

Background: Granzymes have distinct substrate specificities and trigger diverse cell death pathways.
Results: All human granzymes can target hnRNP K, which is essential for tumor cell viability.
Conclusion: Granzyme-mediated cleavage of hnRNP K contributes to the elimination of tumor cells by cytotoxic lymphocytes.
Significance: Elucidating granzyme function provides insights into the role of cytotoxic lymphocytes in tumor immunology.
Keywords: Apoptosis, Cancer, Caspase, Cell Death, Lymphocyte, Tumor Immunology, Granzyme, hnRNP K
Abstract
Granule exocytosis by cytotoxic lymphocytes is the key mechanism to eliminate virus-infected cells and tumor cells. These lytic granules contain the pore-forming protein perforin and a set of five serine proteases called granzymes. All human granzymes display distinct substrate specificities and induce cell death by cleaving critical intracellular death substrates. In the present study, we show that all human granzymes directly cleaved the DNA/RNA-binding protein heterogeneous nuclear ribonucleoprotein K (hnRNP K), designating hnRNP K as the first known pan-granzyme substrate. Cleavage of hnRNP K was more efficient in the presence of RNA and occurred in two apparent proteolysis-sensitive amino acid regions, thereby dissecting the functional DNA/RNA-binding hnRNP K domains. HnRNP K was cleaved under physiological conditions when purified granzymes were delivered into living tumor cells and during lymphokine-activated killer cell-mediated attack. HnRNP K is essential for tumor cell viability, since knockdown of hnRNP K resulted in spontaneous tumor cell apoptosis with caspase activation and reactive oxygen species production. This apoptosis was more pronounced at low tumor cell density where hnRNP K knockdown also triggered a caspase-independent apoptotic pathway. This suggests that hnRNP K promotes tumor cell survival in the absence of cell-cell contact. Silencing of hnRNP K protein expression rendered tumor cells more susceptible to cellular cytotoxicity. We conclude that hnRNP K is indispensable for tumor cell viability and our data suggest that targeting of hnRNP K by granzymes contributes to or reinforces the cell death mechanisms by which cytotoxic lymphocytes eliminate tumor cells.
Introduction
Cytotoxic lymphocytes, including cytotoxic T lymphocytes, NK cells, γδT cells, and NKT cells, are the key effector cells in eliminating virus-infected cells and tumor cells (1, 2). Cell death is induced by ligation of death receptors on the cell surface and predominantly by the release of cytotoxic granules that are directed toward the target cell. These cytotoxic granules contain several proteins, including the membrane-perturbing protein perforin and a family of structurally homologous serine proteases termed granzymes. While perforin facilitates the entry of granzymes into the target cell, granzymes induce cell death by cleaving critical intracellular death substrates. In humans, five granzymes have been identified (GrA, GrB, GrH, GrK, and GrM) that differ on the basis of their substrate specificity (2). All granzymes can induce cell death by cleaving critical intracellular death substrates (1–3).
GrA and GrB have been extensively studied (2), whereas GrH, GrK, and GrM have remained less appreciated (3). GrA initiates cell death through cleavage of the mitochondrial protein NDUFS3 (4). This leads to production of reactive oxygen species (ROS),2 translocation of the SET complex into the nucleus, and GrA-mediated cleavage of SET components (SET, ApeI, and HMG2), resulting in irreversible single-stranded nicking of chromosomal DNA and subsequent cell death (5–9). GrB is a potent inducer of apoptosis through cleavage and activation of pro-caspase-3, leading to activation of the classical caspase cascade (10). GrB also cleaves BID, resulting in increased mitochondrial membrane permeabilization and release of cytochrome c (10). Activation of these GrB pathways leads to DNA fragmentation and apoptosis. Only two studies have addressed the mechanisms by which GrH induces cell death (11, 12). Although both studies show that mitochondria are involved, they demonstrate conflicting results on other hallmarks of GrH cell death, such as caspase activation and cytochrome c release. GrK shares its tryptase-like substrate specificity with GrA and induces similar caspase-independent cell death pathways as GrA, characterized by cleavage of similar substrates (SET, ApeI, and HMG2), and similar cell death hallmarks (single-stranded DNA nicking and ROS production from mitochondria) (13–15). Unlike GrA, GrK also targets unique death substrates, including BID, p53, and valosin-containing protein, to trigger mitochondrial damage, DNA fragmentation, and endoplasmic reticulum stress, respectively (14, 16–18). GrM induces cell death independent of caspase activation and mitochondrial perturbations (19–21). In addition, GrM has been shown to cleave Fas-associated protein with death domain (FADD), leading to pro-caspase-8 activation and subsequent mitochondrial damage and apoptosome formation (22, 23).
Previously, we and others have performed mass spectrometry-based proteomic screens to identify potential human granzyme substrates in tumor cell lysates (24). Interestingly, one protein that has frequently been detected in these proteomic screens is heterogeneous nuclear ribonucleoprotein K (hnRNP K) (17, 25–27). HnRNP K is a multifunctional DNA/RNA-binding protein involved in transcription/translation machinery, including transcription, translation, splicing, and mRNA stability (28). In this study, we determined and validated which granzymes can directly cleave hnRNP K and we addressed the role of hnRNP K during cytotoxic lymphocyte-mediated killing of tumor cells. We showed that hnRNP K is the first known direct pan-granzyme substrate. HnRNP K knockdown rendered tumor cells more susceptible to cellular cytotoxicity and resulted in spontaneous tumor cell apoptosis, indicating that hnRNP K is essential for tumor cell viability. Our data suggest that targeting of hnRNP K by granzymes contributes to or reinforces the cell death mechanisms by which cytotoxic lymphocytes eliminate tumor cells.
EXPERIMENTAL PROCEDURES
Cell Culture and Cell-free Protein Lysates
Cells were cultured in a 5% CO2 atmosphere at 37 °C. HeLa cells were maintained in DMEM (Invitrogen) supplemented with 10% fetal calf serum, 100 units/ml penicillin, and 100 μg/ml streptomycin (Invitrogen). Jurkat and K562 cells were maintained in RPMI 1640 medium (Invitrogen) supplemented with 10% fetal calf serum, 100 units/ml penicillin, and 100 μg/ml streptomycin. Cell-free protein lysates were generated by washing cells three times in PBS and subsequent lysis in PBS by three cycles of freeze-thawing in liquid nitrogen. Samples were centrifuged at 18,000 × g for 10 min at 4 °C, and protein concentration was determined by the method of Bradford (Bio-Rad).
Antibodies and Reagents
Primary antibodies directed against the mid region (rabbit polyclonal, amino acid residues 200–300), N terminus (EP943Y, rabbit monoclonal, amino acid residues near N terminus), and C terminus (F45 P9 C7, mouse monoclonal, amino acid residues 450–463) of hnRNP K were purchased from Abcam. Antibodies against β-tubulin (TUB 2.1, mouse monoclonal) and cleaved caspase-3 (D175, rabbit polyclonal) were obtained from Sigma and Cell Signaling, respectively. Secondary HRP-conjugated goat anti-mouse and goat anti-rabbit antibodies were purchased from BioSource and Jackson, respectively. Immunoblotted proteins were visualized using the ECL detection system (Amersham Biosciences) and ChemiDoc XRS+ (Bio-Rad). Pan-caspase inhibitor zVAD-fmk was obtained from Enzo Life Sciences. RNase A was purchased from Roche and dissolved in 20 mm Tris, pH 7.4, and 150 mm NaCl. Total RNA was isolated using Trizol according to the manufacturer's protocol (Invitrogen).
