

Background: Decreased antioxidant activity is implicated in neurodegenerative diseases, including Huntington disease (HD).
Results: Mutant Huntingtin (mHtt) triggered loss of peroxiredoxin 1 (Prx1) expression in two nerve cell models. Dimercaptopropanol treatment attenuated both mHtt-induced toxicity and loss of Prx1 expression.
Conclusion: Chemical or genetic means of maintaining Prx1 expression counters mHtt toxicity.
Significance: Dithiol compounds may offer new treatment options for neurodegenerative diseases.
Keywords: Disulfide, Neurodegeneration, Neuroprotection, Oxidative Stress, Peroxiredoxin, Polyglutamine Disease, Reactive Oxygen Species (ROS), Redox Signaling, Thiol, Huntington Disease
Abstract
Mitochondrial dysfunction and elevated reactive oxygen species are strongly implicated in both aging and various neurodegenerative disorders, including Huntington disease (HD). Because reactive oxygen species can promote the selective oxidation of protein cysteine sulfhydryl groups to disulfide bonds we examined the spectrum of disulfide-bonded proteins that were specifically altered in a HD context. Protein extracts from PC12 cells overexpressing the amino-terminal fragment of the Huntingtin (Htt) protein with either a nonpathogenic or pathogenic polyglutamine repeat (Htt-103Q) were resolved by redox two-dimensional PAGE followed by mass spectrometry analysis. Several antioxidant proteins were identified that exhibited changes in disulfide bonding unique to Htt-103Q expressing cells. In particular, the antioxidant protein peroxiredoxin 1 (Prx1) exhibited both decreased expression and hyperoxidation in response to mutant Htt expressed in either PC12 cells or immortalized striatal cells exposed to 3-nitropropionic acid. Ectopic expression of Prx1 in PC12 cells attenuated mutant Htt-induced toxicity. In contrast, short hairpin RNA-mediated knockdown of Prx1 potentiated mHtt toxicity. Furthermore, treatment with the dithiol-based compounds dimercaptopropanol and dimercaptosuccinic acid suppressed toxicity in both HD cell models, whereas monothiol compounds were relatively ineffective. Dimercaptopropanol treatment also prevented mutant Htt-induced loss of Prx1 expression in both cell models. Our studies reveal for the first time that pathogenic Htt can affect the expression and redox state of antioxidant proteins; an event countered by specific dithiol-based compounds. These findings should provide a catalyst to explore the use of dithiol-based drugs for the treatment of neurodegenerative diseases.
Introduction
Huntington disease (HD)3 is an inherited adult onset neurodegenerative disorder characterized clinically by chorea, psychiatric disturbances and dementia, and pathologically by the loss of striatal and cortical cell neurons. The disease is caused by an expansion of a CAG trinucleotide repeat region in exon 1 of the HTT gene, which encodes Huntingtin (Htt), a ubiquitously expressed protein in the brain and peripheral tissues with an uncertain molecular function (1). Individuals with HD have a CAG expansion that results in enlargement of the polyglutamine (poly(Q)) tract within the N terminus of Htt to greater than 36 residues. Longer poly(Q) stretches are associated with earlier onset of HD and more severe disease symptoms (2). The precise mechanism of HD pathophysiology is poorly defined but evidence exists that multiple neurodegenerative pathways are involved including mitochondrial impairment, oxidative stress, transcriptional dysregulation, elevated apoptosis, changes in intracellular transport, signaling dysfunction, and altered protein interactions and activity (1).
Mutant Htt (mHtt) containing a poly(Q) repeat greater than 36 has a high predisposition to misfold and disrupt normal processes essential for cellular homeostasis (3). Among these, mitochondrial dysfunction and elevated reactive oxygen species (ROS) production are strongly involved in HD progression (4). Although mitochondria produce most of the cellular ATP, they are also a major source of ROS production via electron leakage from the respiratory chain (especially complexes I and III). Several studies have shown that mHtt is found in association with the outer mitochondrial membrane in brain tissue from HD transgenic mice and in isolated mitochondria from both lymphoblasts and postmortem brain tissue from HD patients (5–7). In addition, isolated mitochondria from HD mice exhibit decreased membrane potential, increased propensity to depolarize at lower calcium loads, and elevated sensitivity to calcium-induced cytochrome c release compared with controls (5, 6). Transcription of peroxisome proliferator-activated receptor, a coactivator 1α (PGC1α), a key transcriptional co-activator that induces expression of genes that regulate mitochondrial respiration and oxidative stress, is repressed in mHtt-expressing neurons (8). Impaired mitochondrial respiration and ATP synthesis have been detected in postmortem brain samples from HD patients and in various HD cell and animal models (9). Collectively these findings strongly indicate that perturbed mitochondrial function contributes to HD pathogenesis.
Expression of mHtt in cultured non-neuronal or neuronal cells has been shown to increase both ROS production and toxicity, which can be rescued by treatment with the thiol-based antioxidants N-acetyl-l-cysteine (NAC) and glutathione (GSH) (10, 11). Antioxidant treatment has also been shown to reduce cytotoxicity associated with the expression of mHtt in transgenic nematode and mouse models of HD (12, 13). However, the exact mechanism by which antioxidants counter mHtt-induced toxicity is poorly defined.
Although ROS have traditionally been viewed as agents that cause nonspecific damage to DNA, lipids, and proteins, recent evidence has shown that ROS can act as second messengers and selectively target proteins leading to a change in their activity or function (14). The specificity of ROS-mediated signaling has become an extremely active area of research over the last decade. Cysteine represents one of the major amino acids modified by ROS in a highly selective and reversible manner. Oxidation of protein cysteine sulfhydryl groups (Cys-SH) can lead to the formation of covalent disulfide bonds (Cys-S-S-Cys) or higher oxidized species such as sulfenic (Cys-SOH), sulfinic (Cys-SO2H), or sulfonic (Cys-SO3H) acids. Functional consequences of Cys-SH oxidation include altered protein interactions, misfolding, catalytic inactivation, and decreased antioxidant capacity (15). The specificity of ROS-mediated oxidation of cysteine residues within proteins that participate in a wide array of cellular processes has made these proteins attractive targets for therapeutic manipulation in diseases strongly linked to oxidative stress (16).
One of the best examples of redox regulation is found in the peroxiredoxin family of proteins. Peroxiredoxins (Prxs) were originally characterized as abundant antioxidant proteins that contain a catalytic cysteine residue and detoxify hydrogen peroxide (H2O2), peroxynitrate, and a range of organic hydroperoxides using reducing equivalents supplied by the thioredoxin system (17). However, recent studies have shown that Prxs participate in a wide array of cellular processes including neuronal differentiation, cell signaling, molecular chaperoning, and mitochondrial function in both a catalytic dependent and independent manner (18–20). For example, Prxs are able to interact with and inactivate protein kinases such as JNK, c-Abl, and ASK1 in a redox-regulated manner (21).
In an effort to determine the spectrum of disulfide-bonded proteins (DSBP) that are altered in an HD context, and hence potential targets of antioxidants, we resolved protein extracts from PC12 cells that inducibly express exon 1 of the HTT gene with either a 25 (nonpathogenic) or 103 (pathogenic) poly(Q) repeat using a novel two-dimensional polyacrylamide gel electrophoresis (PAGE) technique to separate DSBP. Following mass spectrometry analysis, a number of antioxidant proteins were identified that displayed alterations in disulfide bonding only in Htt-103Q expressing cells. In particular, Prx1 was shown to exhibit a progressive decrease in expression and a concomitant increase in protein sulfonylation following induction of mHtt expression. Testing of various thiol-based antioxidants revealed that dimercaptopropanol (DMP) and the structurally related compound dimercaptosuccinic acid (DMSA) were specifically able to rescue mHtt-induced toxicity in PC12 cells, whereas monothiol reducing agents were relatively ineffective. In addition, DMP was able to protect against 3-nitropropionic acid-induced toxicity in a rodent HD striatal cell line. DMP-mediated protection correlated with the maintenance of Prx expression and suppression of Prx1 sulfonylation. These novel findings suggest that dithiol-based compounds can selectively protect against mHtt-induced toxicity.
