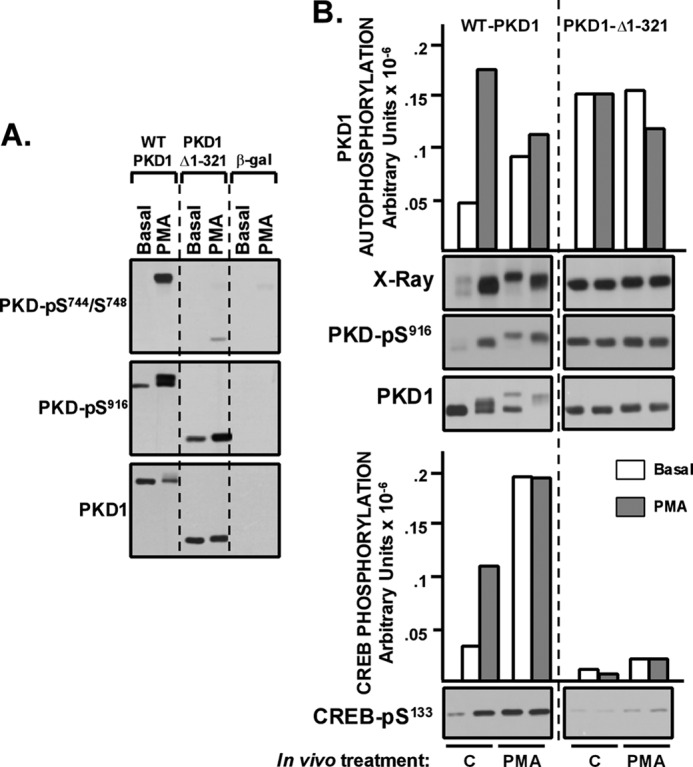
PKD1-Δ1–321 does not acquire CREB kinase activity in vivo in PMA-treated HEK293 cells. HEK293 cells that heterologously overexpress WT-PKD1, PKD1-Δ1–321, or β-galactosidase were challenged for 15 min with vehicle or 300 nm PMA. A, immunoblotting with the indicated antibodies on cell lysates. Endogenous PKD1 expression and phosphorylation is readily detected in β-gal cultures with increased protein loading and more prolonged gel exposure times. B, WT-PKD1 and PKD1-Δ1–321 were immunoprecipitated from resting or PMA-treated cultures and then subjected to IVKAs without (open bars) or with (hatched bars) PMA. 32P incorporation into PKD1 (top) or CREB (bottom) was quantified by PhosphorImager; PKD1-Ser916 and CREB-Ser133 phosphorylation was tracked by immunoblot analysis. The results are from a single experiment and were replicated in two separate experiments.
Fig. 4B shows that WT-PKD1 is recovered from resting HEK293 cells with low basal activity. WT-PKD1 activity is increased by in vitro exposure to PMA. Under these conditions, WT-PKD1 migrates as a doublet, with the slower migrating form of WT-PKD1 displaying a high level of lipid-independent autocatalytic activity (detected as increased 32P incorporation by PhosphorImager). WT-PKD1 is recovered from PMA-treated cells with a high level of lipid-independent CREB kinase activity. In contrast, PKD1-Δ1–321 is recovered from resting or PMA-treated HEK293 cells with a high level of basal autocatalytic activity but only a trivial amount of CREB kinase activity. PKD1-Δ1–321 activity is not influenced by in vitro stimulation with PMA. These results indicate that an N-terminal truncation of PKD1 (that removes the C1 domain) interferes with the in vivo acquisition of CREB kinase activity.
Finally, we examined whether truncation of the N-terminal portion of the regulatory domain leads to a defect in PKD1-dependent cellular responses. Fig. 5 shows that heterologous overexpression of WT-PKD1 leads to an increase in basal phosphorylation of CREB and ERK and that PKD1-Δ1–321 does not substitute for WT-PKD1 as an in vivo activator of CREB or ERK.
FIGURE 5.