Purified Recombinant Proteins
For each human granzyme, the cDNA encoding the mature protease was amplified and cloned into yeast expression vector pPIC9 (Invitrogen). Catalytically inactive control granzymes (GrA-SA, GrB-SA, GrH-SA, GrK-SA, and GrM-SA), in which the Ser195 residue in the catalytic center is mutated into an Ala, were generated by site-directed mutagenesis (Stratagene). All granzymes were expressed and purified as described previously (19). Briefly, Pichia pastoris GS115 cells were transformed with the pPIC9-granzyme expression plasmids and granzymes were expressed for 72 h in conditioned media, according to the manufacturers' protocol (Invitrogen). The granzymes were purified to homogeneity using an SP-Sepharose column (GE Healthcare) with 1 m NaCl for elution and dialyzed against 50 mm Tris pH 7.4 and 150 mm NaCl, and stored at −80 °C. Recombinant granzymes were pure (>98%) as determined by SDS-PAGE. All granzymes were active as determined by small synthetic peptide substrates and by assessing granzyme-specific macromolecular substrate cleavage (data not shown). In contrast to the other granzymes, stock concentrations of GrH and GrH-SA were low because of inefficient recombinant GrH and GrH-SA expression by Pichia pastoris, and therefore lower concentrations of these granzymes had to be used in several experiments.
BL21-CodonPlus competent Escherichia coli cells (Stratagene) were transformed with the pET16-b-His-hnRNP K expression plasmid (a kind gift from Dr. A. Ostareck-Lederer, Martin Luther University Halle-Wittenberg, Germany) and cultured in LB medium. At an A600 between 0.4 and 1.0, cells were stimulated with 0.4 mm IPTG and incubated for 3 h at 37 °C at 225 rpm. Cells were harvested and lysed by sonification. N-terminal His-tagged hnRNP K protein was purified to homogeneity using Talon metal-chelate affinity chromatography (Clontech) with 125 mm imidazole in PBS (pH 7.5) for elution and stored at 4 °C.
Granzyme Killing Assay
HeLa cells were grown to confluence in 96-well plates, washed twice with serum free DMEM and incubated with the perforin-analog streptolysin O (SLO) (Aalto) in serum-free DMEM and purified granzyme in a buffer containing 50 mm Tris and 150 mm NaCl in a final volume of 30 μl for 30 min at 37 °C. Cells treated with GrB were directly lysed by adding reducing sample buffer. For cells treated with the other granzymes, supernatant was removed, and cells were cultured in 100 μl of DMEM containing 10% FCS for 16 h after which cells were washed two times with PBS and directly lysed in reducing sample buffer. Protein expression was analyzed by immunoblotting.
LAK Cell-mediated Cytotoxicity Assay
Lymphokine-activated killer (LAK) cells were obtained by culturing peripheral blood mononuclear cells for 4 days in RPMI 1640 medium supplemented with 5% human AB serum and 1000 units/ml of recombinant interleukin-2 (WAKO, Japan). HeLa cells or K562 cells were incubated with LAK effector cells at the indicated effector:target (E:T) ratios. To measure specific cytotoxicity, cells were labeled with propidium iodide (PI) (Sigma) in 100 μl annexin-V binding buffer (140 mm NaCl, 4 mm KCl, 0.75 mm MgCl2, and 10 mm CaCl2) for 15 min at room temperature after which 500 μl annexin-V binding buffer was added. Cells were analyzed by flow cytometry and target cells were gated based on forward and side scatter analysis. The percentage of (background) cytotoxicity in HeLa cells without LAK cells (1:0) was subtracted from the percentages of cell death in all other E:T ratio conditions to obtain the percentages of specific cytotoxicity. For protein analysis, cells were washed two times with PBS and directly lysed in reducing sample buffer. Protein expression was analyzed by immunoblotting.
siRNA-mediated Knockdown of hnRNP K Protein Expression
HeLa cells were seeded in 6-well plates and left untreated or transfected with control On-Target plus Non-targeting Pool siRNA (Dharmacon) or hnRNP K On-Target plus SMARTpool siRNA (Dharmacon) using Lipofectamine 2000 (Invitrogen) according to the manufacturers' instructions, in the presence or absence of zVAD-fmk (100 μm). To confirm knockdown, cells were lysed 3 days post-infection in PBS by three cycles of freeze/thawing in liquid nitrogen. Samples were centrifuged at 18,000 × g for 10 min at 4 °C, and protein concentration was determined by the method of Bradford (Bio-Rad). Protein expression of hnRNP K was analyzed by immunoblotting. Three days post-transfection, cells were either subjected to xCELLigence or seeded in 96-well plates (1 × 104 cells/well) and measured for cell viability using WST-1 assay (Roche) and cell damage by LDH assay (Roche) according to the manufacturers' protocol. Briefly, cells are incubated with medium supplemented with water-soluble tetrazolium salt (WST-1) for 1 h. Viable cells can cleave the WST-reagent by mitochondrial dehydrogenase into a soluble formazan dye. For the LDH assay, supernatant of cells is incubated with a substrate mixture containing iodotetrazolium chloride (INT). This tetrazolium salt is reduced to a soluble formazan dye by lactate dehydrogenase (LDH), which is released from damaged cells into the supernatant. Both formazan dyes were quantified by measuring the absorbance (A450 for the WST-1 assay, and A492 for the LDH assay) on an Anthos Zenyth 340 rt microtitre plate reader (Anthos). Cells were seeded in 6-well plates (1–2 × 105 cells/well) and cell cycle profiles, apoptosis, and ROS levels were assessed by flow cytometry. To determine caspase-3 and -7 activities, cells were seeded in white 96-well flat bottom plates (3750 cells/well) and subjected to Caspase-Glo 3/7 assay (Promega) according to the manufacturers' protocol. Luciferase activities were assessed in time using a Veritas Microplate Luminometer (Turner Biosystems). Maximum luciferase activity for each condition was determined, normalized for cell numbers as assessed by a WST-1 assay, and the mean luciferase activity from control cells (control siRNA without zVAD-fmk) was set at 1.
xCELLigence
The xCELLigence system (Roche) monitors cellular events in real-time without the incorporation of labels. The system measures electrical impedance across interdigitated micro-electrodes integrated on the bottom of 16-well tissue culture plates (E-plates). Well size of these E-plates is similar to that of common 96-well plates. The impedance measurement depends on the quality of the cell interaction with the electrodes. Cell index (CI) is derived as a relative change in measured electrical impedance and therefore provides quantitative information about the overall biological status of the cells, including cell number, viability, adherence, and morphology. xCELLigence was performed according to the manufacturers' protocol (Roche). Briefly, background CI values for each E-plate well (Roche) were measured using medium only. Medium was removed, and cells were seeded at different cell densities. When cells were treated with zVAD-fmk (100 μm), control cells were treated with vehicle only (DMSO). CI values were measured every 10 min for 6 h and then every 20 min for 72 h.