EXPERIMENTAL PROCEDURES
Reagents
Cell culture reagents including Dulbecco's modified Eagle's medium (DMEM), penicillin/streptomycin, DMEM without phenol red, and Dulbecco's phosphate-buffered saline (DPBS) were purchased from BioWhittaker (Walkersville, MD). Fetal bovine serum (FBS) and horse serum were obtained from PAA Laboratories Inc. (Etobicoke, ON, Canada). Bisbenzimide (Hoechst), poly-d-lysine, 3-(4,5-dimethlythiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT), 3-nitropriopionic acid (3-NP), cysteamine (Cys), penacillamine (Pen), β-mercaptoethanol (β-ME), dithiothreitol (DTT), NAC, glutathione-reduced ethyl ester (eeGSH), DMP, and DMSA were all purchased from Sigma. Opti-MEM media, Lipofectamine 2000, and Mitotracker Red CM-H2XRos (Molecular Probes) was purchased from Invitrogen.
Cell Lines
The PC12 HD nerve cell model was obtained from Dr. Erik Schweitzer (UCLA) and maintained in DMEM supplemented with 10% horse serum, 5% FBS, and 1% penicillin/streptomycin. These cells are stably transfected with a plasmid encoding the entire exon 1 of the HD gene with either a 25 (nonpathogenic) or 103 (pathogenic) poly(Q) repeat fused in-frame to the coding sequence for the enhanced GFP at the carboxyl terminus (22) under control of an ecdysone-regulated promoter. Transcription of the transgene was activated by the addition of 2.5 μm tebufenicide (an ecdysone analog) to the culture medium. These cell lines exhibit extensive cell death (>50%) 48 h after induction of Htt-103Q expression (22). Immortalized striatal cell lines derived from a knock-in transgenic mouse containing homozygous htt loci with a humanized exon 1 with either 7 (STHdhQ7) or 111 (STHdhQ11) poly(Q) repeats (23) were obtained from the Corriel Institute (Camden, NJ) and maintained in DMEM supplemented with 10% FBS and 1% penicillin/streptomycin at 33 °C in a humidified CO2 incubator. Striatal cells were treated with 10 mm 3-nitropriopionic acid for up to 48 h.
Determination of Mitochondrial Produced ROS
Mitochondrial ROS production was visualized using the fluorescent dye Mitotracker Red CM-H2XRos (Invitrogen). PC12 Htt-25Q-GFP and Htt0103Q-GFP cells were seeded at 1 × 105 in polylysine-treated 6-well glass bottom dishes and induced the following day with 2.5 μm tebufenocide. At 24 and 48 h post-induction, medium was aspirated and cells were incubated in DMEM containing 200 nm Mitotracker Red for 20 min at 37.5 °C in a humidified CO2 incubator. The medium was removed and the cells were incubated for 5 min with PBS containing 10 μg/ml of Hoechst stain to stain nuclei, followed by two washes with PBS. Phenol Red-free DMEM was added to the culture prior to visualization using a Zeiss AxioObserver A1 epifluorescent inverted microscope with a ×100 oil objective and equipped with appropriate filters. Pictures were taken of three random fields of view for each experiment using a Q Imaging (Retiga 1300 monochrome 10-bit) camera and Q Capture software. Mitotracker Red fluorescence was quantified with ImageJ software.
Protein Extract Preparation
For two-dimensional PAGE and one-dimensional immunoblot experiments, PC12 cells were seeded at 2.0 × 106 cells in 10-cm dishes and transgene expression was induced 24 h after seeding with 2.5 μm tebufenocide. At various time points after induction, cells were washed with cold phosphate-buffered saline (PBS), incubated for 3 min with PBS containing 40 mm iodoacetamide to prevent postlysis thiol-disulfide exchange, and scraped into 100 μl of Triton X-100 extraction buffer (50 mm Tris, pH 7.5, 1% Triton X-100, 1 mm phenylmethanesulfonyl fluoride, 40 mm iodoacetamide). The cell suspension was rocked on ice for 20 min and centrifuged at 15,000 × g for 10 min. Subsequently the supernatant (containing soluble proteins) was aliquoted and frozen at −80 °C. Striatal cell extracts were prepared by seeding striatal cells at 2.5 × 105 cells in 60-mm dishes and, following various treatments over the course of 48 h, were washed twice with ice-cold PBS and harvested in 100 μl of Triton X-100 extraction buffer as described above. Protein content of cell extracts was determined using the colorimetric Bio-Rad DC Protein Assay Kit.
Redox Two-dimensional PAGE
Separation of DSBP by redox two-dimensional PAGE was performed as described previously (16, 24). In brief, protein extracts (300 μg) from Htt-25Q-GFP or Htt-103Q-GFP expressing PC12 cells were resolved for 5 h at constant current, under nonreducing conditions through a 1.5-mm thick, 12% polyacrylamide gel using a Hoefer SE600 electrophoresis apparatus (San Francisco, CA). Following electrophoresis in the first dimension, gel strips containing resolved proteins were excised and immersed in SDS sample buffer (0.05 m Tris-HCl, pH 6.8, 5% glycerol, and 2% SDS) containing 100 mm dithiothreitol (DTT) for 20 min at room temperature to reduce disulfide bonds. Gel strips were then washed with SDS-running buffer and immersed for 10 min in SDS sample buffer containing 100 mm iodoacetamide. Each gel strip was then applied horizontally to a second gel (1.5 mm thick, 12% polyacrylamide) and run for 14 h at constant current (10 mA/gel). After electrophoresis in the second dimension, gels were fixed in 50% methanol overnight and silver stained as described previously (24).
MS Identification of Disulfide-linked Proteins
Gel spots from redox two-dimensional gels were excised and in-gel digested with trypsin using a MassPREP automated digester station (PerkinElmer Life Sciences). Gel pieces were de-stained of silver using 50 mm sodium thiosulfate 5-hydrate and 15 mm potassium ferricyanide, followed by protein reduction using 10 mm DTT, alkylation using 55 mm iodoacetamide, and finally digested with trypsin. Peptides were extracted using a solution of 1% formic acid and 2% acetonitrile and lyophilized. Prior to mass spectrometry analysis, dried peptide samples were re-dissolved in 10% acetonitrile and 0.1% TFA (trifluoroacetic acid). Aliquots of digested samples were mixed with an equal volume of MALDI matrix (α-cyano-4-hydroxycinnamic acid, prepared as 5 mg/ml in 6 mm ammonium phosphate monobasic, 50% acetonitrile, 0.1% trifluoroacetic acid) at a 1:1 ratio. Mass spectrometry data were obtained using a 4700 Proteomics Analyzer, MALDI TOF-TOF (Applied Biosystems, Foster City, CA). Data acquisition and data processing were, respectively, done using 4000 Series Explorer and Data Explorer (both from Applied Biosystems). The instrument is equipped with a 355-nm Nd:YAG laser and run in the positive reflector mode. Monoisotopic masses were determined after internal calibration and searched against the NCBInr rat protein data base using the Mascot program and a mass tolerance of 25 ppm. Each mass spectrum was collected as a sum of 1000 shots.
RNA Extraction and RT-PCR
RNA was extracted from PC12 cells using TRIzol reagent (Invitrogen) following the manufacturer's protocol. Briefly, 1 μg of total RNA was reverse transcribed into cDNA using a 1st Strand cDNA Synthesis kit for RT-PCR (Quanta BioSciences Inc). PCR was performed on a Mastercycler thermocycler (Eppendorf) for 30 cycles for Prx1 and 27 cycles for GAPDH as a control. Each cycle included denaturation at 94 °C for 20 s, annealing at 57 °C for 45 s, and elongation at 72 °C for 45 s. The primers used for Prx1 were forward, 5′-CCTGTAGCTCGACTCTGCTGATAG3′, reverse, 5′-GCGGCCAACAGGAAGATC-3′ and for GAPDH were forward, 5′-CCTTCATTGACCTCAACTAC-3′, reverse, 5′-GGAAGGCCATGCCAGTGAGC-3′. The size of the PCR products was determined by 1% agarose gel electrophoresis with visualization by ethidium bromide staining.
Immunoblot Analysis
Triton X-100-soluble protein extracts (30 μg) were resolved by 12% nonreducing and reducing PAGE using a Hoefer SE600 electrophoresis apparatus. Extracts resolved under reducing conditions were treated with 100 mm DTT and 2% β-ME and boiled for 5 min. Gels were electroblotted onto Immobilon-P membrane (Millipore, Bedford, MA), blocked with 1% milk and 3% BSA (Sigma) in Tris-buffered saline with 0.5% Tween 20 (TBST). Membranes were incubated with polyclonal rabbit antibodies against Prx1, sulfonylated Prx (Ab Frontier, South Korea), GFP (Sigma), and actin (Cell Signaling) overnight. Following incubation, membranes were washed with TBST and further hybridized with horseradish peroxidase-conjugated secondary antibodies (Bio-Rad). Detection was performed using Pierce ECL Western blotting detection reagents (ThermoScientific) and visualized using a ChemiDoc digital imaging system (Bio-Rad). Densitometric analysis was performed using Quantity One software (Bio-Rad). Data presented are the average of three separate experiments. Statistical analysis of densitometric averages was performed using a one-way ANOVA followed by a post-hoc Tukey test.
