

Background: Mechanisms regulating nuclear translocation of the ErbB4 intracellular domain (ICD) are unclear.
Results: PIAS3 interacts with ErbB4, sequesters ICD into the nucleus, and represses its nuclear signaling.
Conclusion: PIAS3 is a regulator of subcellular localization and nuclear functions of ErbB4.
Significance: A novel mechanism controlling the signaling of an ICD of a receptor tyrosine kinase in the nucleus is described.
Keywords: Nuclear Translocation, Protein-Protein Interactions, Receptor Tyrosine Kinase, Signaling, SUMO, ErbB4/HER4, PIAS3
Abstract
ErbB4 is a receptor tyrosine kinase implicated in the development and homeostasis of the heart, central nervous system, and mammary gland. Cleavable isoforms of ErbB4 release a soluble intracellular domain (ICD) that can translocate to the nucleus and function as a transcriptional coregulator. In search of regulatory mechanisms of ErbB4 ICD function, we identified PIAS3 as a novel interaction partner of ErbB4 ICD. In keeping with the small ubiquitin-like modifier (SUMO) E3 ligase function of protein inhibitor of activated STAT (PIAS) proteins, we showed that the ErbB4 ICD is modified by SUMO, and that PIAS3 stimulates the SUMOylation. Upon overexpression of PIAS3, the ErbB4 ICD generated from the full-length receptor accumulated into the nucleus in a manner that was dependent on the functional nuclear localization signal of ErbB4. In the nucleus, ErbB4 colocalized with PIAS3 and SUMO-1 in promyelocytic leukemia nuclear bodies, nuclear domains involved in regulation of transcription. Accordingly, PIAS3 overexpression had an effect on the transcriptional coregulatory activity of ErbB4, repressing its ability to coactivate transcription with Yes-associated protein. Finally, knockdown of PIAS3 with siRNA partially rescued the inhibitory effect of the ErbB4 ICD on differentiation of MDA-MB-468 breast cancer and HC11 mammary epithelial cells. Our findings illustrate that PIAS3 is a novel regulator of ErbB4 receptor tyrosine kinase, controlling its nuclear sequestration and function.
Introduction
ErbB receptors form the epidermal growth factor receptor/ErbB subfamily of receptor tyrosine kinases, which includes ErbB1/EGF receptor, ErbB2, ErbB3, and ErbB4. ErbB receptors are activated upon binding of EGF-like ligands and regulate gene transcription through the activation of a multilayered network of intracellular signaling molecules. Signaling via ErbB receptors results in various biological outcomes, including proliferation, differentiation, migration, and survival. Aberrant ErbB signaling has been implicated in the initiation and progression of several human malignancies, and drugs targeting the EGF receptor and ErbB2 are currently in clinical use (1).
Unlike other ErbB receptors, ErbB4 is expressed as four isoforms that are generated by alternative mRNA splicing. Two of the isoforms differ structurally in the extracellular juxtamembrane region (JM-a3 and JM-b), and two in the intracellular cytoplasmic domain (CYT-1 and CYT-2) (2, 3). The ErbB4 JM-a isoform was the first receptor tyrosine kinase found to employ a signaling paradigm involving regulated intramembrane proteolysis (RIP) and generation of a soluble intracellular domain (ICD). Upon ligand binding, ErbB4 JM-a is cleaved by tumor necrosis factor-α-converting enzyme, which triggers a second cleavage by γ-secretase activity (4, 5). The released ErbB4 ICD can translocate into the nucleus, where it associates with transcriptional regulators including yes-associated protein (YAP) (6), STAT5 (7), estrogen receptor (8), Eto2 (9), TAB2-N-CoR complex (10), AP-2 (11), and HIF-1α (12), and modifies their activity. In vivo, the soluble ErbB4 ICD has been shown to regulate astrogenesis in the developing brain (10) as well as mammary epithelial cell differentiation and proliferation (13). In addition to nuclear localization and regulation of transcriptional events, the ErbB4 ICD has been reported to localize in the cytosol in mitochondria, where it regulates apoptosis (14). In clinical breast cancer samples, membrane, cytosolic, and nuclear localization of ErbB4 has been described. Interestingly, nuclear localization of ErbB4 epitope is more frequent in malignant cells than in morphologically normal cells (15), and nuclear ErbB4 immunoreactivity has been suggested to associate with different disease outcome in cancer when compared with membranous ErbB4 (16, 17). Although these observations suggest that subcellular localization of the ErbB4 ICD has a role in determining the biological outcome of ErbB4 ICD signaling, little is known about how the subcellular localization and functions of ErbB4 ICD are regulated.
Members of the PIAS (protein inhibitor of activated STAT) family (PIAS1, PIASxα, PIASxβ, PIAS3, and PIASy) interact with and modulate functions of a number of proteins, especially transcriptional regulators. PIAS proteins can function as E3 ligases in the enzymatic reaction that posttranslationally attaches SUMO (small ubiquitin-related modifier) to target proteins. Indeed, most interaction partners of PIAS proteins are also modified by SUMO (18). Many known SUMO targets are nuclear proteins involved in transcriptional regulation. SUMOylation has been reported to regulate several biological processes by altering the activity, stability, or subcellular localization of its targets (19). In addition, PIAS proteins can also regulate their interaction partners in a manner that is independent of their SP-RING finger, a domain that is central for their ability to promote SUMOylation. For example, PIAS proteins can affect the functions of the interacting proteins by altering their subcellular or subnuclear localization (18).
Here, we demonstrate that ErbB4 selectively interacts with members of the PIAS protein family, and show that ErbB4 is modified by SUMO. Our results implicate a role for one of the PIAS proteins, PIAS3, in sequestration of the ErbB4 ICD into the nucleus, repression of its transcriptional coregulatory activity, and in ErbB4 ICD-mediated inhibition of mammary epithelial cell differentiation. Taken together, our data reveal novel mechanisms that control nuclear localization and function of ErbB4 RTK.
EXPERIMENTAL PROCEDURES
Plasmids
The expression plasmids encoding the following inserts have been described: ErbB4 JM-a CYT-2-HA, ErbB4 ICD1-HA, and ErbB4 ICD2-HA (20); ErbB4 ICD2-GST, ErbB4 ICD2-ΔC-GST, and ErbB4 ICD2-ΔN-GST (11); FLAG-PIAS1, FLAG-PIASxα, FLAG-PIAS3, and FLAG-PIASy (21); STAT5a (22); GFP-SUMO-1 and His-SUMO-1 (23); PML-3 (kindly provided by Dr. Thomas G. Hofmann, German Cancer Research Center (DKFZ), Heidelberg, Germany); ErbB4 ICD2-GAL4 (ErbB4 CTF-GAL4, Ref. 6); Wwox-Myc (24); and Omni-YAP2 (25). Point mutations were introduced to plasmids encoding ErbB4 JM-a CYT-2-HA and FLAG-PIAS3 using the QuikChange site-directed mutagenesis kit (Stratagene) and primers E4-V675A 5′-GGGTCTGACATTTGCTGTTTATGCTAGAAGGAAGAGCATCAAAAAG-3′ and PIAS3-C299S 5′-CGTGCCCTCACCTCTGCCCATCTGCAGAG-3′ to generate pcDNA3.1ErbB4JM-aCYT-2V675A-HA and pFlagPIAS3C299S, respectively. Two basic clusters of the tripartite ErbB4 nuclear localization signal (NLS) (676-RRKSIKKKRALRR-688, Ref. 26) were mutated by PCR in two steps to generate pcDNA3.1ErbB4JM-aCYT-2ΔNLSI/II-HA (676-AAASIEMIAALRR-688). First, the second basic cluster (681-KKKR-684) of ErbB4 was mutated with 5′-GAAGGAAGAGCATCGAAATGATAGCGGCCTTGAGAAGATTCTTGG-3′.Next, the first basic cluster (676-RRK-678) was mutated with 5′-GACATTTGCTGTTTATGTTGCAGCGGCGAGCATCGAAATGATAGCGGCC-3′. To generate plasmids for retroviral expression of ErbB4 JM-a CYT-2 and ErbB4 JM-b CYT-2, the XcmI fragment of pcDNA3.1ErbB4JM-aCYT-1 (27), including the JM-a sequence, was swapped to replace a corresponding XcmI fragment in pBABE-puroErbB4JM-bCYT-1 (kindly provided by Dr. Nancy E. Hynes, Friedrich Miescher Institute for Biomedical Research, Basel, Switzerland). Next, the EcoRI fragment of pcDNA3.1ErbB4JM-aCYT-2 (27), including the CYT-2 sequence, was swapped to replace a corresponding EcoRI fragment in pBABE-puroErbB4JM-aCYT-1 and pBABE-puroErbB4JM-bCYT-1 to generate pBABE-puroErbB4JM-aCYT-2 and pBABE-puroErbB4JM-bCYT-2, respectively. All generated constructs were verified by sequencing.
