

Background: The plasminogen system is central in cell migration and is thus involved in many patho/physiological processes.
Results: M6P-IGF2R is a regulatory factor in plasminogen-associated complexes and mediates plasminogen internalization.
Conclusion: The uptake of plasminogen by M6P-IGF2R might be an important pathway to control plasminogen activation in cells.
Significance: M6P-IGF2R restricts plasmin activity and its loss might lead to rampant fibrinolysis.
Keywords: Cell Migration, Endocytosis, Fibrinolysis, Lipid Raft, Proteolytic Enzymes, Blue Native Electrophoresis, Gel Filtration, Internalization, Plasminogen Binding Molecule, Zymography
Abstract
The plasminogen (Plg) activation cascade on the cell surface plays a central role in cell migration and is involved in a plethora of physiological and pathological processes. Its regulation is coordinated by many receptors, in particular the urokinase-type plasminogen activator receptor (uPAR, CD87), receptors that physically interact and functionally cooperate with uPAR, and Plg binding molecules. Here we studied the impact of one of the Plg binding molecules, the mannose 6-phosphate/insulin-like growth factor 2 receptor (M6P-IGF2R, CD222), on cellular Plg activation. By developing both in vitro and in vivo Plg activation assays on size-fractionated lysates of M6P-IGF2R-silenced cells, we identified Plg-associated complexes with M6P-IGF2R as the regulatory factor. Using lipid raft preserving versus dissolving detergents, we found lipid dependence of the Plg regulatory function of these complexes. Furthermore, M6P-IGF2R-silencing in uPAR-positive human cell lines reduced internalization of Plg, resulting in elevated Plg activation. In contrast, the expression of human M6P-IGF2R in mouse embryonic fibroblasts derived from M6P-IGF2R knock-out mice enhanced Plg internalization. Finally, peptide 18–36 derived from the Plg-binding site within M6P-IGF2R enhanced Plg uptake. Thus, by targeting Plg to endocytic pathways, M6P-IGF2R appears to control Plg activation within cells that might be important to restrict plasmin activity to specific sites and substrates.
Introduction
The proteolytic conversion of plasminogen (Plg),2 the central enzyme of fibrinolysis, to the active serine protease plasmin is catalyzed by plasminogen activators including urokinase-type plasminogen activator (uPA), tissue-type plasminogen-activator, and also bacterial proteins such as streptokinase (1–4). uPA, produced as the inactive single-chain glycoprotein pro-urokinase (pro-uPA), is a key activator in the cell surface-mediated proteolysis. The uPA-associated proteolysis requires strict regulation that is controlled by the reorganization of cell surface molecules, in particular, the urokinase-type plasminogen activator receptor (uPAR, CD87) and receptors that physically interact and functionally cooperate with uPAR as well as Plg binding molecules (5). uPAR is a glycoprotein anchored in the plasma membrane by a glycosylphosphatidylinositol moiety and in the cell membrane preferentially associates to cholesterol/glycosphingolipid-enriched membrane microdomains, also called lipid rafts (6, 7). It is present on the surface of cells as a full-length three-domain molecule (D1D2D3) capable of binding integrins and pro-uPA, or a truncated variant (D2D3) lacking the pro-uPA/integrin binding domain D1 (8). Upon binding to uPAR, pro-uPA is proteolytically converted to the active double-chain serine protease uPA that in turn specifically activates cell-bound Plg to the serine protease plasmin (9, 10). Plg/plasmin receptors exert regulatory functions by restraining plasmin activity in space and time (11). Typically, via lysine-binding sites located in its kringle domains, Plg interacts with lysines encompassed in cellular receptors (12, 13). We have found that the mannose 6-phosphate/insulin-like growth factor 2 receptor (M6P-IGF2R, CD222), a multifunctional receptor involved in protein sorting, internalization and degradation (14–16), binds both uPAR and Plg, and regulates Plg activation (17–19). We have mapped a binding site for Plg into the amino-terminal domain 1 of M6P-IGF2R (18). The crystal structure of the three amino-terminal domains of the bovine M6P-IGF2R provided insight into the shape of the lysine-involving binding site (20). In addition, we have shown that M6P-IGF2R accelerates the proteolytic cleavage of uPAR, thereby interfering with the Plg activation cascade and cell invasion, and also controls the expression of αVβ3 integrin (21), another membrane partner molecule of uPAR that is involved in the regulation of the uPAR system (22–24).
Here we investigated the impact of M6P-IGF2R on the reorganization of the cell membrane protein complexes associated with Plg conversion. For this purpose, we silenced M6P-IGF2R in human cell lines by RNA interference, and we expressed human M6P-IGF2R, both the wild type form and a variant containing the lysine 25 point mutation within the Plg binding region, in mouse embryonic fibroblasts derived from M6P-IGF2R knock-out mice. Employing these cells for lipid raft flotation analysis, two-dimensional blue native polyacrylamide gel electrophoresis (BN/SDS-PAGE) followed by zymography, gel filtration analysis combined with cellular proteolysis assays, and internalization assays, we show here that M6P-IGF2R contributes to regulation of pericellular proteolysis by Plg internalization.
EXPERIMENTAL PROCEDURES
Chemicals and Reagents
Tricine, Tris, Coomassie Brilliant Blue (R-250 and G-250), ammonium persulfate, TEMED, sodium dodecyl sulfate (SDS), acrylamide, N,N′-methylenebisacrylamide, BSA were purchased from SERVA (Heidelberg, Germany). All protease inhibitors, beriglobin, puromycin, Polybrene, DAPI, tranexamic acid, monensin, and imidazole were from Sigma. Nonidet P-40, Triton X-100, BCA kit, and N-dodecyl-d-maltoside and EZ-Link Sulfo-NHS-biotin were from Pierce. Glycerol and benzonase were from Merck. Immobilon-P transfer membrane was from Millipore (Bedford, MA). 6-Aminocaproic acid was from Fluka (Buchs, Switzerland). Glu-Plg was purchased from Calbiochem and labeled by us (Plg-AF488). Fluorescently labeled transferrin (Tf-AF546) was from Molecular Probes (Invitrogen).
Antibodies
The monoclonal antibody (mAb) MEM-238 to M6P-IGF2R was generated by us (25). However, unlabeled MEM-238 and MEM-238 conjugated to APC were purchased from EXBIO (Prague, Czech Republic). The biotin conjugate was generated by us. The CD59 mAb MEM-43/5, CD63 (LAMP-3) mAb MEM-259, mAb H902, and mAb AFP-01 were kindly provided by Dr. Václav Hořejší (Institute of Molecular Genetics, Academy of Sciences of the Czech Republic, Prague, Czech Republic). The anti-β3 mAb VIPL-2 was kindly provided by Otto Majdic (Institute of Immunology, Centre for Pathophysiology, Infectiology and Immunology, Medical University of Vienna, Vienna, Austria). The mouse anti-integrin αVβ3 mAb (clone LM609) and anti-integrin αV mAb (clone P3G8) were from Millipore. The mAb H2 and C8 to uPAR were kindly provided by Dr. Ulrich Weidle (Roche Diagnostics, Division Pharma). The mAbs against pro-uPA/uPA (scuPA8 and U5) were from Technoclone (Vienna, Austria). Rabbit polyclonal anti-plasmin and anti-Rab11 Ab were from Abcam (Cambridge, UK), and the rabbit mAb C45B10 to EEA1 was from Cell Signaling Technology (Danvers, MA). As secondary reagents, we used horseradish peroxidase (HRP)-conjugated anti-mouse and anti-rabbit IgG (Sigma), streptavidin-HRP conjugate (GE Healthcare), and goat anti-mouse IgG+IgM (H+L)-FITC conjugate (An der Grub, Kaumberg, Austria).
Peptides
The peptide derived from the amino-terminal part of M6P-IGF2R, peptide 18–36 (pep18–36, TKNNVLYKINICGSVDIVQ), and control scrambled peptide (pepSCR, SVNCAIGSNGKVNYIKVNS) were produced either by Genosphere (Paris, France) or by Peptide 2.0 Inc. (Chantilly, VA).
Cells
The human kidney epithelial tumor cell line TCL-598 was a gift from the Novartis Research Institute (Vienna, Austria). The human monocytic cell line THP-1 was from ATCC. The immortalized M6P-IGF2R negative mouse fibroblasts were kindly provided by E. Wagner (Spanish National Cancer Research Centre, Madrid, Spain). The cells were cultured in RPMI 1640 medium (Sigma) supplemented with 100 μg/ml penicillin, 100 μg/ml streptomycin, 2 mmol/liter l-glutamine, and 10% heat-inactivated fetal calf serum (Sanova Diagnostics, Vienna, Austria). The cells were grown in a humidified atmosphere at 37 °C and 5% CO2 and passaged at least twice a week using trypsin-EDTA solution.