Flow Cytometry Analysis
Cells were trypsinized, harvested, and washed twice with PBS. For analysis of cell death, cells were pelleted and stained with annexin-V-FLUOS (Roche) and PI in 100 μl of annexin-V binding buffer for 15 min at room temperature after which 500 μl of annexin-V binding buffer was added. For analysis of ROS levels, cells were pelleted and stained with 50 μl of 10 μm CM-H2DCFDA for 30 min at 37 °C and resuspended in 250 μl of PBS. As a positive control, cells were resuspended in 500 μm H2O2. To determine cell cycle profiles, cells were fixed (100% ethanol), washed with PBS, treated with RNase A, and stained with PI for 1.5 h at 37 °C. Fluorescence was measured by fluorescence-activated cell sorter (FACS) on a FACS Calibur (BD Biosciences), and data were analyzed with CellQuest Pro software (BD Biosciences).
Statistical Analysis
Data are depicted as mean values ± S.D. All statistical analyses were performed using unpaired t test when comparing two groups or one-way ANOVA with Bonferroni correction when comparing three or more groups. p values less than 0.05 were considered statistically significant.
RESULTS
HnRNP K Is Degraded by All Human Granzymes in Tumor Lysates
To determine whether hnRNP K is cleaved by human granzymes, Jurkat tumor cell lysates were incubated with the individual purified human granzymes or their catalytically inactive control (SA) variants and hnRNP K cleavage was examined by immunoblotting. Cleavage of hnRNP K by all granzymes was demonstrated by the disappearance of full-length hnRNP K (∼62 kDa) in a dose-dependent manner, suggesting that hnRNP K is a pan-granzyme substrate (Fig. 1, A--E). The difference in substrate specificity of granzymes is illustrated by distinct hnRNP K cleavage products that were observed. Interestingly, one hnRNP K cleavage product at ∼45 kDa was detected upon cleavage by GrA, GrB, GrH, and GrM. Also some minor aspecific degradation of hnRNP K was observed when lysates were left untreated or incubated with inactive control granzymes, most likely due to an endogenous protease within lysates. Because GrB activates pro-caspase-3, we determined whether GrB-mediated hnRNP K cleavage is caspase-dependent. To this end, Jurkat tumor cell lysates were incubated with GrB in the absence or presence of pan-caspase inhibitor compound zVAD-fmk (Fig. 1F). Although caspase activity is inhibited by zVAD-fmk, as demonstrated by blocked autocatalytic activity of caspase-3 to process itself into the p17 fragment, hnRNP K was still efficiently cleaved by GrB. This indicates that GrB-mediated hnRNP K cleavage is caspase-independent.
FIGURE 1.

HnRNP K is degraded by all five human granzymes in tumor lysates. A, Jurkat cell lysates (5 μg) were incubated with increasing concentrations of GrA, (B) GrB, (C) GrH, (D) GrK, (E) GrM, or their corresponding catalytically inactive control granzyme (SA) (300 nm) in a buffer containing 50 mm Tris pH 7.4 and 150 mm NaCl for 4 h at 37 °C and immunoblotted using an antibody against the midregion of hnRNP K. F, Jurkat cell lysates (5 μg) were incubated with increasing concentrations of GrB in the absence or presence of the pan-caspase inhibitor zVAD-fmk (100 μm) or GrK (500 nm) for 4 h at 37 °C and immunoblotted using antibodies against the mid region of hnRNP K or active caspase-3 (D175) (*, GrB-mediated hnRNP K cleavage products; **, GrK-mediated hnRNP K cleavage product).
HnRNP K Is a Direct Pan-granzyme Substrate
To determine whether all granzymes directly cleave hnRNP K, purified recombinant His-tagged hnRNP K was incubated with increasing concentrations of individual granzymes or inactive controls and hnRNP K cleavage was monitored by SDS-PAGE analysis. Again, cleavage of hnRNP K was observed upon treatment with all individual granzymes (Fig. 2, A--E). Based on the SDS-PAGE degradation kinetics, GrH and GrK were more efficient in cleaving hnRNP K as compared with GrA, GrB, and GrM (Fig. 2F). These data indicate that hnRNP K is a direct pan-granzyme substrate in vitro. We and others have identified hnRNP K as a potential substrate for several human granzymes in tumor cell lysates using proteomic screens with mass spectrometry (17, 24–27). We took advantage of these screens to gain insight into the granzyme cleavage sites in hnRNP K (Fig. 2G) (25–27, 29, 30). Furthermore, we used monoclonal antibodies that detect the absolute N or C terminus of hnRNP K as well as site-directed mutagenesis of several predicted granzyme cleavage sites (data not shown). This analysis showed that granzyme-mediated cleavage of hnRNP K was mostly confined to two apparent proteolysis-sensitive amino acid regions, located between the functional DNA/RNA binding KH domains (Fig. 2G). Granzyme cleavage sites in hnRNP K include at least Arg37 (GrA), Asp26, Asp93, Asp128, and Asp382 (GrB), Arg305 (GrK), Leu125, Leu133, and Met359 (GrM). Thus, these data indicate that all human granzymes directly cleave hnRNP K, thereby dissecting its functional KH domains.
FIGURE 2.

HnRNP K is a direct pan-granzyme substrate. A, purified His-hnRNP K (1 μg, ∼750 nm) was incubated with increasing concentrations of GrA, (B) GrB, (C) GrH, (D) GrK, (E) GrM, or their corresponding catalytically inactive control granzyme (SA) (300 nm for GrH-SA and 500 nm for the other control granzymes) in a buffer containing 50 mm Tris, pH 7.4, and 150 mm NaCl for 16 h at 37 °C, subjected to SDS-PAGE, and all proteins were stained with Simply Blue. F, band intensities were quantified and plotted with His-hnRNP K without granzyme set at 100%. A non-linear regression curve (one phase exponential decay) was fitted, and the concentration of granzyme required to cleave 50% hnRNP K was determined for each granzyme (GrA, 249 nm; GrB, 146 nm; GrH, 9.5 nm; GrK, 13 nm; GrM, 499 nm). G, schematic overview of granzyme cleavage sites within hnRNP K. Cleavage sites were identified by 1) proteomic screens (25–27, 29, 30), 2) site-directed mutagenesis and validated by immunoblotting, or amino acid regions harboring a potential granzyme cleavage site were predicted by 3) immunoblotting using antibodies directed against the mid region as well as the absolute N or C terminus of hnRNP K. (NLS, nuclear localization signal; KH, K homology domain; KI, K interactive region; KNS, K nuclear shuttling signal; RGG, arginine-glycine-glycine)
RNA Promotes hnRNP K Cleavage by Granzymes
Remarkably, granzyme-mediated cleavage of purified hnRNP K (Fig. 2) necessitated longer incubation times as compared with cleavage in lysates (Fig. 1), suggesting that efficient cleavage of hnRNP K requires cellular factors present within tumor cell lysates. Because hnRNP K is known to bind RNA (28, 31), the effects of RNA on granzyme-mediated hnRNP K cleavage was examined. First, Jurkat tumor cell lysates were either pre-treated with RNase to remove RNA from the lysates or left untreated prior to incubations with granzymes (Fig. 3A). Immunoblot analysis showed that all five human granzymes cleaved hnRNP K less efficiently when lysates were pre-treated with RNase, indicating that RNA promotes granzyme-mediated cleavage of hnRNP K. To further support this finding, purified hnRNP K was pre-treated with or without isolated RNA prior to granzyme incubations, and hnRNP K cleavage was determined by SDS-PAGE analysis. All granzymes more efficiently cleaved purified hnRNP K in the presence of RNA (Fig. 3B). These data indicate that RNA either promotes protease-substrate interaction or renders hnRNP K more sensitive to proteolysis.
