

Background: The adenovirus E4-ORF3 protein disrupts PML nuclear bodies to inhibit antiviral activity.
Results: The WT E4-ORF3 protein forms higher-order multimers, whereas a nonfunctional mutant forms a dimer.
Conclusion: E4-ORF3 protein multimerization likely is required for the activity of this protein.
Significance: These results provide new insight into the properties of the adenovirus E4-ORF3 protein and suggest that higher-order protein multimerization is essential for activity.
Keywords: Adenovirus, Host-pathogen Interactions, Nuclear Organization, Protein Purification, Protein-protein Interactions
Abstract
The early region 4 open reading frame 3 protein (E4-ORF3; UniProt ID P04489) is the most highly conserved of all adenovirus-encoded gene products at the amino acid level. A conserved attribute of the E4-ORF3 proteins of different human adenoviruses is the ability to disrupt PML nuclear bodies from their normally punctate appearance into heterogeneous filamentous structures. This E4-ORF3 activity correlates with the inhibition of PML-mediated antiviral activity. The mechanism of E4-ORF3-mediated reorganization of PML nuclear bodies is unknown. Biophysical analysis of the purified WT E4-ORF3 protein revealed an ordered secondary/tertiary structure and the ability to form heterogeneous higher-order multimers in solution. Importantly, a nonfunctional E4-ORF3 mutant protein, L103A, forms a stable dimer with WT secondary structure content. Because the L103A mutant is incapable of PML reorganization, this result suggests that higher-order multimerization of E4-ORF3 may be required for the activity of the protein. In support of this hypothesis, we demonstrate that the E4-ORF3 L103A mutant protein acts as a dominant-negative effector when coexpressed with the WT E4-ORF3 in mammalian cells. It prevents WT E4-ORF3-mediated PML track formation presumably by binding to the WT protein and inhibiting the formation of higher-order multimers. In vitro protein binding studies support this conclusion as demonstrated by copurification of coexpressed WT and L103A proteins in Escherichia coli and coimmunoprecipitation of WT·L103A E4-ORF3 complexes in mammalian cells. These results provide new insight into the properties of the Ad E4-ORF3 protein and suggest that higher-order protein multimerization is essential for E4-ORF3 activity.
Introduction
PML nuclear bodies (PML-NB)5 are punctate multiprotein complexes linked to a variety of important cellular processes, including transcriptional regulation, cellular growth control, DNA damage repair, apoptosis, and response to interferons (1). A number of DNA and RNA viruses disrupt PML-NB organization upon virus infection to inhibit the antiviral activities of these structures. The mechanisms by which this is accomplished involves different processes including proteasome-dependent degradation of the PML protein (herpes simplex virus type 1) or relocalization/reorganization of the PML protein (adenovirus (Ad) and human cytomegalovirus) (2). Following Ad infection, PML-NB are relocalized within the nucleus by the E4-ORF3 protein into heterogeneous filamentous structures termed “tracks” (3). The E4-ORF3 protein recruits a number of cellular proteins into nuclear tracks (e.g. PML and Daxx) to inhibit their antiviral effects (4, 5). E4-ORF3 also relocalizes two PML-NB-associated cellular transcription factors, TIF1α and TIF1γ, into tracks perhaps related to the regulation of cellular gene expression (6–8). Although the E4-ORF3 protein shares a common localization with PML in the nuclear matrix fraction isolated from Ad-infected cells (9–11), the mechanism underlying PML-NB reorganization by E4-ORF3 is not known and it is not understood how E4-ORF3 recruits other cellular proteins into these structures. The size (>1 μm) and appearance of the E4-ORF3-dependent tracks are consistent with the hypothesis that the E4-ORF3 protein multimerizes into higher-order structures.