Site Mutagenesis; Generation of M6P-IGF2RK25T Mutant Variant
The construct M6P-IGF2R-pTag/BMN-Z was used for the site-directed mutagenesis of Lys-25. This construct contains the complete gene sequence of the full-length M6P-IGF2R fused at the carboxyl terminus to the pTag sequence in the retroviral vector pBMNZ (18). The pTag encompasses an epitope for mAb H902 enabling purification of the recombinant protein (26). The PCR site-directed mutagenesis was done as follows. Lys-25 coded by three adenine nucleotides lies partially in a recognition site of the PsiI restriction enzyme (TTATAA). The forward primer (5′-TGTACTGTATACAATCAACATCTGT-3′) was designed to contain a cytosine instead of an adenine, resulting in a change from the AAA codon for lysine-to-threonine ACA codon. In addition, CTT was changed to CTG so that finally the PsiI recognition site TTATAA was changed to GTATAC, the recognition site for SnaI. The reverse primer was designed to contain an EcoRI recognition site (5′-TGTTGAATTCCAGGAGAGATCTGGT-3′). Because the desired amplification area contained a high portion of guanine and cytosine nucleotides known to hinder the PCR reaction, we used a GC-rich resolution kit (Roche Diagnostics) to provide sufficient amplification. The corresponding PCR product was purified using Wizard SV Gel and PCR Clean-Up System (Promega), cleaved by SnaI and EcoRI, and inserted by ligation into the matching vector generated by a cleavage of M6P-IGF2R-pTag/BMN-Z PsiI and EcoRI. The resulting construct M6P-IGF2RK25T was analyzed by digestion with BglII and PsiI. Plasmids containing mutations were cleaved by BglII only, whereas non-mutated plasmids were cleaved by both. The presence of the desired mutation in these plasmids was verified by DNA sequencing analysis (VBC Genomics Bioscience Research GmbH, Vienna, Austria).
Transfection and Transduction Procedures
All transfection and transduction procedures were performed as described earlier (27). Briefly, the retroviral plasmids were used for transient transfection of the ecotropic packaging cell line Phoenix by the Superfect Transfection method (Qiagen, Hilden, Germany). Three days after transfection, the supernatants containing viral particles were mixed with Polybrene (4 μg/ml), and the mixtures were used to transduce target cells. M6P-IGF2R-negative mouse fibroblasts expressing human uPAR (18) were transduced with the pTag-tagged full-length normal and mutated forms of human M6P-IGF2R. The expression of the recombinant proteins was verified by flow cytometry with specific mAb; positive cells were selected by limiting dilution for further study.
Purification of Receptors
Transductants (5 × 107) were lysed for 30 min on ice in lysis buffer (20 mmol/liter Tris-HCl, 140 mmol/liter NaCl, pH 8.2) containing 1% Nonidet P-40 as a detergent and a mixture of protease inhibitors (5 μmol/liter aprotinin, 5 μmol/liter leupeptin, 5 μmol/liter pepstatin, 1 mmol/liter PMSF). Lysates were centrifuged for 2 min at 10,000 × g at 4 °C, and the supernatants were subjected to affinity chromatography using anti-pTag mAb H902 coupled to CNBr-Sepharose. The columns were washed with lysis buffer, and purified receptor proteins were eluted from the column with elution buffer (20 mmol/liter Tris-HCl, 140 mmol/liter NaCl, pH 11.7). The eluted fractions were adjusted to pH 7.5 using HCl. A uPA-conjugated agarose column was used to isolate soluble uPAR from the conditioned medium of CHO cells expressing the recombinant human soluble uPAR as described previously (28).
Flow Cytometry and Internalization Assay
Cells were detached using 5 mmol/liter EDTA, PBS. For surface staining, the cells were washed with PBS containing 1% BSA and afterward incubated on ice for 30 min with specific mAb. In the case of non-labeled antibodies, the cells were washed again, and a second step staining was done with FITC-conjugated F(ab′)2 anti-mouse IgG+IgM antibodies (An der Grub, Kaumberg, Austria). Before analysis, the cells were washed with 1% BSA/PBS. Dead cells were excluded by staining with 7-amino-actinomycin D. Flow cytometry was performed on a LSR II flow cytometer (BD Biosciences). Data acquisition was executed with the FACS DIVA software. Data analysis was accomplished with the FlowJo software (Treestar Inc., Ashland, OR).
For the internalization assay, the cells were first detached (see above) and then incubated in suspension for at least 1 h after the EDTA treatment in the incubator with or without monensin (30 μmol/ml). Then the cells were pre-cooled and incubated for 20 min on ice with fluorescently labeled Plg-AF488 (100 nmol/liter) with or without the fluorescently labeled specific mAb MEM-238 (5 μg/ml), peptides (20 μmol/ml), or inhibitors. Afterward, the cells were incubated for various time intervals at 37 °C. After acid stripping (50 mmol/liter glycine, 150 mmol/liter NaCl, pH 3.0), the cells were analyzed by flow cytometry for remaining (stripping-resistant) fluorescent Plg.
RNA Interference
RNAi was performed as described previously (21). Briefly, stable M6P-IGF2R knockdown cell lines TCL-598 and THP-1 were generated by the delivery of a short hairpin RNA (shRNA) expression cassette cloned in the lentiviral vector pLKOpuro1 (provided by S. Steward; Washington University School of Medicine, St. Louis, MO). Two different short hairpin sequences specific for M6P-IGF2R were used: sh4525 and sh6588 described previously (21). Both target positions were equally efficient (more than 80% silencing efficiency) and yielded similar results. As a control construct we used the MISSIONTM non-target shRNA control vector (pLKOpuro1) from Sigma. Lentiviral particles were generated as previously described (29). Briefly, HEK-293 cells were co-transfected by the calcium phosphate precipitation method with vector DNA, the pseudo-typing plasmid (pCMV_VSV.G) and the packaging construct (pHR′8–2ΔR) in a ratio of 2/1/1.5. Forty-eight hours after transfection, viral supernatants were harvested, filtered, and used fresh for the transduction of target cell lines. The transduction was done overnight with the addition of 5 μg/ml Polybrene. Stably transduced cells, both the control (shCTR) and with silenced expression of M6P-IGF2R (shM6P-IGF2R), were cultured permanently with puromycin (3 μg/ml for TCL-598, 1.5 μg/ml for THP-1).
In Vitro Binding Assay and Immunoblotting Analysis
For the binding assay, 5 μg/ml various molecules were coated on wells of a 96-well BD FalconTM plate in PBS, pH 8.7, for 2 h at 37 °C. Then the wells were blocked with 1% BSA in PBS for 1 h at room temperature and washed 2 times with binding buffer (20 mmol/liter Tris-HCl, 140 mmol/liter NaCl, pH 7.5). Afterward, the wells were incubated for 4 h on ice with binding buffer supplemented with 5 μg/ml purified assayed proteins and washed 2 times with ice-cold binding buffer. Bound material was analyzed by SDS/polyacrylamide gel electrophoresis (SDS-PAGE) followed by transfer to Immobilon polyvinylidene difluoride membranes (Millipore). The membranes were blocked using 4% nonfat milk and immunostained with a specific (optionally biotinylated) mAb followed by the incubation with the anti-mouse IgG HRP-conjugate Ab (optionally with streptavidin-HRP conjugate). For visualization of the proteins, either the chemiluminescence image analyzer FUJIFILM/LAS-4000 was used or the membranes were exposed to light-sensitive Eastman Kodak Co films. The films were developed by using the AGFA Curix 60 developing machine (Agfa Deutschland Vertriebsgesellschaft mbH and Co. KG, Cologne, Germany).
Sucrose Density Gradient Centrifugation Analysis
Cells (5 × 107) were lysed directly in culture flasks using lysis buffer containing a mixture of protease inhibitors as above and detergent 1% Brij-58. The cell lysates were then mixed with 80% sucrose solution to get a final concentration of 40% sucrose. This fraction was placed onto a 60% sucrose fraction in a centrifuge tube and then overlaid gradually with 20, 15, 10, and 5% sucrose fractions (each fraction of 500 μl) to establish a linear gradient. All the layers contained 0.5% Brij-58 and protease inhibitors. After ultracentrifugation at 200,000 × g for 18 h at 4 °C, 10 fractions were gradually taken from the top to bottom of the gradient, and the aliquots were analyzed by immunoblotting analysis as described above.
BN/SDS-PAGE and Zymography
Based on previously described techniques (30, 31), we washed the cells (5 × 107) twice with solubilization buffer (50 mmol/liter NaCl, 5 mmol/liter aminocaproic acid, 1 mmol/liter EDTA, 50 mmol/liter imidazole-HCl, pH 7.0) and harvested them from the flask in the same buffer by scraping. The cells were centrifuged for 10 min at 360 × g and 4 °C. Afterward, the cells were resuspended in solubilization buffer and homogenized by a Teflon pestle (types S20 and S21, Glas-Col, Terre Haute, IN). The nuclei and debris were removed by spinning for 30 s at 10,000 × g and 4 °C. The supernatants were then centrifuged for another 45 min at 100,000 × g and 4 °C. The pellets containing membrane proteins were solubilized in Triton X-100 (1 g/1 g of total protein) in solubilization buffer and incubated for 15 min on ice. The protein amount was measured with the BCA protein kit (Pierce).
To run the BN/SDS-PAGE, a 3–13% gradient polyacrylamide gel was used as a separation gel, and a 2.5% native gel was used as a stacking gel. The samples were loaded, and deep blue cathode buffer (50 mmol/liter Tricine, 7.5 mmol/liter imidazole pH 7.0 and 0.02% Coomassie Blue G-250) was poured on top of the samples and into the upper (cathode) chamber. The anode buffer (25 mmol/liter imidazole-HCl; pH 7.0) was poured into the lower (anode) chamber, and electrophoresis was performed at 5 V/cm and 4 °C overnight. The following day, after the samples entered the first third of the gel, the deep blue cathode buffer B was replaced with the slightly blue cathode buffer (50 mmol/liter Tricine, 7.5 mmol/liter imidazole, pH 7.0, and 0.002% Coomassie Blue G-250), and the voltage was increased to 13 V/cm. Ferritin and BSA were used as markers. Lanes were cut from the gel and either used immediately in the second-dimension run or frozen at −20 °C for a later usage. The gel strips from the first dimension were equilibrated in SDS-PAGE running buffer for 5 min and then put on the top of a 7.5% SDS-polyacrylamide gel for the second dimension separation. The gel was run at 1 V/cm at 4 °C overnight. The next day the voltage was increased to 13 V/cm. The gel was blotted at constant voltage (15 V), and the membrane was analyzed as described above. In some experiments, we applied zymographic SDS-PAGE as the second dimension.