FIGURE 3.

RNA promotes granzyme-mediated cleavage of hnRNP K. A, Jurkat cell lysates (5 μg) were pre-treated with RNase (100 μg/ml) or left untreated for 30 min at 37 °C followed by incubation with GrA (200 nm), GrB (200 nm), GrH (25 nm), GrK (25 nm), GrM (200 nm), or their corresponding concentration-matched catalytically inactive control granzyme (SA) in a buffer containing 50 mm Tris pH 7.4 and 150 mm NaCl for 4 h at 37 °C and immunoblotted using an antibody against the mid region of hnRNP K. B, purified His-hnRNP K (1 μg, ∼1 μm) was pre-treated with RNA (100 ng) or left untreated for 30 min at 37 °C followed by incubations with GrA (249 nm), GrB (146 nm), GrH (9.5 nm), GrK (13 nm), GrM (499 nm), or their corresponding concentration-matched catalytically inactive control granzyme (SA) in a buffer containing 50 mm Tris, pH 7.4, and 150 mm NaCl for 16 h at 37 °C, subjected to SDS-PAGE, and all proteins were stained with Instant Blue.
HnRNP K Is Cleaved under Physiological Conditions
To assess hnRNP K cleavage under physiological conditions, intact HeLa tumor cells were treated with the perforin-analog SLO in combination with an individual granzyme (Fig. 4A). Cells that were treated with inactive control granzymes did not show any cleavage of hnRNP K. Except for GrA, all granzymes cleaved hnRNP K when delivered into target cells as illustrated by the appearance of cleavage products in cells treated with both granzyme and SLO. These hnRNP K cleavage fragments were at the same size as detected in cellular lysates (Fig. 1). This indicates that at least GrB, GrH, GrK, and GrM target hnRNP K within living tumor cells. Cleavage of hnRNP K was further analyzed by challenging HeLa cells with lymphokine-activated killer (LAK) cells, which contain all granzymes, for 4 h, after which cell lysates were subjected to immunoblotting. As expected, no decrease in full-length hnRNP K could be observed, because hnRNP K originating from LAK cells was also present in all samples (data not shown). However, a cleavage fragment of ∼45 kDa appeared with increasing effector:target ratios (Fig. 4B, upper panel), indicating that LAK cells mediate cleavage of hnRNP K inside tumor target cells. Whereas multiple cleavage fragments were observed when hnRNP K was cleaved by individual granzymes in cell lysates (Figs. 1 and 3) and living cells (Fig. 4A), the ∼45 kDa cleavage fragment was the only cleavage product observed when living cells were challenged with LAK cells (Fig. 4B). This is most likely due to increased degradation of cleavage fragments in living cells, differences in granzyme concentration between conditions, or the net result of granzyme-mediated hnRNP K cleavage at multiple distinct sites following delivery of all granzymes by LAK cells. Similar results were observed when K562 tumor cells were challenged with LAK cells (data not shown). Antibodies against the absolute N or C terminus of hnRNP K identified this ∼45 kDa hnRNP K cleavage fragment as the N-terminal part of hnRNP K (Fig. 4B, middle panel) and a smaller ∼20 kDa fragment as the C-terminal part of hnRNP K (Fig. 4B, lower panel). These data indicate that intracellular hnRNP K in living tumor cells is cleaved by individual granzymes and during cytotoxic lymphocyte-mediated killing.
FIGURE 4.

HnRNP K is a physiological granzyme substrate. A, HeLa cells were incubated with GrA (1 μm), GrB (1 μm), GrH (0.2 μm), GrK (1 μm), GrM (1 μm) or their corresponding catalytically inactive control granzyme (SA) in the presence of SLO (500 ng/ml) for 30 min (GrB) or 16 h (other granzymes) at 37 °C. Lysates were subjected to immunoblotting using an antibody against the mid region of hnRNP K. B, HeLa cells were challenged with increasing E:T ratios of LAK cells for 4 h at 37 °C. Lysates were subjected to immunoblotting using antibodies directed against the mid region, N terminus, or C terminus of hnRNP K.
HnRNP K Is Required for Tumor Cell Survival
Granzyme-mediated cleavage and inactivation of hnRNP K might be directly involved in the induction of tumor cell death. To investigate whether hnRNP K is required for tumor cell survival, HeLa cells were treated with control siRNA or siRNA directed against hnRNP K, and knockdown was confirmed by immunoblotting to be ∼80–90% (Fig. 5A). Cells were seeded at different cell densities, and overall cell behavior was monitored real-time for 72 h using xCELLigence. Here, an increase of CI values in time is a consequence of increased cell numbers. Therefore, a lower increase of CI values in time represents a reduced rate of cell proliferation, increased cell death, or both. Cells treated with hnRNP K siRNA showed dramatically decreased CI values compared with control cells at low cell densities (below 5,000 cells/well) (Fig. 5B). Microscopic analysis of these cells revealed that this decreased CI value coincided with fewer adherent HeLa cells (Fig. 5C). Interestingly, this hnRNP K knockdown phenotype became less apparent when cells were seeded at higher cell densities (above 5,000 cells/well) (Fig. 5B). To examine hnRNP K knockdown in more detail, control and hnRNP K-deficient tumor cells were evaluated in several biological assays. Knockdown of hnRNP K resulted in decreased cell numbers (Fig. 5D), which was not explained by decreased proliferation, since cell cycle profiles were similar between hnRNP K knockdown and control cells (Fig. 5E). However, cells treated with hnRNP K siRNA showed low cell viability (WST-1 assay) (Fig. 5F) and high cell damage (LDH assay) (Fig. 5G) as compared with control cells. FACS analysis showed increased annexin-V-positive and PI-positive cells when cells were treated with hnRNP K siRNA as compared with control cells (Fig. 5H). Finally, hnRNP K knockdown resulted in increased ROS production (Fig. 5I). Taken together, these data indicate that down-regulation of hnRNP K leads to spontaneous apoptosis, which is most pronounced at low tumor cell density.
FIGURE 5.