Prx1 Expression Plasmids and Transfection Procedure
Expression constructs containing FLAG-tagged versions of both wild type and cysteine mutant (C52S) Prx cDNAs were generously supplied by Dr. Hyunjung Ha (Chungbuk National University, Cheonju Korea). PC12-Htt-103Q-GFP cells were seeded in 35-mm dishes at 3 × 105 cells/dish and co-transfected with either pcDNA or FLAG-tagged Prx expression constructs along with a mCherry Fluorescent Protein reporter plasmid at a 3:1 molar ratio for a total of 4 μg of DNA per dish. Plasmid DNA was mixed with 4 μl of Lipofectamine 2000 (Invitrogen) in Opti-MEM, added to PC12 cell cultures, and 6 h later the transfection medium was replaced with DMEM containing 10% horse serum and 5% FBS. The following day transfected cells were treated with 2.5 μm tebufenocide to induce mHtt expression and at 48 and 72 h post-induction, cells were treated Hoechst stain (10 μg/ml) in PBS for 10 min, washed twice with PBS, and trypsinized. Cells were counted using a hemocytometer by both light and epifluorescent microscopy. Cell viability was assessed by comparing the average number of Hoescht-stained, mCherry-positive cells following tebufenocide induction versus cells transfected with the same plasmid but without tebufenocide treatment. Mean cell viability ratios from three separate experiments were analyzed using a one-way ANOVA and post hoc Tukey test.
shRNA-mediated Knockdown of Prx1 Expression
HuSH-29-mer shRNA pRS plasmids (Origene, Rockville, MD) directed at rat Prx1 were used to knockdown endogenous Prx1 expression in PC12 Htt-103Q-GFP cells. Two shRNA containing expression cassettes, targeting different regions of Prx1 mRNA (Prx1-shRNA3, ATACCATCAAGCCTGATGTCAATAAGAGT; Prx1-shRNA4, TCAACTGCCAAGTGATTGGAGCTTCTGTG) were used. A scrambled (SCR) shRNA plasmid, which does not target any endogenous mRNA, was used as a negative control. Prx1 and SCR shRNA plasmids were separately transfected into PC12 Htt-103Q-GFP cells followed by selection with 2 μg/ml of puromycin for 2 weeks. Cell viability assays were performed as described below.
MTT and Trypan Blue Cell Viability Assays
For MTT cell viability assays PC12 Htt-103Q-GFP cells were seeded in quadruplicate at 7,000 cells/well in 96-well plates (100 μl total culture volume). Thiol-based compounds were added at various concentrations (1, 5, 10, 50, 100, 500, and 1000 μm) to 96-well plates 24 h after seeding. One hour after compound exposure, transgene expression was induced with 2.5 μm tebufenocide. 48 h after induction of Htt-103Q-GFP expression 10 μl of 2 mg/ml of MTT solution prepared in PBS was added to the culture media and incubated at 37 °C for 2 h. Media was thoroughly aspirated from the wells and the purple formazan product of MTT was released from cells by the addition of 100 μl of DMSO. Absorbance was measured at a test wavelength of 595 nm and reference wavelength of 655 nm using a Bio-Rad 3550 microplate reader.
Due to interference of 3-nitropropionic acid with the MTT assay, cell viability in striatal cells was measured by trypan blue dye exclusion. In brief, STHdhQ7 and STHdhQ111 cells were seeded in triplicate in 12-well plates at 2.5 × 104 cells/well and treated with 10 mm 3-NP the following day. 24 h after 3-NP treatment, cells were detached with 100 μl of trypsin solution and an equal volume of trypan blue was added. Cells were counted using a hemocytometer and cell viability was determined by comparing the number of trypan blue excluding cells in treated versus nontreated cells. Mean cell viability ratios from three separate experiments were analyzed using a one-way ANOVA and post hoc Tukey test.
RESULTS
Increased Mitochondrial ROS in mHtt Expressing PC12 Cells
We utilized a PC12 nerve cell model that has been stably transfected with a plasmid encoding the entire exon 1 of the HTT gene with either a 25 (nonpathogenic) or 103 (pathogenic) poly(Q) repeat fused in-frame to the coding sequence for the enhanced GFP at the carboxyl terminus (22). Transcription of the transgene is driven by an ecdysone-regulated promoter that can be turned on by the addition of ecdysone or tebufenocide (an ecdysone analog). Although several studies have shown elevated ROS formation in mHtt expressing mammalian cells (10, 11, 25), the exact site of ROS production is poorly defined. We therefore examined mitochondrial derived ROS levels in Htt expressing PC12 cells using MitoTracker Red CM-H2XROS, a dye that is specifically targeted to mitochondria and only fluoresces in the presence of ROS (26). Induction of Htt-25Q-GFP expression in PC12 cells resulted in diffuse green fluorescence, whereas Htt-103Q-GFP expressing cells exhibited large perinuclear protein aggregates (Fig. 1A). Elevated mitochondrial ROS were detected at 2 days post-induction only in Htt-103Q-GFP expressing cells, whereas mitochondrial ROS levels remained constant in either uninduced or induced Htt-25Q-GFP expressing cells (Fig. 1B). Therefore, expression of Htt containing a pathogenic poly(Q) repeat results in elevated mitochondrial ROS production.
FIGURE 1.

Expression of mHtt promotes an increase in mitochondrial ROS. A, induction of mutant Huntingtin (HTT-103Q-GFP) in PC12 cells for 48 h resulted in the formation of large aggregates (green) and increased mitochondrial ROS production (red), whereas induced expression of wild type HTT (HTT-25Q-GFP) resulted in diffuse Htt expression and no increase in ROS production compared with uninduced cells. B, quantification of fluorescence intensity revealed a significant (*, p < 0.01) increase in mitochondrial ROS only in HTT-103Q-GFP expressing cells at 48 h. Cells were stained with Mitotracker Red CM-H2XRos and nuclei were stained with Hoescht (blue) followed by visualization with fluorescence microscopy at ×400 magnification and quantification with ImageJ software. Data represent the average ± S.D. of three independent experiments. Data were analyzed by a one-way ANOVA followed by a Tukey test.
mHtt Expression Promotes Changes in Protein Disulfide Bonding
To determine the spectrum of proteins that specifically undergo changes in disulfide bonding in response to mHtt-induced ROS production, cell lysates were analyzed by redox two-dimensional PAGE. This technique, also known as diagonal electrophoresis, allows for the separation of disulfide-linked proteins that appear as distinct spots in silver-stained gels above and below a strong diagonal line (16). Lysates were processed from cells cultured for 48 h in either the presence of tebufenocide or a vehicle control (DMSO). Redox two-dimensional PAGE analysis of protein extracts revealed a number of proteins that exhibited both increased and decreased disulfide bonding unique to Htt-103Q expressing cells (Fig. 2). Specifically, spots 2 and 3 exhibited a diminishment in disulfide bonding, whereas spots 1 and 4–8 displayed increased disulfide bonding. In contrast, minimal alterations were observed in DSBP following induction of nonpathogenic Htt-25Q expression. To ensure that the redox two-dimensional PAGE technique did not artificially induce disulfide bonding, protein extracts were treated in both dimensions with the reducing agent DTT. As expected, a clear diagonal line was revealed with no spots in the off-diagonal zones (supplemental Fig. 1).
FIGURE 2.

Redox two-dimensional PAGE analysis of protein extracts from PC12 cells expressing nonpathogenic and pathogenic Htt. Triton X-100-soluble proteins were isolated from PC12 cells following tebufenocide-induced expression of an amino-terminal Huntingtin (Htt) protein containing either a nonpathogenic (25Q) or pathogenic (103Q) polyglutamine repeat. Following 2 days of induction, extracts were resolved by sequential nonreducing/reducing SDS-PAGE (redox two-dimensional PAGE) and DSBP, spots in off-diagonal zones, were visualized by silver staining. PC12 cells expressing Htt-25Q (A) showed little or no increase in DSBP following induced expression. In contrast, PC12 cells expressing Htt-103Q (B) displayed a pronounced increase in some DSBP (spots 1 and 4–6) and reduction in others (spots 2 and 3). The numbered spots were subsequently identified by mass spectrometery (see Table 1). The gels presented are representative of three separate experiments.
