

Background: TGF-β1 suppresses growth of B-cell lymphoma cells.
Results: TGF-β1-induced down-regulation of mutant p53 via p14ARF renders B-cell lymphoma cells sensitive to TGF-β1.
Conclusion: Overexpression of p14ARF possibly causes TGF-β1 resistance.
Significance: p14ARF is a potential therapeutic target for B-cell lymphoma.
Keywords: Cytokine, Lymphoma, p53, Smad Transcription Factor, Transforming Growth Factor β (TGF-β)
Abstract
Previously we reported that TGF-β1-induced growth suppression was associated with a decrease in mutant p53 levels in B-cell lymphoma cells. The goal of the present study was to understand the mechanism involved in TGF-β1-mediated down-regulation of mutant p53. In RL and CA46, two B-cell lymphoma cell lines, TGF-β1 treatment caused down-regulation of E2F-1 transcription factor resulting in the down-regulation of both p14ARF and mutant p53, leading to growth arrest. Experimental overexpression of E2F-1 increased p14ARF level and blocked TGF-β1-induced down-regulation of p14ARF. Overexpression of p14ARF blocked the down-regulation of mutant p53 and prevented growth arrest. p14ARF also attenuated TGF-β1-induced p21Cip1/WAF1 induction, which was reversible by p53 siRNA, indicating the involvement of mutant p53 in controlling the TGF-β1-induced expression of p21Cip1/WAF1. The interaction observed between phospho-Smad2 and mutant p53 in the nucleus could be the mechanism responsible for blocking the growth-suppressive effects of TGF-β1. In RL cells, p14ARF is present in a trimer consisting of mutant p53-Mdm2-p14ARF and in a dimer consisting of Mdm2-p14ARF. Because it is known that Mdm2 can degrade p53, it is possible that, in its trimeric form, p14ARF is able to stabilize mutant p53 by inhibiting Mdm2. In its dimeric form, p14ARF may be sequestering Mdm2, limiting its ability to degrade p53. Collectively, these data demonstrate a unique mechanism in which the inhibition of TGF-β1-mediated growth suppression by mutant p53 can be reversed by the down-regulation of its stabilizing protein p14ARF. This work suggests that the high levels of p14ARF often found in tumor cells could be a potential therapeutic target.
Introduction
Members of the TGF-β1 family have pleiotropic functions, including proliferation, differentiation, migration, and apoptosis, in a broad range of cell lineages (1, 2). TGF-β1 signals through a heteromeric receptor complex on the cell surface and downstream intracellular signal-transducing Smad complexes activated by phosphorylation. Activated Smad complexes translocate into the nucleus and, in conjunction with other nuclear cofactors, regulate the transcription of target genes (3). The signaling of TGF-β1 is finely regulated by negative feedback, including inhibitory Smads (4, 5) and PPM1A phosphatase (6).
p53 is a transcription factor that mediates several cellular processes, including regulation of the cell cycle, apoptosis, DNA damage repair, and angiogenesis (7). Approximately 50% of human cancers have inactivating mutations of p53, and most of the remaining malignancies deactivate the p53 pathway either by blunting its activity, reducing its activators, or inactivating its downstream targets (8). It has been shown that mutant p53-expressing tumors are aggressive and associated with poor prognosis (9). Various p53 mutants confer different gain-of-function phenotypes such as increased cell growth (10, 11), enhanced tumorigenicity (11–15) and invasiveness (11, 13, 16), disturbed spindle checkpoint (17, 18), and resistance to cytotoxic agents (19).
p53 can have different effects on TGF-β1-induced growth suppression depending on its wild type or mutant status. In mink Mv1Lu epithelial cells, TGF-β1-mediated G1 growth arrest is heavily dependent on wild type p53 inhibiting the translation of CDK42 (20). It is also reported that p53 is required for TGF-β1-mediated growth arrest in certain mammalian cell types (21). It appears that p53 functions in this regard by cooperating with Smads to up-regulate the expression of the CDK inhibitor p21Cip1/WAF1. However, it was shown that the expression of mutant p53 caused cells to become resistant to TGF-β1-mediated growth suppression (12, 22). Recently, it was shown that a mutant p53-Smad complex contributes to TGF-β1-induced cell migration, invasion, and metastasis (23), confirming that p53 and Smad physically interact.
In primary tissues, p53 is expressed at very low levels because of its rapid degradation. For p53 to effectively function in growth arrest, its level must be stabilized. The primary mechanism involved in stabilizing p53 is through inhibition of the interaction between p53 and Mdm2, which targets p53 for degradation (24–26). Among several mechanisms involved in disrupting the Mdm2-p53 interaction, up-regulation of the tumor-suppressor protein p14ARF is an important one (27). Activation of p14ARF disrupts the physical interaction between p53 and Mdm2 resulting in the rapid degradation of Mdm2 and consequently more stable expression of p53. p14ARF tumor suppressor is the product of the alternative reading frame of the Ink4a locus, which also codes for p16Ink4a, an inhibitor for cyclin D-dependent kinases (28–30). In primary tissue, p14ARF (p19ARF in the mouse) is expressed at low levels. However, it can be induced by oncogenes such as Ras (31), Myc (32), and v-Abl (33) to cause p53-dependent growth arrest or apoptosis. In addition, p14ARF is able to inhibit cell growth through p53-independent pathways. For example, it has been shown that p14ARF is able to inhibit DNA synthesis in p53-null cells (34, 35). NFκB activity has been shown to be inhibited by p14ARF through interacting with RelA and repressing its transcriptional activity (36). p14ARF is also involved in inhibiting the function of proproliferative factor B23 through direct interaction with B23 and promoting its polyubiquitinylation and proteosomal degradation (37). It has been suggested that p53-independent functions of p14ARF may include its ability to promote sumoylation of several p14ARF-interacting proteins (38).
We have previously reported the effect of TGF-β1 on a human B-lymphoma cell line, RL, which expresses a mutant form of p53 having a single point mutation, A138P. We found that TGF-β1 causes growth inhibition in these cells that occurs simultaneously with a decrease in the level of mutant p53 (39). In this study, we examine the role and mechanism of the down-regulation of mutant p53 level caused by TGF-β1 treatment. We provide evidence suggesting that the decrease in mutant p53 level upon exposure to TGF-β1 mediates the growth-suppressive effect of this cytokine in B-cell lymphoma cell lines RL and CA46. The decrease in mutant p53 level is likely to be the result of a reduction in p14ARF levels because overexpression of p14ARF blocked TGF-β1-induced down-regulation of mutant p53 and subsequent growth arrest. Moreover, siRNA-mediated knockdown of p14ARF resulted in the down-regulation of mutant p53 and rendered cells more sensitive to TGF-β1-mediated growth suppression. Collectively these data demonstrate a unique mechanism in which the inhibition of TGF-β1-mediated growth suppression by mutant p53 is relieved by a TGF-β1-mediated signaling pathway that results in the down-regulation of the p53-stabilizing protein p14ARF. They also suggest that p14ARF antagonists may have an inhibitory effect on lymphoma proliferation.
EXPERIMENTAL PROCEDURES
Reagents
For Western blot analysis and immunoprecipitation, anti-TGF-β1 receptor II (TβRII) (sc-400), Smad2 (sc-6200), p14ARF (sc-8613), and p53 (sc-126) were obtained from Santa Cruz Biotechnology Inc. (Santa Cruz, CA); rabbit polyclonal phospho-Smad2 and anti-E2F-1 antibodies were purchased from Cell Signaling Technology Inc. (Beverly, MA); mouse monoclonal anti-p21Cip1/WAF1 antibody was from Upstate (Charlottesville, VA); anti-β-actin was purchased from Abcam (Cambridge, UK); and anti-Nucleoporin p62 was from BD Biosciences. All HRP-conjugated secondary antibodies were purchased from GE Healthcare. Recombinant TGF-β1 (240-B) was purchased from R&D Systems (Minneapolis, MN). Phorbol 12-myristate 13-acetate (PMA) was from Sigma. Anti-IgM was purchased from Jackson ImmunoResearch Laboratories (West Grove, PA).