Viruses with linear, dsDNA genomes, such as Ad, encounter a number of host cell responses that may severely inhibit virus replication (12). The open ends of the linear viral genomes are sensed by the host cell as broken DNA, which triggers a cellular DNA damage response (DDR) (13). The DDR severely inhibits Ad DNA replication if unabated (13) because the ends of the viral genome are ligated to one another via the non-homologous end-joining pathway. This results in the loss of DNA sequences at the multimeric junctions (14, 15), which contain the Ad origins of DNA replication. In addition, cytoplasmic sensors recognize dsDNA in the cytoplasm of infected cells early after virus infection and activate an IFN response (16). The IFN response can block virus replication by multiple mechanisms, including the inhibition of viral gene transcription and viral DNA replication. It is very important for dsDNA viruses to counteract these antiviral host cell responses early after virus infection in order for a productive replication cycle to ensue.
Ad has evolved two redundant mechanisms to inhibit a cellular DDR (13). The Ad5 E4-ORF3 protein relocalizes nuclear proteins involved in a DDR, including Mre11, Rad50, and Nbs1 (the MRN complex) into the track structures (17, 18). The available evidence suggests that E4-ORF3 inhibits the functions of these DNA repair proteins by a sequestration mechanism to block their access to the viral genome (17–21). The Ad5 E1B-55K·E4-ORF6 complex functions as an adaptor molecule in an E3 ubiquitin ligase complex (22, 23). E1B-55K·E4-ORF6 target the MRN proteins, as well as DNA ligase IV and Blm helicase, for inactivation via ubiquitin-mediated, proteasome-dependent degradation (18, 24, 25). The E4-ORF3 protein facilitates this process by promoting the transport of the MRN proteins to cytoplasmic aggresomes (26, 27). Either mechanism alone is sufficient to inhibit a DDR and allow efficient Ad DNA replication to occur. Finally, the E4-ORF3 protein inhibits p53-induced gene expression by establishing heterochromatin at p53-responsive cellular promoter regions (28). The E4-ORF3 and E1B-55K·E4-ORF6 proteins are multifunctional and play additional roles in the viral life cycle, including the promotion of cell cycle-independent viral replication, the regulation of viral late mRNA splicing and cytoplasmic mRNA accumulation, and the regulation of late protein translation (29–37). Mechanisms underlying the roles of these Ad proteins in the aforementioned processes are poorly understood.
To probe the properties of the E4-ORF3 protein, we established a method to express soluble E4-ORF3 protein in Escherichia coli and purify the protein to near-homogeneity utilizing the solubility properties of the protein in buffer containing sodium chloride. CD spectroscopy and coprecipitation experiments were used to demonstrate that the recombinant protein is both stably and correctly folded. We examined the biophysical properties of the WT E4-ORF3 protein and a non-functional mutant protein, L103A (17), in vitro. Our results demonstrate that the WT E4-ORF3 protein forms heterogeneous multimers in solution. In contrast, the L103A mutant protein forms a stable dimer. Both proteins are folded similarly and demonstrate equivalent secondary and tertiary structural characteristics, as demonstrated by CD and fluorescence emission spectroscopy. In addition, the presence of a well folded tertiary structure was confirmed by near-UV CD spectroscopy for both proteins, and by one-dimensional NMR of the L103A mutant protein. These data demonstrate that the L103A loss of function is not due to improper folding and is consistent with the idea that the dimer formed by the L103A protein represents an intermediate in higher-order protein assembly. We propose that the L103A mutant protein is nonfunctional because it is largely trapped in a dimeric state and deficient in higher-order protein multimerization required for track formation. A prediction is that the L103A mutant protein could dimerize with WT E4-ORF3 and prevent further multimerization. In support of this hypothesis, expression of the L103A protein in vivo was found to block nuclear track formation and PML-NB reorganization by the WT E4-ORF3 protein and the WT and L103A protein were found to interact in vitro. In addition, the L103A mutant proteins interfered with the interaction of WT E4-ORF3 with the cellular binding partner TIF1γ. These results provide new insight into the properties of the multifunctional E4-ORF3 protein and suggest that higher-order multimerization may be essential for E4-ORF3 activity.