Knockdown of hnRNP K in tumor cells results in spontaneous cell death. HeLa cells were transfected with control siRNA, hnRNP K siRNA, or left untreated. A, knockdown of hnRNP K was validated by subjecting cell lysates to immunoblotting using antibodies against the mid region of hnRNP K and β-tubulin. B, three days post-transfection, cells were seeded in E-plates at different cell densities and overall cell behavior was monitored real-time using xCELLigence. The number of seeded cells per well is indicated, and data depicted represent the mean ± S.D. of triplicates. C, cells were assessed by phase contrast microscopy 5 days post-transfection and (D) cell numbers were counted using a counter chamber 7 days post-transfection. E, cells (2 × 105 cells/well) were seeded in 6-well plates 3 days post-transfection, and cell cycle profiles were assessed by staining fixed and permeabilized cells with PI 6 days post-transfection. F, cells were seeded in 96-well plates, and cell viability was determined by WST-1 assay, and (G) cell damage was assessed by LDH assay 6 days post-transfection. H, cells (2 × 105 cells/well) were seeded in 6-well plates, and apoptosis was assessed by staining cells with annexin-V-FLUOS and PI followed by flow cytometry analysis 6 days post-transfection. I, ROS levels in cells were determined by staining cells with CM-H2DCFDA followed by flow cytometry analysis 6 days post-transfection. Cells treated with H2O2 were used as a positive control. Bars represent the mean ± S.D. of at least two individual experiments performed in triplicate. *, p < 0.01; **, p < 0.001.
Loss of hnRNP K Leads to Caspase-dependent and Caspase-independent Cell Death
To examine whether caspases are involved in apoptosis caused by loss of hnRNP K, HeLa cells were treated with hnRNP K or control siRNA, seeded at low cell density, and activation of caspase-3 and -7 was determined. Knockdown of hnRNP K resulted in increased caspase-3 and -7 activation, which was completely blocked by the pan-caspase inhibitor zVAD-fmk (Fig. 6A). Knockdown of hnRNP K in the presence of zVAD-fmk at least partially inhibited apoptosis (Fig. 6B). This indicates that loss of hnRNP K results in caspase activation. Because the hnRNP K knockdown phenotype was highly dependent on cell density as observed by xCELLigence (Fig. 5B), we examined the role of caspases during hnRNP K knockdown at different cell densities. To this end, cells were treated with either hnRNP K or control siRNA in the presence or absence of zVAD-fmk, seeded at low and high cell density, and the hnRNP K knockdown phenotype was assessed using xCELLigence. Inhibition of caspases had a minor effect at low cell density, whereas inhibition of caspases completely restored the hnRNP K knockdown phenotype at high cell density (Fig. 6C). These data indicate that cell death due to loss of hnRNP K is mediated through activation of caspases, but also through a caspase-independent pathway that only occurs at low tumor cell density, likely when there is loss of cell-cell contact.
FIGURE 6.

Loss of hnRNP K leads to caspase-dependent and caspase-independent cell death. HeLa cells were transfected with control or hnRNP K siRNA in the presence or absence of zVAD-fmk (100 μm). A, 3 days post-transfection, cells (3,750 cells/well) were seeded in 96-well plates and 5 days post-transfection, activity of caspase-3 and -7 was assessed by subjecting cell lysates to Caspase-Glo 3/7 Assay. Bars represent the mean ± S.D. of triplicates. B, cells (1 × 105 cells/well) were seeded in 6-well plates 3 days post-transfection, and apoptosis was assessed by staining cells with annexin-V-FLUOS and PI followed by flow cytometry analysis 5 days post-transfection. Background apoptotic values of control siRNA-treated cells were subtracted and the apoptotic value of hnRNP K siRNA-treated cells without zVAD-fmk was set to 100%. Bars represent the mean with range of two individual experiments. C, 3 days post-transfection, cells were seeded in E-plates at different cell densities (3,750 or 7,500 cells/well), and overall cell behavior was monitored real-time using xCELLigence. Bars represent the mean ± S.D. of at least two individual experiments performed in triplicate. *, p < 0.01; **, p < 0.001.
HnRNP K Knockdown Sensitizes Tumor Cells to Cytotoxic Lymphocyte-mediated Killing
To determine whether inactivation of hnRNP K plays an additional role during cytotoxic lymphocyte-mediated attack next to the induction of spontaneous cell death, we examined the susceptibility of hnRNP K-deficient tumor cells to cytotoxic lymphocyte-mediated killing. HnRNP K-deficient or control tumor cell numbers were matched (30,000 cells/well) and incubated with LAK cells for 8 h at different effector:target ratios, and specific LAK cell-mediated cytotoxicity was determined. Interestingly, knockdown of hnRNP K rendered tumor cells more sensitive to LAK cell-mediated killing (Fig. 7). These effects were dependent on the presence of LAK cells and could not be explained by increased apoptosis in tumor target cells due to loss of hnRNP K since these data have been corrected for cell death of target cells that were not challenged with LAK cells (0:1 ratio). Of note, spontaneous cell death was <5% in hnRNP K-deficient tumor cells compared with control cells (data not shown), which is expected when hnRNP K-deficient tumor cells are seeded at high cell density. These data indicate that hnRNP K knockdown in tumor cells facilitates cell death induced by LAK cells.
FIGURE 7.
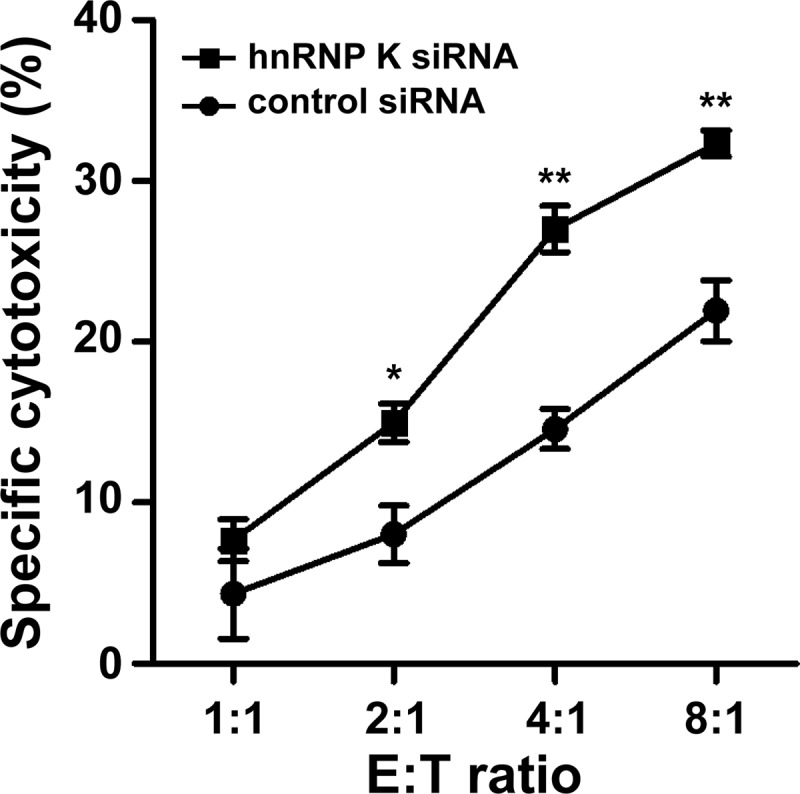
HnRNP K protects tumor cells from LAK cell-mediated killing. HeLa cells were transfected with control siRNA or hnRNP K siRNA. Three days post-transfections, cells (3 × 104 cells/well) were challenged with increasing E:T ratios of LAK cells for 8 h at 37 °C. Cells were stained with PI, and cell death of gated HeLa cells was analyzed by flow cytometry. LAK cell-specific cytotoxicity was calculated and corrected for background cytotoxicity as measured in HeLa cells at E:T ratio of 1:0. These background values were 7.41 ± 0.72% and 12.05 ± 0.36% for control tumor cells and hnRNP K-deficient tumor cells, respectively, and were subtracted from all E:T ratios. Data depicted represent the mean ± S.D. of triplicates. *, p < 0.01; **, p < 0.001.