Cell Culture and Transfections
COS-7, MDA-MB-468, HEK 293, and WM-266-4 cells were cultured in DMEM supplemented with 10% fetal calf serum (Promocell) and 1% l-glutamine-penicillin-streptomycin solution (Sigma-Aldrich). MCF-7 and HC11 (a kind gift from Dr. Lars-Arne Haldosén, Karolinska Institutet, Huddinge, Sweden) cells were cultured in RPMI 1640 supplemented with 10% fetal calf serum and 1% l-glutamine-penicillin-streptomycin solution. The culture medium of MCF-7 cells was further supplemented with 1 μg/ml insulin (Roche) and 1 nm 17-β-estradiol (Sigma-Aldrich) and the medium of HC11 cells with 5 μg/ml insulin and 10 ng/ml epidermal growth factor (Sigma-Aldrich). COS-7 and HEK 293 cells were transfected with expression plasmids by using FuGENE HD transfection reagent (Roche) according to the instructions of the manufacturer.
Glutathione S-transferase Pull-down Assay
GST fusion proteins have been described (11). FLAG-tagged PIAS proteins, STAT5a, or Wwox-Myc were transiently expressed in COS-7 cells, and the GST pull-down assay was performed as described previously (28). The interaction between FLAG-tagged PIAS proteins, STAT5a, or Wwox-Myc and GST fusion proteins was detected by Western blotting using anti-FLAG (M2, Sigma-Aldrich), anti-STAT5 (sc-835, Santa Cruz Biotechnology), or anti-Myc (Zymed Laboratories, Inc.) antibodies, and GST fusion proteins with an anti-GST antibody (GE Healthcare).
Coimmunoprecipitation of ErbB4 and PIAS3
COS-7 cells were transiently transfected with indicated expression plasmids and lysed 24 h after transfection. Coimmunoprecipitation was carried out as described previously (28). Precipitates were analyzed by Western blotting using anti-FLAG and anti-ErbB4 (sc-283, Santa Cruz Biotechnology) antibodies.
RNA Interference
siRNA oligonucleotides targeting human (PIAS3 siRNA#1 ID 3048, 5′-GGUCGAAGUUAUUGACUUGTT-3′ and PIAS3 siRNA#2 ID 2956, 5′-GGUGCAGCUAAGGUUCUGUTT-3′) and mouse (PIAS3 siRNA#3 ID 90899 5′-GGGACCCUUCUACAAAAACTT-3′ and PIAS3 siRNA#4 ID 90805, 5′-GGUUAAUGGGAAACUCUGCTT-3′) PIAS3 were purchased from Ambion, and siRNAs targeting human PML (PML siRNA#1 Hs_PML_4, 5′-CCAAGAUCUAAACCGAGAATT-3′ and PML siRNA#2 Hs_PML_8, 5′-GGAGCAGGAUAGUGCCUUUTT-3′) from Qiagen. Cells were transfected with siRNAs at final concentrations of 20–50 nm using Lipofectamine 2000 (Invitrogen) according to the instructions of the manufacturer. Experiments were controlled with AllStars negative control siRNA (Qiagen) or Silencer negative control #1 siRNA (Ambion). Knockdown efficacy was determined by Western blotting with anti-PIAS3 (sc-46682, Santa Cruz Biotechnology) and anti-PML (sc-5621, Santa Cruz Biotechnology) antibodies (supplemental Fig. S1) and quantified as signal intensities relative to actin (anti-actin, sc-1616, Santa Cruz Biotechnology) using the ImageJ software. In immunofluorescence analysis of HC11 cells, PIAS3 knockdown efficacy was determined by quantifying fluorescence intensities (anti-PIAS3, ab22856, Abcam) using the ImageJ software.
In Situ Proximity Ligation Assay (PLA)
MCF-7 cells cultured on coverslips were transfected with control siRNA (Ambion) or siRNAs targeting PIAS3 at a final concentration of 30 nm. Cells were starved without serum overnight, treated with 50 ng/ml neuregulin-1 (NRG-1-β1 EGF domain, R&D Systems) for 15 min, and fixed with methanol. Samples were incubated with anti-ErbB4 (HFR-1, Abcam) and anti-PIAS3 (ab22856) for 90 min. Proximity ligation was carried out with the Duolink II in situ PLA kit (Olink Biosciences) according to the instructions of the manufacturer. PLA signals were detected with an Axiovert 200 M confocal microscope with LSM510 Meta (Zeiss). Two confocal images of each sample were acquired with a ×63 objective, and all signals were counted in five sections separated by 1.5 μm to quantify the number of interactions per cell. The interactions were classified as cytosolic or nuclear depending on their colocalization with DAPI.
Analysis of ErbB4 SUMOylation
COS-7 cells were transiently transfected with HA-tagged ErbB4 ICD2 with or without His-SUMO-1, GFP-SUMO-1, FLAG-PIAS3, or FLAG-PIAS3C299S. Cells were lysed in lysis buffer (29), supplemented with 20 mm N-ethylmaleimide (Sigma-Aldrich). SUMO-modified ErbB4 ICD2 was detected by immunoprecipitation with anti-HA (Roche) or anti-ErbB4 antibodies (HFR-1), followed by Western blotting with anti-HA antibody.
Immunofluorescence and Confocal Microscopy
Samples were prepared and analyzed as described previously (28).When indicated, cells were treated with GSI IX (Calbiochem), leptomycin B (Sigma-Aldrich), or NRG-1. Fixed cells were stained with the following primary antibodies: rat anti-HA, mouse anti-ErbB4 (HFR-1), mouse anti-PIAS3 (sc-46682), rabbit anti-PIAS3 (ab22856), mouse anti-PML (sc-966), rabbit anti-PML (sc-5621), and rabbit anti-SUMO-1 (ab11672, Abcam). Alexa Fluor 488 goat anti-mouse, 488 goat anti-rabbit, 555 goat anti-rabbit, 568 goat anti-mouse, 568 goat anti-rat, and 633 goat anti-mouse secondary antibodies (Molecular Probes) were used. The nuclei were visualized with DAPI (Sigma-Aldrich). Quantification of nuclear ErbB4 in HC11 cells was performed using ImageJ. COS-7 cells expressing ErbB4 JM-a CYT-2, ErbB4 JM-a CYT-2 V675A, or ErbB4 JM-a CYT-2 NLSI/II-mutant were scored for cytosolic or nuclear staining in 200 randomly selected cells.