Cell Culture
Lymphoma cells were cultured in RPMI 1640 medium supplemented with 10% fetal bovine serum (FBS), 2 mm l-glutamine, 1,000 units of penicillin/ml, and 100 μg of streptomycin/ml. No exogenous growth factors were added. Cells were grown at 37 °C in 5% CO2. Fresh growth medium was added to cells every 3–4 days. For growth inhibition, cells were stimulated with 2 ng/ml recombinant TGF-β1 in RPMI 1640 medium with 5% FBS.
Cell Lysis and Western Blot Analysis
Cytoplasmic and nuclear extracts were prepared according to the procedure reported earlier (40). Whole cell lysates were prepared according to the following procedure. Harvested cells were resuspended in lysis buffer (10 mm Tris-Cl (pH 8.0), 150 mm NaCl, 1 mm MgCl2, 5 μg/ml E-64, 1 mm PMSF, 2.5 mm sodium pyrophosphate, 1 mm β-glycerophosphate, 1 mm Na3VO4, 1 mm NaF, and 1% Triton X-100) and incubated on ice for 30 min. Samples were then homogenized and centrifuged, and the supernatants were collected. Protein concentrations were measured with Bio-Rad Protein Assay Dye reagent. Gel electrophoresis was carried out using 4–12% SDS-PAGE under reducing conditions. After membrane transfer, bound antibodies were detected using a chemiluminescence detection system (GE Healthcare).
WST-1 Assay
The quantification of cell proliferation was evaluated by WST-1 assay (Roche Applied Science), a colorimetric assay based on the cleavage of tetrazolium salt WST-1 by mitochondrial dehydrogenases in viable cells. 1 × 104 cells in 100 μl of culture medium were loaded in each well of a 96-well plate and incubated with 2 ng/ml TGF-β1 for 48 h at 37 °C in 5% CO2. 10 μl of WST-1 reagent was added to each well, and cells were incubated for 1 h at 37 °C. The absorbance was measured with a microplate reader (Bio-Rad Model 680XR) with a 450-nm wavelength filter and a 655-nm reference wavelength filter.
Immunoprecipitation
Lysates were precleared with protein A/G Plus-agarose (sc-2003, Santa Cruz Biotechnology Inc.) for 30 min at 4 °C, and the precleared lysates were incubated overnight with the primary antibody and A/G Plus-agarose beads. The agarose beads were washed three times with extraction buffer containing 25 mm MOPS (pH 7.2), 15 mm MgCl2, 137 mm NaCl, 1 mm PMSF, 15 mm EGTA, 5 μg/ml E-64, 1 mm Na3OV4, 1 mm NaF, and 0.1% Triton X-100. The immune complexes were dissociated with lithium dodecyl sulfate buffer and boiled for 5 min. Electrophoresis was carried out by 4–12% SDS-PAGE under reducing conditions.
Northern Blotting
Total RNA was extracted from RL cells with an RNeasy Mini kit (Qiagen, Valencia, CA) and purified according to the manufacturer's specifications. Up to 25 μg of total RNA from each sample was electrophoresed under denaturing conditions on a 1% agarose gel in glyoxal (Ambion, Austin, TX), blotted onto Hybond-N+ membrane (Amersham Biosciences), and cross-linked by UV irradiation. The membrane was prehybridized overnight at 42 °C in Hybrisol I (Chemicon, Millipore) followed by overnight hybridization with 1 × 106 cpm/ml specific cDNA probe labeled with 32P by random priming of a 450-bp BamHI/EcoRV fragment from the p14ARF/pcDNA3 using High Primer DNA labeling reagent (Roche Diagnostics). Probes were purified by a G-50 column (Amersham Biosciences) before use. The membranes were then washed and exposed to x-ray film at −70 °C.
Statistical Analysis
Values were obtained from three independent experiments and were expressed as means ± S.D. Statistical analysis was performed using Student's t test and analyzed by two-tailed test of paired samples. Values were considered significant (*) if p values were <0.05.
DNA Constructs and Transfection
pSuper/p14ARFsi is from Dr. Sonia Lain, Ninewells Hospital, University of Dundee, Scotland, UK; pCMV-Bam-Neo/p53 was obtained from Dr. Bert Vogelstein, The Johns Hopkins University, Baltimore, MD; and pCMV-Bam-Neo/p53 A138P was reported earlier (41). The E2F-1 expression vector was kindly provided by Dr. Asish Lal, National Cancer Institute, National Institutes of Health, Bethesda, MD. Exponentially growing RL cells were resuspended in 100 μl of Kit V solution (Lonza, Gaithersburg, MD) containing 3 μg of plasmid. Cells were then exposed to electroporation (program S-18) using a Nucleofector device (Lonza). After 12 h, cells were transferred to fresh medium and were cultured for 24 h.
Virus Production and Transduction
To generate adenovirus constructs, sequences corresponding to the wild type TβRII and p14ARF were amplified from DNA constructs (generous gifts from Drs. Joan Massague from Memorial Sloan-Kettering Cancer Center, New York and Kevin Ryan from the National Cancer Institute, Frederick, MD, respectively) and subcloned into pENTRY/SD/D-TOPO vector (Invitrogen). Recombination between p14ARF from pENTRY/SD/D and pAD/CMV/V5-DEST vector was carried out by LR Clonase II Enzyme Mix (Invitrogen), and preparation of adenoviruses bearing p14ARF was done according to the manufacturer's instructions. To introduce the human coxsackie adenovirus receptor (hCAR) to the B-cell lymphoma cell line, pHRCMV/hCAR-EGFP (generously provide by Dr. Mikko Mättö, University of Kuopio, Finland) was used. RL/hCAR-EGFP cells were infected with adenovirus harboring p14ARF with a multiplicity of infection (m.o.i.) equal to 200. To reduce mutant p53 expression in RL cells, a lentiviral construct, pWPXL-p53si (kindly provided by Dr. Radek C. Skoda, Basel University Hospital, Switzerland), was used. Lentiviral production with pWPXL-p53si and pHRCMV/hCAR-EGFP using the envelope vector pMD.G and the packaging vector pCMVR8.91 (kindly provided by Dr. Didier Trono, Global Health Institute, Switzerland) was carried out as described before (42). RL cells were seeded in a 12-well plate at 1×106 cells/well, and virus was added at an m.o.i. equal to 10. EGFP-positive cells were isolated by limiting dilution and checked by FACScan (BD Biosciences).
p21Cip1/WAF1 Promoter-Reporter Assay
RL/hCAR-EGFP cells were infected with TβRII/pAD and p14ARF/pAD for 24 h. Then the cells were transfected with p53 expression construct (a gift from Dr. Bert Vogelstein, The Johns Hopkins University), p21Cip1/WAF1 promoter-luciferase construct pWWP-Luc (p21Cip1/WAF1) (a gift from Dr. Weiguo Zhu, Peking University Health Science Center, China), and β-galactosidase expression construct by electroporation as described previously (43). For mutant p53 knockdown experiments, RL cells were infected with pWPXL-p53si for 24 h before adenovirus infection. Transfected cells were further cultured for 24 h before activation with TGF-β1 for 16 h. β-Galactosidase and luciferase activity was measured with the Beta-Glo and Bright-Glo (Promega, Madison, WI) assays according to the instructions of the manufacturer. p21Cip1/WAF1 promoter-luciferase activity was normalized with the β-galactosidase assay.
Chromatin Immunoprecipitation (ChIP) Assay
This assay was carried out according to the protocol described previously with minor modification (44). After cross-linking with formaldehyde, soluble chromatin from the samples was obtained by sonication (Bronson sonicator 350). The chromatin was incubated overnight with 2 μg of either normal IgG or anti-Smad2/3 or anti-E2F-1 antibody. After washing and elution, the immunocomplexed DNA was isolated with Chelex beads (Bio-Rad) and analyzed by PCR. Primers used to amplify DNA fragments corresponding to a Smad binding region (SBR1) on human p21Cip1/WAF1 promoter were 5′-GAGGAAAAGCATCTTGGAG-3′ (forward) and 5′-AATAGACGGGAGCAACG-3′ (reverse) (45). Primers for p14ARF promoters were 5′-GCTGAGGGTGGGAAGATGG-3′ (forward) and 5′-AGACTGGGACCCACGCACC-3′ (reverse).