Zymography was performed according to the published protocols (32, 33) with some modifications. A 10% SDS-polyacrylamide gel was impregnated with fibrinogen (3 mg/ml) as a plasmin substrate. The zymogen Plg was added into the gel (30 μg/ml) to be activated to plasmin by cellular uPA during the assay. The electrophoresis was run at 1 V/cm and 4 °C overnight. The next day the voltage was increased to 13 V/cm. Afterward, the gel was first incubated twice for 15 min in 2.5% Triton X-100 (in 0.1 m Tris; pH 8.1) to withdraw SDS from the gel and then incubated overnight (in 10 mmol/liter CaCl2 in 0.1 mol/liter Tris; pH 8.1) at room temperature to allow an enzymatic reaction inside the gel. Finally, the gel was stained with Coomassie Brilliant Blue and destained in 25% (v/v) methanol with 10% (v/v) acetic acid in water. The staining of the gel revealed areas of proteolytic activity as transparent bands in a dark blue background. The gel was scanned with an Epson Perfection 1200 photo scanning system, and images were analyzed by the Adobe Photoshop CS3 software (extended Version 10.0.1). The images presented here are in a grayscale.
Gel Filtration and Plg Activation Assay
The GE Healthcare AKTA FPLC System was used for the gel filtration experiments. Cell lysates were prepared as described above by solubilization either with 1% Triton X-100 or 1% N-dodecyl-d-maltoside. Samples were loaded at 4 °C in a volume of 500 μl onto a Superose 6 HR 10/300 GL column (GE Healthcare) equilibrated with Triton X-100 (0.5%)/PBS. The absorbance at 280 nm was monitored, and 500-μl fractions were collected and analyzed by immunoblotting and in vitro Plg activation assay. The following standards of molecular mass (all from GE Healthcare or Sigma) were used: blue dextran (2000 kDa), thyroglobulin (669 kDa), ferritin (440 kDa), catalase (232 kDa), aldolase (158 kDa), BSA (66 kDa), and carbonic anhydrase (29 kDa). In some experiments the cells were pretreated with Plg.
Individual gel filtration fractions (50 μl) were loaded into wells of a 96-well plastic plate and incubated at 37 °C with or without (when the cells were pretreated with Plg) human Glu-Plg (50 nmol/liter). Plasmin activities were analyzed by adding the chromogenic plasmin (S-2251) substrate (0.8 mmol/liter). The absorbance change at 405 nm was monitored by using a 96-well plate reader (SpectraMax M5, Molecular Devices, Sunnyvale, CA).
Confocal Microscopy
Cells were seeded into chamber slides (5 × 103 cells/well; Nalgene/Nunc, Wiesbaden, Germany) and cultured under standard conditions. The directly conjugated proteins Transferrin-AF546 (20 μg/ml), Plg-AF488 (50 nmol/liter), and the mAb MEM-238-AF647-labeled antibody directed against the M6P-IGF2R (20 μg/ml) were added directly into the culture medium, and the cells were incubated for 20 min at 37 °C under normal culture conditions protected from light. Then medium was discarded, and cells were washed with PBS and fixed with Fixing Solution (Invitrogen) for 15 min at room temperature and permeabilized. The cells were shortly rehydrated with 1× PBS and afterward incubated for 10 min with DAPI solution to visualize the nuclei. The chambers were removed, and the slides were washed three times for 5 min in PBS before mounting with fluorescent mounting medium (Dako, Copenhagen, DK)). Pictures were captured with a confocal laser scanning microscope (LSM 700, Zeiss, GER) and evaluated with the software ZEN 2009 (Lite version, Zeiss, GER).
Statistical Analysis
All experiments were performed at least three times in at least triplicates. The data were expressed as mean values with S.D. Statistical significance was evaluated by using Student's t test; a value of p <0.05 (as indicated) was considered to be significant.
RESULTS
Lipid rafts are described as platforms controlling receptor effector and signaling functions at and across the plasma membrane (34). Therefore, we tested whether M6P-IGF2R influenced lipid raft partitioning of uPAR and uPAR/uPA-mediated Plg activation. We used the human kidney carcinoma cell line TCL-598 because of its high expression of both uPAR and its ligand pro-uPA (21). We lysed control-silenced versus M6P-IGF2R-silenced TCL-598 cells with the lipid raft preserving detergent Brij-58 and subjected the cell lysates to a sucrose density gradient centrifugation. We tested individual fractions for the presence of uPAR, β3 integrin, M6P-IGF2R, and CD59, another glycosylphosphatidylinositol-anchored protein frequently used as a lipid raft marker. We found parts of the full-length form of uPAR (D1D2D3), the truncated D2D3 form, and pro-uPA present in light density fractions corresponding to detergent-resistant lipid rafts (Fig. 1). This was particularly true for the D2D3 form of uPAR that was in line with the previous observation that the uPAR cleavage was accelerated in lipid rafts (6). Interestingly, M6P-IGF2R showed a similar distribution as the D2D3 form, and moreover, full-length uPAR (D1D2D3) was stronger expressed in the M6P-IGF2R-silenced cells, which was in agreement with our previous finding that M6P-IGF2R facilitated uPAR cleavage (21). However, M6P-IGF2R-silencing did not result in any significant effect on the density distribution of the analyzed molecules. Likewise in the control cells, the majorities of full-length uPAR together with uPA and β3 integrin molecules were seen in the heavy density fractions corresponding to detergent-soluble proteins. Alternatively, they could also be part of the recently described heavy rafts (35).
FIGURE 1.
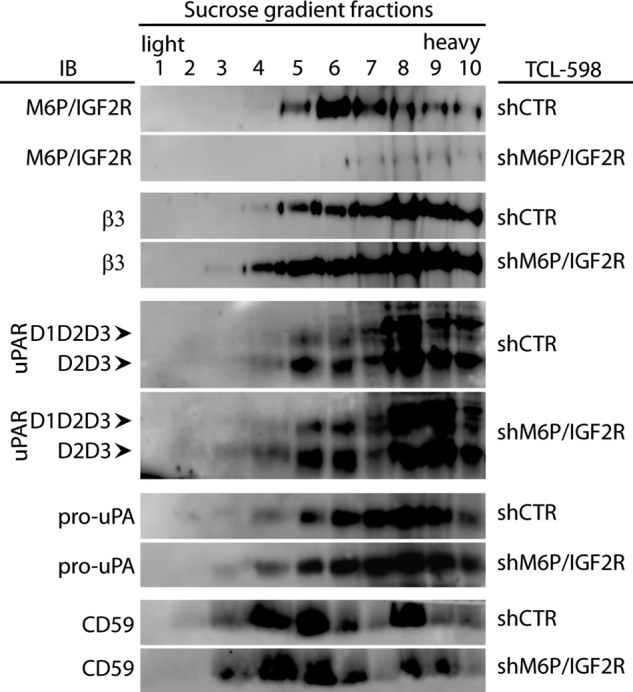
Effect of M6P-IGF2R silencing on the lipid raft partitioning of uPAR. Control (shCTR)- and M6P-IGF2R-silenced (shM6P-IGF2R) TCL-598 cells were lysed by using the detergent Brij-58, and the cell lysates were subjected to sucrose gradient density centrifugation. Ten individual gradient fractions were taken gradually from the top (light fractions corresponding to detergent-resistant lipid rafts) to the bottom (heavy fractions corresponding to soluble proteins). The samples were analyzed by SDS-PAGE followed by immunoblotting (IB) with mAb specific to M6P-IGF2R (mAb MEM-238), β3 integrin (mAb VIPL-2), uPAR (mAb H2), pro-uPA (mAb U5), and CD59 (mAb MEM-43/5) as a control of lipid raft fractionation. Chemiluminescence was used for visualization. D1D2D3 points to the band corresponding to the full-length uPAR, and D2D3 points to the band corresponding to the truncated form. Results are representative for three independent experiments.
Next, we isolated cell membrane fractions from control- and M6P-IGF2R-silenced cells, lysed them with Triton X-100, and subjected the cell membrane lysates to BN/SDS-PAGE. This technique allowed us to isolate native cell membrane protein complexes in the first dimension and dissect individual components of the complexes by SDS-PAGE in the second dimension. In the control cell membrane fractions, M6P-IGF2R was present in a high molecular mass complex of the apparent size in the range from 440 to 880 kDa. In addition, αV integrin and partially also full-length uPAR (D1D2D3) migrated to the similar position, indicating that they might be components of the same complex (dotted frame in Fig. 2A). D1D2D3 and αV integrin were further detected together in two smaller complexes of the apparent sizes of ∼440 and 200 kDa, respectively (dashed frames in Fig. 2A). We also observed uPAR bands (both D1D2D3 and D2D3) of ∼100 kDa probably corresponding to dimeric forms (36). Pro-uPA mirrored the position of uPAR D1D2D3 (Fig. 2A) in line with its restricted ability to bind only to full-length uPAR (8). Upon M6P-IGF2R silencing, both the full-length uPAR (D1D2D3) and the pro-uPA bands got enriched and concentrated in the 200-kDa region. This finding was in accord with our previous results showing their up-regulation on the cell surface of silenced cells (21). To analyze which of these complexes possessed proteolytic activity, we combined BN-PAGE in the first dimension with zymography in the second dimension (Fig. 2B). Although we detected clear zones corresponding to the pro-uPA bands both in control- and in M6P-IGF2R-silenced cells (compare Fig. 2, A and B), the intensity was so weak that no quantitative evaluation was possible. This revealed the limit of combining native gel electrophoresis with zymography, yet this method can be useful for studying other cellular proteolytic systems.