Identification of Proteins That Undergo Changes in Disulfide Bonding in Response to mHtt Expression
Protein spots that exhibited reproducible migration following redox two-dimensional PAGE were excised, in-gel digested with trypsin, and identified by matrix-assisted laser desorption/ionization time-of-flight (MALDI-TOF) mass spectrometry. Analysis of the protein spots revealed that half of the proteins identified (Table 1) were categorized as antioxidant proteins. Specifically the peroxiredoxin (Prx) family of antioxidant proteins Prx1, -2, and -4 were identified as proteins that exhibited changes in disulfide bonding following mHtt expression. In addition, the antioxidant Cu/Zn-superoxide dismutase was shown to have increased disulfide bonding. The four other proteins that were identified (Table 1 and Fig. 2, spots 5–7) have various intracellular roles including molecular chaperoning, cytoskeletal remodeling, and vesicular trafficking.
TABLE 1.
MS identification of DSBP altered in PC12 cells expressing mutant Huntingtin
Silver-stained DSBP that were altered in a mHtt-dependent manner in Fig. 2 were picked, in-gel digested with trypsin, and identified by MALDI-TOF mass spectrometry. The relative change in disulfide bonding (↑ increased, ↓ decreased) of induced versus uninduced cells is indicated. A probability based MOWSE score greater than 50 was considered significant.
| Spot No. | Protein | Accession No. | Molecular mass | MOWSE score | Relative change | Function |
|---|---|---|---|---|---|---|
| kDa | ||||||
| 1 | Cu/Zn-SOD | 203,658 | 15.7 | 57 | ↑ | Antioxidant |
| 2 | Peroxiredoxin 2 | 34,849,738 | 21.8 | 95 | ↓ | Antioxidant |
| 3 | Peroxiredoxin 1 | 16,923,958 | 22.3 | 183 | ↓ | Antioxidant |
| 4 | Peroxiredoxin 4 | 16,758,274 | 31 | 52 | ↑ | Antioxidant |
| 5 | Annexin-A2 | 9,845,234 | 39 | 72 | ↑ | Vesicle trafficking |
| 6 | β-Actin | 71,620 | 42 | 72 | ↑ | Cytoskeletal |
| 7 | Calreticulin | 11,693,172 | 48.1 | 180 | ↑ | Chaperone |
| 8 | Hypothetical | 66,730,523 | 51.1 | 167 | ↑ | Unknown |
mHtt Promotes Decrease in Prx1 Expression
Prx1 is an abundant intracellular protein within mammalian cells and plays an essential role in protecting against oxidative stress (17). In this study, Prx1 also exhibited the most robust decrease in disulfide bonding following redox two-dimensional PAGE analysis. Thus, we chose to examine Prx1 expression in the PC12 cell model using one-dimensional immunoblot analysis under both nonreducing and reducing conditions. Under normal cellular conditions, Prx1 predominately exists as a disulfide-linked homodimer. During its normal catalytic cycle, the homodimeric form of Prx1 can be recycled back to its monomeric form via thioredoxin (17). However, exposure to high intracellular ROS can overoxidize the catalytic cysteine residue of Prx1 to a sulfonic acid thereby rendering the protein unable to form a disulfide bond (27). Immunoblot analysis of nonreduced protein extracts revealed a decrease in the homodimeric disulfide-linked form of Prx1 at the 2- and 3-day time points following induction of mHtt expression (Fig. 3A). Also apparent under nonreduced conditions was a monomeric form of Prx1 (Fig. 3A). Blotting using an antibody that specifically recognizes sulfinic and sulfonic acid derivatives of Prxs (Prx-SO2–3) revealed that the monomeric form of Prx1 was indeed a hyperoxidized form of the protein (Fig. 3A). In contrast, Prx1 expression levels and the redox state remained unaltered in cells expressing Htt-25Q. RT-PCR analysis revealed that levels of the Prx1 mRNA transcript decreased dramatically 48 h after induction of mHtt expression (Fig. 3B). To rule out the possibility that decreased Prx1 expression following mHtt induction was simply due to nonspecific degradation of protein or mRNA in dying cells, a live-dead cell assay was performed on the adherent cells used in expression analysis studies. As expected, an overall decrease in the total number of cells was observed following induction of mHtt expression compared with nontreated cells. However, under induced conditions the remaining adherent cells showed greater that 90% viability for up to 3 days (supplemental Fig. 2). These findings indicate that mHtt specifically promotes the loss of Prx1 expression in PC12 cells.
FIGURE 3.
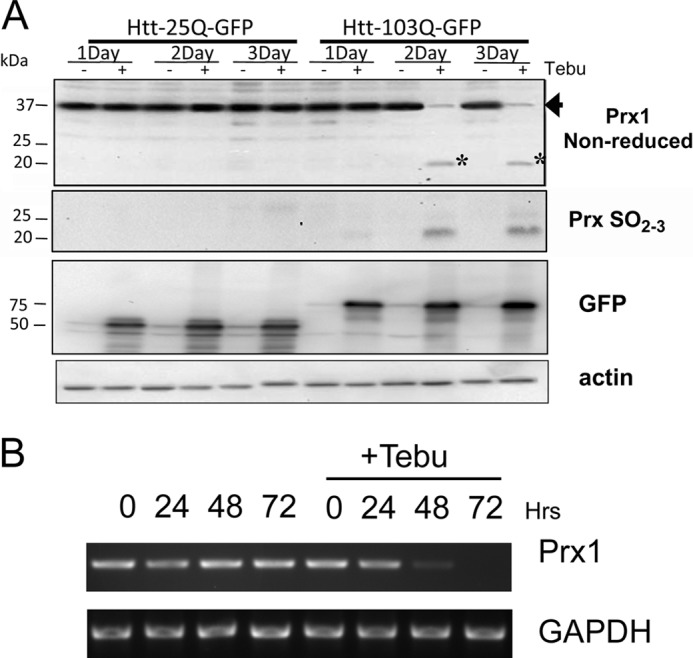
Mutant Huntingtin promotes a decrease in peroxiredoxin 1 expression. A, cell lysates from PC12 cells treated with tebufenocide (+) to induce expression of either nonpathogenic (Htt-25Q-GFP) or pathogenic (Htt-103Q-GFP) forms of Htt were analyzed by immunoblotting with the indicated antibodies. Cell extracts resolved under nonreducing conditions (top panel) revealed that the disulfide-linked homodimeric form of Prx1 (spot 3 in Fig. 2) was markedly decreased (arrow) after 2 and 3 days of mHtt induction. In addition, mHtt expression promoted the accumulation of a potentially overoxidized monomeric form Prx1 (asterisk). Reprobing the above blot with an anti-PrxSO2–3 antibody revealed that the monomeric Prx1 represented a sulfonylated species. Detection of transgene expression with an anti-GFP antibody revealed similar levels of Htt-25Q-GFP and Htt-103Q-GFP expression throughout the course of induction. As a control, actin expression (bottom panel) revealed equal loading for both uninduced and induced conditions. Presented blots are representative of three separate experiments. B, RT-PCR analysis of RNA extracted from PC12 treated with tebufenocide to induce expression of Htt-103Q-GFP revealed dramatically decreased expression of Prx1 by 48 h compared with uninduced controls. Data are representative of three separate experiments.
Overexpression of Wild-type Prx1 Suppresses mHtt-induced Toxicity
Because mHtt expression promotes the loss of endogenous Prx1, it was asked if ectopic expression of Prx1 can attenuate mHtt-induced toxicity. PC12 cells were transfected with cDNA expression constructs that allow high level expression of FLAG-tagged versions of either wild type or a catalytically inactive version (C52S) of Prx1. Unlike endogenous Prx1, levels of ectopically expressed wild type or mutant Prx1 were maintained following induction of mHtt expression over the course of 3 days (Fig. 4A). Moreover, cell viability analysis revealed that elevated expression of wild type Prx1 attenuated mHtt-induced toxicity at 48 and 72 h (Fig. 4, B and C). In contrast, mutant Prx1 exhibited only modest protection against mHtt-induced toxicity at 48 h (Fig. 4B) and had no protective effect at 72 h (Fig. 4C). These findings indicate that maintenance of Prx1 levels can protect against toxicity initiated by mHtt expression.