RESULTS
Down-regulation of p14ARF and Mutant p53 in B-cell Lymphoma Cells in Response to TGF-β1
We have shown previously that RL cells are unresponsive to TGF-β1-mediated growth suppression, whereas in the presence of a low dose of PMA, RL cells can be rendered responsive to TGF-β1 (43) (Fig. 1A). We have also reported that treatment of RL cells with PMA/TGF-β1 results in the down-regulation of mutant p53 (Ala-138 to Pro), the only form of p53 present in RL cells (i.e. wild type p53 is absent) (39, 46) (Fig. 1B). Because p14ARF is involved in the stability of p53 protein, we wanted to determine the status of p14ARF in PMA/TGF-β1-treated cells. As shown in Fig. 1B, down-regulation of the p14ARF level was observed only with PMA/TGF-β1 treatment, conditions that also decrease mutant p53 levels, but PMA or TGF-β1 treatment alone had no effect on p14ARF or mutant p53 levels. TGF-β1-induced down-regulation of p14ARF and mutant p53 was also observed in the presence of physiologically relevant stimulation delivered by anti-IgM treatment (Fig. 1C). To further elucidate the relative kinetics of the down-regulation of both p14ARF and mutant p53, RL cells were treated with either PMA alone, TGF-β1 alone, or with PMA/TGF-β1 for various periods of time, and the levels of p14ARF, mutant p53, and Mdm2 were measured by Western blot analysis. As shown in Fig. 1D, a reduction in p14ARF and mutant p53 levels occurred at 16 h (lane 7 versus lane 5 or lane 6), although the degree of down-regulation was greater in the case of p14ARF compared with mutant p53. The level of Mdm2 was unaffected at the 16-h time point. We next wanted to assess whether the down-regulation of p14ARF was linked to the down-regulation of mutant p53.
FIGURE 1.

Down-regulation of p14ARF and mutant p53 in B-cell lymphoma cells in response to TGF-β1. A, RL cells were plated at 0.1 × 106 cells/ml and treated with either medium alone, TGF-β1 (2 ng/ml), PMA (0.15 ng/ml), or PMA plus TGF-β1 for various time periods. At the end of each time point, cell counts were performed. * indicates that the growth suppression is statistically significant (p < 0.01). B, RL cells were treated with either medium alone or TGF-β1 in the presence or absence of PMA for 72 h, and equal amounts of whole cell lysates were analyzed by Western blot analysis. C, RL cells were treated with either medium alone or TGF-β1 in the presence or absence of anti-IgM (1 μg/ml) for 24 h, and equal amounts of whole cell lysates were analyzed by Western blot analysis. D, RL cells were treated with either medium alone or TGF-β1 in the presence or absence of PMA for various time periods, and equal amounts of whole cell lysates were analyzed by Western blot analysis.
Effect of p14ARF on TGF-β1-induced Down-regulation of Mutant p53
We have observed that the resistance of RL cells to TGF-β1-induced growth arrest was due to the ligand-induced down-regulation of TβRII, which can be prevented by low dose PMA treatment (47). In accordance with this finding, transfection of TβRII DNA leading to forced receptor expression rendered RL cells responsive to TGF-β1-induced growth suppression in the absence of PMA (Fig. 2A, left panel). However, transfection of p14ARF into TβRII-overexpressing cells rendered these cells resistant to TGF-β1-induced growth suppression (Fig. 2A, right panel). To investigate the relationship between the status of p14ARF and mutant p53, levels of p14ARF and mutant p53 in cell lysates from TβRII-overexpressing cells were analyzed by Western blot analysis in the presence or absence of experimentally overexpressed p14ARF. As shown in Fig. 2B, TGF-β1-induced down-regulation of p14ARF coincided with the down-regulation of mutant p53 (lane 2 versus lane 1), and this down-regulation was blocked by overexpression of p14ARF (lane 2 versus lane 4). Interestingly, down-regulation of mutant p53 coincided with the up-regulation of p21Cip1/WAF1 (lane 1 versus lane 2), and the induction of p21Cip1/WAF1 was blocked in the presence of p14ARF (lane 2 versus lane 4). The inhibition of TGF-β1-induced up-regulation of p21Cip1/WAF1 by overexpressed p14ARF was not due to the blockage in TGF-β1 signaling because TGF-β1-induced phosphorylation of Smad2 was unaffected by the p14ARF overexpression (lane 2 versus lane 4). These data suggest that p14ARF is involved in TGF-β1-induced down-regulation of mutant p53 and subsequent growth suppression of RL cells.
FIGURE 2.


Effect of p14ARF overexpression on TGF-β1-mediated response. A, left panel, RL/hCAR-EGFP cells were infected with pAD/CMV/TβRII (RII.pAD) (m.o.i., 200). After 48 h, cells were treated with either medium alone or TGF-β1 for various time periods. Right panel, RL/hCAR-EGFP cells were infected with pAD/CMV/TβRII and pAD/CMV/p14ARF (p14.pAD) (m.o.i., 200). After 48 h, cells were treated with either medium alone or TGF-β1 for various time periods, and at the end of each time point, cell proliferation was analyzed by WST-1 assay. * indicates that the growth suppression is statistically significant (p < 0.01). B, left panel, RL/hCAR-EGFP cells were infected with pAD/CMV/TβRII in the presence or absence of pAD/CMV/p14ARF. After 48 h, cells were treated with either medium alone or TGF-β1 for 48 h, and equal amounts of cell lysates were analyzed by Western blot analysis. Right panel, quantitation of the bands is presented as -fold change. C, upper left panel, RL.hCAR cells were transfected with either vector alone (pSuper) or p14ARF siRNA construct (pSuper/p14si). After 48 h, whole cell lysates were prepared, and equal amounts of lysates were analyzed by Western blot analysis. Lower left panel, RL.hCAR cells were transfected with either vector alone (pSuper) or p14ARF siRNA construct in the absence (pSuper/p14si) or presence of p14ARF expression vector (pcDNA3/p14ARF). After 48 h, whole cell lysates were prepared, and equal amount of lysates were analyzed by Western blot analysis. Upper right panel, RL.hCAR cells were infected with RII.pAD (m.o.i., 200). After 24 h, cells were transfected with either pSuper vector or pSuper/p14si and incubated for an additional 24 h. Cells were then treated with either medium alone or TGF-β for different periods of time. At the end of each time point, cell counts were performed. * indicates that the difference between the two TGF-β-treated samples is statistically significant (p < 0.05). Lower right panel, RL.hCAR cells were infected with RII.pAD (m.o.i., 200). After 24 h, cells were transfected with either p14ARF siRNA construct (p14si) or p14ARF siRNA construct in the presence of p14ARF expression vector pcDNA3/p14ARF (p14si/p14) and incubated for an additional 24 h. Cells were then treated with either medium alone or TGF-β for different periods of time. At the end of each time point, cell counts were performed. D, CA46 cells were transfected with either pcDNA3 vector or pcDNA3/p14ARF. After 48 h, cells were treated with either medium alone or TGF-β1 for different periods of time, and at the end of each time point, cell counts were performed (left panel). Results are representative of experiments done in triplicate. * indicates that the growth suppression is statistically significant (p < 0.01). Right panel, after 48 h of transfection, cells were treated with either medium alone or TGF-β1 for 48 h, and equal amounts of nuclear extracts were analyzed by Western blot analysis. One representative experiment of two independent experiments is shown here. E, RL/hCAR-EGFP cells were infected with pAD/CMV/TβRII in the presence or absence of pAD/CMV/p14ARF. After 24 h, cells were transiently transfected with pWWP-Luc (p21Cip1/WAF1) reporter construct and β-galactosidase plasmid as well as either wild type p53 or A138P mutant p53 construct. After 24-h incubation, cells were treated with either medium alone or TGF-β1 for an additional 16 h. Promoter-luciferase activity was normalized with β-galactosidase activity. Standard deviation was calculated from triplicate samples. Results are representative of two independent experiments. pSmad2, phospho-Smad2.