FIGURE 2.

BN/SDS-PAGE analysis of the effect of M6P-IGF2R silencing on reorganization of membrane protein complexes associated with Plg activation. A, cell membrane fractions derived from control (shCTR)- and M6P-IGF2R-silenced (shM6P-IGF2R) TCL-598 cells were prepared by solubilization with Triton X-100 as described under “Experimental Procedures.” The samples were then analyzed by BN-PAGE in the first dimension followed by SDS-PAGE in the second dimension. For the immunoblotting analysis (IB), the mAbs to M6P-IGF2R (mAb MEM-238), αV integrin (mAb P3G8), uPAR (mAb H2), and pro-uPA (mAb scuPA8) were used followed by an HRP-conjugated anti-mouse Ab and chemiluminescence detection. D1D2D3 points to the band corresponding to the full-length uPAR, and D2D3 points to the band corresponding to the truncated form. Molecular mass markers are shown on the top for the first dimension (dimers and monomers of Ferritin (880, 440 kDa) and BSA (130, 66 kDa)) and along the panel border for the second dimension (standard SDS-PAGE molecular mass markers). The dotted frame indicates the complex of M6P-IGF2R, αV integrin, and uPAR (D1D2D3) in shCTR cells. The dashed frame indicates the complex of αV integrin and uPAR. B, the gel strips from the first dimension of BN/SDS-PAGE were analyzed by zymographic SDS-PAGE as described under “Experimental Procedures.” Afterward, the gel was scanned, and the images were analyzed. The images are presented in a grayscale mode so that the clear zones represent the areas of proteolytic activation. Standard molecular masses are shown along the panel border. Results are representative for two independent experiments.
To overcome this limitation and get better insight into the ability of the identified protein complexes to activate Plg, we solubilized control- and M6P-IGF2R-silenced cells and subjected the cell lysates to gel filtration fractionation through a Superose 6 column. Individual fractions were subjected to in vitro proteolysis assay in the presence of Plg to measure their ability to generate plasmin. When Triton X-100 was used as a detergent, we detected three peaks of plasmin activity (Fig. 3A). The first matched the high molecular mass molecules in the void volume of the column in fractions 4–7 (≥2000 kDa), the second corresponded to intermediate molecular mass molecular complexes in fractions 19–22 (∼100–440 kDa), and the third was in fractions 25–28 (low molecular mass range less than 66 kDa). Remarkably, fractions 25–28 derived from the M6P-IGF2R-silenced cell lysate displayed higher plasmin activity (Fig. 3A), which might be likely linked with the higher expression of pro-uPA on the surface of these cells (21). Then we used the detergent N-dodecyl-d-maltoside that in contrast to Triton X-100 solubilizes also lipid rafts. We detected the similar three major peaks of plasmin activity, but interestingly, in the high molecular mass complex derived from M6P-IGF2R-deficient cells, higher activity was apparent. Furthermore, the activity in the intermediate molecular mass proteolytic fractions was higher compared with the Triton X-100 solubilization, but similar for both control- and M6P-IGF2R-silenced cells (Fig. 3A).
FIGURE 3.

Analysis of M6P-IGF2R complexes activating Plg in vitro. A, cell lysates derived from control (shCTR)- and M6P-IGF2R-silenced (shM6P-IGF2R) TCL-598 cells were prepared as described under “Experimental Procedures” by solubilization with either detergent Triton X-100 (TX) or detergent N-dodecyl-d-maltoside (DM). The samples were loaded onto a Superose 6 column, and individual fractions were collected. The fraction samples were transferred into wells of a 96-well plastic plate and incubated at 37 °C with human Glu-Plg (50 nmol/liter). Plasmin activities were analyzed by adding the chromogenic plasmin substrate (S-2251; 0.8 mmol/liter). The absorbance change at 405 nm was monitored after 4 h by using a 96-well plate reader. B, the fraction samples from A were analyzed by SDS-PAGE followed by immunoblotting (IB) with mAbs specific to M6P-IGF2R (mAb MEM-238), β3 integrin (mAb VIPL-2), uPAR (mAb H2), pro-uPA (mAb U5), Rab11 (polyclonal), LAMP-3 (mAb MEM-259), and EEA1 (mAb C45B10). Chemiluminescence was used for visualization. D1D2D3 points to the band corresponding to the full-length uPAR, and D2D3 points to the band corresponding to the truncated form. Results are representative for three independent experiments. Molecular mass standards used in A are specified under “Experimental Procedures.”
Next, we analyzed the individual fractions from the gel filtration analysis by immunoblotting (Fig. 3B). In control cells, we found M6P-IGF2R in the molecular mass range from 440 kDa to above 669 kDa, and β3 integrin similarly to uPAR ranging approximately from 400 to 600 kDa. In M6P-IGF2R-silenced cells, we did not observe any major differences compared with control cells except higher expression of pro-uPA and relatively higher full-length uPAR (D1D2D3), again in line with our previous results (21). These data corresponded well with the data obtained by BN/SDS-PAGE analysis (compare Figs. 3B and 2A). According to the protein distribution in the gel filtration fractions, we reckoned that 1) the low molecular mass peak of detected plasmin activity corresponded to the single uPA molecule and probably also to truncated proteolytically active uPA molecules (37), and 2) the intermediate molecular mass peak of plasmin activity was linked with uPA bound to full-length uPAR complexed with other membrane partners, such as e.g. αVβ3 integrin and in particular M6P-IGF2R. In the Plg activating high molecular mass fractions 4–7, we repeatedly observed very weak yet detectable bands of M6P-IGF2R (Fig. 3B).
It has been shown that upon rapid internalization, M6P-IGF2R traffics through late endosomes and delivers its cargo to lysosomes, but a large fraction returns to the cell surface through the early endosomal compartment (16, 38, 39). Therefore, we tested the fractions also for the early endosome antigen 1 (EEA1), a well established marker of endosomal compartments, Rab11, a marker of recycling endosomes, and LAMP-3, a lysosomal marker. We detected EEA1 and a very slight portion of Rab11 in the high molecular mass fractions, suggesting that Plg was internalized through endosomal compartments. The vast majority of 25-kDa Rab11 was found in fractions 20–24 (∼66–200 kDa), indicating its participation in other protein complexes (Fig. 3A).
We then were curious to test which molecular complexes were responsible for activation of Plg in vivo in the living cell. We incubated live cells for 30 min with Plg on ice to allow Plg binding onto the cell surface and then for an additional 60 min without Plg at 37 °C to allow its activation. Afterward, we solubilized the cells and subjected the cell lysates to size-exclusion chromatography. We tested the individual fractions for plasmin activity in the presence of a chromogenic plasmin substrate. In M6P-IGF2R-silenced cells, we again detected three areas of plasmin activity; the first co-eluted with the EEA1-containing high molecular mass molecules in the void volume of the column in fractions 4–7, the second eluted with fractions 14–18, and the third eluted in fractions 21–24 of the low-molecular mass range (∼160–66 kDa). Interestingly, the latter plasmin activity overlapped with Rab11, which might indicate that internalized plasmin was disseminated via endosomes to different compartments of the cell (compare Figs. 4 and 3B). Strikingly, when we pretreated the cells for 60 min with the carboxylic ionophore monensin (30 μmol/liter), an established inhibitor of receptor trafficking and recycling, the pattern of plasmin activation in the different fractions, was identical to that seen upon M6P-IGF2R-silencing: reduced plasmin content within both the EEA-1-containing high molecular mass fractions and the Rab11-containing low molecular mass fractions corresponding to endosomal membrane complexes. The intermediate molecular mass plasmin activity was stronger and corresponded well to the higher appearance of full-length uPAR and αVβ3 integrin in these fractions (compare Figs. 4A with 3B).
FIGURE 4.

Analysis of M6P-IGF2R complexes activating Plg in vivo. A, control (shCTR)- and M6P-IGF2R-silenced (shM6P-IGF2R) TCL-598 cells were incubated for 30 min with human Glu-Plg (50 nmol/liter) on ice followed by an additional 60-min incubation without Plg at 37 °C. Then the cells were solubilized in detergent Triton X-100, and the cell lysates were subjected to size exclusion using a Superose 6 column. Individual fractions were assayed for plasmin activity in the presence of the chromogenic plasmin substrate S-2251 (0.8 mmol/liter) as described in Fig. 3A. The absorbance change at 405 nm was monitored after 18 h by using a 96-well plate reader. B, the fraction samples were analyzed by SDS-PAGE followed by immunoblotting (IB) with rabbit polyclonal anti-plasmin Ab. In some settings the cells were preincubated with monensin (30 μmol/liter).