EXPERIMENTAL PROCEDURES
Plasmid Constructs and Purification of E4-ORF3
The complete experimental details are provided in supplemental “Experimental Procedures.”
In Vitro Binding Analyses
Whole cell extracts of N52.E6 cells (a cell line that expresses the Ad5 E1A and E1B proteins (38); and HeLa cells were prepared by cell lysis in F lysis buffer (10 mm Tris-HCl, 50 mm NaCl, 10% glycerol, 0.5% Triton X-100, 5 μm ZnCl2, pH 7.5) on ice for 20 min followed by clarification for 30 min at 20,000 × g at 4 °C. 2 mg whole cell extract was incubated in 650 μl of F lysis buffer with E4-ORF3 WT or L103A protein at 7.5 μm on ice for 3 h, followed by the addition of anti-E1B-55K (39); N52.E6 cell extract or anti-TIF1γ (7); HeLa cell extract rabbit polyclonal antiserum and protein A-agarose beads. The samples were rotated at 4 °C overnight and then washed five times with F lysis buffer at 4 °C. The final protein A precipitates were boiled in SDS sample buffer containing 50 mm dithiothreitol for 10 min. Samples were separated by SDS-PAGE and analyzed by Western blot using anti-E1B-55K (39), anti-TIF1γ (7), or anti-E4-ORF3 (40) antibodies. Western blots were analyzed by enhanced chemiluminescence (Amersham Biosciences) according to the manufacturer's instructions. The same approach was used to characterize the binding of WT E4-ORF3 and the L103A mutant protein to TIF1γ following Ad infection. HeLa cells were infected with recombinant Ad5 E1-replacement vectors (41) that expressed either FLAG-tagged WT E4-ORF3 protein, HA-tagged L103A mutant E4-ORF3 protein, or with both viruses simultaneously, at the multiplicity of infection described in the text. Whole cell extracts were prepared 24 h after infection using F lysis buffer and sonication. WT E4-ORF3 was immunoprecipitated using anti-FLAG monoclonal antibody M2 (Sigma-Aldrich); TIF1γ was immunoprecitated using anti-TIF1γ antibody (7). WT E4-ORF3 and L103A coimmunoprecipitation was analyzed using anti-E4-ORF3 antibody (40), as described above.
Analytical Ultracentrifugation
Sedimentation equilibrium experiments were conducted at 25 °C at three rotor speeds (12,000, 18,000, and 32,000 rpm) on a Beckman Optima XL-A analytical ultracentrifuge. Typically, three samples at concentrations of 40, 20, and 10 μm (A280 of 0.6, 0.3, 0.15) in 10 mm Tris-HCl, 0.5 mm TCEP (buffer A) were prepared. The partial specific volume of 0.7403 cm3/g was calculated based on the amino acid composition, and the solvent density of 0.9985 g/cm3 was estimated from standard tables (10 mm Tris-HCl). Data were analyzed using HeteroAnalysis software (University of Connecticut Analytical Ultracentrifugation Facility). The data were fit globally across samples and speeds to yield the apparent molar mass.
Dynamic Light Scattering
Measurements were carried out using a Brookhaven Instruments 90Plus Particle Size Analyzer at a wavelength of 659 nm. For the L103A mutant protein, the NMR sample of ∼400 μm protein concentration was used (see below). For WT E4-ORF3, fractions A, B, and C eluting near the void volume of the Superdex 75 26/60 column were studied (see “Results”); the protein concentration of these samples was estimated to be 10, 15, and 10 μm, respectively (A280 of 0.142, 0.223, and 0.154). Ten data collection runs of 1–2 min each were acquired and combined; data were analyzed with the method of cumulants to yield a log-normal distribution of particle sizes, per the manufacturer's instructions.