Next we wanted to investigate the relationship between p14ARF and mutant p53 by knocking down p14ARF with an siRNA construct and test whether down-regulation of p53 by p14ARF was able to cause growth suppression in the absence of TGF-β1 treatment. As shown in Fig. 2C (upper left panel), down-regulation of p14ARF by the siRNA construct caused down-regulation of the mutant p53 level, and this down-regulation was specific to the siRNA construct because we were able to rescue the mutant p53 by overexpressing p14ARF in the presence of p14ARF siRNA (Fig. 2C, lower left panel). Although the p14ARF siRNA construct down-regulated mutant p53, this siRNA construct alone did not affect the growth of RL cells (right panel). TGF-β1 signaling was still required for growth suppression. Interestingly, the degree of growth suppression was higher in the presence of the p14ARF siRNA construct as compared with the vector control, suggesting that growth inhibition is enhanced if p14ARF is suppressed before exposure to TGF-β1. We have also shown functionally the specificity of the p14ARF siRNA by rescuing the TGF-β1-mediated growth suppression in the presence of both p14ARF siRNA and overexpressed p14ARF (Fig. 2C, lower right panel).
It was of interest to determine whether this novel down-regulation of p14ARF and mutant p53 by TGF-β1 occurs in other B-cell lymphoma cell lines. As shown in Fig. 2D (right panel), both p14ARF and mutant p53 were also down-regulated upon TGF-β1 treatment in the TGF-β1-sensitive Burkitt lymphoma cell line CA46 (lane 2 versus lane 1), and the down-regulation of mutant p53 was blocked by overexpression of p14ARF (lane 2 versus lane 4). The down-regulation of p14ARF and mutant p53 by TGF-β1 correlated with the induction of p21Cip1/WAF1 expression and growth suppression, which were blocked by overexpression of p14ARF. Both RL and CA46 are derived from tumors of germinal center B-cells.
Next we wanted to investigate how p14ARF and mutant p53 were connected to TGF-β1-induced p21Cip1/WAF1 expression in RL cells. There are binding sites for Smads and p53 in the promoter region of p21Cip1/WAF1 (21, 48). Because the alanine to proline mutation at position 138 of p53 in RL cells is in the DNA binding domain, the mutant lacks transcriptional activity (supplemental Fig. 1) (41). To explore whether the A138P p53 mutant affected p21Cip1/WAF1 transcription induced by Smad complexes, we analyzed p21Cip1/WAF1 promoter activity in the presence of either p14ARF, wild type p53, or A138P mutant p53. As shown in Fig. 2E, TGF-β1-induced promoter activity of p21Cip1/WAF1 was inhibited by p14ARF and mutant p53, whereas wild type p53 expression caused a slightly higher activity compared with TGF-β1 treatment alone. Collectively, these data suggest that the inhibition of TGF-β1-induced p21Cip1/WAF1 promoter activity by p14ARF could be mediated by mutant p53.
Inhibition of p21Cip1/WAF1 Promoter Activity Mediated by p14ARF Can Be Blocked by p53 siRNA
If the inhibitory effect of p14ARF on p21Cip1/WAF1 promoter activity was mediated by the mutant p53, knocking down mutant p53 in the presence of overexpressed p14ARF should reverse the inhibitory effect of p14ARF. As shown in Fig. 3A, the siRNA construct against p53 (p53si) induced down-regulation of mutant p53 in RL cells in a time-dependent manner. Using this siRNA, we were able to show that the inhibition of p21Cip1/WAF1 promoter activity by p14ARF was reversed by the down-regulation of mutant p53 (Fig. 3B). Consistent with these data, lysates from the cells harboring different expression constructs showed the TGF-β1-induced up-regulation of p21Cip1/WAF1 expression upon down-regulation of mutant p53 (Fig. 3C, compare lane 5 with lane 4). These data strongly suggest that p14ARF plays an important role in blocking TGF-β1-induced growth suppression by controlling the expression of mutant p53 in these B-cell lymphomas.
FIGURE 3.

Role of mutant p53 in p14ARF-mediated down-regulation of TGF-β1-induced p21Cip1/WAF1 expression. A, RL cells were infected with pWPXL-p53si (m.o.i., 20). Cells were harvested at various time points. Equal amounts of cell lysates were analyzed by Western blot analysis. B, RL/hCAR-EGFP cells were infected with pWPXL-p53si followed by infection with pAD/CMV/TβRII in the presence or absence of pAD/CMV/p14ARF. After 24 h, cells were transiently transfected with pWWP-Luc (p21Cip1/WAF1) reporter construct and β-galactosidase plasmid. Cells were then treated with either medium alone or TGF-β1 for an additional 16 h. Promoter-luciferase activity was normalized with β-galactosidase activity. Standard deviation was calculated from experiments done in triplicate. Results are representative of two independent experiments. C, left panel, RL/hCAR-EGFP cells were infected with pWPXL-p53si followed by infection with pAD/CMV/TβRII in the presence or absence of pAD/CMV/p14ARF. After 48 h of incubation, cells were treated with either medium alone or TGF-β1 for an additional 48 h. Equal amounts of cell lysates were analyzed by Western blot analysis. Right panel, quantitation of the bands is presented as -fold change. D, recruitment of Smad2/3 to p21Cip1/WAF1 gene. As described under “Experimental Procedures,” immunoprecipitated soluble chromatin complexes were isolated from cells infected with either pWPXL-p53si (p53si) or control vector that were treated with either medium alone or PMA/TGF-β1 for the indicated time points. The level of DNA enrichment was assessed by PCR followed by analysis of equal volumes of PCRs on an agarose gel. One representative experiment of two is shown here. pSmad2, phospho-Smad2; IP, immunoprecipitation.
To further examine the involvement of mutant p53 and TGF-β1 signaling in controlling p21Cip1/WAF1 expression, we performed ChIP analysis using anti-Smad2/3 antibody to detect the recruitment of Smad2/3 to p21Cip1/WAF1 promoter. As shown in Fig. 3D, recruitment of Smad2/3 to p21Cip1/WAF1 promoter was observed as early as 1 h and increased substantially after 24 h of PMA/TGF-β1 treatment. The increase in Smad2/3 recruitment at the 24-h time point correlated with the down-regulation of mutant p53 upon PMA/TGF-β1 treatment (Fig. 1D). To examine whether mutant p53 is involved in blocking TGF-β1-induced Smad2/3 recruitment to p21Cip1/WAF1 promoter, a ChIP assay was performed in the cells where mutant p53 was down-regulated by siRNA. Interestingly, an increased level of Smad2/3 recruitment was observed only after 1 h of PMA/TGF-β1 treatment when mutant p53 was down-regulated by siRNA, suggesting a role of mutant p53 in blocking TGF-β1 signaling.
Mutant p53 Physically Interacts with Phospho-Smad2 in the Nucleus
To gain more insight into the mechanism underlying mutant p53-mediated blockage of TGF-β1-mediated p21Cip1/WAF1 promoter activity, we investigated whether mutant p53 interacts with phospho-Smads and prevents phospho-Smads from binding to the promoter. RL cells were treated with TGF-β1 for different time periods, and the nuclear extracts were used for immunoprecipitation with anti-Smad2 antibody. As shown in Fig. 4, anti-Smad2 antibody was able to pull down mutant p53, and the interaction was increased upon TGF-β1 treatment in a time-dependent manner. This experiment suggests that by interacting with phospho-Smad2 mutant p53 might have interfered in the TGF-β1-induced activation of p21Cip1/WAF1 promoter.