Cell Fractionation
Cytosolic and nuclear fractions were prepared with a NE-PER kit (Thermo Scientific) as described previously (20) and analyzed by Western blotting with anti-HA, anti-FLAG, anti-ErbB4 (E200, Abcam), and anti-PIAS3 (sc-46682) as indicated. Antibodies against Lamin B (sc-6217, Santa Cruz Biotechnology) and Mek1/2 (Cell Signaling) were used to control the fractionation.
GAL4 Transactivation Assay
HEK 293 cells were cotransfected with ErbB4 ICD2 fused to the GAL4 DNA-binding domain (ErbB4 ICD2-GAL4) and a Firefly luciferase reporter construct pFR-luc (Stratagene) driven by GAL4 binding sites with or without Omni-YAP2 and FLAG-tagged PIAS proteins. A cotransfected Renilla luciferase construct pTK-RL (Promega) was used as an internal control. Luciferase activity was measured and analyzed as described previously (30). Expression of PIAS proteins and YAP2 were controlled by Western blotting with anti-FLAG and anti-Omni (sc-7270, Santa Cruz Biotechnology), respectively, and loading with anti-HSP90 (Calbiochem).
Differentiation of MDA-MB-468 and HC11 cells in Three-dimensional Cultures
Stable MDA-MB-468 cell lines expressing ErbB4 JM-a CYT-2 have been described (31). HC11 cells were infected with retroviruses encoding pBABE-puroErbB4JM-aCYT-2 or pBABE-puroErbB4JM-bCYT-2 to generate stable cell lines (31). The protocol for three-dimensional cultures of HC11 cells was modified from the protocol described for MDA-MB-468 cells by Tvorogov et al. (31). Briefly, MDA-MB-468 transfectants were suspended into Matrigel (BD Biosciences) on duplicate 96-well plates, supplemented with or without 50 ng/ml NRG-1 and 5 μm GSI IX, and maintained at 37 °C for 6 days. HC11 cells were suspended into growth factor-reduced Matrigel (BD Biosciences), supplemented with 50 ng/ml NRG-1, and maintained at 37 °C for 14 or 15 days as indicated in the figures. For siRNA knockdown experiments, cells were transfected with siRNAs targeting PIAS3 or PML or with negative control siRNAs and suspended into Matrigel 24 h after siRNA transfection. One hundred colonies from three independent views through the whole thickness of Matrigel were counted and classified as undifferentiated colonies or differentiated acini on the basis of their morphology (31).
Statistical Analyses
Student's t test was used for all statistical analyses.
RESULTS
ErbB4 Interacts with PIAS3 and PIASy and Is Modified by SUMO
Ligand-induced RIP of ErbB4 is of biological significance and results in nuclear translocation of the soluble ErbB4 ICD, which has transcriptional coregulatory activity (5, 10). PIAS proteins have been implicated as regulators of a variety of proteins, particularly transcription factors (18). To address the putative interactions between ErbB4 ICD and PIAS proteins, the GST-tagged ICD of CYT-2 type (ICD2) or C- or N-terminal deletion of ICD2 (ICD2-ΔC or ICD2-ΔN, respectively) (Fig. 1, A and B) were purified from bacteria and incubated together with lysate from COS-7 cells expressing FLAG-tagged PIAS proteins. PIAS3 and PIASy interacted with ICD2-GST and ICD2-ΔC-GST containing the ErbB4 tyrosine kinase domain but not with ICD2-ΔN-GST containing the C-terminal tail of ErbB4 (Fig. 1C). In contrast, no interaction between ErbB4 and PIAS1 or PIASxα was observed (Fig. 1C). To control the integrity of both deletion constructs, GST pull-down assays were carried out with STAT5a and WW domain-containing oxidoreductase (Wwox), the expected interaction partners of ICD-ΔC and ICD-ΔN, respectively (30, 32) (Fig. 1C). Reciprocal coimmunoprecipitation analyses with either anti-ErbB4 (Fig. 1D) or anti-FLAG (data not shown) confirmed the interaction between transiently expressed FLAG-tagged PIAS3 and ErbB4 ICD of CYT-1 type (ICD1) in COS-7 cells. Finally, the interaction between endogenous PIAS3 and ErbB4 in MCF-7 breast cancer cells was analyzed using PLA, which allows for detection of endogenous protein complexes in situ (33). Positive PLA signals (red dots) were detected in both the cytosol and the nucleus (Fig. 1, E and F). Knockdown of PIAS3 with siRNA significantly reduced the number of signals in the nucleus, controlling the specificity of observed interactions (Fig. 1, E and F).
FIGURE 1.

Interaction of ErbB4 with PIAS3. A, schematic structure of full-length ErbB4 JM-a CYT-2, and GST fusion proteins containing the whole ErbB4 intracellular domain of CYT-2-type (ICD2), ICD2 with C- (ΔC), or N-terminal (ΔN) deletion. TK, tyrosine kinase. B, GST fusion proteins (A) were pulled down with glutathione-Sepharose and analyzed by Western blotting (W) with anti-GST. C, GST-fusion proteins (A) were incubated with lysates of COS-7 cells transiently expressing FLAG-tagged PIAS proteins, STAT5A, or Myc-tagged Wwox. Pull-downs were analyzed by Western blotting with indicated antibodies. D, COS-7 cells were transfected with indicated constructs. Lysates were immunoprecipitated (IP) with anti-ErbB4 (HFR-1) followed by Western blotting with anti-FLAG to detect PIAS3. The membrane was reblotted with anti-ErbB4 (sc-283). Expression of PIAS3 was controlled by Western blotting with anti-FLAG. E, MCF-7 cells were transfected with the indicated control siRNA (Ambion) or siRNAs targeting PIAS3, stimulated for 15 min with 50 ng/ml NRG-1, and fixed. Complexes of ErbB4 and PIAS3 were visualized with anti-ErbB4 (HFR-1) and anti-PIAS3 (ab22856) antibodies using in situ PLA. Each red dot represents a single interaction. The nuclei were stained with DAPI (blue). F, quantification of PLA signals per cell. Signals were classified as cytosolic or nuclear on the basis of their colocalization with DAPI. Data of two independent experiments are shown (mean ± S.D.). *, p < 0.05. Densitometric data of PIAS3 protein levels relative to control knockdown as detected by Western blotting are indicated (protein (%)). G, COS-7 cells transfected with the indicated HA-tagged ErbB4 ICD2 and either His- or GFP-tagged SUMO-1 constructs were lysed in the presence of 20 mm N-ethylmaleimide. Lysates were analyzed by immunoprecipitation and Western blotting with anti-HA. H, COS-7 cells were transfected with HA-tagged ErbB4 ICD2 and the indicated FLAG-tagged PIAS3 and His-tagged SUMO-1 constructs. Lysates prepared as in G were immunoprecipitated with anti-ErbB4 (HFR-1) followed by Western blotting with anti-HA. Expression of PIAS3 was controlled by Western blotting with anti-FLAG. The band marked with an asterisk presumably represents another SUMO modification site.
Most interaction partners of PIAS proteins are modified by SUMO, and PIAS proteins can function as SUMO E3 ligases (18). This prompted us to examine whether ErbB4 could be SUMOylated. COS-7 cells expressing ErbB4 ICD2 with or without His- or GFP-tagged SUMO-1 were lysed in the presence of N-ethylmaleimide to inhibit the activity of SUMO isopeptidases. In addition to the unmodified ErbB4 ICD2 (∼80 kDa), Western blot analyses revealed higher molecular weight species of ErbB4, corresponding to the size of ErbB4 ICD2 modified with His- or GFP-tagged SUMO-1 (∼90 kDa and 120 kDa, respectively) (Fig. 1G). Similar results showing ErbB4 ICD SUMOylation were obtained when ErbB4 ICD2 and SUMO-1 or SUMO-3 were overexpressed in HEK 293T, WM-266–4, or MCF-7 cells (supplemental Fig. S2). In accordance with the requirement of intact SP-RING for the SUMO E3 ligase function of PIAS proteins, overexpression of PIAS3, but not a SP-RING-disrupted mutant of PIAS3 (C299S), further increased SUMOylation of the ErbB4 ICD (Fig. 1H). Overexpression of PIAS3 could also promote SUMOylation of ErbB4 ICD in MCF-7 cells (supplemental Fig. S2D), whereas PIAS3 knockdown with siRNAs inhibited ICD SUMOylation (supplemental Fig. S2E). These data show that ErbB4 interacts with PIAS3 and that PIAS3 can stimulate the SUMOylation of ErbB4.