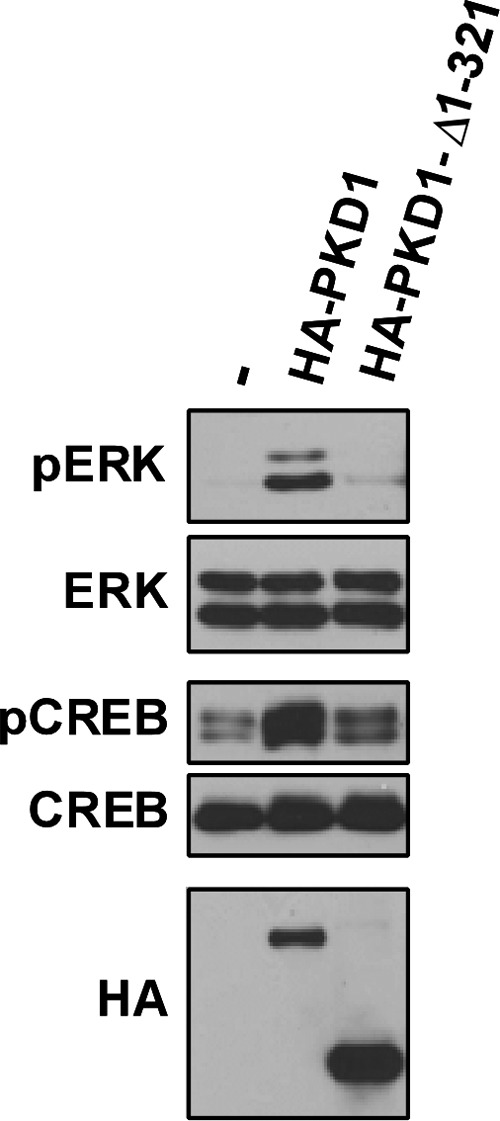
PKD1-Δ1–321 does not mimic the in vivo actions of WT-PKD1 to increase basal ERK and CREB phosphorylation in NIH3T3 cells. PKD1 signaling responses were examined in NIH3T3 cells because WT-PKD1 overexpression did not increase the already high levels of CREB and ERK phosphorylation in the HEK293 cell cultures used in our studies. Lysates from NIH3T3 cells that heterologously overexpress WT-PKD1 or PKD1-Δ1–321 were subjected to immunoblot analysis with the indicated antibodies. Similar results were obtained in three separate experiments.
DISCUSSION
PKD1 has recently emerged as a key signaling enzyme that is activated by many physiologically relevant stimuli. Current dogma holds that PKD1 activity is maintained at low basal levels in resting cells as a result of intramolecular autoinhibitory interactions involving both the C1 and PH domains. The observation that the PKD1-ΔPH mutant is recovered from resting COS7 cells as a constitutively Ser744-, Ser748-, and Ser916-phosphorylated enzyme that displays a high level of basal catalytic activity toward protein and peptide substrates is consistent with the notion that the PH domain acts as an autoinhibitory regulator of PKD1 activity. In contrast, the activity profile of the PKD1-Δ1–321 mutant (which harbors a C1 domain deletion and is predicted to mimic the signaling phenotype of the PKD1-ΔC1 mutant) is quite different. PKD1-Δ1–321 is recovered from resting cells as a Ser916-phosphorylated enzyme with enhanced CREBtide kinase activity, but it lacks key characteristics of a constitutively active enzyme; the N-terminal deletion that removes the C1 domain and flanking sequence disrupts activation loop autocatalytic activity, and it prevents phosphorylation of protein substrates, such as CREB and cTnI. Combined deletion of both the C1 and PH domains in the PKD1-CD construct leads to a similar activity profile; PKD1-CD retains basal CREBtide kinase activity, but it displays decreased basal activation loop autocatalytic activity and no basal activity toward full-length CREB or cTnI. Of note, PKD1-Δ1–321 and PKD1-CD acquire CREB and cTnI kinase activity (but no activation loop autocatalytic activity) when assays are performed with dextran sulfate, an agonist that activates PKD1 via a mechanism that bypasses the requirement for activation loop phosphorylation. Collectively, these results argue that the C1 domain is required for activation loop autocatalytic activity and that the activation loop autophosphorylation defects displayed by PKD1-Δ1–321 and PKD1-CD mutants contribute to their defects in CREB and cTnI kinase activity. Of note, a previous study described a similar role for the C1 domain as a positive regulator of PKD2 catalytic activity (14).