Fluorescence Spectroscopy
Intrinsic tryptophan fluorescence spectra were collected at 25 °C on a Photon Technology International spectrofluorometer. The protein concentration was 12 μm (A280 of 0.18) in buffer A. Fluorescence emission spectra (310–400 nm) were collected upon selective excitation of the Trp fluorophore at 290 nm. Fluorescence excitation spectra (emission monitored at 350 nm) were collected upon excitation at a range of 250–330 nm. Quenching studies were carried out as a titration of increasing concentration of acrylamide dissolved in buffer A; the final fluorescence intensity was corrected for dilution effects. The excitation wavelength was set to 295 nm. Data were normalized by the fluorescence intensity in the absence of quencher and analyzed by linear regression to give the Stern-Volmer constant.
Circular Dichroism Spectroscopy
Measurements were carried out at 25 °C using a Chiroscan spectrometer (Applied Photophysics) equipped with a Peltier temperature controller. Far-ultraviolet spectra were collected from 190–260 nm with sample concentrations of 10 μm and path length of 1 mm. Near-ultraviolet spectra were collected from 260–340 nm on samples at 60–100 μm concentration and path length of 10 mm. In both cases, five spectra were collected (1 nm increments, 2 s/nm), averaged, and smoothed using a Savitzky-Golay filter to yield the final result.
NMR Spectroscopy
NMR spectra were recorded at 293 K on a Varian Inova 600 MHz spectrometer using a presaturation pulse sequence. The dimeric fraction of L103A protein was concentrated to ∼400 μm by ultrafiltration, and D2O was added to 10%. The spectra were visualized using MestReNova NMR software.
SAXS Experiments
Scattering experiments were performed at beamline X9 at Brookhaven National Laboratory, National Synchrotron Light Source I (Upton, New York). Protein samples were injected to flow through a 1-mm diameter capillary continuously during the measurement, at a rate of 0.67 μl/s to avoid radiation damage. The exposure time for each measurement was 30 s. Each sample was measured three times, and the results were averaged before data analysis. The program PRIMUS (42) was used for buffer subtraction, and the radius of gyration (Rg) was obtained by the Guinier approximation (43) I(q) = I(0) exp(−Rg2q2/3), where I is the intensity at scattering angle q.
In Vivo Analyses of E4-ORF3 Localization
A pcDNA3 vector that expresses WT E4-ORF3 fused to an N-terminal His6 tag plus TEV protease cleavage site was transfected into HeLa cells (ATCC) on glass coverslips for immunofluorescence (IF) assays using Nanojuice (Novagen) according to the manufacturer's instructions. Transfected cells were washed with PBS 24 h after transfection, fixed using 100% methanol at −20 °C for 5 min, washed with PBS, and blocked with PBS containing 10% goat serum for 1 h at room temperature. The coverslips were incubated with anti-E4-ORF3 rat monoclonal primary antibody (40) for 1 h at room temperature. The coverslips were then washed with PBS, incubated with FITC-conjugated anti-rat secondary antibody (Molecular Probes) for 45 min, and washed with PBS, and the coverslips were mounted on slides using Immumount (Thermo Shandon). For E4-ORF3 localization analyses following virus infection, HeLa cells on glass coverslips were infected with recombinant Ad5 E1-replacement vectors (41) that expressed either FLAG epitope-tagged WT E4-ORF3 protein, HA epitope-tagged L103A mutant E4-ORF3 protein, or with both viruses simultaneously, at the multiplicity of infection described in the text. 24 h after infection, cells were processed for IF as described above using antibodies directed against the epitope tags (rabbit anti-HA, Rockland Immunochemicals, and mouse anti-FLAG M2, Sigma-Aldrich), or anti-PML rabbit antibody (Santa Cruz Biotechnology) and anti-E4-ORF3 monoclonal antibody 6A11 (40), followed by the addition of FITC- and TRITC-conjugated secondary antibodies. Images were captured with a Zeiss digital deconvolution microscope equipped with Apotome and Axiovision software (version 4.5).