PIAS3 Promotes Accumulation of the Nuclear ErbB4 ICD
To visualize ErbB4 in cells expressing PIAS proteins, COS-7 cells were transfected with constructs encoding PIAS1, PIASxα, PIAS3, or PIASy together with full-length, HA-tagged ErbB4 JM-a CYT-2. Notably, coexpression of PIAS3 and PIASy, but not PIAS1 or PIASxα, resulted in a primarily nuclear staining pattern of ErbB4 (Fig. 2A). PIAS protein expression levels were controlled by Western blotting (Fig. 2B). In accordance with the increased nuclear staining of ErbB4 upon PIAS3 overexpression, Western blot analysis demonstrated an increased protein level of ErbB4 ICD2 in the nuclear fraction when PIAS3 was coexpressed (Fig. 2C). Inhibition of the RIP of the full-length receptor at the cell membrane by the γ-secretase inhibitor IX (GSI IX) efficiently abolished nuclear ErbB4 staining (87% of untreated cells and 40% of GSI IX-treated cells were scored positive for nuclear staining, n = 100, Fig. 2D), indicating that overexpression of PIAS3 resulted in accumulation of the ICD and not the full-length ErbB4 receptor into the nucleus. Consistently, the nuclear accumulation of a mutant ErbB4 JM-a CYT-2 not susceptible to RIP (V675A) (34) was reduced significantly compared with wild-type ErbB4 (Fig. 2E and supplemental Fig. S3).
FIGURE 2.

Regulation of ErbB4 nuclear localization by PIAS3. A, COS-7 cells expressing HA-tagged ErbB4 JM-a CYT-2 with or without FLAG-tagged PIAS proteins were stained with anti-HA and photographed under a fluorescence microscope with a ×40 objective. B, expression of FLAG-tagged PIAS proteins in COS-7 cells was controlled by Western blotting with anti-FLAG. C, COS-7 cells expressing HA-tagged ErbB4 ICD2 and FLAG-tagged PIAS3 were subjected to subcellular fractionation. Nuclear and cytosolic fractions were analyzed by Western blotting (W) with indicated antibodies. Lamin B, nuclear marker; Mek1/2, cytosolic marker. D, COS-7 cells expressing HA-tagged ErbB4 JM-a CYT-2 and FLAG-tagged PIAS3 were treated for 4 h with 0 or 5 μm GSI IX and stained with anti-HA (red) and anti-PIAS3 (sc-46682, green). The nuclei were stained with DAPI (blue). The cells were visualized by confocal microscopy with a ×63 objective. E, COS-7 cells were transfected with constructs encoding HA-tagged ErbB4 JM-a CYT-2, ErbB4 JM-a CYT-2-V675A, or ErbB4 JM-a CYT-2-NLSI/II mutant and FLAG-tagged PIAS3 and stained with anti-HA. Cells were visualized by fluorescence microscopy and scored for cytosolic or nuclear staining. Columns (mean ± S.D.) show representative data from one of three independent experiments (*, p < 0.05). F, MCF-7 cells transfected with non-targeting (Qiagen) or PIAS3 siRNA were treated for 3 h with 25 ng/ml leptomycin B and for 45 min with 50 ng/ml NRG-1 and stained with anti-ErbB4 (HFR-1, red). The nuclei were stained with DAPI (blue). The cells were visualized by confocal microscopy with a ×40 objective. G, MCF-7 cells transfected with the indicated control siRNA (Ambion) or siRNAs targeting PIAS3 were subjected to subcellular fractionation. Nuclear and cytosolic fractions were analyzed by Western blotting with anti-ErbB4 (E200), anti-PIAS3 (sc-46682), anti-lamin B, and anti-Mek1/2. Loading was controlled by reblotting with anti-actin.
ErbB4 has been shown to contain a tripartite NLS at the juxtamembrane intracellular region (7, 26). To further characterize the role of PIAS3 in increased nuclear localization of the ErbB4 ICD, an NLSI/II mutant of full-length ErbB4 JM-a CYT-2, with a disrupted first (I) and second (II) part of the tripartite NLS, was generated. Compared with cells coexpressing wild-type ErbB4 and PIAS3, significantly less nuclear ErbB4 staining was detected in cells coexpressing the ErbB4 NLSI/II mutant together with PIAS3 (Fig. 2E and supplemental Fig. S3), implying that the functional NLS is critical for PIAS3 induced nuclear accumulation of the ErbB4 ICD.
The requirement of PIAS3 on the localization of endogenous ErbB4 was also assessed. ErbB4 staining was detected in the nuclei of MCF-7 breast cancer cells when cells were stimulated with an ErbB4 ligand NRG-1, and nuclear export was inhibited by leptomycin B treatment (supplemental Fig. S4). Consistent with our data on the nuclear accumulation of ErbB4 upon PIAS3 overexpression, knockdown of PIAS3 with siRNA resulted in reduced nuclear staining of ErbB4 (Fig. 2F). In addition, a Western analysis of nuclear fraction of MCF-7 cells demonstrated a decrease in ErbB4 ICD protein level upon PIAS3 knockdown with siRNAs (Fig. 2G). Taken together, these data demonstrate that PIAS3 sequesters the soluble ErbB4 ICD into the nucleus in a manner that depends on γ-secretase cleavage and the functional nuclear localization signal of ErbB4.
ErbB4 Colocalizes with PIAS3, SUMO-1, and PML in Nuclear Bodies
Several transcriptional regulators, as well as SUMOylated proteins localize in promyelocytic leukemia (PML) nuclear bodies (35). To test whether this would be the case for ErbB4, full-length HA-tagged ErbB4 JM-a CYT-2 was coexpressed together with FLAG-PIAS3, PML-3, and GFP-tagged SUMO-1 in COS-7 cells, and the proteins were visualized by immunofluorescence and confocal microscopy. In the presence of PIAS3, ErbB4 colocalized with SUMO-1 and PIAS3 (Fig. 3A) in PML-immunoreactive nuclear bodies (B). However, SUMO E3 ligase activity of PIAS3 was not necessary for efficient recruitment of ErbB4 into the PML bodies because overexpression of the C299S mutant of PIAS3 could also induce colocalization of ErbB4 with SUMO-1 (Fig. 3A) and PML (B). In addition to COS-7 cells, stable transfectants of HC11 mouse mammary epithelial cells expressing cleavable ErbB4 JM-a CYT-2 were analyzed for colocalization of ErbB4 with endogenous PIAS3 and PML. In a quantitative colocalization analysis, ∼11% of the PIAS3 signal and 19% of the PML signal were also positive for ErbB4 (supplemental Fig. S5).
FIGURE 3.

Colocalization of ErbB4 with SUMO-1, PIAS3, and PML. A, COS-7 cells expressing HA-tagged ErbB4 JM-a CYT-2, GFP-SUMO-1, and PML-3 in the presence or absence of PIAS3 or PIAS3C299S were stained with anti-HA (red) and anti-PIAS3 (sc-46682, blue). B, COS-7 cells expressing HA-tagged ErbB4 JM-a CYT-2, GFP-SUMO-1, and PML-3 in the presence or absence of PIAS3 or PIAS3C299S were stained with anti-HA (red) and anti-PML (sc-966, blue) antibodies. The cells were visualized by confocal microscopy with a ×63 objective.