FIGURE 4.

Overexpression of wild type Prx1 suppresses mHtt-induced toxicity. A, FLAG-tagged Prx1 expression plasmids transfected into PC12–103Q-GFP cells exhibited constitutive Prx1-FLAG expression (upper band) 48 and 72 h after tebufenocide (+)-induced mHtt expression, whereas endogenous Prx1 levels (lower band) were decreased. Cell viability of PC12-Htt103Q cells transfected with the indicated plasmids was assessed after 48 (B) or 72 h (C) exposure to tebufenocide to induce expression of mHtt. Data presented are the average of three independent experiments. Wild type Prx1 conferred significant protection against mHtt-induced toxicity compared with the vector control (*, p < 0.05; **, p < 0.01; n = 3).
Dimercaptopropanol Suppresses mHtt-induced Toxicity, Prx1 Hyperoxidation, and Loss of Prx1 Expression
Because mHtt expression was shown to induce both loss of Prx1 expression and cell death in PC12 cells it was asked if thiol-based antioxidants could counter these effects. Various thiol compounds were examined for their ability to counter mHtt toxicity including β-ME, DTT, cysteamine (Cys), NAC, reduced eeGSH, DMP, DMSA, and penicillimine. DMP, and the related compound DMSA, exhibited the most robust protection (greater than 60% viability) against mHtt-induced toxicity in PC12 cells (Fig. 5). In contrast, all the other thiol-based compounds either exhibited very modest protection (i.e. DTT) or had no effect on cell viability at the concentrations tested. Because DMP may have antioxidant effects, we examined mitochondrial ROS production in Htt-103Q expressing PC12 cells. DMP treatment failed to attenuate mitochondrial ROS production in mHtt expressing cells (supplemental Fig. 3). Therefore, DMP is unlikely to confer neuroprotection against mHtt toxicity by nonspecific antioxidant effects.
FIGURE 5.
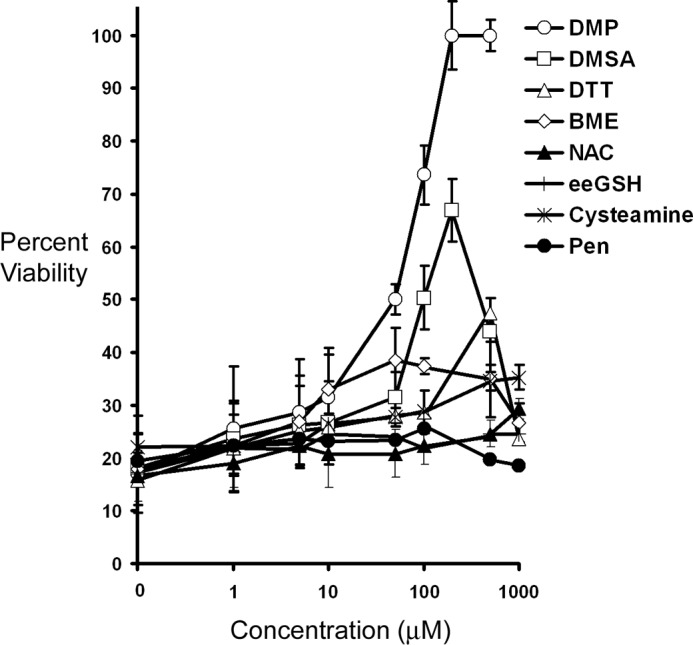
DMP treatment confers protection against mHtt-induced toxicity. PC12–103Q cells were treated with various thiol-based compounds including β-mercaptoethanol (BME), DTT, cysteamine (Cys), NAC, eeGSH, DMP, DMSA, and penicillimine (Pen) for 1 h before induction of mHtt expression with 2 μm tebufenocide. After 48 h cell viability was measured by reduction of MTT. DMP elicited the highest degree of protection against mHtt-induced toxicity.
We next sought to determine whether the neuroprotective properties of DMP also correlated with suppression of altered disulfide bonding initiated by mHtt expression using redox two-dimensional PAGE. Exposure of Htt-103Q expressing cells to 100 μm DMP, a concentration that is strongly neuroprotective, resulted in minimal changes in disulfide bonding following induction of mHtt expression (Fig. 6A). In particular, the mHtt-induced loss of disulfide-linked homodimers of peroxiredoxin 1 and 2 (spots 3 and 2, respectively, in Fig. 2) was completely prevented by pretreatment with DMP. RT-PCR and immunoblot analysis revealed that DMP pretreatment maintained both Prx1 mRNA and protein levels following induction of mHtt expression (Fig. 6, B and C).
FIGURE 6.

DMP treatment protects against mHtt-induced aberrant disulfide bonding and loss of Prx1 expression. A, redox two-dimensional PAGE analysis of extracts from PC12-Htt-103Q cells treated with either DMP alone (control) or DMP followed by tebufenocide treatment to induce mHtt expression (induced). DMP pre-treatment prevented the aberrant formation or loss of DSBP following mHtt expression. B, RT-PCR analysis of PC12-Htt-103Q cells either treated with 100 μm DMP alone or before Htt induction revealed that DMP treatment prevented loss of Prx1 expression even after 3 days of mHtt induction. C, top, Western blot analysis of tebufenocide-treated PC12-Htt-103Q cells revealed that mHtt expression promoted increased sulfonylation and a concomitant loss of Prx1 expression. DMP pre-treatment prevented mHtt-induced loss of Prx1 expression and suppressed sulfonylation. Bottom, quantification of Western blots revealed a significantly lower ratio of sulfonylated Prx1 to total Prx1 in DMP-treated PC12 cells after induction of Htt-103Q, particularly at 72 h (*, p < 0.05, n = 3).
Recent studies have indicated that hyperoxidation (sulfonylation) of peroxiredoxins not only leads to their catalytic inactivation but also renders the proteins more susceptible to degradation (28, 29). Immunoblot analysis using an anti-Prx-SO2–3 antibody revealed that induction of mHtt expression resulted in increased sulfonylation of Prx1 relative to total Prx1 levels (Fig. 6C). In contrast, DMP pretreatment prevented both mHtt-induced loss of Prx1 expression and sulfonylation.
Knockdown of Endogenous Prx1 Potentiates mHtt Toxicity
To further determine the ability of Prx1 to attenuate mHtt toxicity, endogenous Prx1 expression was knocked down using shRNAs specific to Prx1. Western blot analysis revealed an ∼65% knockdown in Prx1 protein levels in PC12-Htt-103Q cells stably transfected with two shRNA expressing constructs targeting different regions of the Prx1 mRNA compared with control shRNA-transfected cells (Fig. 7A). A significant decrease in viability was observed in Prx1-shRNA expressing cells compared with the control cells following induction of mHtt expression at both 24 and 48 h (Fig. 7, B and C). Furthermore, DMP treatment was unable to fully restore viability in Prx1-shRNA expressing cells when compared with control cells. These findings suggest that DMP-mediated suppression of mHtt toxicity works, in part, by maintaining expression of Prx1.
FIGURE 7.

Knockdown of Prx1 potentiates mHtt-induced toxicity. A, PC12 Htt-103Q cells transfected with shRNA constructs targeting Prx1 mRNA exhibited decreased Prx1 expression compared with control SCR-transfected cells. B, densitometric analysis of immunoblots revealed at least a 65% reduction of Prx1 expression in Prx1-shRNA stably transfected cells compared with SCR cells. Cells stably expressing Prx1-shRNA exhibited significantly decreased viability following induction of mHtt expression compared with SCR control cells at both 24 (C) and 48 h (D). DMP treatment (100 μm) was unable to fully rescue mHtt-induced toxicity in Prx1-shRNA expressing cells compared with the scrambled control. Data presented are the average of three independent experiments (*, p < 0.05; **, p < 0.01).