RESULTS
Solubility of Recombinant E4-ORF3 Protein Is Affected by Ionic Strength and May Be Exploited for Purification
Initial E4-ORF3 protein purification approaches utilized the IMPACT-CN system where the WT E4-ORF3 protein was expressed in E. coli as a bipartite fusion protein containing a self-splicing intein and a chitin binding domain. E4-ORF3 protein was largely insoluble when extracts were prepared using buffers that contained ≥500 mm NaCl. A significant portion of WT E4-ORF3 protein, however, was soluble in buffer containing only 20 mm sodium phosphate, 10 mm 2-mercaptoethanol, pH 7.5 (Fig. 1A). Intermediate levels of E4-ORF3 protein solubility were observed in buffers containing 50–500 mm NaCl (Fig. 1A). Given these results, a novel purification strategy was implemented. WT E4-ORF3 was cloned into bacterial vectors for the expression of His6-tagged protein. Recombinant E4-ORF3 expression was induced and E. coli were cultured overnight at 25 °C. E. coli extracts were prepared by sonication in buffer containing 10 mm Tris-HCl, 10 mm 2-mercaptoethanol, pH 7.5. E4-ORF3 protein was salted out by the addition of NaCl to 100 mm and centrifugation. Upon resuspension in lysis buffer at pH 8.5, a significant amount of the E4-ORF3 protein was resolubilized (Fig. 1B). Given the low salt conditions used for E4-ORF3 precipitation, very few bacterial proteins were precipitated. Once resuspended, the His6-tagged protein was purified using a Ni2+-NTA column (Fig. 1C) and subsequently by gel filtration. The addition of NaCl provided a simple and efficient purification step for E4-ORF3 proteins from E. coli cell extracts. The solubility profile of the recombinantly expressed, and purified WT E4-ORF3 protein was very similar to that observed with E4-ORF3 in E. coli cell extracts (Fig. 1D). However, the solubility profile of recombinant E4-ORF3 differed from that found when the protein was expressed in mammalian cells; WT E4-ORF3 protein expressed in HeLa cells was equally soluble in buffer containing 0–150 mm NaCl following sonication as observed by Western blot analysis (data not shown). This may be due to lower levels of expression achieved in mammalian cells.
FIGURE 1.
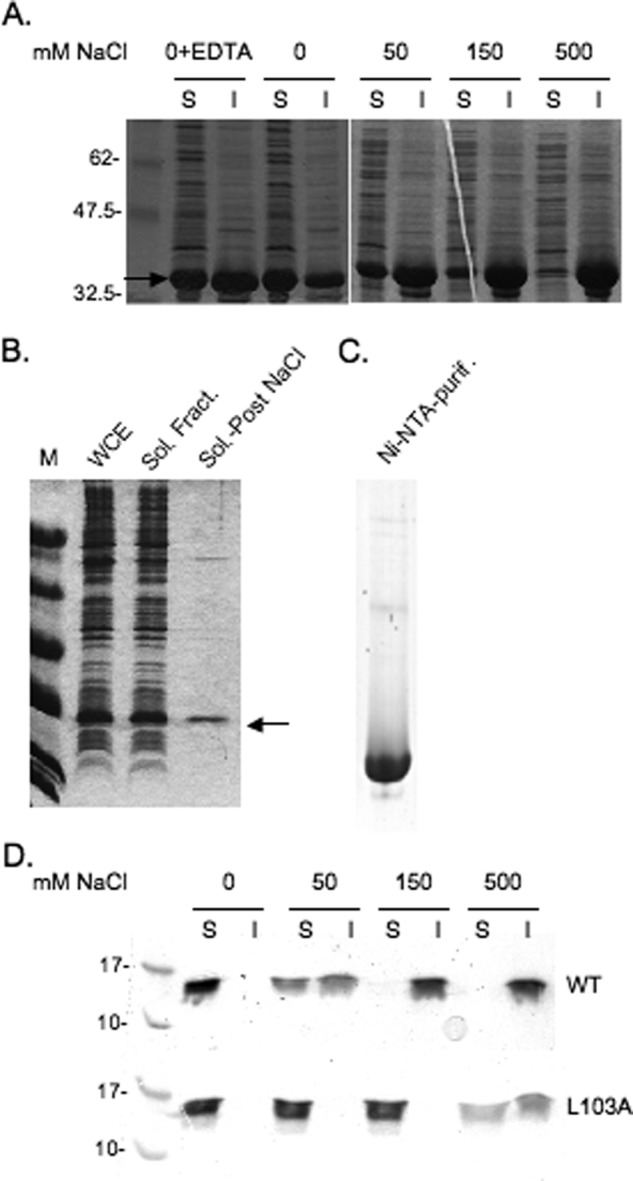
Recombinant E4-ORF3 protein solubility and purification. A, cell pellets from 10-ml cultures of E. coli induced for the expression of recombinant WT E4-ORF3 protein were resuspended in lysis buffer containing 20 mm sodium phosphate, pH 7.5, and different concentrations of NaCl, or containing no NaCl and 10 mm EDTA, as indicated in the figure. Cells were lysed by sonication and clarified by centrifugation prior to analysis by SDS-PAGE. The soluble fraction (S) was mixed 1:1 with SDS sample buffer and boiled. The insoluble fraction (I) was