Finally, the subnuclear distribution of endogenous ErbB4 was analyzed in WM-266-4 metastatic melanoma cells naturally expressing the cleavable JM-a CYT-2 isoform of ErbB4 (data not shown). When WM-266–4 cells were treated with NRG-1 and leptomycin B, ErbB4 colocalized together with SUMO-1 (Fig. 4A) and PML (Fig. 4B) in nuclear bodies. These data demonstrate that ErbB4 localizes in PML bodies with PIAS3 and SUMO-1.
FIGURE 4.

Colocalization of endogenous ErbB4 with SUMO-1 and PML. WM-266-4 cells were treated for 3 h with 0 or 25 ng/ml leptomycin B and for 45 min with 0 or 50 ng/ml NRG-1. Cells were stained with anti-ErbB4 (HFR-1, red) and anti-SUMO-1 (green) (A) or anti-ErbB4 (HFR-1, red) and anti-PML (sc-5621, green) (B). The nuclei were stained with DAPI (blue). The cells were visualized by confocal microscopy with a ×63 objective.
PIAS3 Represses Nuclear Functions of the ErbB4 ICD
Because PIAS proteins often regulate the transcriptional activity of their interaction partners (18), the effect of PIAS3 overexpression on transcriptional coregulatory activity of ErbB4 was examined. One system described previously to measure the transcriptional coregulatory activity of ErbB4 is a transactivation assay where ErbB4 ICD fused to the GAL4 DNA-binding domain (ErbB4 ICD2-GAL4) and YAP coactivate a luciferase gene driven by GAL4 binding sites (6). Consistently, we observed a robust increase in luciferase activity when ErbB4 ICD2-GAL4 was expressed together with YAP2 in HEK 293 cells (Fig. 5). Coexpression of PIAS3 in this system decreased the luciferase activity by ∼70%, whereas PIASxα or PIASy could not repress ICD-YAP2-mediated coactivation. Interestingly, disrupting the PIAS3 SP-RING domain with the C299S point mutation partially removed the repressive effect of PIAS3 (Fig. 5). These data suggest specificity for PIAS3 in this system and a role for its SUMO E3 ligase activity.
FIGURE 5.
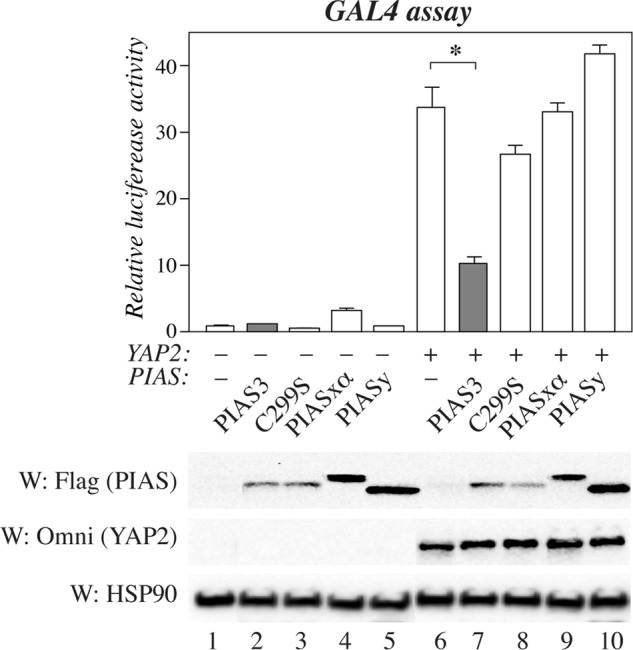
PIAS proteins in ErbB4-regulated transcription. HEK 293 cells were transfected with a construct encoding ErbB4 ICD2 fused to the GAL4 DNA-binding domain and pFR-luc together with constructs encoding Omni-YAP2 and FLAG-tagged PIAS proteins as indicated. Columns represent relative luciferase activity normalized to signal from the cotransfected Renilla luciferase construct (mean ± S.D.). *, p < 0.05. Expression of PIAS proteins and YAP2 were controlled with anti-FLAG and anti-Omni, respectively. Loading was controlled by reblotting with anti-HSP90.W, Western blot.
We and others have previously utilized three-dimensional cultures of MDA-MB-468 human breast cancer (31) and HC11 mouse mammary epithelial cells (13, 34) as models to address the role of ErbB4 isoforms in mammary differentiation. In a reconstituted basement membrane matrix (Matrigel), mammary epithelial cells are able to differentiate morphologically and functionally, forming small, organized colonies that resemble mammary acini, whereas transformed cells grow in large, disorganized colonies (36). To characterize the function of soluble ErbB4 ICD2 in three-dimensional growth of MDA-MB-468 human breast cancer cells, stable transfectants expressing ErbB4 JM-a CYT-2 or an empty vector (31) were embedded in Matrigel and treated with or without GSI IX to inhibit ErbB4 cleavage (Fig. 6A). Roughly half of cells expressing empty vector but only ∼25% of cells expressing ErbB4 grew in small spheres resembling mammary acini (Fig. 6B). When ErbB4 cleavage was inhibited by GSI IX, cells expressing ErbB4 showed a shift toward a more differentiated phenotype (Fig. 6B), indicating that the soluble ErbB4 ICD2 had an inhibitory effect on differentiation, consistent with previous findings (13). To address the effect of PIAS3 and PML on ErbB4 ICD function in this system, MDA-MB-468 cells expressing ErbB4 JM-a CYT-2 and the vector control cells were transfected with either non-targeting siRNA or two independent siRNAs targeting PIAS3 or PML. Knockdown of PIAS3 (Fig. 6C) or PML (D) had a negative effect on the basal differentiation of the vector control cells. However, knockdown of PIAS3 or PML in cells expressing ErbB4 increased the formation of differentiated acini (Fig. 6, C and D).
FIGURE 6.

PIAS3 in ErbB4-regulated differentiation. A, MDA-MB-468 cells expressing ErbB4 JM-a CYT-2 and the vector control cells were cultured for 6 h in the presence of 0, 1, or 5 μm GSI IX. Cell lysates were analyzed by Western blotting with anti-ErbB4 (sc-283). n.s., non-specific. B, MDA-MB-468 cells expressing ErbB4 JM-a CYT-2 and the vector control cells were suspended into Matrigel and cultured in the presence of 50 ng/ml NRG-1 and in the presence or absence of 5 μm GSI IX. On day 6 the colonies were counted. The columns represent the percentage of acini of all colonies in two independent experiments (mean ± S.D.). *, p < 0.05. C, MDA-MB-468 cells expressing ErbB4 JM-a CYT-2 and the vector control cells were treated with non-targeting (Qiagen) or the indicated PIAS3-specific siRNAs for 24 h, suspended into Matrigel, and cultured for 6 days. The columns represent the percentage of acini of all colonies in three independent experiments (mean ± S.D.). *, p < 0.05. Densitometric data of PIAS3 protein levels relative to control knockdown as detected by Western blotting are indicated (protein (%)). D, MDA-MB-468 cells expressing ErbB4 JM-a CYT-2 and the vector control cells were treated with non-targeting (Qiagen) or the indicated PML-specific siRNAs for 24 h, suspended into Matrigel, and cultured for 6 days. The columns represent the percentage of acini of all colonies in four independent experiments (mean ± S.D.). *, p < 0.05. Densitometric data of PML protein levels relative to control knockdown as detected by Western blotting are indicated (protein (%)). E, HC11 cells expressing ErbB4 JM-a CYT-2 or JM-b CYT-2 and the vector control cells were treated for 15 min with 50 ng/ml NRG-1 and analyzed by Western blotting with anti-ErbB4 (E200). The membrane was reprobed with anti-actin to control loading. F, HC11 cells expressing the indicated constructs were suspended into growth factor-reduced Matrigel and cultured for 14 days in the presence of 50 ng/ml NRG-1. The columns represent the percentage of acini of all colonies in three independent experiments (mean ± S.D.). *, p < 0.05. G, HC11 cells expressing ErbB4 JM-a CYT-2 and the vector control cells were treated with non-targeting (Ambion) or the indicated PIAS3-specific siRNAs for 24 h, suspended into Matrigel, and cultured for 15 days. The columns represent the percentage of acini of all colonies in two independent experiments (mean ± S.D.). *, p < 0.05. Densitometric data of PIAS3 protein levels relative to control knockdown as detected by Western blotting are indicated (protein (%)). H, representative confocal images (visualized with a ×63 objective) of HC11 cells expressing ErbB4 JM-a CYT-2 treated with non-targeting (Ambion) or PIAS3-specific siRNAs as in G. Cells were stained with anti-ErbB4 (HFR-1, green) and anti-PIAS3 (ab22856, red). The nuclei were stained with DAPI (blue). I, Quantification of cytosolic and nuclear ErbB4 immunoreactivity (mean ± S.D.). *, p < 0.05. Densitometric data of PIAS3 protein levels relative to control knockdown as detected by Western blotting are indicated (protein (%)).