DMP Prevents 3-NP-induced Cell Death and Prx1 Hyperoxidation in a Striatal Cell Model of HD
To determine whether DMP exerted similar protective effects in another cell model of HD, we examined immortalized striatal cell lines derived from a knock-in transgenic mouse in which either 7 glutamine repeats (STHdHQ7) or 111 glutamine repeats (STHdHQ111) were introduced into exon 1 of the mouse Htt gene (23). Treatment with 3-NP, a mitochondrial complex II toxin, induces mitochondrial depolarization and cell death at significantly higher levels in STHdhQ111 cells compared with STHdhQ7 cells (30). Pre-treatment of STHdhQ111 cells with 10 μm DMP before 3-NP exposure restored cell viability to a level comparable with STHdhQ7 cells treated with 3-NP alone (Fig. 8A). Immunoblot analysis revealed that 3-NP treatment promoted a dramatic loss of Prx1 expression in STHdHQ111 cells but had little effect on Prx1 levels in 3-NP-treated STHdHQ7 cells. In addition, increased sulfonylation of Prx1 and another Prx isoform of ∼22 kDa was observed in both STHdhQ7 and STHdhQ111 cells following 3-NP treatment (Fig. 8B). However, the degree or Prx1 oxidation, relative to total Prx1 levels, was higher in 3-NP-treated STHdhQ111 cells compared with STHdhQ7 cells (Fig. 8C). Moreover, DMP pre-treatment prevented Prx1 sulfonylation and the overall diminishment of Prx1 protein levels in STHdhQ111 cells following 3-NP exposure. Therefore, DMP treatment can prevent loss of Prx1 expression, Prx1 hyperoxidation, and cell death associated with either elevated mHtt expression or exposure to a mitochondrial toxin that exacerbates HD pathology.
FIGURE 8.

DMP treatment promotes viability and prevents loss of Prx1 expression in 3-NP-treated HD mouse striatal cells. A, DMP attenuated 3-NP toxicity, following 24 h, in mouse striatal cells derived from mice with a knock-in of either 7 (STHdHQ7) or 111 (STHdHQ111) polyglutamine repeats. DMP-treated STHdHQ111 mouse striatal cells exhibited levels of viability similar to STHdHQ7 cells treated with 3-NP alone (*, p < 0.05, n = 3). B, Western blot analysis of protein extracts from striatal cells revealed that 3-NP treatment promoted both loss of Prx1 expression and increased sulfonylation in STHdHQ111 cells but not in STHdHQ7 cells. DMP pre-treatment prevented 3-NP-mediated loss of Prx1 expression and elevated sulfonylation. C, densitometric analysis of Western blots revealed a significantly higher ratio (*, p < 0.05, n = 3) of sulfonylated Prx1 to total Prx1 following a 48-h 3-NP treatment in STHdHQ111 cells. In contrast, the sulfonylated to total Prx1 ratio in DMP-treated STHdHQ111 cells was markedly lower following 3-NP exposure.
DISCUSSION
ROS have traditionally been viewed as deleterious molecules that nonspecifically damage cellular macromolecules and contribute to various forms of neurodegeneration and aging. Elevated oxidation of DNA, protein, and lipids has frequently been detected in postmortem brain tissue from patients afflicted with various neurodegenerative diseases. Whether the elevation in oxidative damage associated with disease states is a causal or correlative event has long been the subject of debate. However, over the last decade numerous studies have shown that ROS-mediated oxidation of cysteine residues acts as a key signaling mechanism controlling multiple cellular processes including protein phosphorylation, gene expression, chromatin remodeling, protein turnover, molecular chaperoning, metabolism, autophagy, antioxidant defense, and mitochondrial function in a specific and reversible manner (31). In fact, it is now recognized that under physiological conditions transient low level generation of ROS appears to be essential to maintain cellular homeostasis (32). However, relatively few studies have attempted to detect oxidative modifications to cysteine residues in a disease context. One study revealed that glucose-6-phosphate dehydrogenase, a key enzyme that generates NADPH reducing equivalents in cells, is elevated in Alzheimer disease brain along with a concomitant increase in reactive sulfhydryls in the neuronal cytoplasm (33). However, the identity of proteins with reactive sulfhydryl groups that are altered in neurodegenerative diseases is poorly defined.
Here we show for the first time that increased expression of pathogenic Htt promotes aberrant disulfide bonding in nerve cell lines. In particular, mHtt expression promotes increased disulfide bonding of the antioxidant proteins Cu/Zn-superoxide dismutase 1 (SOD1) and Prx4 while decreasing disulfide bonding of Prx1 and Prx2. SOD1 is a cytosolic protein responsible for the enzymatic conversion of the superoxide anion to hydrogen peroxide. Prx1 and Prx2 are predominately cytosolic, whereas Prx4 is a secreted peroxiredoxin isoform that is also found in the endoplasmic reticulum. All three Prx isoforms belong to the typical 2-cysteine family of peroxiredoxins that undergo a catalytic cycle in which the N-terminal cysteine is oxidized by H2O2 to a sulfenic acid that then reacts with the C-terminal cysteine of another subunit to produce an intermolecular disulfide (17). This disulfide is then reduced by the Trx/TrxR system, completing the catalytic cycle. During oxidative stress the sulfenic intermediate is hyperoxidized to a sulfinic acid or a sulfonic acid, which is incapable of forming a disulfide bond leading to inactivation of Prx peroxidase activity (27). Oxidation of the catalytic cysteine to a sulfinic acid in Prxs 1–4 is reversed by a reaction catalyzed by sulfiredoxin (34). However, overoxidation of all Prx isoforms to a sulfonic state is irreversible.
In this study, we observed that mHtt expression promoted a decrease in Prx1 expression and a concurrent increase in Prx1 sulfonylation. Hyperoxidation of Prxs has been show to render these proteins susceptible to ubiquitin-mediated proteolysis (28, 29). Therefore, mHtt may affect Prx1 expression at both transcriptional and post-translational levels. In contrast, DMP treatment prevented both mHtt-induced loss of Prx1 expression and increased sulfonylation. Prx1 expression is controlled by the transcription factor NF-E2 related factor-2 (Nrf2) (35). Elevated ROS can promote oxidative modifications to Kelch-like ECH-associated protein 1 (Keap1), a negative regulator of Nrf2, leading to the stabilization and nuclear translocalization of Nrf2 (36). Once in the nucleus, Nrf2 dimerizes with a small Maf protein and the heterodimer then binds antioxidant response element sequences within the promoters of antioxidant and phase II detoxifying genes. Besides activating transcription of Prx1, Nrf2 can also activate expression of sulfiredoxin, an enzyme that can reverse oxidation of Prx sulfinic acids to sulfhydryl groups and various other antioxidant enzymes (37). The observation that DMP exposure can partially rescue mHtt-induced toxicity even when Prx1 expression is knocked down (Fig. 7) suggests that this compound is likely to alter expression of other neuroprotective genes. It is possible that DMP may affect Nrf2 activation via a redox-dependent mechanism leading to increased expression of Prx1, sulfiredoxin, and other antioxidant enzymes even in the presence of mHtt. Future studies will evaluate this hypothesis.
We have previously shown that Prx1 levels are elevated and overoxidized Prx2 isoforms predominate in Alzheimer diseased brain tissue (38). Overexpression of Prx1 alone can protect against amyloid-β toxicity in nerve cells. Furthermore, amyloid-β-resistant nerve cells up-regulate multiple Prx isoforms, in addition to the reductive enzymes required to maintain Prxs in an active state, thereby countering toxicity initiated by amyloid-β exposure (38). A recent proteomic study showed that the Prx isoforms 1, 2, and 6 are elevated in the striatum and cortex of HD patients compared with unaffected individuals (39). However, this study also detected the accumulation of acidic (i.e. sulfonylated) forms of Prx1 and Prx6 in HD patients as well. Collectively these findings indicate that maintenance of Prxs at a high level and in an active state is key to ensuring optimal neuronal cell survival in the presence of pathogenic proteins associated with various neurodegenerative diseases.
The observation that oxidative damage to macromolecules is increased in HD has spurred interest in evaluating the ability of various antioxidants to attenuate mHtt-induced toxicity. A number of studies have focused on the use of the molecule Coenzyme Q10 (CoQ10) for the treatment of HD (40). CoQ10 functions in the inner membrane of mitochondria as an electron carrier from enzyme complex I and complex II to complex III and can also act as a lipid soluble antioxidant, particularly in its reduced form (ubiquinol, CoQ10H2). CoQ10 has been shown to significantly increase survival and delay motor symptoms in transgenic HD mouse models (13). However, clinical trials testing various formulations of CoQ10 in HD patients have, so far, failed to fully recapitulate the positive effects of this compound observed in animal models (41, 42). Whether CoQ10 is able to completely target affected brain regions in HD patients and either increase mitochondrial function or detoxify elevated ROS is uncertain. The modest effect of this antioxidant in HD patients may be related to its relative lack of specificity in targeting oxidized intracellular targets. Although, CoQ10 can detoxify lipid peroxides, it does so to varying extents in different tissues and in a relatively nonselective manner. Furthermore, it is now recognized that under physiological conditions, the transient generation of ROS, within certain limits, appears to be essential to maintain cellular homeostasis, whereas countering low level ROS with broad-acting antioxidants can actually be detrimental (32). For example, administration of antioxidants has been shown to inhibit autophagy and increase toxicity of poly(Q) proteins (43). Therefore, identification of compounds that possess good bioavailability and display some degree of specificity in targeting oxidized proteins may be key factors for the treatment of neurodegenerative diseases.