The N-terminal PKD1-Δ1–321 truncation mutant used in this study models a PKD1 cleavage product that accumulates during apoptosis. Although the precise site for caspase-3-dependent cleavage of PKD1 has been disputed, there is general consensus that cleavage is in the C1-PH interdomain; caspase-3-dependent cleavage has variably been mapped to CQND378↓S (a sequence in human PKD1 that is not conserved in rodent or other PKD1s and does not conform to a consensus DXXD caspase-3 recognition motif (4)) or the more evolutionarily conserved caspase-3 recognition sites at DDND355S and DQED397S (5, 6). The functional consequences of PKD1 cleavage remain more ambiguous because proteolysis has variably been implicated as a mechanism to activate or inactivate PKD1 (5, 9). Our studies provide a framework to reconcile the seemingly conflicting data from previous studies; we show that conclusions regarding the molecular controls of PKD1 activity can be heavily swayed by the choice of end point used for the analysis. For example, the PKD1-Δ1–321 truncation mutant displays constitutive lipid-independent autocatalytic and CREBtide kinase activity, but it does not phosphorylate physiologically relevant protein substrates. Cell-based studies provide further evidence that cleavage limits the maximal activity of PKD1, showing that PKD1-Δ1–321 does not substitute for WT-PKD1 as an in vivo activator of CREB or ERK. These results are consistent with the recent finding that the action of PKD1 to regulate lipoprotein lipase-mediated triglyceride accumulation in cardiomyocytes is lost during apoptosis, under conditions associated with the activation of caspase-3 and caspase-3-dependent cleavage of PKD1 (15).
Studies reported herein emphasize that the molecular controls of PKD1-Ser916 autophosphorylation, PKD1 phosphorylation of model peptides, and PKD1 phosphorylation of heterologous protein substrates differ considerably. Several factors may contribute to the discrepant results obtained in these assays. First, there is growing evidence that certain cis autophosphorylations are “simple” reactions that proceed under conditions that do not support trans phosphorylation of target substrates. We previously reported that PKD1-Ser916 autophosphorylation is mediated by a cis autocatalytic reaction that proceeds under conditions (such as at exceedingly low ATP concentrations or in the absence of activation loop phosphorylation) that do not support the phosphorylation of heterologous substrates (2). Second, assays that track phosphorylation of peptides (syntide-2 or CREBtide) versus proteins (CREB or cTnI) might yield divergent results due to a generally overlooked difference in assay conditions. PKD1-dependent phosphorylation of peptides typically is tracked in assays containing 50–200 μm syntide-2 or CREBtide, concentrations at or above the Km for substrate. In contrast, protein phosphorylation is monitored in assays containing ∼0.1 μm full-length CREB-MBP because practical issues related to cost and protein insolubility preclude the use of comparably high concentrations of full-length CREB-MBP in kinase assays. A difference in substrate concentration could be pertinent for at least two reasons. First, a regulatory event (such as an intramolecular interaction involving the C1 domain) that decreases PKD1 catalytic efficiency or affinity for substrate would be most apparent in assays with limiting amounts of substrate; it might be obscured in assays performed with saturating concentrations of substrate. Alternatively, a docking interaction with a high concentration of CREBtide (either at the substrate binding site or an allosteric regulatory site) might in theory stabilize the active conformation of PKD1 and rescue a catalytic defect resulting from an inactivating mutation (such as a C1 domain deletion). In fact, the observation that CREBtide and dextran sulfate act in a similar manner to rescue PKD1-Δ1–321 activity could suggest that these compounds share a similar (activation loop-independent) allosteric mechanism to increase PKD1 activity.
This study identifies elaborate and generally underappreciated complexities in the regulation of PKD1 activity that are exposed only when studies of enzyme activity are broadened to examine activity toward physiologically relevant protein substrates. Our results emphasize that various assays typically used to track PKD1 activity may provide conflicting information and that assays that track autophosphorylation or phosphorylation of peptide substrates are not necessarily valid surrogate readouts of PKD1 activity toward more physiologically relevant protein substrates. The approach used in this study to define the biochemical and cellular actions of regulatory domain truncation mutants of PKD1 should provide a useful road map for future research designed to elucidate the molecular controls of PKD1 activity.
This work was supported, in whole or in part, by National Institutes of Health (NIH), NHLBI, Grant HL 77860 and National Center for Research Resources (NCRR) (a component of the NIH and NIH Roadmap for Medical Research) Grant TL1 RR024158.
- PKD
- protein kinase D
- PH
- pleckstrin homology
- PMA
- phorbol 12-myristate 13-acetate
- CREB
- cAMP-response element-binding protein
- cTnI
- cardiac troponin I
- MBP
- maltose-binding protein
- CD
- catalytic domain
- IVKA
- in vitro kinase assay
- DS
- dextran sulfate.