To further characterize the functional significance of ErbB4 regulation by PIAS3, stable transfectants of HC11 mouse mammary epithelial cells expressing cleavable ErbB4 JM-a CYT-2, non-cleavable JM-b CYT-2, or an empty vector were generated (Fig. 6E) and analyzed for their three-dimensional growth in Matrigel. Similar to our observations with MDA-MB-468 cells, HC11 cells expressing ErbB4 JM-a CYT-2 demonstrated less differentiation than the vector control cells (Fig. 6F). In contrast, cells expressing non-cleavable ErbB4 JM-b CYT-2 did not differ from control cells in their ability to form differentiated acini (Fig. 6F). Next, HC11 vector control cells and cells expressing ErbB4 JM-a CYT-2 were transfected with non-targeting siRNA or two independent siRNA oligonucleotides targeting PIAS3. Again, knockdown of PIAS3 could partially rescue the reduced differentiation of the cells expressing cleavable ErbB4 JM-a CYT-2 (Fig. 6G). Together with the role of PIAS3 as a regulator of nuclear localization of ErbB4 ICD (Fig. 2), these data prompted us to hypothesize that the outcome of ErbB4 JM-a CYT-2 signaling in MDA-MB-468 and HC11 cells is associated with the subcellular distribution of the soluble ICD2. Indeed, confocal immunofluorescence analyses of HC11 transfectants revealed reduced nuclear ErbB4 staining upon PIAS3 knockdown (Fig. 6, H and I). Taken together, these data indicate that PIAS3 and PML have repressive effects on nuclear signaling functions of ErbB4 ICD.
DISCUSSION
ErbB4 is a receptor tyrosine kinase that has been demonstrated to undergo RIP upon ligand binding (5). Functionally, ErbB4 ICD plays critical roles in regulating astrogenesis in the developing brain (10) and epithelial differentiation in the mammary gland (13). ErbB4 ICD localizes in the nucleus but also in the cytosol (15) and exhibits differential functional outcomes depending on its localization (14, 16, 37). Although previous studies have reported nuclear localization and putative nuclear export signals within the ErbB4 sequence (5, 7, 15, 26), the factors determining subcellular distribution of ErbB4 ICD have remained largely unknown. Here, we characterized PIAS3 as a novel interaction partner and regulator of nuclear localization of ErbB4 ICD. PIAS3 also stimulated posttranslational SUMO modification of ErbB4. Interaction of ErbB4 ICD by PIAS3 had functional consequences, demonstrated by inhibition of ErbB4 ICD nuclear signaling functions.
The nuclear ErbB4 ICD interacts with a number of transcriptional regulators and modulates their activity (6–12). PIAS proteins and SUMOylation both regulate transcriptional processes (18–19), and ErbB4 ICD was found to both interact with and become SUMOylated by PIAS3. PIAS3, but not a SP-RING mutant incapable of SUMOylating ErbB4, repressed the transcriptional coregulatory activity of ErbB4 ICD in the GAL4 assay where ErbB4 and YAP2 coactivate transcription. In contrast, both the wild type and the SP-RING mutant of PIAS3 were equally efficient in promoting the localization of ErbB4 in PML nuclear bodies, nuclear subdomains that are considered to have a central role in transcriptional regulation (35). It is interesting to note that N-CoR and YAP2, transcriptional regulators that associate with ErbB4, have also been reported to localize in PML bodies (38, 39). PML bodies could thus regulate nuclear signaling of the ErbB4 ICD by providing a site for formation of transcriptional regulatory complexes, including ErbB4. Although the functional SP-RING domain of PIAS3 was not necessary in promoting colocalization of ErbB4 with PML, the fraction of ErbB4 in PML bodies may still be SUMOylated, as many other proteins are (35). Interestingly, our colocalization experiments indicated that PIAS3 also partially localizes in PML bodies. Future experiments will elucidate the functional significance of PIAS3 promoted SUMOylation versus nuclear sequestration for ErbB4 signaling.
ErbB4 has an important role in the mammary gland, regulating lobuloalveolar differentiation during lactation (40–42). Studies that employ ErbB4 constructs with a mutated γ-secretase cleavage site, mutated nuclear localization signal, or ectopic expression of ErbB4 ICD suggest a role for the soluble ErbB4 ICD in promoting mammary epithelial cell differentiation (7, 34, 43). It is notable that these studies specifically describe the effects of the CYT-1 isoform of ErbB4. Interestingly, in a recent study that compared the effects of ectopic expression of the ErbB4 ICD of the CYT-1 and CYT-2 types, CYT-1 promoted but CYT-2 suppressed mammary gland differentiation both in vitro and in vivo (13). Here, we studied the differentiation of MDA-MB-468 breast cancer and HC11 mammary epithelial cells expressing either an empty vector or ErbB4 JM-a CYT-2 in three-dimensional Matrigel cultures. Expression of the cleavable JM-a CYT-2 isoform had inhibitory effects on differentiation. However, when the formation of soluble ErbB4 ICD was inhibited either chemically with a γ-secretase inhibitor or genetically by expressing the non-cleavable JM-b isoform, the number of differentiated acinus-like structures increased. siRNA-mediated knockdown of PIAS3 or PML resulted in increased differentiation of cells expressing ErbB4, likely reflecting altered subcellular localization of the ErbB4 ICD. Although these data suggest a role for PML bodies in regulating ErbB4 function, it is unclear whether the sequestration of ErbB4 into PML bodies is in fact necessary for PIAS3 to affect nuclear signaling of ErbB4. Together, these findings suggest that the outcome of ErbB4 signaling in MDA-MB-468 and HC11 cells is dependent on the subcellular distribution of soluble ErbB4 ICD2.
Nuclear localization of the ErbB4 epitope has been detected by immunohistochemical analyses in breast cancer in several studies (15–16, 44). Compared with malignant cells, nuclear ErbB4 is detected in only a small proportion of cells representing normal morphology (15). Interestingly, the prognostic significance of nuclear ErbB4 immunoreactivity in breast cancer cells also seems to be different from the prognostic significance of ErbB4 at the cell surface or in the cytosol (16, 17). The role of PIAS3 in cancer has not been extensively studied, but increased PIAS3 expression in breast cancer has been reported (45, 46). Therefore, it is interesting to speculate whether the increased expression of PIAS3 in breast cancer could provide a mechanism for nuclear accumulation of ErbB4 ICD with functional consequences on ErbB4 signaling.