To further test whether M6P-IGF2R functioned in Plg internalization, we analyzed the Plg-binding site within the amino-terminal region of M6P-IGF2R; we and others previously showed that a Plg-binding site within M6P-IGF2R was situated in its amino-terminal region (18, 20). Because M6P-IGF2R bound Plg in a lysine-dependent manner (17) and Lys-25 of M6P-IGF2R had been shown to be encompassed within the Plg binding region (18, 20), we tested whether this residue was directly implicated in Plg binding and internalization. First, Lys-25 was changed into threonine by site-directed mutagenesis, and both human wild type M6P-IGF2R (M6P-IGF2Rwt) and M6P-IGF2R bearing the lysine-to-threonine mutation (M6P-IGF2RK25T) were expressed in the M6P-IGF2R-negative mouse fibroblasts expressing human uPAR (18). However, M6P-IGF2RK25T was expressed in a much lower amount compared with M6P-IGF2Rwt (Fig. 5A); we observed this phenomenon with all characterized M6P-IGF2RK25T clones (data not shown). Despite the poor expression we were able to purify sufficient amounts of the mutant to perform a Plg binding assay; compared with the wild type receptor M6P-IGF2Rwt, the purified M6P-IGF2RK25T displayed reduced binding to both Plg and uPAR (Fig. 5B) suggesting that Lys-25 within the Plg-binding site was involved in Plg and uPAR binding. Next, we performed an internalization assay with M6P-IGF2R knock-out mouse fibroblasts expressing human uPAR with or without both variants of human M6P-IGF2R. We incubated the cells on ice with fluorescently labeled Plg to enable its binding onto the cell surface, then we incubated the cells for various time intervals at 37 °C to allow Plg internalization. After stripping the surface-bound Plg, the cells were subjected to flow cytometry to detect intracellular Plg. We observed ∼60% elevated Plg internalization in cells expressing wild type human M6P-IGF2R (M6P-IGF2Rwt) (Fig. 5C). Expression of mutant M6P-IGF2RK25T did not stimulate a comparable internalization rate. However, the mutant displayed as well a markedly reduced internalization of the fluorescently labeled anti-M6P-IGF2R mAb MEM-238 (Fig. 5C), similarly to the M6P-IGF2R-silenced cell lines THP-1 and TCL-598 (Fig. 6, B and C). Thus, the failure of the mutant receptor to facilitate Plg internalization could have been likely due to insufficient receptor expression and not insufficient Plg binding.
FIGURE 5.

Effect of M6P-IGF2R silencing on Plg internalization in mouse fibroblasts. A, shown is characterization of uPAR/M6P-IGF2R mouse fibroblast (MF) transductants. A retroviral system was used for gene transfer and expression of human M6P-IGF2Rwt and human M6P-IGF2RK25T in murine M6P-IGF2R-knock out mouse fibroblasts (M6P-IGF2RKOMF) expressing human uPAR. Cell surface expression of the receptors was verified by flow cytometry using mAb specific to M6P-IGF2R (mAb MEM-238-APC) and uPAR (H2-AF647). B, for the binding assay, 5 μg/ml purified Glu-Plg or soluble uPAR were coated on wells of a plastic plate in PBS. Then the wells were blocked with BSA and incubated for 4 h on ice with binding buffer supplemented with 5 μg/ml purified M6P-IGF2Rwt or M6P-IGF2RK25T. Bound material was analyzed by SDS-PAGE, and immunoblotting (IB) with mAb specific to M6P-IGF2R (mAb MEM-238) is shown. C, pre-cooled murine M6P-IGF2R-negative mouse fibroblasts expressing human uPAR and M6P-IGF2Rwt or M6P-IGF2RK25T were incubated for 20 min on ice with fluorescently labeled Plg-AF488 (100 nmol/liter) and the anti-M6P-IGF2R MEM-238-APC (5 μg/ml) and then incubated for various time intervals at 37 °C. After acid stripping, the cells were analyzed by flow cytometry for the remaining molecules. Geometric mean fluorescence intensity values are shown as a measure of internalization.
FIGURE 6.

Effect of M6P-IGF2R silencing on Plg internalization in human cell lines. A, shown is expression of M6P-IGF2R on the surface of M6P-IGF2R-silenced (shM6P-IGF2R) versus control-silenced (shCTR) TCL-598 and THP-1 cells. mAb MEM-238 conjugated to APC was used as a specific mAb. B, precooled control-silenced and M6P-IGF2R-silenced TCL-598 and THP-1 cells were subjected to the internalization assay with Plg conjugated to AF488 (100 nmol/liter) and the anti-M6P-IGF2R MEM-238-APC (5 μg/ml). C, confocal microscopy was used to investigate Plg uptake in TCL-598 cells silenced for M6P-IGF2R (shM6P-IGF2R) and in control silenced cells (shCTR). The cells were incubated for 20 min at 37 °C with labeled Plg (Plg-AF488), transferrin (Tf-AF546), and the mAb MEM-238-AF647. Subsequently, medium was discarded, the cells were rinsed, fixed, and permeabilized, and nuclei were visualized with DAPI. The left panels show the phase contrast images with the inset panels showing the confocal fluorescent images of the anti-M6P-IGF2R mAb MEM-238-AF647 (blue). The middle panels show the merged confocal fluorescent images of Plg-AF488 (green) and transferrin-AF546 (red); colocalization areas appear yellow (arrows); the arrowheads point to Plg aggregates on the cell surface. The right panels show the magnified insets from the middle panels. DAPI is shown in gray. Scale bars = 20 μm.
We also subjected to the internalization assay the M6P-IGF2R-silenced and control-silenced human TCL-598 cells. We found that upon M6P-IGF2R silencing, Plg internalization was reduced by about 40% (Fig. 6, A and B). A similar result we obtained with M6P-IGF2R-silenced THP-1 cells (Fig. 6, A and B). We confirmed these results also by confocal microscopy; we preincubated control- and M6P-IGF2R-silenced cells at 37 °C for 20 min with fluorescently labeled Plg, transferrin (Tf), and the mAb MEM-238 to M6P-IGF2R to allow their internalization. We detected large Plg aggregates concentrated on the cell surface of both control- and M6P-IGF2R-silenced cells, probably representing cell surface-bound Plg (arrowheads in Fig. 6C). On the other hand, we found partially overlapping intracellular punctuate accumulations of Plg and Tf, a marker for early sorting and recycling endosomes (40), indicating partially common endocytic pathways. Notably, upon M6P-IGF2R-silencing these intracellular colocalization areas markedly decreased (arrows in Fig. 6C), which further supports the role of M6P-IGF2R in Plg uptake.
Finally, we tested on Plg uptake the effect of peptide 18–36 (pep18–36) derived from the Plg-binding site within the amino-terminal region of M6P-IGF2R. Previously we showed that pep18–36 blocked binding of Plg to uPA and inhibited Plg activation (41). Surprisingly, pep18–36 enhanced Plg internalization by about 114% in control- and 85% in M6P-IGF2R-silenced cells. Upon treatment with monensin, the enhancement in Plg uptake was only 52% in control-silenced cells and blunted completely in M6P-IGF2R-silenced cells (Fig. 7). Also, tranexamic acid, an inhibitor of lysine-dependent binding of Plg to its receptors, blunted Plg internalization. These results suggest that binding of Plg via the amino-terminal region of M6P-IGF2R induces further binding so that its internalization is enhanced.
FIGURE 7.
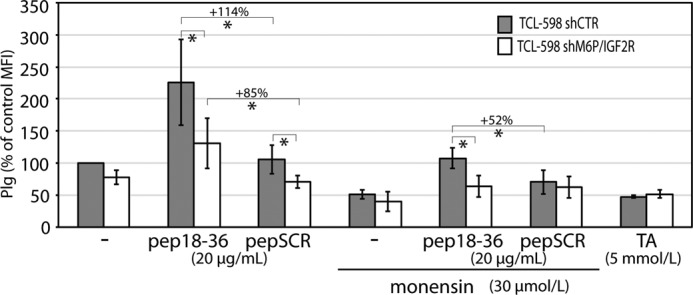
Effect of inhibitors and M6P-IGF2R-derived peptide 18–36 on Plg internalization in the TCL-598 cell line. Control-silenced (shCTR) and M6P-IGF2R-silenced (shM6P-IGF2R) TCL-598 cells were preincubated at 37 °C for 60 min with or without monensin (30 μmol/liter) and then subjected for 90 min to the internalization assay with Plg-AF488 (100 nmol/liter) as described in Fig. 6 in the presence of either peptide 18–36 (pep18–36; 20 μmol/liter), control scrambled peptide (pepSCR; 20 μmol/liter), or tranexamic acid (TA; 5 mmol/liter). Values of internalization were calculated as percentage differences in the geometric mean fluorescence intensity values relative to the geometric mean fluorescence intensity value (MFI) obtained for control cells without any additives (100%).
DISCUSSION
Previously, we have provided evidence that M6P-IGF2R serves as a regulator of pericellular proteolysis. The expression of M6P-IGF2R both facilitates the proteolytic cleavage of uPAR, thus leading to the loss of uPA binding and subsequently to the blunted Plg activation and cell migration (21), and down-regulates αVβ3 integrin expression (21). Furthermore, we have demonstrated that soluble M6P-IGF2R also dampens Plg activation on the cell surface (41). Here, we complement these data by showing that M6P-IGF2R mediates Plg internalization.