FIGURE 4.

Mutant p53 physically interacts with Smad2. RL cells were treated with either medium alone or TGF-β1 for 1 or 6 h. Nuclear extracts were prepared, and equal amounts of extracts were used for immunoprecipitation (IP) with anti-Smad2. Left panel, the immunocomplexes were analyzed by Western blot analysis. Right panel, equal amounts of nuclear extracts were analyzed by Western blot analysis. One representative experiment of two is shown here. The quality of nuclear extracts was shown by probing with β-actin. pSmad2, phospho-Smad2.
Down-regulation of p14ARF Protein by TGF-β1 Was Due to a Reduction in Its mRNA Level
To investigate whether the down-regulation of p14ARF and mutant p53 protein levels by TGF-β1 was due to the reduction in mRNA expression, RL cells were treated with TGF-β1 for different time periods, and the total RNAs were analyzed by Northern blot analysis. As shown in Fig. 5, whereas TGF-β1 treatment decreased the p14ARF mRNA level in a time-dependent manner, the mRNA level for mutant p53 was unaffected by TGF-β1 throughout the time points tested. These data along with the data shown in Fig. 1C indicate that TGF-β1 down-regulated p14ARF and mutant p53 expression by two different mechanisms: transcriptional or mRNA stability in the case of p14ARF and at the protein level in the case of mutant p53.
FIGURE 5.

Down-regulation of p14ARF, but not mutant p53, by TGF-β1 was due to a reduction in its mRNA level. A, RL Cells were treated with either medium alone or with TGF-β1 in the presence of PMA for various time periods. Total RNA was extracted, and the mRNA levels of p14ARF and p53 were determined by Northern blot analysis. One representative experiment of two is shown here. B, left panel, RL cells were treated with either medium alone or PMA/TGF-β1 for various time periods, and equal amounts of nuclear extracts were analyzed by Western blot analysis. Right panel, quantitation of the bands is presented as -fold change. C, TβRII-overexpressing RL cells (RL/pWPI/HA-TβRII) and CA46 cells were treated with either medium alone or TGF-β1, and equal amounts of nuclear extracts were analyzed by Western blot analysis. D, RL cells were transfected with either the empty vector or the E2F-1 expression vector, and after 48 h, cells were treated with either medium alone or PMA/TGF-β1 for 24 h. Equal amounts of nuclear extracts were analyzed by Western blot analysis. E, as described under “Experimental Procedures,” immunoprecipitated soluble chromatin complexes were isolated from RL cells treated with either medium alone or PMA/TGF-β1 for the indicated time points. The level of DNA enrichment was assessed by PCR followed by analysis of equal volumes of PCRs on an agarose gel. One representative experiment of two is shown here. IP, immunoprecipitation.
Next we wanted to understand the mechanism underlying the down-regulation of p14ARF upon TGF-β1 treatment. It has been shown that TGF-β1-mediated growth arrest in CA46 cells is regulated by transcriptional repression of E2F-1 (49). It also has been reported previously that E2F-1 transcriptionally activates p14ARF expression (50). So we tested whether E2F-1 is involved in TGF-β1-mediated down-regulation of p14ARF expression in RL cells. To elucidate the relative kinetics of the down-regulation of E2F-1, p14ARF, and mutant p53, RL cells were treated with PMA/TGF-β1 for various periods of time, and the levels of all three proteins were analyzed by Western blot analysis. The time points were chosen based on the slow kinetics of E2F-1 down-regulation observed in CA46 cells after TGF-β1 treatment (49). As shown in Fig. 5B, PMA/TGF-β1-induced down-regulation of E2F-1 was observed as early as 6 h, and this down-regulation preceded the down-regulation of p14ARF and mutant p53, indicating the possible involvement of E2F-1 in PMA/TGF-β1-induced down-regulation of p14ARF. We also observed that TGF-β1 treatments of TβRII-overexpressing RL cells and CA46 cells caused down-regulation of E2F-1 levels (Fig. 5C). To test the effect of E2F-1 expression on the levels of p14ARF and mutant p53, RL cells were transfected with either the empty vector or the E2F-1 expression vector, and the cells were treated with medium alone or with PMA/TGF-β1. As shown in Fig. 5D, ectopic expression of E2F-1 resulted in increased levels of both p14ARF and mutant p53 (lane 2 versus lane 1). Moreover, overexpression of E2F-1 blocked the down-regulation of p14ARF and mutant p53 by PMA/TGF-β1 treatment (lane 3 versus lane 2). To further demonstrate the role of E2F-1 in controlling the expression of p14ARF, we performed ChIP analysis to assess the binding of E2F-1 to the p14ARF promoter. As shown in Fig. 5E, E2F-1 was found to constitutively occupy the p14ARF promoter, and this promoter binding of E2F-1 was inhibited upon TGF-β1 treatment in a time-dependant manner. Collectively, our data suggest that PMA/TGF-β-induced down-regulation of p14ARF was mediated via E2F-1.
The relation between p14ARF, p53, and Mdm2 is well established (27). To monitor the interaction among these proteins in response to TGF-β treatment, TβRII-overexpressing cells (RL/pWPI/HA-TβRII) were treated with TGF-β1 for 24 h in the presence or absence of a proteasome inhibitor, MG132, for the last 8 h. Nuclear extracts were prepared and subjected to immunoprecipitation analysis using anti-Mdm2 antibody. As shown in Fig. 6, whereas anti-Mdm2 antibody was able to pull down both p14ARF and mutant p53 from untreated cells (lane 2), no p14ARF and mutant p53 were observed in the immunoprecipitated complex from TGF-β1-treated samples (lane 3 versus lane 2). This was due to the down-regulation of p14ARF and mutant p53 expression upon TGF-β1 treatment (Input panel, lane 2). Blocking of TGF-β1-induced degradation of mutant p53, but not p14ARF, by MG132 resulted in an immunoprecipitated complex that contained mutant p53 (lane 5 versus lane 3). These data suggest that TGF-β1-induced down-regulation of mutant p53 might be via Mdm2-mediated degradation, whereas down-regulation of p14ARF was not at the protein level.
FIGURE 6.

TGF-β1-induced down-regulation of mutant p53 was via Mdm2-mediated degradation. TβRII-overexpressing cells (RL/pWPI/HA-TβRII) were treated with either medium alone or TGF-β1. After 24 h, cells were treated either with or without MG132 for an additional 8 h, and nuclear extracts were prepared. Left panel, equal amounts of extracts were used for immunoprecipitation (IP) with anti-Mdm2 antiserum, and the immunocomplexes were analyzed by Western blot analysis. Right panel, equal amounts of nuclear lysates were analyzed by Western blot analysis. One representative experiment of two is shown here.
DISCUSSION
TGF-β1 blocks growth in a large number of cell types. Mutants of the tumor suppressor p53 have been shown to increase cell proliferation and resistance to TGF-β1-mediated growth inhibition (12, 22). In this study, we have shown a unique mechanism whereby TGF-β1 treatment down-regulated the E2F-1 level, leading to the down-regulation of p14ARF and mutant p53 to allow the expression of p21Cip1/WAF1 and subsequent growth suppression of B-cell lymphoma cell lines (Fig. 7). Experimental overexpression of E2F-1 not only up-regulated p14ARF and mutant p53 but also blocked TGF-β1-induced down-regulation of p14ARF and mutant p53. Overexpression of p14ARF blocked TGF-β1-induced growth suppression by preventing down-regulation of mutant p53 and up-regulation of p21Cip1/WAF1. p14ARF-mediated blockage of p21Cip1/WAF1 expression was reversed by siRNA-mediated reduction of mutant p53, indicating the involvement of mutant p53 in controlling TGF-β1-induced expression of p21Cip1/WAF1. We have also shown by ChIP analysis that TGF-β1-induced Smad2/3 recruitment to p21Cip1/WAF1 promoter was increased when the mutant p53 level was down-regulated by siRNA (Fig. 3D). Collectively, these data indicate a critical role for p14ARF in stabilizing mutant p53 and possibly conferring TGF-β1 resistance in B-cell lymphoma cell lines.