In conclusion, we have demonstrated that PIAS3 is a novel interaction partner of ErbB4 that regulates the SUMOylation, nuclear localization, and functions of the ErbB4 ICD. Our findings highlight the importance of ErbB4 proteolytic cleavage and subcellular distribution of the ErbB4 ICD in determining the outcome of ErbB4 signaling.
Supplementary Material
Acknowledgments
We thank Minna Santanen, Mika Savisalo, and Maria Tuominen for excellent technical assistance; Dr. Ville Veikkolainen for help with designing the ErbB4 NLSI/II mutant; Ilkka Paatero and Johannes Merilahti for help with the colocalization analysis; and Dr. Erik Meulmeester for valuable discussions.
This study was supported by the Academy of Finland, the EU-FP7 Marie Curie Grant, Finnish Cancer Organizations, the Finnish Cultural Foundation, the Jusélius Foundation, the Turku Doctoral Program of Biomedical Sciences, Turku University Central Hospital, and the Turku University Foundation.

This article contains supplemental Figs. S1–S5.
- JM
- juxtamembrane
- RIP
- regulated intramembrane proteolysis
- ICD
- intracellular domain
- YAP
- Yes-associated protein
- PIAS
- protein inhibitor of activated STAT
- SUMO
- small ubiquitin-like modifier
- SP-RING
- Siz/PIAS-RING
- NLS
- nuclear localization signal
- PLA
- proximity ligation assay
- Wwox
- WW domain-containing oxidoreductase
- PML
- promyelocytic leukemia.
REFERENCES
- 1. Hynes N. E., Lane H. A. (2005) ERBB receptors and cancer. The complexity of targeted inhibitors. Nat. Rev. Cancer 5, 341–354 [DOI] [PubMed] [Google Scholar]
- 2. Elenius K., Corfas G., Paul S., Choi C. J., Rio C., Plowman G. D., Klagsbrun M. (1997) A novel juxtamembrane domain isoform of HER4/ErbB4. Isoform-specific tissue distribution and differential processing in response to phorbol ester. J. Biol. Chem. 272, 26761–26768 [DOI] [PubMed] [Google Scholar]
- 3. Elenius K., Choi C. J., Paul S., Santiestevan E., Nishi E., Klagsbrun M. (1999) Characterization of a naturally occurring ErbB4 isoform that does not bind or activate phosphatidyl inositol 3-kinase. Oncogene 18, 2607–2615 [DOI] [PubMed] [Google Scholar]
- 4. Rio C., Buxbaum J. D., Peschon J. J., Corfas G. (2000) Tumor necrosis factor α-converting enzyme is required for cleavage of erbB4/HER4. J. Biol. Chem. 275, 10379–10387 [DOI] [PubMed] [Google Scholar]
- 5. Ni C. Y., Murphy M. P., Golde T. E., Carpenter G. (2001) γ-Secretase cleavage and nuclear localization of ErbB-4 receptor tyrosine kinase. Science 294, 2179–2181 [DOI] [PubMed] [Google Scholar]
- 6. Komuro A., Nagai M., Navin N. E., Sudol M. (2003) WW domain-containing protein YAP associates with ErbB-4 and acts as a cotranscriptional activator for the carboxyl-terminal fragment of ErbB-4 that translocates to the nucleus. J. Biol. Chem. 278, 33334–33341 [DOI] [PubMed] [Google Scholar]
- 7. Williams C. C., Allison J. G., Vidal G. A., Burow M. E., Beckman B. S., Marrero L., Jones F. E. (2004) The ERBB4/HER4 receptor tyrosine kinase regulates gene expression by functioning as a STAT5A nuclear chaperone. J. Cell Biol. 167, 469–478 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Zhu Y., Sullivan L. L., Nair S. S., Williams C. C., Pandey A. K., Marrero L., Vadlamudi R. K., Jones F. E. (2006) Coregulation of estrogen receptor by ERBB4/HER4 establishes a growth-promoting autocrine signal in breast tumor cells. Cancer Res. 66, 7991–7998 [DOI] [PubMed] [Google Scholar]
- 9. Linggi B., Carpenter G. (2006) ErbB-4 s80 intracellular domain abrogates ETO2-dependent transcriptional repression. J. Biol. Chem. 281, 25373–25380 [DOI] [PubMed] [Google Scholar]
- 10. Sardi S. P., Murtie J., Koirala S., Patten B. A., Corfas G. (2006) Presenilin-dependent ErbB4 nuclear signaling regulates the timing of astrogenesis in the developing brain. Cell 127, 185–197 [DOI] [PubMed] [Google Scholar]
- 11. Sundvall M., Veikkolainen V., Kurppa K., Salah Z., Tvorogov D., van Zoelen E. J., Aqeilan R., Elenius K. (2010) Cell death or survival promoted by alternative isoforms of ErbB4. Mol. Biol. Cell 21, 4275–4286 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Paatero I., Jokilammi A., Heikkinen P. T., Iljin K., Kallioniemi O. P., Jones F. E., Jaakkola P. M., Elenius K. (2012) Interaction with ErbB4 promotes hypoxia-inducible factor-1α signaling. J. Biol. Chem. 287, 9659–9671 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Muraoka-Cook R. S., Sandahl M. A., Strunk K. E., Miraglia L. C., Husted C., Hunter D. M., Elenius K., Chodosh L. A., Earp H. S. (2009) ErbB4 splice variants Cyt1 and Cyt2 differ by 16 amino acids and exert opposing effects on the mammary epithelium in vivo. Mol. Cell. Biol. 29, 4935–4948 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Naresh A., Long W., Vidal G. A., Wimley W. C., Marrero L., Sartor C. I., Tovey S., Cooke T. G., Bartlett J. M., Jones F. E. (2006) The ERBB4/HER4 intracellular domain 4ICD is a BH3-only protein promoting apoptosis of breast cancer cells. Cancer Res. 66, 6412–6420 [DOI] [PubMed] [Google Scholar]
- 15. Srinivasan R., Gillett C. E., Barnes D. M., Gullick W. J. (2000) Nuclear expression of the c-erbB-4/HER-4 growth factor receptor in invasive breast cancers. Cancer Res. 60, 1483–1487 [PubMed] [Google Scholar]
- 16. Junttila T. T., Sundvall M., Lundin M., Lundin J., Tanner M., Härkönen P., Joensuu H., Isola J., Elenius K. (2005) Cleavable ErbB4 isoform in estrogen receptor-regulated growth of breast cancer cells. Cancer Res. 65, 1384–1393 [DOI] [PubMed] [Google Scholar]
- 17. Thor A. D., Edgerton S. M., Jones F. E. (2009) Subcellular localization of the HER4 intracellular domain, 4ICD, identifies distinct prognostic outcomes for breast cancer patients. Am. J. Pathol. 175, 1802–1809 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Rytinki M. M., Kaikkonen S., Pehkonen P., Jääskeläinen T., Palvimo J. J. (2009) PIAS proteins. Pleiotropic interactors associated with SUMO. Cell. Mol. Life Sci. 66, 3029–3041 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Geiss-Friedlander R., Melchior F. (2007) Concepts in SUMOylation. A decade on. Nat. Rev. Mol. Cell Biol. 8, 947–956 [DOI] [PubMed] [Google Scholar]
- 20. Sundvall M., Peri L., Määttä J. A., Tvorogov D., Paatero I., Savisalo M., Silvennoinen O., Yarden Y., Elenius K. (2007) Differential nuclear localization and kinase activity of alternative ErbB4 intracellular domains. Oncogene 26, 6905–6914 [DOI] [PubMed] [Google Scholar]