Our results obtained from lipid raft flotation analysis indicate that M6P-IGF2R is not implicated in the uPAR partitioning into lipid rafts (Fig. 1). The distribution of M6P-IGF2R after sucrose density gradient centrifugation is similar to the truncated form of uPAR (D2D3), which is in line with our previous finding that M6P-IGF2R facilitates the cleavage of uPAR yielding the truncated D2D3 fragment incapable of binding most uPAR ligands (21). Furthermore we show by size-exclusion chromatography of in vivo activated plasmin (Fig. 4) that the intermediate molecular mass complex-linked plasmin activity correlates with full-length uPAR seen in the M6P-IGF2R-silenced cells; in the corresponding fractions obtained from M6P-IGF2R-positive cells both uPAR was more cleaved and Plg activation reduced. These results support our previous data and demonstrate that within this molecular environment M6P-IGF2R approximates plasmin to uPAR for D1 release as negative feedback regulation of Plg activation (21). Gel filtration fractions obtained with N-dodecyl-d-maltoside, a detergent that solubilizes lipid rafts, display the elevated capability of Plg activation in comparison to the fractions derived from the lipid raft preserving detergent Triton X-100 (Fig. 3A), which falls into line with recent findings indicating that lipid rafts are small entities preserving certain lipid-modified receptors in lipid shells, thereby preventing their interaction with potential other partners (42, 43).
Upon M6P-IGF2R silencing, plasmin activated in vivo shifts both from the EEA1-containing high molecular mass and from the Rab11-containing lower molecular mass fractions to the intermediate molecular mass fractions. The latter are enriched in uPAR and αVβ3 integrin, suggesting that they mostly represent parts of the plasma membrane. Strikingly, we found the same redistribution of plasmin activation from high as well as low molecular mass endosomal complexes to the intermediate plasma membrane fractions when we treated the cells with monensin, an inhibitor of protein transport and recycling (Figs. 3 and 4). The similar effects on plasmin distribution of both M6P-IGF2R-silencing and monensin treatment suggest that M6P-IGF2R functions as a receptor for Plg internalization. It is possible that these complexes might be scaffolds to control the activity of the highly potent plasmin by either internalization and/or by mediating intracellular activation (44). Notably, endosomes not only represent an important compartment in endocytosis, but also they have been identified as a compartment wherein physiological and specific proteolysis could occur (45–47); an intriguing possibility that Plg could be proteolytically converted to plasmin inside specific endosomal compartments, its physiological relevance, and a molecular mechanism whereby M6P-IGF2R would control this conversion remain to be followed.
Our results obtained from BN/SDS-PAGE and in particular from gel filtration analysis underscore a crucial role of the cell membrane protein assemblies associated to uPAR in Plg activation and propose a function for M6P-IGF2R in controlling Plg uptake (Figs. 3 and 4). Hitherto uPAR has been known to be internalized through endocytic compartments either in a LDL receptor-related protein-1-dependent or independent manner (48). In the former pathway, uPAR undergoes internalization when engaged with its ligand uPA complexed with plasminogen activator inhibitor-1 (49–51). For the latter pathway several direct uPAR membrane partners have been suggested to internalize uPAR including the urokinase receptor-associated protein uPARAP/endo180 (52) and also M6P-IGF2R (53), although we and others have previously found that M6P-IGF2R did not significantly participate in the internalization of uPAR (21, 54). M6P-IGF2R-dependent Plg internalization would comprise an additional level to control pericellular Plg activation. However, our internalization assays clearly reveal that, first, RNAi with M6P-IGF2R expression in TCL-598 and THP-1 has led to no more than an ∼40% decrease in Plg internalization and, second, re-expression of human M6P-IGF2R in M6P-IGF2R-negative mouse fibroblasts has resulted in a comparable increase over the basal Plg internalization. Thus Plg is constitutively endocytosed also independently of M6P-IGF2R.
Many heterogeneous Plg receptors have been characterized up to now, including cytokeratin 8 (55), annexin A2 (56), dipeptidyl peptidase IV (57), S100A10 (58), Plg-R(KT) (5), prion (59), and others (for reviews, see Refs. 10, 13, and 60). A vast majority of these receptors potentiate plasmin generation. On the other hand, little is known about negative regulators on the cell membrane. It has been shown that a direct interaction between uPA and Plg is necessary for Plg activation on cells (61, 62); hence, LDL receptor-related protein-1 that internalizes the uPA·plasminogen activator inhibitor-1·uPAR complex and recycles back to the plasma membrane (51) might be involved in Plg clearance as well. Another feasible candidate could be VLDL receptor, which has been shown to mediate endocytosis of lipoprotein(a) whose component apolipoprotein(a) is highly similar to Plg (63). Future studies should identify receptors that, in addition to M6P-IGF2R, do play a role in the endocytosis of Plg.
Finally, by using truncated variants of M6P-IGF2R we have previously identified a sequence within domain 1 of M6P-IGF2R responsible for Plg binding (18). This sequence is similar to the Plg binding region of streptokinase (18), and a related epitope was found also in human fibronectin (64). Because we have shown that M6P-IGF2R binds Plg in a lysine-dependent manner (17), we suggested that lysines encompassed within this region might be determinants for Plg binding. There are several evolutionary conserved lysines in this region. Here, we particularly scrutinized Lys-25 of human M6P-IGF2R and assayed its role in Plg binding and internalization. In contrast to Bohnsack et al. (65) who used in a site-directed mutagenesis study soluble recombinant truncated variants of bovine M6P-IGF2R, we applied the full-length human M6P-IGF2R expressed in M6P-IGF2R-negative mouse fibroblasts stably expressing human uPAR. M6P-IGF2RK25T was nevertheless expressed at the much lower level than M6P-IGF2Rwt (Fig. 5B), which could be due to the inferior stability of the mutant receptor. It is possible that a missfolding of the protein caused by the mutation leads to a higher degradation rate. Bohnsack et al. (65) have demonstrated that Lys-53 located in domain 1 together with Lys-125 located in the loop connecting domains 1 and 2 of bovine M6P-IGF2R are crucially implicated in Plg binding. Lysines 53 and 125 in bovine M6P-IGF2R correspond to lysines 49 and 121 in human M6P-IGF2R. Nevertheless, crystal structure studies demonstrate that Lys-29 (human Lys-25) located on β1-sheet strand of domain 1 together with two lysines, Lys-132 (human Lys-128) and Lys-215 (human Lys-211), in domain 2 of bovine M6P-IGF2R are situated on a same crevice of the molecule, presumably interacting with Plg (20). Indeed, although we show a reduction in M6P-IGF2RK25T binding to Plg (Fig. 5C), the Lys-25 mutation was not sufficient to abolish the binding completely, which is in line with the aforementioned observations made by Bohnsack et al. (65). It is, therefore, reasonable that multiple clefts within M6P-IGF2R are implicated in Plg binding, and several lysines in this region are necessary for recognition by Plg via lysine-binding sites. Irrespective of this, according to our results M6P-IGF2RK25T does not render cells with the capacity to facilitate Plg internalization due to the low expression level rather than the impaired binding to Plg.
The notion that multiple binding sites might be involved in Plg uptake is further strengthened by our finding that the peptide derived from the Plg-binding site in domain 1 of M6P-IGF2R, peptide 18–36 (pep18–36), induces Plg internalization. Upon monensin treatment the enhanced Plg uptake by pep18–36 was blunted in M6P-IGF2R-silenced cells (Fig. 7). This finding provides another strong indication for involvement of M6P-IGF2R in Plg uptake and also widens our view on the pharmacological potential of pep18–36 because its inhibitory effect on neovascularization and tumor growth is obviously not restricted to a blockade of binding of Plg to uPA as originally described by us (41). We hypothesize that the binding of Plg via this particular region changes the conformation of Plg so that the latter binds subsequently to further sites encompassed either within M6P-IGF2R itself or other Plg binding receptors, which ultimately amplifies its internalization. We will test this possibility in future studies.
Taken together, direct interactions of M6P-IGF2R with uPAR and with Plg, as shown by us elsewhere (18, 21, 41) and in the present study, might provide a control to restrict plasmin activity to specific time and sites and thus to prevent unfavorable degradation of the surrounding structures during cell migration. Furthermore, these interactions might be important for the processing of specific substrates, such as latent transforming growth factor β (17, 19) or uPAR itself (21). On the other hand, in the absence of M6P-IGF2R, Plg activation might occur uncontrolled, and thus the loss of proper M6P-IGF2R function would lead to disabled regulation of fibrinolysis.
Acknowledgments
We thank Václav Hořejší for the anti-M6P-IGF2R mAb MEM-240, the CD59 mAb MEM-43/5, the CD63 (LAMP-3) mAbs MEM-259, H902, and AFP-01, Ulrich Weidle for the mAb H2 against uPAR, and Otto Majdic for the mAb VIPL-2 to αVβ3 integrin. S. Stewart is acknowledged for the lentiviral vector pLKOpuro1.
This work was supported by Austrian Science Fund Grants P19014-B13, P22908, and I00300 and grants from the Science and Technology Assistance Agency of the Slovak Republic (APVT-51-026204), the Slovak Grant Agency VEGA (2/5119/25), and the Bilateral Cooperation Slovakia-Austria (SK-11-AUT-9).
- Plg
- plasminogen
- uPA
- urokinase
- uPAR
- uPA-type plasminogen activator receptor
- AF
- Alexa Fluor
- BN
- blue native
- EEA1
- early endosome antigen 1
- M6P-IGF2R
- mannose 6-phosphate/insulin-like growth factor 2 receptor
- mAb
- monoclonal antibody
- Tf
- transferrin
- TEMED
- N,N,N′,N′'-tetramethylethylenediamine.