resuspended in SDS sample buffer and boiled. Equal amounts of the soluble and insoluble fractions were analyzed by SDS-PAGE and Coomassie Blue staining. The E4-ORF3 protein is indicated by an arrow. B, cell pellets from a 2-liter culture of E. coli induced for the expression of recombinant E4-ORF3 WT protein were extracted as described under the supplemental “Experimental Procedures.” Equal amounts of the whole cell extract (WCE), soluble fraction (Sol. Fract.), and soluble fraction following a salting out step (Sol.-Post NaCl) were analyzed by SDS-PAGE and Coomassie Blue staining. The E4-ORF3 protein is indicated by an arrow. C, 6.5 μg of E4-ORF3 WT protein purified using a Ni2+-NTA column (Ni-NTA-purif.) was analyzed by SDS-PAGE and Coomassie Blue staining. D, purified WT E4-ORF3 and the L103A mutant protein were incubated at 20 μm in buffer, pH 7.5, containing different concentrations of NaCl, as indicated in the figure, on ice for 1 h. Samples were clarified by centrifugation prior to analysis by SDS-PAGE, as described in A.
His6-TEV-tagged E4-ORF3 Protein Is Functional in Vivo and in Vitro
During Ad infection, the WT E4-ORF3 protein forms filamentous nuclear structures referred to as tracks; these structures are heterogenous in an infected cell population (3). To examine whether the His6-TEV tags present at the N terminus of the recombinant E4-ORF3 protein expressed in E. coli affected E4-ORF3 activity in mammalian cells, the localization of untagged E4-ORF3 expressed during Ad5 infection was compared with that of His6-TEV-tagged E4-ORF3 protein expressed by transfection. Both proteins displayed comparable nuclear tracks with the characteristic filamentous structures formed by the E4-ORF3 protein when examined by IF microscopy (Fig. 2, A and B). We conclude that an N-terminal His6-TEV tag on WT E4-ORF3 does not interfere with protein localization in vivo. To determine whether recombinant WT E4-ORF3 was properly folded to allow for bona fide protein-protein interaction, coimmunoprecipitation assays were performed. Recombinant WT and the non-functional mutant L103A (17) E4-ORF3 proteins were purified and added individually to cellular extracts prepared from HeLa cells or N52.E6 cells (as sources of the known E4-ORF3 binding partners TIF1γ (6–8) and Ad5 E1B-55K (40), respectively). The TIF1γ and E1B-55K proteins were immunoprecipitated using specific antibodies, and coprecipitation of recombinant E4-ORF3 proteins was examined by Western blot (Fig. 2C, IP). These results demonstrated that WT E4-ORF3 protein purified by these methods coprecipitated with the E4-ORF3 binding partners TIF1γ and E1B-55K, indicating that the protein is properly folded. In contrast, the L103A mutant protein did not interact with TIF1γ and E1B-55K in vitro.