In this study we demonstrate for the first time that specific dithiol-based compounds confer selective resistance to mHtt-induced neurotoxicity. Although many commonly used monothiol antioxidants such as β-ME, NAC, and eeGSH were tested in the mHtt-inducible PC12 cell model, only the dithiols DMP, DMSA, and to a lesser extent DTT, showed neuroprotection. This observation suggests that the protective effect of DMP cannot fully be attributed to the nonspecific and widespread reduction in intracellular disulfide bonding. DMP was previously shown in an unbiased blind screen of over 1000 FDA approved compounds to be neuroprotective against mHtt toxicity in the same PC12 model used in this study (22). In addition, DMP, also known as British anti-Lewisite (BAL), was shown to reduce pathophysiological symptoms of HD and halt disease progression in a long term study of two patients conducted in 1955 (44). At the time DMP was believed to work by chelating metals but subsequent studies using the clinically more tolerable metal chelator penicillamine failed to alleviate HD clinical symptoms (45). It is intriguing to note that penacillamine did not confer protection against mHtt expression in PC12 cells (Fig. 5) suggesting that the protective effect of DMP is unlikely to be mediated by metal chelation. Although DMP exhibited the highest level of protection against mHtt toxicity, it has poor bioavailability properties and requires intramuscular injections. DMSA also exhibited protection in this study, albeit to a lesser extent than DMP. DMSA is orally active and can cross the blood-brain barrier (46, 47) making it an excellent candidate drug for further testing as a therapy for HD. Moreover, the structural relationship between the dithiols DMP and DMSA and their strong neuroprotective effects in HD cell culture models suggest that these molecules may be a good starting point for further development of dithiol-based compounds to treat HD and other neurodegenerative disorders.
Supplementary Material
This work was supported by Natural Sciences and Engineering Research Council (NSERC) of Canada Grant 355806-2008 and the Canada Foundation of Innovation Leaders Opportunity Fund (CFI-LOF).

This article contains supplemental Figs. S1–S3.
- HD
- Huntington disease
- β-ME
- β-mercaptoethanol
- CoQ10
- coenzyme Q10
- Cys
- cysteamine
- Cys-SH
- cysteine sulfhydryl
- DMP
- dimercaptopropanol
- DMSA
- dimercaptosuccinic acid
- DSBP
- disulfide-bonded protein
- eeGSH
- glutathione-reduced ethyl ester
- Htt
- Huntingtin protein
- Htt-103Q
- Huntingtin with a pathogenic 103 polyglutamine repeat
- mHtt
- mutant Huntingtin protein
- NAC
- N-acetylcysteine
- Prx
- peroxiredoxin
- ROS
- reactive oxygen species
- SOD
- superoxide dismutase
- MTT
- 3-(4,5-dimethlythiazol-2-yl)-2,5-diphenyltetrazolium bromide
- 3-NP
- 3-nitropropionic acid
- Nrf2
- NF-E2 related factor-2
- GFP
- green fluorescent protein
- ANOVA
- analysis of variance
- SCR
- scrambled.
REFERENCES
- 1. Imarisio S., Carmichael J., Korolchuk V., Chen C. W., Saiki S., Rose C., Krishna G., Davies J. E., Ttofi E., Underwood B. R., Rubinsztein D. C. (2008) Huntington disease. From pathology and genetics to potential therapies. Biochem. J. 412, 191–209 [DOI] [PubMed] [Google Scholar]
- 2. Ross C. A. (1995) When more is less. Pathogenesis of glutamine repeat neurodegenerative diseases. Neuron 15, 493–496 [DOI] [PubMed] [Google Scholar]
- 3. Ross C. A., Tabrizi S. J. (2011) Huntington disease. From molecular pathogenesis to clinical treatment. Lancet Neurol. 10, 83–98 [DOI] [PubMed] [Google Scholar]
- 4. Reddy P. H., Mao P., Manczak M. (2009) Mitochondrial structural and functional dynamics in Huntington disease. Brain Res. Rev. 61, 33–48 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Choo Y. S., Johnson G. V., MacDonald M., Detloff P. J., Lesort M. (2004) Mutant huntingtin directly increases susceptibility of mitochondria to the calcium-induced permeability transition and cytochrome c release. Hum. Mol. Genet. 13, 1407–1420 [DOI] [PubMed] [Google Scholar]
- 6. Panov A. V., Gutekunst C. A., Leavitt B. R., Hayden M. R., Burke J. R., Strittmatter W. J., Greenamyre J. T. (2002) Early mitochondrial calcium defects in Huntington disease are a direct effect of polyglutamines. Nat. Neurosci. 5, 731–736 [DOI] [PubMed] [Google Scholar]
- 7. Shirendeb U., Reddy A. P., Manczak M., Calkins M. J., Mao P., Tagle D. A., Reddy P. H. (2011) Abnormal mitochondrial dynamics, mitochondrial loss, and mutant huntingtin oligomers in Huntington disease. Implications for selective neuronal damage. Hum. Mol. Genet. 20, 1438–1455 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Cui L., Jeong H., Borovecki F., Parkhurst C. N., Tanese N., Krainc D. (2006) Transcriptional repression of PGC-1α by mutant huntingtin leads to mitochondrial dysfunction and neurodegeneration. Cell 127, 59–69 [DOI] [PubMed] [Google Scholar]
- 9. Bossy-Wetzel E., Petrilli A., Knott A. B. (2008) Mutant huntingtin and mitochondrial dysfunction. Trends Neurosci. 31, 609–616 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Wyttenbach A., Sauvageot O., Carmichael J., Diaz-Latoud C., Arrigo A. P., Rubinsztein D. C. (2002) Heat shock protein 27 prevents cellular polyglutamine toxicity and suppresses the increase of reactive oxygen species caused by huntingtin. Hum. Mol. Genet. 11, 1137–1151 [DOI] [PubMed] [Google Scholar]
- 11. Firdaus W. J., Wyttenbach A., Diaz-Latoud C., Currie R. W., Arrigo A. P. (2006) Analysis of oxidative events induced by expanded polyglutamine huntingtin exon 1 that are differentially restored by expression of heat shock proteins or treatment with an antioxidant. FEBS J. 273, 3076–3093 [DOI] [PubMed] [Google Scholar]
- 12. Parker J. A., Arango M., Abderrahmane S., Lambert E., Tourette C., Catoire H., Néri C. (2005) Resveratrol rescues mutant polyglutamine cytotoxicity in nematode and mammalian neurons. Nat. Genet. 37, 349–350 [DOI] [PubMed] [Google Scholar]
- 13. Ferrante R. J., Andreassen O. A., Dedeoglu A., Ferrante K. L., Jenkins B. G., Hersch S. M., Beal M. F. (2002) Therapeutic effects of coenzyme Q10 and remacemide in transgenic mouse models of Huntington disease. J. Neurosci. 22, 1592–1599 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Finkel T. (2011) Signal transduction by reactive oxygen species. J. Cell Biol. 194, 7–15 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Sitia R., Molteni S. N. (2004) Stress, protein (mis)folding, and signaling. The redox connection. Sci. STKE 2004, pe27. [DOI] [PubMed] [Google Scholar]
- 16. Cumming R. C., Andon N. L., Haynes P. A., Park M., Fischer W. H., Schubert D. (2004) Protein disulfide bond formation in the cytoplasm during oxidative stress. J. Biol. Chem. 279, 21749–21758 [DOI] [PubMed] [Google Scholar]
- 17. Rhee S. G., Chae H. Z., Kim K. (2005) Peroxiredoxins. A historical overview and speculative preview of novel mechanisms and emerging concepts in cell signaling. Free Radic. Biol. Med. 38, 1543–1552 [DOI] [PubMed] [Google Scholar]
- 18. Woo H. A., Yim S. H., Shin D. H., Kang D., Yu D. Y., Rhee S. G. (2010) Inactivation of peroxiredoxin 1 by phosphorylation allows localized H2O2 accumulation for cell signaling. Cell 140, 517–528 [DOI] [PubMed] [Google Scholar]