REFERENCES
- 1. Bugge T. H. (2008) The Cancer Degradome: Proteases and Cancer Biology, pp. 183–202, Springer-Verlag New York Inc., New York [Google Scholar]
- 2. Castellino F. J., Ploplis V. A. (2005) Structure and function of the plasminogen/plasmin system. Thromb. Haemost. 93, 647–654 [DOI] [PubMed] [Google Scholar]
- 3. Zorio E., Gilabert-Estellés J., España F., Ramón L. A., Cosín R., Estellés A. (2008) Fibrinolysis. The key to new pathogenetic mechanisms. Curr. Med. Chem. 15, 923–929 [DOI] [PubMed] [Google Scholar]
- 4. Young K. C., Shi G. Y., Wu D. H., Chang L. C., Chang B. I., Ou C. P., Wu H. L. (1998) Plasminogen activation by streptokinase via a unique mechanism. J. Biol. Chem. 273, 3110–3116 [DOI] [PubMed] [Google Scholar]
- 5. Andronicos N. M., Chen E. I., Baik N., Bai H., Parmer C. M., Kiosses W. B., Kamps M. P., Yates J. R., 3rd, Parmer R. J., Miles L. A. (2010) Proteomics-based discovery of a novel, structurally unique, and developmentally regulated plasminogen receptor, Plg-RKT, a major regulator of cell surface plasminogen activation. Blood 115, 1319–1330 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Cunningham O., Andolfo A., Santovito M. L., Iuzzolino L., Blasi F., Sidenius N. (2003) Dimerization controls the lipid raft partitioning of uPAR/CD87 and regulates its biological functions. EMBO J. 22, 5994–6003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Ragno P. (2006) The urokinase receptor. A ligand or a receptor? Story of a sociable molecule. Cell. Mol. Life Sci. 63, 1028–1037 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Montuori N., Carriero M. V., Salzano S., Rossi G., Ragno P. (2002) The cleavage of the urokinase receptor regulates its multiple functions. J. Biol. Chem. 277, 46932–46939 [DOI] [PubMed] [Google Scholar]
- 9. Andronicos N. M., Ranson M. (2001) The topology of plasminogen binding and activation on the surface of human breast cancer cells. Br. J. Cancer 85, 909–916 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Ellis V. (2003) Plasminogen activation at the cell surface. Curr. Top. Dev. Biol. 54, 263–312 [DOI] [PubMed] [Google Scholar]
- 11. Herren T., Swaisgood C., Plow E. F. (2003) Regulation of plasminogen receptors. Front. Biosci. 8, d1–8 [DOI] [PubMed] [Google Scholar]
- 12. Kudinov S. A. (1985) Ukr. Biokhim. Zh. 57, 23–35 [PubMed] [Google Scholar]
- 13. Miles L. A., Hawley S. B., Baik N., Andronicos N. M., Castellino F. J., Parmer R. J. (2005) Plasminogen receptors. The sine qua non of cell surface plasminogen activation. Front. Biosci. 10, 1754–1762 [DOI] [PubMed] [Google Scholar]
- 14. Brown J., Jones E. Y., Forbes B. E. (2009) Interactions of IGF-II with the IGF2R/cation-independent mannose 6-phosphate receptor mechanism and biological outcomes. Vitam. Horm. 80, 699–719 [DOI] [PubMed] [Google Scholar]
- 15. El-Shewy H. M., Luttrell L. M. (2009) Insulin-like growth factor-2/mannose-6 phosphate receptors. Vitam. Horm. 80, 667–697 [DOI] [PubMed] [Google Scholar]
- 16. Ghosh P., Dahms N. M., Kornfeld S. (2003) Mannose 6-phosphate receptors. New twists in the tale. Nat. Rev. Mol. Cell Biol. 4, 202–212 [DOI] [PubMed] [Google Scholar]
- 17. Godár S., Horejsi V., Weidle U. H., Binder B. R., Hansmann C., Stockinger H. (1999) M6P/IGFII receptor complexes urokinase receptor and plasminogen for activation of transforming growth factor-β1. Eur. J. Immunol. 29, 1004–1013 [DOI] [PubMed] [Google Scholar]
- 18. Leksa V., Godár S., Cebecauer M., Hilgert I., Breuss J., Weidle U. H., Horejsí V., Binder B. R., Stockinger H. (2002) The N terminus of mannose 6-phosphate/insulin-like growth factor 2 receptor in regulation of fibrinolysis and cell migration. J. Biol. Chem. 277, 40575–40582 [DOI] [PubMed] [Google Scholar]
- 19. Leksa V., Godar S., Schiller H. B., Fuertbauer E., Muhammad A., Slezakova K., Horejsi V., Steinlein P., Weidle U. H., Binder B. R., Stockinger H. (2005) TGF-β-induced apoptosis in endothelial cells mediated by M6P/IGFII-R and mini-plasminogen. J. Cell Sci. 118, 4577–4586 [DOI] [PubMed] [Google Scholar]
- 20. Olson L. J., Yammani R. D., Dahms N. M., Kim J. J. (2004) Structure of uPAR, plasminogen, and sugar-binding sites of the 300-kDa mannose 6-phosphate receptor. EMBO J. 23, 2019–2028 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Schiller H. B., Szekeres A., Binder B. R., Stockinger H., Leksa V. (2009) Mannose 6-phosphate/insulin-like growth factor 2 receptor limits cell invasion by controlling αVβ3 integrin expression and proteolytic processing of urokinase-type plasminogen activator receptor. Mol. Biol. Cell 20, 745–756 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Khatib A. M., Nip J., Fallavollita L., Lehmann M., Jensen G., Brodt P. (2001) Regulation of urokinase plasminogen activator/plasmin-mediated invasion of melanoma cells by the integrin vitronectin receptor αVβ3. Int. J. Cancer 91, 300–308 [DOI] [PubMed] [Google Scholar]
- 23. Tarui T., Akakura N., Majumdar M., Andronicos N., Takagi J., Mazar A. P., Bdeir K., Kuo A., Yarovoi S. V., Cines D. B., Takada Y. (2006) Direct interaction of the kringle domain of urokinase-type plasminogen activator (uPA) and integrin αvβ3 induces signal transduction and enhances plasminogen activation. Thromb. Haemost. 95, 524–534 [DOI] [PubMed] [Google Scholar]
- 24. Tarui T., Majumdar M., Miles L. A., Ruf W., Takada Y. (2002) Plasmin-induced migration of endothelial cells. A potential target for the anti-angiogenic action of angiostatin. J. Biol. Chem. 277, 33564–33570 [DOI] [PubMed] [Google Scholar]
- 25. Godár S., Leksa V., Cebecauer M., Hilgert I., Horejsi V., Stockinger H. (2002) in Leukocyte Typing VII (Mason D., Andre P., Bensussan A., Buckley C., Civin C., Clark E., de Haas M., Goyert S., Hadam M., Hart D., Horejsi V., Jones Y., Meuer S., Morrissey J., Schwarz-Albiez R., Shaw S., Simmons D., Turni L., Uguccioni M., van der Schoot E., Vivier E., Zola H., eds.) pp. 482–485, Oxford University Press, Oxford, New York, Tokyo [Google Scholar]
- 26. Baier G., Baier-Bitterlich G., Couture C., Telford D., Giampa L., Altman A. (1994) An efficient expression, purification, and immunodetection system for recombinant gene products Biotechniques 17, 94–99 [PubMed] [Google Scholar]
- 27. Swift S., Lorens J., Achacoso P., Nolan G. P. (1999) in Current Protocols in Immunology (Coligan J. E., Kruisbeek A. M., Margulies D. H., Shevach E. M., Strober W., eds) pp. 10.17.14–10.17.29, Green Publishing Associates and Wiley-Interscience, New York [Google Scholar]
- 28. Magdolen V., Rettenberger P., Lopens A., Oi H., Lottspeich F., Kellermann J., Creutzburg S., Goretzki L., Weidle U. H., Wilhelm O. (1995) Expression of the human urokinase-type plasminogen activator receptor in E. coli and Chinese hamster ovary cells. Purification of the recombinant proteins and generation of polyclonal antibodies in chicken. Electrophoresis 16, 813–816 [DOI] [PubMed] [Google Scholar]
- 29. Naldini L., Blömer U., Gallay P., Ory D., Mulligan R., Gage F. H., Verma I. M., Trono D. (1996) In vivo gene delivery and stable transduction of nondividing cells by a lentiviral vector. Science 272, 263–267 [DOI] [PubMed] [Google Scholar]
- 30. Schägger H., Cramer W. A., von Jagow G. (1994) Analysis of molecular masses and oligomeric states of protein complexes by blue native electrophoresis and isolation of membrane protein complexes by two-dimensional native electrophoresis. Anal. Biochem. 217, 220–230 [DOI] [PubMed] [Google Scholar]
- 31. Camacho-Carvajal M. M., Wollscheid B., Aebersold R., Steimle V., Schamel W. W. (2004) Two-dimensional blue native/SDS gel electrophoresis of multi-protein complexes from whole cellular lysates. A proteomics approach. Mol. Cell Proteomics 3, 176–182 [DOI] [PubMed] [Google Scholar]
- 32. McGuire P. G., Orkin R. W. (1992) Urokinase activity in the developing avian heart. A spatial and temporal analysis. Dev. Dyn. 193, 24–33 [DOI] [PubMed] [Google Scholar]
- 33. Zhang X., Chaudhry A., Chintala S. K. (2003) Inhibition of plasminogen activation protects against ganglion cell loss in a mouse model of retinal damage. Mol. Vis. 9, 238–248 [PubMed] [Google Scholar]