- 21. Rytinki M. M., Palvimo J. J. (2009) SUMOylation attenuates the function of PGC-1alpha. J. Biol. Chem. 284, 26184–26193 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Pircher T. J., Flores-Morales A., Mui A. L., Saltiel A. R., Norstedt G., Gustafsson J. A., Haldosén L. A. (1997) Mitogen-activated protein kinase kinase inhibition decreases growth hormone-stimulated transcription mediated by STAT5. Mol. Cell. Endocrinol. 133, 169–176 [DOI] [PubMed] [Google Scholar]
- 23. Hietakangas V., Ahlskog J. K., Jakobsson A. M., Hellesuo M., Sahlberg N. M., Holmberg C. I., Mikhailov A., Palvimo J. J., Pirkkala L., Sistonen L. (2003) Phosphorylation of serine 303 is a prerequisite for the stress-inducible SUMO modification of heat shock factor 1. Mol. Cell. Biol. 23, 2953–2968 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Aqeilan R. I., Palamarchuk A., Weigel R. J., Herrero J. J., Pekarsky Y., Croce C. M. (2004) Physical and functional interactions between the Wwox tumor suppressor protein and the AP-2γ transcription factor. Cancer Res. 64, 8256–8261 [DOI] [PubMed] [Google Scholar]
- 25. Salah Z., Melino G., Aqeilan R. I. (2011) Negative regulation of the Hippo pathway by E3 ubiquitin ligase ITCH is sufficient to promote tumorigenicity. Cancer Res. 71, 2010–2020 [DOI] [PubMed] [Google Scholar]
- 26. Hsu S. C., Hung M. C. (2007) Characterization of a novel tripartite nuclear localization sequence in the EGFR family. J. Biol. Chem. 282, 10432–10440 [DOI] [PubMed] [Google Scholar]
- 27. Määttä J. A., Sundvall M., Junttila T. T., Peri L., Laine V. J., Isola J., Egeblad M., Elenius K. (2006) Proteolytic cleavage and phosphorylation of a tumor-associated ErbB4 isoform promote ligand-independent survival and cancer cell growth. Mol. Biol. Cell 17, 67–79 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Sundvall M., Korhonen A., Paatero I., Gaudio E., Melino G., Croce C. M., Aqeilan R. I., Elenius K. (2008) Isoform-specific monoubiquitination, endocytosis, and degradation of alternatively spliced ErbB4 isoforms. Proc. Natl. Acad. Sci. U.S.A. 105, 4162–4167 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Kainulainen V., Sundvall M., Määttä J. A., Santiestevan E., Klagsbrun M., Elenius K. (2000) A natural ErbB4 isoform that does not activate phosphoinositide 3-kinase mediates proliferation but not survival or chemotaxis. J. Biol. Chem. 275, 8641–8649 [DOI] [PubMed] [Google Scholar]
- 30. Aqeilan R. I., Donati V., Palamarchuk A., Trapasso F., Kaou M., Pekarsky Y., Sudol M., Croce C. M. (2005) WW domain-containing proteins, WWOX and YAP, compete for interaction with ErbB-4 and modulate its transcriptional function. Cancer Res. 65, 6764–6772 [DOI] [PubMed] [Google Scholar]
- 31. Tvorogov D., Sundvall M., Kurppa K., Hollmén M., Repo S., Johnson M. S., Elenius K. (2009) Somatic mutations of ErbB4. Selective loss-of-function phenotype affecting signal transduction pathways in cancer. J. Biol. Chem. 284, 5582–5591 [DOI] [PubMed] [Google Scholar]
- 32. Schulze W. X., Deng L., Mann M. (2005) Phosphotyrosine interactome of the ErbB-receptor kinase family. Mol. Syst. Biol. 1, E1-E13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Söderberg O., Leuchowius K. J., Gullberg M., Jarvius M., Weibrecht I., Larsson L. G., Landegren U. (2008) Characterizing proteins and their interactions in cells and tissues using the in situ proximity ligation assay. Methods 45, 227–232 [DOI] [PubMed] [Google Scholar]
- 34. Muraoka-Cook R. S., Sandahl M., Husted C., Hunter D., Miraglia L., Feng S. M., Elenius K., Earp H. S., 3rd (2006) The intracellular domain of ErbB4 induces differentiation of mammary epithelial cells. Mol. Biol. Cell 17, 4118–4129 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Bernardi R., Pandolfi P. P. (2007) Structure, dynamics and functions of promyelocytic leukaemia nuclear bodies. Nat. Rev. Mol. Cell Biol. 8, 1006–1016 [DOI] [PubMed] [Google Scholar]
- 36. Petersen O. W., Rønnov-Jessen L., Howlett A. R., Bissell M. J. (1992) Interaction with basement membrane serves to rapidly distinguish growth and differentiation pattern of normal and malignant human breast epithelial cells. Proc. Natl. Acad. Sci. U.S.A. 89, 9064–9068 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Naresh A., Thor A. D., Edgerton S. M., Torkko K. C., Kumar R., Jones F. E. (2008) The HER4/4ICD estrogen receptor coactivator and BH3-only protein is an effector of tamoxifen-induced apoptosis. Cancer Res. 68, 6387–6395 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Khan M. M., Nomura T., Kim H., Kaul S. C., Wadhwa R., Shinagawa T., Ichikawa-Iwata E., Zhong S., Pandolfi P. P., Ishii S. (2001) Role of PML and PML-RARα in Mad-mediated transcriptional repression. Mol. Cell 7, 1233–1243 [DOI] [PubMed] [Google Scholar]
- 39. Lapi E., Di Agostino S., Donzelli S., Gal H., Domany E., Rechavi G., Pandolfi P. P., Givol D., Strano S., Lu X., Blandino G. (2008) PML, YAP, and p73 are components of a proapoptotic autoregulatory feedback loop. Mol. Cell 32, 803–814 [DOI] [PubMed] [Google Scholar]
- 40. Jones F. E., Welte T., Fu X. Y., Stern D. F. (1999) ErbB4 signaling in the mammary gland is required for lobuloalveolar development and Stat5 activation during lactation. J. Cell Biol. 147, 77–88 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Long W., Wagner K. U., Lloyd K. C., Binart N., Shillingford J. M., Hennighausen L., Jones F. E. (2003) Impaired differentiation and lactational failure of Erbb4-deficient mammary glands identify ERBB4 as an obligate mediator of STAT5. Development 130, 5257–5268 [DOI] [PubMed] [Google Scholar]
- 42. Tidcombe H., Jackson-Fisher A., Mathers K., Stern D. F., Gassmann M., Golding J. P. (2003) Neural and mammary gland defects in ErbB4 knockout mice genetically rescued from embryonic lethality. Proc. Natl. Acad. Sci. U.S.A. 100, 8281–8286 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Vidal G. A., Naresh A., Marrero L., Jones F. E. (2005) Presenilin-dependent γ-secretase processing regulates multiple ERBB4/HER4 activities. J. Biol. Chem. 280, 19777–19783 [DOI] [PubMed] [Google Scholar]
- 44. Kew T. Y., Bell J. A., Pinder S. E., Denley H., Srinivasan R., Gullick W. J., Nicholson R. I., Blamey R. W., Ellis I. O. (2000) c-erbB-4 protein expression in human breast cancer. Br. J. Cancer 82, 1163–1170 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. McHale K., Tomaszewski J. E., Puthiyaveettil R., Livolsi V. A., Clevenger C. V. (2008) Altered expression of prolactin receptor-associated signaling proteins in human breast carcinoma. Mod. Pathol. 21, 565–571 [DOI] [PubMed] [Google Scholar]
- 46. Wang L., Banerjee S. (2004) Differential PIAS3 expression in human malignancy. Oncol. Rep. 11, 1319–1324 [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.