- 19. Yan Y., Sabharwal P., Rao M., Sockanathan S. (2009) The antioxidant enzyme Prdx1 controls neuronal differentiation by thiol-redox-dependent activation of GDE2. Cell 138, 1209–1221 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Jang H. H., Lee K. O., Chi Y. H., Jung B. G., Park S. K., Park J. H., Lee J. R., Lee S. S., Moon J. C., Yun J. W., Choi Y. O., Kim W. Y., Kang J. S., Cheong G. W., Yun D. J., Rhee S. G., Cho M. J., Lee S. Y. (2004) Two enzymes in one. Two yeast peroxiredoxins display oxidative stress-dependent switching from a peroxidase to a molecular chaperone function. Cell 117, 625–635 [DOI] [PubMed] [Google Scholar]
- 21. Neumann C. A., Cao J., Manevich Y. (2009) Peroxiredoxin 1 and its role in cell signaling. Cell Cycle 8, 4072–4078 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Aiken C. T., Tobin A. J., Schweitzer E. S. (2004) A cell-based screen for drugs to treat Huntington disease. Neurobiol. Dis. 16, 546–555 [DOI] [PubMed] [Google Scholar]
- 23. Trettel F., Rigamonti D., Hilditch-Maguire P., Wheeler V. C., Sharp A. H., Persichetti F., Cattaneo E., MacDonald M. E. (2000) Dominant phenotypes produced by the HD mutation in STHdh(Q111) striatal cells. Hum. Mol. Genet. 9, 2799–2809 [DOI] [PubMed] [Google Scholar]
- 24. Cumming R. C. (2008) Analysis of global and specific changes in the disulfide proteome using redox two-dimensional polyacrylamide gel electrophoresis. Methods Mol. Biol. 476, 165–179 [DOI] [PubMed] [Google Scholar]
- 25. Li X., Valencia A., Sapp E., Masso N., Alexander J., Reeves P., Kegel K. B., Aronin N., Difiglia M. (2010) Aberrant Rab11-dependent trafficking of the neuronal glutamate transporter EAAC1 causes oxidative stress and cell death in Huntington disease. J. Neurosci. 30, 4552–4561 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Degli Esposti M. (2002) Measuring mitochondrial reactive oxygen species. Methods 26, 335–340 [DOI] [PubMed] [Google Scholar]
- 27. Yang K. S., Kang S. W., Woo H. A., Hwang S. C., Chae H. Z., Kim K., Rhee S. G. (2002) Inactivation of human peroxiredoxin I during catalysis as the result of the oxidation of the catalytic site cysteine to cysteine-sulfinic acid. J. Biol. Chem. 277, 38029–38036 [DOI] [PubMed] [Google Scholar]
- 28. Bae S. H., Woo H. A., Sung S. H., Lee H. E., Lee S. K., Kil I. S., Rhee S. G. (2009) Induction of sulfiredoxin via an Nrf2-dependent pathway and hyperoxidation of peroxiredoxin III in the lungs of mice exposed to hyperoxia. Antioxid. Redox Signal. 11, 937–948 [DOI] [PubMed] [Google Scholar]
- 29. Kim B. J., Hood B. L., Aragon R. A., Hardwick J. P., Conrads T. P., Veenstra T. D., Song B. J. (2006) Increased oxidation and degradation of cytosolic proteins in alcohol-exposed mouse liver and hepatoma cells. Proteomics 6, 1250–1260 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Ruan Q., Lesort M., MacDonald M. E., Johnson G. V. (2004) Striatal cells from mutant huntingtin knock-in mice are selectively vulnerable to mitochondrial complex II inhibitor-induced cell death through a nonapoptotic pathway. Hum. Mol. Genet. 13, 669–681 [DOI] [PubMed] [Google Scholar]
- 31. Paulsen C. E., Carroll K. S. (2010) Orchestrating redox signaling networks through regulatory cysteine switches. ACS Chem. Biol. 5, 47–62 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Trachootham D., Lu W., Ogasawara M. A., Nilsa R. D., Huang P. (2008) Redox regulation of cell survival. Antioxid. Redox Signal. 10, 1343–1374 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Russell R. L., Siedlak S. L., Raina A. K., Bautista J. M., Smith M. A., Perry G. (1999) Increased neuronal glucose-6-phosphate dehydrogenase and sulfhydryl levels indicate reductive compensation to oxidative stress in Alzheimer disease. Arch. Biochem. Biophys. 370, 236–239 [DOI] [PubMed] [Google Scholar]
- 34. Jeong W., Park S. J., Chang T. S., Lee D. Y., Rhee S. G. (2006) Molecular mechanism of the reduction of cysteine sulfinic acid of peroxiredoxin to cysteine by mammalian sulfiredoxin. J. Biol. Chem. 281, 14400–14407 [DOI] [PubMed] [Google Scholar]
- 35. Kim Y. J., Ahn J. Y., Liang P., Ip C., Zhang Y., Park Y. M. (2007) Human prx1 gene is a target of Nrf2 and is up-regulated by hypoxia/reoxygenation. Implication to tumor biology. Cancer Res. 67, 546–554 [DOI] [PubMed] [Google Scholar]
- 36. Dinkova-Kostova A. T., Holtzclaw W. D., Cole R. N., Itoh K., Wakabayashi N., Katoh Y., Yamamoto M., Talalay P. (2002) Direct evidence that sulfhydryl groups of Keap1 are the sensors regulating induction of phase 2 enzymes that protect against carcinogens and oxidants. Proc. Natl. Acad. Sci. U.S.A. 99, 11908–11913 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Soriano F. X., Léveillé F., Papadia S., Higgins L. G., Varley J., Baxter P., Hayes J. D., Hardingham G. E. (2008) Induction of sulfiredoxin expression and reduction of peroxiredoxin hyperoxidation by the neuroprotective Nrf2 activator [3H]1,2-dithiole-3-thione. J. Neurochem. 107, 533–543 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Cumming R. C., Dargusch R., Fischer W. H., Schubert D. (2007) Increase in expression levels and resistance to sulfhydryl oxidation of peroxiredoxin isoforms in amyloid β-resistant nerve cells. J. Biol. Chem. 282, 30523–30534 [DOI] [PubMed] [Google Scholar]
- 39. Sorolla M. A., Reverter-Branchat G., Tamarit J., Ferrer I., Ros J., Cabiscol E. (2008) Proteomic and oxidative stress analysis in human brain samples of Huntington disease. Free Radic. Biol. Med. 45, 667–678 [DOI] [PubMed] [Google Scholar]
- 40. Chaturvedi R. K., Beal M. F. (2008) Mitochondrial approaches for neuroprotection. Ann. N.Y. Acad. Sci. 1147, 395–412 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Huntington Study Group (2001) A randomized, placebo-controlled trial of coenzyme Q10 and remacemide in Huntington disease. Neurology 57, 397–404 [DOI] [PubMed] [Google Scholar]
- 42. Verbessem P., Lemiere J., Eijnde B. O., Swinnen S., Vanhees L., Van Leemputte M., Hespel P., Dom R. (2003) Creatine supplementation in Huntington disease. A placebo-controlled pilot trial. Neurology 61, 925–930 [DOI] [PubMed] [Google Scholar]
- 43. Underwood B. R., Imarisio S., Fleming A., Rose C., Krishna G., Heard P., Quick M., Korolchuk V. I., Renna M., Sarkar S., García-Arencibia M., O'Kane C. J., Murphy M. P., Rubinsztein D. C. (2010) Antioxidants can inhibit basal autophagy and enhance neurodegeneration in models of polyglutamine disease. Hum. Mol. Genet. 19, 3413–3429 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Nielsen J. M., Butt E. M. (1955) Treatment of Huntington chorea with BAL. Bull. Los Angel Neuro. Soc. 20, 38–39 [PubMed] [Google Scholar]
- 45. Haslam M. T. (1967) Cellular magnesium levels and the use of penicillamine in the treatment of Huntington chorea. J. Neurol. Neurosurg. Psychiatry 30, 185–188 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Cory-Slechta D. A. (1988) Mobilization of lead over the course of DMSA chelation therapy and long-term efficacy. J. Pharmacol. Exp. Ther. 246, 84–91 [PubMed] [Google Scholar]
- 47. Aaseth J., Jacobsen D., Andersen O., Wickstrøm E. (1995) Treatment of mercury and lead poisonings with dimercaptosuccinic acid and sodium dimercaptopropanesulfonate. A review. Analyst 120, 853–854 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.