DISCUSSION
Granzymes are the key molecules that cytotoxic lymphocytes use to eliminate virus-infected cells and tumor cells. In humans, five granzymes exist that all trigger cell death and differ on the basis of their substrate specificity (1–3). These differences in granzyme substrate specificities allow distinct mechanisms to induce cell death, thereby likely bypassing potential inhibitors of apoptosis that target cells often express. Despite the distinct substrate specificities, we showed in the present study for the first time that all five human granzymes directly targeted the same substrate in vitro (Figs. 1 and 2), i.e. hnRNP K, a protein that is involved in a variety of RNA and DNA processes (28). HnRNP K was cleaved under physiological conditions when purified GrB, GrH, GrK, or GrM was delivered into living tumor cells and during LAK cell-mediated attack (Fig. 4). HnRNP K is essential for tumor cell viability, since knockdown of hnRNP K resulted in spontaneous tumor cell apoptosis with caspase activation and ROS production (Figs. 5 and 6). This apoptosis was most pronounced at low tumor cell density, where hnRNP K knockdown also triggered a caspase-independent apoptotic pathway (Figs. 5 and 6). Finally, silencing of hnRNP K protein expression rendered tumor cells more susceptible to LAK cell-mediated cytotoxicity (Fig. 7). Cleavage of hnRNP K by multiple granzymes provides a high functional redundancy, most likely reflecting the importance of hnRNP K as a target during cytotoxic lymphocyte-mediated killing.
The main function described for hnRNP K is its involvement in DNA and RNA processes, including chromatin remodeling, transcription, splicing, mRNA stability, and translation (28). HnRNP K function is predominantly mediated through its three DNA/RNA binding KH domains (Fig. 2G) (28). The functional importance of these domains is supported by the high evolutionary conservation of the primary structure of the three KH domains between hnRNP K and orthologs in a wide range of other species, including Saccharomyces cerevisiae, Caenorhabditis elegans, Drosophila melanogaster, and Xenopus laevis (28). KH domains cooperate in a synergistic manner, since individual or combinations of two KH domains bind dramatically less efficient to RNA as compared with full-length hnRNP K (31). Apparently, full cooperation between KH domains is required in a three-pronged interaction with DNA/RNA to create high affinity binding and increased specificity. We identified two proteolysis-sensitive amino acid regions in hnRNP K that were attacked by multiple human granzymes, thereby dissecting all three functional KH domains (Figs. 1 and 2). Therefore, it seems conceivable that proteolytic dissociation of a single or multiple KH domains by granzymes abolishes both affinity and specificity of hnRNP K to bind RNA and DNA structures. Interestingly, granzymes more efficiently cleaved hnRNP K in the presence of RNA (Fig. 3). This suggests that RNA facilitates the interaction between hnRNP K and granzymes or that granzymes cleave hnRNP K more efficiently in its functional RNA-bound conformation.
We have demonstrated that functional inactivation of hnRNP K by RNA-interference led to spontaneous apoptosis in tumor cells (Fig. 5). This is consistent with two recent studies that show a role of hnRNP K in cell survival (32, 33). In contrast, several other studies have demonstrated that hnRNP K depletion from virus-infected cells does not affect cell survival or apoptosis (34–36). Several observations could explain this discrepancy. First, we found that spontaneous apoptosis following hnRNP K knockdown was most efficient at low cell density (Fig. 5), suggesting that loss of cell-cell contact enhances the apoptotic signal initiated in hnRNP K-deficient tumor cells. This may also explain why elevated hnRNP K expression is beneficial for tumor cell migration and metastasis (37). Second, Chen et al. have demonstrated that knockdown of hnRNP K does not result in spontaneous cell death, but instead sensitizes tumor cells to death receptor (TRAIL)-induced apoptosis (38). Likewise, we demonstrated that loss of hnRNP K sensitizes tumor cells to cytotoxic lymphocyte-mediated killing (Fig. 7). Taken together, these data suggest that loss of hnRNP K leads to cell death under conditions of stress depending on other pro-apoptotic factors in the biological environment. In this model, granzyme-mediated cleavage of hnRNP K could reinforce the multiple cell death pathways that all granzymes and/or death receptors trigger, thereby contributing to the mechanism by which cytotoxic lymphocytes eliminate tumor cells.
The precise mechanism by which hnRNP K inactivation or knockdown triggers apoptosis remains unclear. Knockdown of hnRNP K shifts the splicing of the Bcl-x pre-mRNA from the anti-apoptotic Bcl-xl toward the pro-apoptotic Bcl-xs isoform (39). HnRNP K also directly binds to the promotor region of the caspase-8 inhibitor FLIP gene, thereby increasing FLIP expression (38). Although these studies do not show a direct correlation between hnRNP K and apoptosis or cell viability, it could explain how hnRNP K inactivation triggers caspase activation and apoptosis. Consistent with these data, we found that cell death due to loss of hnRNP K was completely caspase-dependent at high cell density (Fig. 6). At low cell density, however, we also found a caspase-independent pathway that mediates cell death following hnRNP K depletion from tumor cells (Fig. 6). Further studies are required to address the mechanism by which hnRNP K positively regulates cell survival independent of caspases.
Overexpression or altered subcellular expression of hnRNP K in numerous tumors has been positively correlated with poor clinical outcome (40–45). This further supports a role of hnRNP K in tumorigenesis that can be attributed to improved tumor cell survival (Figs. 5 and 6) and/or improved tumor cell migration and metastasis (37). Our data show that hnRNP K also protected tumor cells from cytotoxic lymphocyte-mediated attack (Fig. 7). This raises the interesting possibility that tumor cells up-regulate hnRNP K expression to evade granzyme-mediated cytotoxic immune response. Thus, hnRNP K would be an interesting therapeutic target to inhibit tumorigenesis and tumor progression.
Acknowledgments
We thank Dr. A. Ostareck-Lederer (Martin Luther University Halle-Wittenberg, Germany) for providing the pET16-b-His-hnRNP K expression plasmid, R. L. Tatsis for technical assistance, and Dr. S. A. H. de Poot and Dr. P. J. Coffer for critical reading of the manuscript.