FIGURE 7.
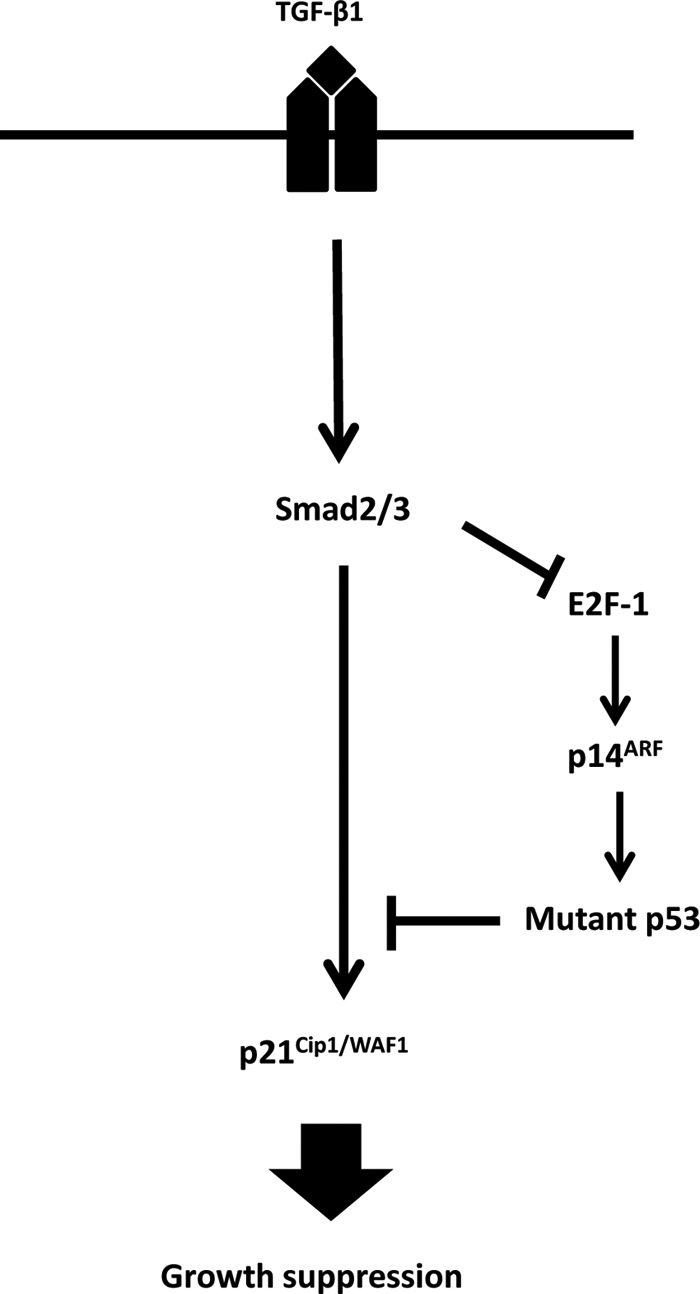
Schematic representation of the TGF-β signaling in RL cells. In RL cells, the basal level of p14ARF is maintained by E2F-1 transcription factor. This p14ARF level maintains the basal level of mutant p53. Experimental overexpression of p14ARF increases the levels of mutant p53, which in turn interferes with the TGF-β1-induced recruitment of phospho-Smad2/3 to the p21Cip1/WAF1 promoter and thus blocks p21Cip1/WAF1 expression and inhibition of growth suppression. Upon TGF-β1 treatment, the basal expression level of E2F-1 is down-regulated, which in turn causes the down-regulation of p14ARF and mutant p53 levels, resulting in the up-regulation of p21Cip1/WAF1 and subsequent growth arrest.
In a gene expression profile study of diffuse large B-cell lymphomas, Lenburg et al. (51) observed increased levels of p14ARF transcript expression in four of seven lymphomas classified as aggressive lymphomas. The levels of p14ARF transcript expression in these four aggressive lymphomas ranged from ∼18- to 140-fold greater than the average level of transcript expression observed in normal resting and activated B-cells. When the levels of p14ARF transcript expression in these four aggressive lymphomas were compared with the average level observed in the marginal zone lymphomas or transitional lymphomas in the study, the level of expression in the four aggressive lymphomas ranged from ∼3- to 19-fold higher. A similar observation was made by Sánchez-Aguilera et al. (52), who noted that a group of aggressive non-Hodgkin lymphomas showed both elevated levels of p14ARF protein expression and an uncharacteristic non-nucleolar distribution versus the almost exclusive nucleolar localization of p14ARF protein observed in normal cells. Overexpression of p14ARF was associated with high levels of p53, and over 50% of these cases carried a mutation in p53. We show here that the high levels of p14ARF can contribute to the proliferative state of these cells.
p14ARF was initially characterized for its ability to cause G1 arrest in response to elevated mitogenic signals by stabilizing p53 (29, 53). The role of p14ARF in TGF-β1-mediated growth inhibition has not been elucidated. Recently it has been shown that the expression of p19ARF (murine homologue of human p14ARF) induced by TGF-β1 contributes to growth arrest in mouse keratinocytes (54). In addition, it was reported that TGF-β2 is required for p19ARF transcription in mouse embryo fibroblasts during development (55).
In primary cells, the level of wild type 53 is usually very low, whereas the level of mutant p53 is abundant in some tumor cell lines, including RL and CA46. These high levels of mutant p53 not only give these tumor cells a proliferative advantage but also confer resistance to TGF-β1-induced growth suppression. We have shown here that mutant p53 by interacting with phospho-Smad2 interferes with TGF-β1-induced p21Cip1/WAF1 promoter activity. The level of p14ARF is critical in maintaining the level of mutant p53 and subsequent blockage in TGF-β1-induced p21Cip1/WAF1 expression because siRNA knockdown of mutant p53 reverses not only the p14ARF-mediated down-regulation of p21Cip1/WAF1 promoter activity but also increases p21Cip1/WAF1 protein expression.
These changes in the tumor cells are not restricted to TGF-β signaling. A similar sequence of cellular events occurs after signal transduction through the surface immunoglobulin molecule. Prior work has demonstrated that anti-idiotypic antibodies may kill tumor cells in vivo through cell signaling events (56). It is likely that activation-induced cell death from a variety of stimuli require the down-regulation of mutant p53 through this or a closely related mechanism.
In summary, we have demonstrated that in RL and CA46 lymphoma cells an altered TGF-β1-induced signaling pathway has developed that reduces levels of p14ARF via transcriptional repression or mRNA stability. In normal cells, wild type p53 and p14ARF are expressed at very low levels. When p53 is mutated, it cannot feed back and down-regulate p14ARF levels as the wild type protein does (57). Therefore, mutant p53 and p14ARF levels build to high levels. These high p14ARF and mutant p53 levels block TGF-β1-mediated growth suppression by attenuating p21Cip1/WAF1 expression. The data presented here also demonstrate that TGF-β1 reduces levels of p14ARF via down-regulating E2F-1 transcription factor. It will be interesting to see whether E2F-1 overexpression is also observed in aggressive non-Hodgkin lymphomas where elevated levels of p14ARF expression have been reported (52). Our data suggest that the high levels of p14ARF often found in tumor cells may be a potential therapeutic target in that reducing them may make other treatments more effective by compromising the capacity of mutant p53 to promote proliferation.
Supplementary Material
This work was supported, in whole or in part, by the NIA, National Institutes of Health Intramural Research Program.

This article contains supplemental Fig. 1.
- CDK
- cyclin-dependent kinase
- TβRII
- TGF-β1 receptor II
- PMA
- phorbol 12-myristate 13-acetate
- hCAR
- human coxsackie adenovirus receptor
- m.o.i.
- multiplicity of infection
- EGFP
- enhanced GFP.