- 34. Simons K., Gerl M. J. (2010) Revitalizing membrane rafts. New tools and insights. Nat. Rev. Mol. Cell Biol. 11, 688–699 [DOI] [PubMed] [Google Scholar]
- 35. Otáhal P., Angelisová P., Hrdinka M., Brdicka T., Novák P., Drbal K., Horejsí V. (2010) A new type of membrane raft-like microdomains and their possible involvement in TCR signaling. J. Immunol. 184, 3689–3696 [DOI] [PubMed] [Google Scholar]
- 36. Caiolfa V. R., Zamai M., Malengo G., Andolfo A., Madsen C. D., Sutin J., Digman M. A., Gratton E., Blasi F., Sidenius N. (2007) Monomer dimer dynamics and distribution of GPI-anchored uPAR are determined by cell surface protein assemblies. J. Cell Biol. 179, 1067–1082 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Stepanova V. V., Tkachuk V. A. (2002) Urokinase as a multidomain protein and polyfunctional cell regulator. Biochemistry 67, 109–118 [DOI] [PubMed] [Google Scholar]
- 38. Damke H., Klumperman J., von Figura K., Braulke T. (1991) Effects of brefeldin A on the endocytic route. Redistribution of mannose 6-phosphate/insulin-like growth factor II receptors to the cell surface. J. Biol. Chem. 266, 24829–24833 [PubMed] [Google Scholar]
- 39. Lin S. X., Mallet W. G., Huang A. Y., Maxfield F. R. (2004) Endocytosed cation-independent mannose 6-phosphate receptor traffics via the endocytic recycling compartment en route to the trans-Golgi network and a subpopulation of late endosomes. Mol. Biol. Cell 15, 721–733 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Ghosh R. N., Gelman D. L., Maxfield F. R. (1994) Quantification of low density lipoprotein and transferrin endocytic sorting HEp2 cells using confocal microscopy. J. Cell Sci. 107, 2177–2189 [DOI] [PubMed] [Google Scholar]
- 41. Leksa V., Loewe R., Binder B., Schiller H. B., Eckerstorfer P., Forster F., Soler-Cardona A., Ondrovicová G., Kutejová E., Steinhuber E., Breuss J., Drach J., Petzelbauer P., Binder B. R., Stockinger H. (2011) Soluble M6P-IGF2R released by TACE controls angiogenesis via blocking plasminogen activation. Circ. Res. 108, 676–685 [DOI] [PubMed] [Google Scholar]
- 42. Brameshuber M., Weghuber J., Ruprecht V., Gombos I., Horváth I., Vigh L., Eckerstorfer P., Kiss E., Stockinger H., Schütz G. J. (2010) Imaging of mobile long-lived nanoplatforms in the live cell plasma membrane. J. Biol. Chem. 285, 41765–41771 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Drbal K., Moertelmaier M., Holzhauser C., Muhammad A., Fuertbauer E., Howorka S., Hinterberger M., Stockinger H., Schütz G. J. (2007) Single-molecule microscopy reveals heterogeneous dynamics of lipid raft components upon TCR engagement. Int. Immunol. 19, 675–684 [DOI] [PubMed] [Google Scholar]
- 44. Rubino M., Miaczynska M., Lippé R., Zerial M. (2000) Selective membrane recruitment of EEA1 suggests a role in directional transport of clathrin-coated vesicles to early endosomes. J. Biol. Chem. 275, 3745–3748 [DOI] [PubMed] [Google Scholar]
- 45. Claus V., Jahraus A., Tjelle T., Berg T., Kirschke H., Faulstich H., Griffiths G. (1998) Lysosomal enzyme trafficking between phagosomes, endosomes, and lysosomes in J774 macrophages. Enrichment of cathepsin H in early endosomes. J. Biol. Chem. 273, 9842–9851 [DOI] [PubMed] [Google Scholar]
- 46. Pillay C. S., Elliott E., Dennison C. (2002) Endolysosomal proteolysis and its regulation. Biochem. J. 363, 417–429 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Tjelle T. E., Brech A., Juvet L. K., Griffiths G., Berg T. (1996) Isolation and characterization of early endosomes, late endosomes, and terminal lysosomes. Their role in protein degradation. J. Cell Sci. 109, 2905–2914 [DOI] [PubMed] [Google Scholar]
- 48. Cortese K., Sahores M., Madsen C. D., Tacchetti C., Blasi F. (2008) Clathrin and LRP-1-independent constitutive endocytosis and recycling of uPAR. PLoS One 3, e3730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Czekay R. P., Kuemmel T. A., Orlando R. A., Farquhar M. G. (2001) Direct binding of occupied urokinase receptor (uPAR) to LDL receptor-related protein is required for endocytosis of uPAR and regulation of cell surface urokinase activity. Mol. Biol. Cell 12, 1467–1479 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Herz J., Strickland D. K. (2001) LRP. A multifunctional scavenger and signaling receptor. J. Clin. Invest. 108, 779–784 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Gonias S. L., Gaultier A., Jo M. (2011) Regulation of the urokinase receptor (uPAR) by LDL receptor-related protein-1 (LRP1). Curr. Pharm. Des. 17, 1962–1969 [DOI] [PubMed] [Google Scholar]
- 52. Engelholm L. H., Nielsen B. S., Danø K., Behrendt N. (2001) The urokinase receptor associated protein (uPARAP/endo180). A novel internalization receptor connected to the plasminogen activation system. Trends Cardiovasc. Med. 11, 7–13 [DOI] [PubMed] [Google Scholar]
- 53. Nykjaer A., Christensen E. I., Vorum H., Hager H., Petersen C. M., Røigaard H., Min H. Y., Vilhardt F., Møller L. B., Kornfeld S., Gliemann J. (1998) Mannose 6-phosphate/insulin-like growth factor-II receptor targets the urokinase receptor to lysosomes via a novel binding interaction. J. Cell Biol. 141, 815–828 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Prager G. W., Breuss J. M., Steurer S., Olcaydu D., Mihaly J., Brunner P. M., Stockinger H., Binder B. R. (2004) Vascular endothelial growth factor receptor-2-induced initial endothelial cell migration depends on the presence of the urokinase receptor. Circ. Res. 94, 1562–1570 [DOI] [PubMed] [Google Scholar]
- 55. Gonias S. L., Hembrough T. A., Sankovic M. (2001) Cytokeratin 8 functions as a major plasminogen receptor in select epithelial and carcinoma cells. Front. Biosci. 6, D1403–D1411 [DOI] [PubMed] [Google Scholar]
- 56. Hajjar K. A., Jacovina A. T., Chacko J. (1994) An endothelial cell receptor for plasminogen/tissue plasminogen activator. I. Identity with annexin II. J. Biol. Chem. 269, 21191–21197 [PubMed] [Google Scholar]
- 57. Gonzalez-Gronow M., Kaczowka S., Gawdi G., Pizzo S. V. (2008) Dipeptidyl peptidase IV (DPP IV/CD26) is a cell-surface plasminogen receptor. Front. Biosci. 13, 1610–1618 [DOI] [PubMed] [Google Scholar]
- 58. Phipps K. D., Surette A. P., O'Connell P. A., Waisman D. M. (2011) Plasminogen receptor S100A10 is essential for the migration of tumor-promoting macrophages into tumor sites. Cancer Res. 71, 6676–6683 [DOI] [PubMed] [Google Scholar]
- 59. Fischer M. B., Roeckl C., Parizek P., Schwarz H. P., Aguzzi A. (2000) Binding of disease-associated prion protein to plasminogen. Nature 408, 479–483 [DOI] [PubMed] [Google Scholar]
- 60. Rijken D. C., Lijnen H. R. (2009) New insights into the molecular mechanisms of the fibrinolytic system. J. Thromb. Haemost. 7, 4–13 [DOI] [PubMed] [Google Scholar]
- 61. Ellis V., Whawell S. A., Werner F., Deadman J. J. (1999) Assembly of urokinase receptor-mediated plasminogen activation complexes involves direct, non-active-site interactions between urokinase and plasminogen. Biochemistry 38, 651–659 [DOI] [PubMed] [Google Scholar]
- 62. Stillfried G. E., Saunders D. N., Ranson M. (2007) Plasminogen binding and activation at the breast cancer cell surface. The integral role of urokinase activity. Breast Cancer Res. 9, R14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Argraves K. M., Kozarsky K. F., Fallon J. T., Harpel P. C., Strickland D. K. (1997) The atherogenic lipoprotein Lp(a) is internalized and degraded in a process mediated by the VLDL receptor. J. Clin. Invest. 100, 2170–2181 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Gonzalez-Gronow M., Enghild J. J., Pizzo S. V. (1993) Streptokinase and human fibronectin share a common epitope. Implications for regulation of fibrinolysis and rheumatoid arthritis. Biochim. Biophys. Acta 1180, 283–288 [DOI] [PubMed] [Google Scholar]
- 65. Bohnsack R. N., Patel M., Olson L. J., Twining S. S., Dahms N. M. (2010) Residues essential for plasminogen binding by the cation-independent mannose 6-phosphate receptor. Biochemistry 49, 635–644 [DOI] [PMC free article] [PubMed] [Google Scholar]