FIGURE 2.
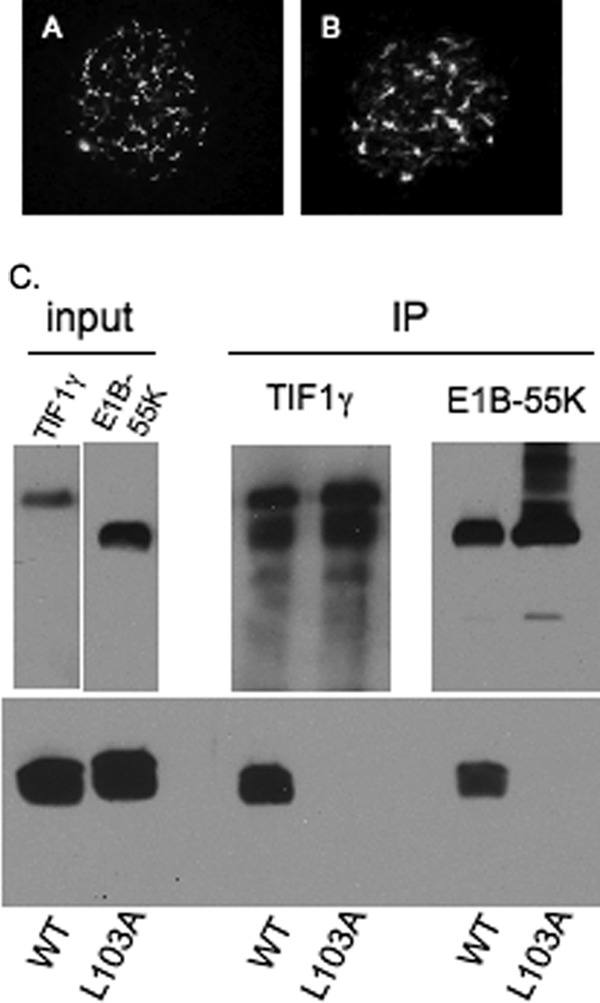
His6-TEV-tagged E4-ORF3 protein is functional. A, untagged WT E4-ORF3 protein was expressed by Ad5 infection of HeLa cells, and subcellular localization was analyzed by IF microscopy. The image is of one cell with E4-ORF3 tracks evident within the nucleus as filamentous structures. B, His6-TEV-tagged WT E4-ORF3 protein was expressed by transient transfection of HeLa cells and subcellular localization analyzed by IF microscopy as in A. C, the binding of E4-ORF3 WT and L103A proteins to TIF1γ and E1B-55K was analyzed by coimmunoprecipitation. Purified recombinant E4-ORF3 WT and L103A proteins were added to cell extracts prepared from HeLa cells (for TIF1γ) or N52.E6 cells (for E1B-55K), and immunoprecipitations were performed using anti-TIF1γ- or anti-E1B-55K-specific antibodies. TIF1γ, E1B-55K, and the E4-ORF3 proteins were analyzed by Western blot using specific antibodies. Input, TIF1γ present in HeLa cell extract, and E1B-55K present in N52.E6 cell extract (upper panels, left), and recombinant, purified E4-ORF3 WT and L103A proteins (lower panel, left). IP, immunoprecipitated TIF1γ and E1B-55K (upper panels, right) and coimmunoprecipitated E4-ORF3 WT and L103A proteins (lower panel, right). Due to its small size, E4-ORF3 in some SDS-PAGE gels smears toward the gel front.