REFERENCES
- 1. Feng X. H., Derynck R. (2005) Specificity and versatility in TGF-β signaling through Smads. Annu. Rev. Cell Dev. Biol. 21, 659–693 [DOI] [PubMed] [Google Scholar]
- 2. Shi Y., Massagué J. (2003) Mechanisms of TGF-β signaling from cell membrane to the nucleus. Cell 113, 685–700 [DOI] [PubMed] [Google Scholar]
- 3. Miyazono K., ten Dijke P., Heldin C. (2000) TGF-β signaling by Smad proteins. Adv. Immunol. 75, 115–157 [DOI] [PubMed] [Google Scholar]
- 4. Nakao A., Afrakhte M., Morén A., Nakayama T., Christian J. L., Heuchel R., Itoh S., Kawabata M., Heldin N. E., Heldin C. H., ten Dijke P. (1997) Identification of Smad7, a TGFβ-inducible antagonist of TGF-β signalling. Nature 389, 631–635 [DOI] [PubMed] [Google Scholar]
- 5. Hayashi H., Abdollah S., Qiu Y., Cai J., Xu Y. Y., Grinnell B. W., Richardson M. A., Topper J. N., Gimbrone M. A., Jr., Wrana J. L., Falb D. (1997) The MAD-related protein Smad7 associates with the TGFβ receptor and functions as an antagonist of TGFβ signaling. Cell 89, 1165–1173 [DOI] [PubMed] [Google Scholar]
- 6. Lin X., Duan X., Liang Y. Y., Su Y., Wrighton K. H., Long J., Hu M., Davis C. M., Wang J., Brunicardi F. C., Shi Y., Chen Y. G., Meng A., Feng X. H. (2006) PPM1A functions as a Smad phosphatase to terminate TGFβ signaling. Cell 125, 915–928 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Oren M. (2003) Decision making by p53: life, death and cancer. Cell Death Differ. 10, 431–442 [DOI] [PubMed] [Google Scholar]
- 8. Green D. R., Kroemer G. (2009) Cytoplasmic functions of the tumour suppressor p53. Nature 458, 1127–1130 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Soussi T., Béroud C. (2001) Assessing TP53 status in human tumours to evaluate clinical outcome. Nat. Rev. Cancer 1, 233–240 [DOI] [PubMed] [Google Scholar]
- 10. Chen Y., Chen P. L., Lee W. H. (1994) Hot-spot p53 mutants interact specifically with two cellular proteins during progression of the cell cycle. Mol. Cell. Biol. 14, 6764–6772 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Hsiao M., Low J., Dorn E., Ku D., Pattengale P., Yeargin J., Haas M. (1994) Gain-of-function mutations of the p53 gene induce lymphohematopoietic metastatic potential and tissue invasiveness. Am. J. Pathol. 145, 702–714 [PMC free article] [PubMed] [Google Scholar]
- 12. Gerwin B. I., Spillare E., Forrester K., Lehman T. A., Kispert J., Welsh J. A., Pfeifer A. M., Lechner J. F., Baker S. J., Vogelstein B. (1992) Mutant p53 can induce tumorigenic conversion of human bronchial epithelial cells and reduce their responsiveness to a negative growth factor, transforming growth factor β1. Proc. Natl. Acad. Sci. U.S.A. 89, 2759–2763 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Wang X. J., Greenhalgh D. A., Jiang A., He D., Zhong L., Brinkley B. R., Roop D. R. (1998) Analysis of centrosome abnormalities and angiogenesis in epidermal-targeted p53172H mutant and p53-knockout mice after chemical carcinogenesis: evidence for a gain of function. Mol. Carcinog. 23, 185–192 [DOI] [PubMed] [Google Scholar]
- 14. Lányi A., Deb D., Seymour R. C., Ludes-Meyers J. H., Subler M. A., Deb S. (1998) ‘Gain of function’ phenotype of tumor-derived mutant p53 requires the oligomerization/nonsequence-specific nucleic acid-binding domain. Oncogene 16, 3169–3176 [DOI] [PubMed] [Google Scholar]
- 15. Shaulsky G., Goldfinger N., Rotter V. (1991) Alterations in tumor development in vivo mediated by expression of wild type or mutant p53 proteins. Cancer Res. 51, 5232–5237 [PubMed] [Google Scholar]
- 16. Cardinali M., Kratochvil F. J., Ensley J. F., Robbins K. C., Yeudall W. A. (1997) Functional characterization in vivo of mutant p53 molecules derived from squamous cell carcinomas of the head and neck. Mol. Carcinog 18, 78–88 [DOI] [PubMed] [Google Scholar]
- 17. Hixon M. L., Flores A., Wagner M., Gualberto A. (2000) Gain of function properties of mutant p53 proteins at the mitotic spindle cell cycle checkpoint. Histol. Histopathol. 15, 551–556 [DOI] [PubMed] [Google Scholar]
- 18. Gualberto A., Aldape K., Kozakiewicz K., Tlsty T. D. (1998) An oncogenic form of p53 confers a dominant, gain-of-function phenotype that disrupts spindle checkpoint control. Proc. Natl. Acad. Sci. U.S.A. 95, 5166–5171 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Blandino G., Levine A. J., Oren M. (1999) Mutant p53 gain of function: differential effects of different p53 mutants on resistance of cultured cells to chemotherapy. Oncogene 18, 477–485 [DOI] [PubMed] [Google Scholar]
- 20. Ewen M. E., Oliver C. J., Sluss H. K., Miller S. J., Peeper D. S. (1995) p53-dependent repression of CDK4 translation in TGF-β-induced G1 cell-cycle arrest. Genes Dev. 9, 204–217 [DOI] [PubMed] [Google Scholar]
- 21. Cordenonsi M., Dupont S., Maretto S., Insinga A., Imbriano C., Piccolo S. (2003) Links between tumor suppressors: p53 is required for TGF-β gene responses by cooperating with Smads. Cell 113, 301–314 [DOI] [PubMed] [Google Scholar]
- 22. Reiss M., Vellucci V. F., Zhou Z. L. (1993) Mutant p53 tumor suppressor gene causes resistance to transforming growth factor β1 in murine keratinocytes. Cancer Res. 53, 899–904 [PubMed] [Google Scholar]
- 23. Adorno M., Cordenonsi M., Montagner M., Dupont S., Wong C., Hann B., Solari A., Bobisse S., Rondina M. B., Guzzardo V., Parenti A. R., Rosato A., Bicciato S., Balmain A., Piccolo S. (2009) A mutant-p53/Smad complex opposes p63 to empower TGFβ-induced metastasis. Cell 137, 87–98 [DOI] [PubMed] [Google Scholar]
- 24. Kubbutat M. H., Jones S. N., Vousden K. H. (1997) Regulation of p53 stability by Mdm2. Nature 387, 299–303 [DOI] [PubMed] [Google Scholar]
- 25. Haupt Y., Maya R., Kazaz A., Oren M. (1997) Mdm2 promotes the rapid degradation of p53. Nature 387, 296–299 [DOI] [PubMed] [Google Scholar]
- 26. Vogelstein B., Lane D., Levine A. J. (2000) Surfing the p53 network. Nature 408, 307–310 [DOI] [PubMed] [Google Scholar]
- 27. Lowe S. W., Sherr C. J. (2003) Tumor suppression by Ink4a-Arf: progress and puzzles. Curr. Opin. Genet. Dev. 13, 77–83 [DOI] [PubMed] [Google Scholar]
- 28. Kamijo T., Zindy F., Roussel M. F., Quelle D. E., Downing J. R., Ashmun R. A., Grosveld G., Sherr C. J. (1997) Tumor suppression at the mouse INK4a locus mediated by the alternative reading frame product p19ARF. Cell 91, 649–659 [DOI] [PubMed] [Google Scholar]
- 29. Quelle D. E., Cheng M., Ashmun R. A., Sherr C. J. (1997) Cancer-associated mutations at the INK4a locus cancel cell cycle arrest by p16INK4a but not by the alternative reading frame protein p19ARF. Proc. Natl. Acad. Sci. U.S.A. 94, 669–673 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Roussel M. F. (1999) The INK4 family of cell cycle inhibitors in cancer. Oncogene 18, 5311–5317 [DOI] [PubMed] [Google Scholar]