This study was supported by The Netherlands Organization for Scientific Research (NWO) (Grant 916.66.044) and the Dutch Cancer Society (UU-2009-4302) (to N. B.).
- ROS
- reactive oxygen species
- CI
- cell index
- hnRNP K
- heterogeneous nuclear ribonucleoprotein K
- LAK
- lymphokine-activated killer
- PI
- propidium iodide
- SLO
- streptolysin O.
REFERENCES
- 1. Barry M., Bleackley R. C. (2002) Cytotoxic T lymphocytes: all roads lead to death. Nat. Rev. Immunol. 2, 401–409 [DOI] [PubMed] [Google Scholar]
- 2. Chowdhury D., Lieberman J. (2008) Death by a thousand cuts: granzyme pathways of programmed cell death. Annu. Rev. Immunol. 26, 389–420 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Bovenschen N., Kummer J. A. (2010) Orphan granzymes find a home. Immunol. Rev. 235, 117–127 [DOI] [PubMed] [Google Scholar]
- 4. Martinvalet D., Dykxhoorn D. M., Ferrini R., Lieberman J. (2008) Granzyme A cleaves a mitochondrial complex I protein to initiate caspase-independent cell death. Cell 133, 681–692 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Martinvalet D., Zhu P., Lieberman J. (2005) Granzyme A induces caspase-independent mitochondrial damage, a required first step for apoptosis. Immunity 22, 355–370 [DOI] [PubMed] [Google Scholar]
- 6. Beresford P. J., Zhang D., Oh D. Y., Fan Z., Greer E. L., Russo M. L., Jaju M., Lieberman J. (2001) Granzyme A activates an endoplasmic reticulum-associated caspase-independent nuclease to induce single-stranded DNA nicks. J. Biol. Chem. 276, 43285–43293 [DOI] [PubMed] [Google Scholar]
- 7. Fan Z., Beresford P. J., Zhang D., Lieberman J. (2002) HMG2 interacts with the nucleosome assembly protein SET and is a target of the cytotoxic T-lymphocyte protease granzyme A. Mol. Cell Biol. 22, 2810–2820 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Fan Z., Beresford P. J., Zhang D., Xu Z., Novina C. D., Yoshida A., Pommier Y., Lieberman J. (2003) Cleaving the oxidative repair protein Ape1 enhances cell death mediated by granzyme A. Nat. Immunol. 4, 145–153 [DOI] [PubMed] [Google Scholar]
- 9. Zhu P., Zhang D., Chowdhury D., Martinvalet D., Keefe D., Shi L., Lieberman J. (2006) Granzyme A, which causes single-stranded DNA damage, targets the double-strand break repair protein Ku70. EMBO Reports 7, 431–437 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Afonina I. S., Cullen S. P., Martin S. J. (2010) Cytotoxic and non-cytotoxic roles of the CTL/NK protease granzyme B. Immunol. Rev. 235, 105–116 [DOI] [PubMed] [Google Scholar]
- 11. Fellows E., Gil-Parrado S., Jenne D. E., Kurschus F. C. (2007) Natural killer cell-derived human granzyme H induces an alternative, caspase-independent cell-death program. Blood 110, 544–552 [DOI] [PubMed] [Google Scholar]
- 12. Hou Q., Zhao T., Zhang H., Lu H., Zhang Q., Sun L., Fan Z. (2008) Granzyme H induces apoptosis of target tumor cells characterized by DNA fragmentation and Bid-dependent mitochondrial damage. Mol. Immunol. 45, 1044–1055 [DOI] [PubMed] [Google Scholar]
- 13. Guo Y., Chen J., Zhao T., Fan Z. (2008) Granzyme K degrades the redox/DNA repair enzyme Ape1 to trigger oxidative stress of target cells leading to cytotoxicity. Mol. Immunol. 45, 2225–2235 [DOI] [PubMed] [Google Scholar]
- 14. Zhao T., Zhang H., Guo Y., Fan Z. (2007) Granzyme K directly processes bid to release cytochrome c and endonuclease G leading to mitochondria-dependent cell death. J. Biol. Chem. 282, 12104–12111 [DOI] [PubMed] [Google Scholar]
- 15. Zhao T., Zhang H., Guo Y., Zhang Q., Hua G., Lu H., Hou Q., Liu H., Fan Z. (2007) Granzyme K cleaves the nucleosome assembly protein SET to induce single-stranded DNA nicks of target cells. Cell Death Differ. 14, 489–499 [DOI] [PubMed] [Google Scholar]
- 16. Guo Y., Chen J., Shi L., Fan Z. (2010) Valosin-containing protein cleavage by granzyme K accelerates an endoplasmic reticulum stress leading to caspase-independent cytotoxicity of target tumor cells. J. Immunol. 185, 5348–5359 [DOI] [PubMed] [Google Scholar]
- 17. Bovenschen N., Quadir R., van den Berg A. L., Brenkman A. B., Vandenberghe I., Devreese B., Joore J., Kummer J. A. (2009) Granzyme K displays highly restricted substrate specificity that only partially overlaps with granzyme A. J. Biol. Chem. 284, 3504–3512 [DOI] [PubMed] [Google Scholar]
- 18. Hua G., Wang S., Zhong C., Xue P., Fan Z. (2009) Ignition of p53 bomb sensitizes tumor cells to granzyme K-mediated cytolysis. J. Immunol. 182, 2152–2159 [DOI] [PubMed] [Google Scholar]
- 19. Bovenschen N., de Koning P. J., Quadir R., Broekhuizen R., Damen J. M., Froelich C. J., Slijper M., Kummer J. A. (2008) NK cell protease granzyme M targets α-tubulin and disorganizes the microtubule network. J. Immunol. 180, 8184–8191 [DOI] [PubMed] [Google Scholar]
- 20. Cullen S. P., Afonina I. S., Donadini R., Lüthi A. U., Medema J. P., Bird P. I., Martin S. J. (2009) Nucleophosmin is cleaved and inactivated by the cytotoxic granule protease granzyme M during natural killer cell-mediated killing. J. Biol. Chem. 284, 5137–5147 [DOI] [PubMed] [Google Scholar]
- 21. Kelly J. M., Waterhouse N. J., Cretney E., Browne K. A., Ellis S., Trapani J. A., Smyth M. J. (2004) Granzyme M mediates a novel form of perforin-dependent cell death. J. Biol. Chem. 279, 22236–22242 [DOI] [PubMed] [Google Scholar]
- 22. Hua G., Zhang Q., Fan Z. (2007) Heat shock protein 75 (TRAP1) antagonizes reactive oxygen species generation and protects cells from granzyme M-mediated apoptosis. J. Biol. Chem. 282, 20553–20560 [DOI] [PubMed] [Google Scholar]
- 23. Wang S., Xia P., Shi L., Fan Z. (2012) FADD cleavage by NK cell granzyme M enhances its self-association to facilitate procaspase-8 recruitment for auto-processing leading to caspase cascade. Cell Death Differ. 19, 605–615 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. van Domselaar R., de Poot S. A., Bovenschen N. (2010) Proteomic profiling of proteases: tools for granzyme degradomics. Expert. Rev. Proteomics 7, 347–359 [DOI] [PubMed] [Google Scholar]