REFERENCES
- 1. Avkiran M., Rowland A. J., Cuello F., Haworth R. S. (2008) Protein kinase D in the cardiovascular system. Emerging roles in health and disease. Circ. Res. 102, 157–163 [DOI] [PubMed] [Google Scholar]
- 2. Rybin V. O., Guo J., Steinberg S. F. (2009) Protein kinase D1 autophosphorylation via distinct mechanisms at Ser744/Ser748 and Ser916. J. Biol. Chem. 284, 2332–2343 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Jacamo R., Sinnett-Smith J., Rey O., Waldron R. T., Rozengurt E. (2008) Sequential protein kinase C (PKC)-dependent and PKC-independent protein kinase D catalytic activation via Gq-coupled receptors. Differential regulation of activation loop Ser744 and Ser748 phosphorylation. J. Biol. Chem. 283, 12877–12887 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Endo K., Oki E., Biedermann V., Kojima H., Yoshida K., Johannes F. J., Kufe D., Datta R. (2000) Proteolytic cleavage and activation of protein kinase Cμ by caspase-3 in the apoptotic response of cells to 1-β-d-arabinofuranosylcytosine and other genotoxic agents. J. Biol. Chem. 275, 18476–18481 [DOI] [PubMed] [Google Scholar]
- 5. Vántus T., Vertommen D., Saelens X., Rykx A., De Kimpe L., Vancauwenbergh S., Mikhalap S., Waelkens E., Kéri G., Seufferlein T., Vandenabeele P., Rider M. H., Vandenheede J. R., Van Lint J. (2004) Doxorubicin-induced activation of protein kinase D1 through caspase-mediated proteolytic cleavage. Identification of two cleavage sites by microsequencing. Cell Signal 16, 703–709 [DOI] [PubMed] [Google Scholar]
- 6. Cryns V., Yuan J. (1998) Proteases to die for. Genes Dev. 12, 1551–1570 [DOI] [PubMed] [Google Scholar]
- 7. Iglesias T., Rozengurt E. (1999) Protein kinase D activation by deletion of its cysteine-rich motifs. FEBS Lett. 454, 53–56 [DOI] [PubMed] [Google Scholar]
- 8. Iglesias T., Rozengurt E. (1998) Protein kinase D activation by mutations within its pleckstrin homology domain. J. Biol. Chem. 273, 410–416 [DOI] [PubMed] [Google Scholar]
- 9. Häussermann S., Kittstein W., Rincke G., Johannes F. J., Marks F., Gschwendt M. (1999) Proteolytic cleavage of protein kinase Cμ upon induction of apoptosis in U937 cells. FEBS Lett. 462, 442–446 [DOI] [PubMed] [Google Scholar]
- 10. Guo J., Gertsberg Z., Ozgen N., Sabri A., Steinberg S. F. (2011) Protein kinase D isoforms are activated in an agonist-specific manner in cardiomyocytes. J. Biol. Chem. 286, 6500–6509 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Gschwendt M., Johannes F. J., Kittstein W., Marks F. (1997) Regulation of protein kinase Cμ by basic peptides and heparin. Putative role of an acidic domain in the activation of the kinase. J. Biol. Chem. 272, 20742–20746 [DOI] [PubMed] [Google Scholar]
- 12. Hausser A., Link G., Bamberg L., Burzlaff A., Lutz S., Pfizenmaier K., Johannes F. J. (2002) Structural requirements for localization and activation of protein kinase C mu (PKC μ) at the Golgi compartment. J. Cell Biol. 156, 65–74 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Oancea E., Bezzerides V. J., Greka A., Clapham D. E. (2003) Mechanism of persistent protein kinase D1 translocation and activation. Dev. Cell 4, 561–574 [DOI] [PubMed] [Google Scholar]
- 14. Auer A., von Blume J., Sturany S., von Wichert G., Van Lint J., Vandenheede J., Adler G., Seufferlein T. (2005) Role of the regulatory domain of protein kinase D2 in phorbol ester binding, catalytic activity, and nucleocytoplasmic shuttling. Mol. Biol. Cell 16, 4375–4385 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Kim M. S., Wang F., Puthanveetil P., Kewalramani G., Innis S., Marzban L., Steinberg S. F., Webber T. D., Kieffer T. J., Abrahani A., Rodrigues B. (2009) Cleavage of protein kinase D after acute hypoinsulinemia prevents excessive lipoprotein lipase-mediated cardiac triglyceride accumulation. Diabetes 58, 2464–2475 [DOI] [PMC free article] [PubMed] [Google Scholar]