- 31. Palmero I., Pantoja C., Serrano M. (1998) p19ARF links the tumour suppressor p53 to Ras. Nature 395, 125–126 [DOI] [PubMed] [Google Scholar]
- 32. Zindy F., Eischen C. M., Randle D. H., Kamijo T., Cleveland J. L., Sherr C. J., Roussel M. F. (1998) Myc signaling via the ARF tumor suppressor regulates p53-dependent apoptosis and immortalization. Genes Dev. 12, 2424–2433 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Cong F., Zou X., Hinrichs K., Calame K., Goff S. P. (1999) Inhibition of v-Abl transformation by p53 and p19ARF. Oncogene 18, 7731–7739 [DOI] [PubMed] [Google Scholar]
- 34. Korgaonkar C., Zhao L., Modestou M., Quelle D. E. (2002) ARF function does not require p53 stabilization or Mdm2 relocalization. Mol. Cell. Biol. 22, 196–206 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Yarbrough W. G., Bessho M., Zanation A., Bisi J. E., Xiong Y. (2002) Human tumor suppressor ARF impedes S-phase progression independent of p53. Cancer Res. 62, 1171–1177 [PubMed] [Google Scholar]
- 36. Rocha S., Campbell K. J., Perkins N. D. (2003) p53- and Mdm2-independent repression of NF-κB transactivation by the ARF tumor suppressor. Mol. Cell 12, 15–25 [DOI] [PubMed] [Google Scholar]
- 37. Itahana K., Bhat K. P., Jin A., Itahana Y., Hawke D., Kobayashi R., Zhang Y. (2003) Tumor suppressor ARF degrades B23, a nucleolar protein involved in ribosome biogenesis and cell proliferation. Mol. Cell 12, 1151–1164 [DOI] [PubMed] [Google Scholar]
- 38. Sherr C. J., Bertwistle D., DEN Besten W., Kuo M. L., Sugimoto M., Tago K., Williams R. T., Zindy F., Roussel M. F. (2005) p53-dependent and -independent functions of the Arf tumor suppressor. Cold Spring Harb. Symp. Quant. Biol. 70, 129–137 [DOI] [PubMed] [Google Scholar]
- 39. Beckwith M., Ruscetti F. W., Sing G. K., Urba W. J., Longo D. L. (1995) Anti-IgM induces transforming growth factor-β sensitivity in a human B-lymphoma cell line: inhibition of growth is associated with a downregulation of mutant p53. Blood 85, 2461–2470 [PubMed] [Google Scholar]
- 40. Uzzo R. G., Rayman P., Kolenko V., Clark P. E., Cathcart M. K., Bloom T., Novick A. C., Bukowski R. M., Hamilton T., Finke J. H. (1999) Renal cell carcinoma-derived gangliosides suppress nuclear factor-κB activation in T cells. J. Clin. Investig. 104, 769–776 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. O'Farrell T. J., Ghosh P., Dobashi N., Sasaki C. Y., Longo D. L. (2004) Comparison of the effect of mutant and wild-type p53 on global gene expression. Cancer Res. 64, 8199–8207 [DOI] [PubMed] [Google Scholar]
- 42. Salmon P., Kindler V., Ducrey O., Chapuis B., Zubler R. H., Trono D. (2000) High-level transgene expression in human hematopoietic progenitors and differentiated blood lineages after transduction with improved lentiviral vectors. Blood 96, 3392–3398 [PubMed] [Google Scholar]
- 43. Chen G., Ghosh P., Osawa H., Sasaki C. Y., Rezanka L., Yang J., O'Farrell T. J., Longo D. L. (2007) Resistance to TGF-β1 correlates with aberrant expression of TGF-β receptor II in human B-cell lymphoma cell lines. Blood 109, 5301–5307 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Sasaki C. Y., Slemenda C. F., Ghosh P., Barberi T. J., Longo D. L. (2007) Traf1 induction and protection from tumor necrosis factor by nuclear factor-κB p65 is independent of serine 536 phosphorylation. Cancer Res. 67, 11218–11225 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Koinuma D., Tsutsumi S., Kamimura N., Taniguchi H., Miyazawa K., Sunamura M., Imamura T., Miyazono K., Aburatani H. (2009) Chromatin immunoprecipitation on microarray analysis of Smad2/3 binding sites reveals roles of ETS1 and TFAP2A in transforming growth factor β signaling. Mol. Cell. Biol. 29, 172–186 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Li C. C., O'Connell C. D., Beckwith M., Longo D. L. (1995) Detection of p53 mutations in B cell non-Hodgkin's lymphoma cell lines. Leukemia 9, 650–655 [PubMed] [Google Scholar]
- 47. Chen G., Ghosh P., Longo D. L. (2011) Distinctive mechanism for sustained TGF-β signaling and growth inhibition: MEK1 activation-dependent stabilization of type II TGF-β receptors. Mol. Cancer Res. 9, 78–89 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Datto M. B., Yu Y., Wang X. F. (1995) Functional analysis of the transforming growth factor β responsive elements in the WAF1/Cip1/p21 promoter. J. Biol. Chem. 270, 28623–28628 [DOI] [PubMed] [Google Scholar]
- 49. Spender L. C., Inman G. J. (2009) TGF-β induces growth arrest in Burkitt lymphoma cells via transcriptional repression of E2F-1. J. Biol. Chem. 284, 1435–1442 [DOI] [PubMed] [Google Scholar]
- 50. Bates S., Phillips A. C., Clark P. A., Stott F., Peters G., Ludwig R. L., Vousden K. H. (1998) p14ARF links the tumour suppressors RB and p53. Nature 395, 124–125 [DOI] [PubMed] [Google Scholar]
- 51. Lenburg M. E., Sinha A., Faller D. V., Denis G. V. (2007) Tumor-specific and proliferation-specific gene expression typifies murine transgenic B cell lymphomagenesis. J. Biol. Chem. 282, 4803–4811 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Sánchez-Aguilera A., Sánchez-Beato M., García J. F., Prieto I., Pollan M., Piris M. A. (2002) p14ARF nuclear overexpression in aggressive B-cell lymphomas is a sensor of malfunction of the common tumor suppressor pathways. Blood 99, 1411–1418 [DOI] [PubMed] [Google Scholar]
- 53. Honda R., Yasuda H. (1999) Association of p19ARF with Mdm2 inhibits ubiquitin ligase activity of Mdm2 for tumor suppressor p53. EMBO J. 18, 22–27 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Vijayachandra K., Higgins W., Lee J., Glick A. (2009) Induction of p16ink4a and p19ARF by TGFβ1 contributes to growth arrest and senescence response in mouse keratinocytes. Mol. Carcinog. 48, 181–186 [DOI] [PubMed] [Google Scholar]
- 55. Freeman-Anderson N. E., Zheng Y., McCalla-Martin A. C., Treanor L. M., Zhao Y. D., Garfin P. M., He T. C., Mary M. N., Thornton J. D., Anderson C., Gibbons M., Saab R., Baumer S. H., Cunningham J. M., Skapek S. X. (2009) Expression of the Arf tumor suppressor gene is controlled by Tgfβ2 during development. Development 136, 2081–2089 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Renschler M. F., Wada H. G., Fok K. S., Levy R. (1995) B-lymphoma cells are activated by peptide ligands of the antigen binding receptor or by anti-idiotypic antibody to induce extracellular acidification. Cancer Res. 55, 5642–5647 [PubMed] [Google Scholar]
- 57. Stott F. J., Bates S., James M. C., McConnell B. B., Starborg M., Brookes S., Palmero I., Ryan K., Hara E., Vousden K. H., Peters G. (1998) The alternative product from the human CDKN2A locus, p14ARF, participates in a regulatory feedback loop with p53 and MDM2. EMBO J. 17, 5001–5014 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.