- 25. de Poot S. A., Westgeest M., Hostetter D. R., Van Damme P., Plasman K., Demeyer K., Broekhuizen R., Gevaert K., Craik C. S., Bovenschen N. (2011) Human and mouse granzyme M display divergent and species-specific substrate specificities. Biochem. J. 437, 431–442 [DOI] [PubMed] [Google Scholar]
- 26. Van Damme P., Maurer-Stroh S., Hao H., Colaert N., Timmerman E., Eisenhaber F., Vandekerckhove J., Gevaert K. (2010) The substrate specificity profile of human granzyme A. Biol. Chem. 391, 983–997 [DOI] [PubMed] [Google Scholar]
- 27. Van Damme P., Staes A., Bronsoms S., Helsens K., Colaert N., Timmerman E., Aviles F. X., Vandekerckhove J., Gevaert K. (2010) Complementary positional proteomics for screening substrates of endo- and exoproteases. Nat. Methods 7, 512–515 [DOI] [PubMed] [Google Scholar]
- 28. Bomsztyk K., Denisenko O., Ostrowski J. (2004) hnRNP K: one protein multiple processes. Bioessays 26, 629–638 [DOI] [PubMed] [Google Scholar]
- 29. Plasman K., Van Damme P., Kaiserman D., Impens F., Demeyer K., Helsens K., Goethals M., Bird P. I., Vandekerckhove J., Gevaert K. (2011) Probing the efficiency of proteolytic events by positional proteomics. Mol. Cell Proteomics, in press [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Van Damme P., Maurer-Stroh S., Plasman K., Van Durme J., Colaert N., Timmerman E., De Bock P. J., Goethals M., Rousseau F., Schymkowitz J., Vandekerckhove J., Gevaert K. (2009) Analysis of protein processing by N-terminal proteomics reveals novel species-specific substrate determinants of granzyme B orthologs. Mol. Cell Proteomics 8, 258–272 [DOI] [PubMed] [Google Scholar]
- 31. Paziewska A., Wyrwicz L. S., Bujnicki J. M., Bomsztyk K., Ostrowski J. (2004) Cooperative binding of the hnRNP K three KH domains to mRNA targets. FEBS Lett. 577, 134–140 [DOI] [PubMed] [Google Scholar]
- 32. White M. C., Gao R., Xu W., Mandal S. M., Lim J. G., Hazra T. K., Wakamiya M., Edwards S. F., Raskin S., Teive H. A., Zoghbi H. Y., Sarkar P. S., Ashizawa T. (2010) Inactivation of hnRNP K by expanded intronic AUUCU repeat induces apoptosis via translocation of PKCδ to mitochondria in spinocerebellar ataxia 10. PLoS Genetics 6, e1000984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Gao F. H., Wu Y. L., Zhao M., Liu C. X., Wang L. S., Chen G. Q. (2009) Protein kinase C-delta mediates down-regulation of heterogeneous nuclear ribonucleoprotein K protein: involvement in apoptosis induction. Exp. Cell Res. 315, 3250–3258 [DOI] [PubMed] [Google Scholar]
- 34. Lin J. Y., Li M. L., Huang P. N., Chien K. Y., Horng J. T., Shih S. R. (2008) Heterogeneous nuclear ribonuclear protein K interacts with the enterovirus 71 5' untranslated region and participates in virus replication. J. Gen. Virol. 89, 2540–2549 [DOI] [PubMed] [Google Scholar]
- 35. Ng L. F., Chan M., Chan S. H., Cheng P. C., Leung E. H., Chen W. N., Ren E. C. (2005) Host heterogeneous ribonucleoprotein K (hnRNP K) as a potential target to suppress hepatitis B virus replication. PLoS Medicine 2, e163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Schmidt T., Striebinger H., Haas J., Bailer S. M. (2010) The heterogeneous nuclear ribonucleoprotein K is important for Herpes simplex virus-1 propagation. FEBS Lett. 584, 4361–4365 [DOI] [PubMed] [Google Scholar]
- 37. Inoue A., Sawata S. Y., Taira K., Wadhwa R. (2007) Loss-of-function screening by randomized intracellular antibodies: identification of hnRNP-K as a potential target for metastasis. Proc. Natl. Acad. Sci. U.S.A. 104, 8983–8988 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Chen L. C., Chung I. C., Hsueh C., Tsang N. M., Chi L. M., Liang Y., Chen C. C., Wang L. J., Chang Y. S. (2010) The antiapoptotic protein, FLIP, is regulated by heterogeneous nuclear ribonucleoprotein K and correlates with poor overall survival of nasopharyngeal carcinoma patients. [DOI] [PubMed] [Google Scholar]
- 39. Revil T., Pelletier J., Toutant J., Cloutier A., Chabot B. (2009) Heterogeneous nuclear ribonucleoprotein K represses the production of pro-apoptotic Bcl-xS splice isoform. J. Biol. Chem. 284, 21458–21467 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Zhou R., Shanas R., Nelson M. A., Bhattacharyya A., Shi J. (2010) Increased expression of the heterogeneous nuclear ribonucleoprotein K in pancreatic cancer and its association with the mutant p53. Int. J. Cancer 126, 395–404 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Matta A., Tripathi S. C., DeSouza L. V., Grigull J., Kaur J., Chauhan S. S., Srivastava A., Thakar A., Shukla N. K., Duggal R., DattaGupta S., Ralhan R., Michael Siu K. W. (2009) Heterogeneous ribonucleoprotein K is a marker of oral leukoplakia and correlates with poor prognosis of squamous cell carcinoma. Int. J. Cancer 125, 1398–1406 [DOI] [PubMed] [Google Scholar]
- 42. Barboro P., Repaci E., Rubagotti A., Salvi S., Boccardo S., Spina B., Truini M., Introini C., Puppo P., Ferrari N., Carmignani G., Boccardo F., Balbi C. (2009) Heterogeneous nuclear ribonucleoprotein K: altered pattern of expression associated with diagnosis and prognosis of prostate cancer. Br. J. Cancer 100, 1608–1616 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Chen L. C., Hsueh C., Tsang N. M., Liang Y., Chang K. P., Hao S. P., Yu J. S., Chang Y. S. (2008) Heterogeneous ribonucleoprotein k and thymidine phosphorylase are independent prognostic and therapeutic markers for nasopharyngeal carcinoma. Clin. Cancer Res. 14, 3807–3813 [DOI] [PubMed] [Google Scholar]
- 44. Carpenter B., McKay M., Dundas S. R., Lawrie L. C., Telfer C., Murray G. I. (2006) Heterogeneous nuclear ribonucleoprotein K is over expressed, aberrantly localized and is associated with poor prognosis in colorectal cancer. Br. J. Cancer 95, 921–927 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Roychoudhury P., Chaudhuri K. (2007) Evidence for heterogeneous nuclear ribonucleoprotein K overexpression in oral squamous cell carcinoma. Br. J. Cancer 97, 574–575 [DOI] [PMC free article] [PubMed] [Google Scholar]