Hydrodynamic Characterization of E4-ORF3 Protein
We noticed that the purified L103A protein was more resistant to NaCl precipitation than WT E4-ORF3 (Fig. 1D). This observation, in addition to the inability of L103A to bind to TIF1γ and E1B-55K in vitro, suggested that the L103A mutation alters the biophysical properties of recombinant E4-ORF3 protein. To investigate this further, we turned to a detailed biophysical investigation of the WT and L103A E4-ORF3 proteins. Comparison of the recombinant WT E4-ORF3 and the L103A proteins revealed that the altered functional properties of the two proteins in vivo may be attributable to a difference in their oligomerization state. We observed that the Superdex 200 size exclusion elution profiles of the two proteins differed significantly (supplemental Fig. 1A). The WT protein elution profile was broad while L103A eluted as a well-separated and highly-abundant symmetric peak at 75 ml, suggesting that L103A may exist as a monodisperse molecule. When chromatographed on a Superdex 75 column, the WT E4-ORF3 protein eluted as an asymmetric peak near the column void volume of 110 ml (supplemental Fig. 1, A and B). Both proteins eluted earlier than is expected for a 16.2-kDa globular protein, suggesting that the proteins oligomerize. In addition, the earlier elution times of WT E4-ORF3 suggested that it forms larger oligomeric assemblies than the mutant L103A protein. Three fractions from the WT E4-ORF3 near-void peak were chosen for further characterization (see below; fractions A, 112–116 ml; B, 120–124 ml; and C, 128–132 ml; supplemental Fig. 1B). To test the hypothesis that recombinant WT and L103A E4-ORF3 proteins form oligomers, we set out to quantitatively evaluate their association state. Sedimentation equilibrium analytical ultracentrifugation (AUC) experiments showed that the WT E4-ORF3 protein is heterogeneous in solution (Fig. 3A). The equilibrium heterogeneity is consistent with the size exclusion profiles of the protein eluting from both Superdex 200 and Superdex 75 columns (supplemental Fig. 1A). Attempts to deconvolve the sedimentation profiles using an equilibrium model of discrete oligomeric assembly (monomer/N-mer or monomer/M-mer/N-mer) were not successful. In contrast to WT E4-ORF3, the equilibrium sedimentation of the L103A mutant protein was well described as a single ideal solute with apparent molecular weight of 30.3 kDa, as judged by the randomness of the residuals and the linearity of the transformed data (Fig. 3A). The theoretical mass of L103A calculated from its sequence is 16.2 kDa; the AUC-estimated molecular weight is within 7% of a homodimer (32.4 kDa), within the experimental error of our instrument. This indicated that L103A exists as a dimer at μm concentrations. In addition, when L103A was chromatographed at high concentrations, we noticed an appearance of a left shoulder to the dimeric peak. Using sedimentation equilibrium AUC, we found that fractions in this shoulder were well fit to an ideal solute model with molecular weights corresponding to dimer-of-dimers and trimer-of-dimers species (data not shown).
FIGURE 3.

Hydrodynamic characterization of the E4-ORF3 WT and L103A proteins. A, sedimentation equilibrium data of the E4-ORF3 WT and L103A proteins. Molecular heterogeneity is evidenced by curvature of the sedimentation profile when log(A280) is plotted versus squared radius. A least-squares regression line (gray) is displayed for each data set; the residuals for the L103A protein are plotted in gray near y = 0. The residuals of WT E4-ORF3 are clearly not random. B, dynamic light scattering properties of the E4-ORF3 WT (fractions A, B, and C; supplemental Fig. 1B) and L103A proteins. Distribution function intensities versus effective particle diameter, derived from dynamic light scattering, indicate that WT E4-ORF3 assembles into larger oligomers than L103A. Note the logarithmic spacing of the x axis. C, Guinier analysis of SAXS of WT and L103A. Least-squares linear regression fits of the linear portions of the two data sets are shown as solid gray lines. D, Kratky plots of the data in C show a peak in their profiles, indicating globular structures for both WT and L103A E4-ORF3 proteins.
Although size exclusion chromatography can be used to estimate the size of proteins, ionic interactions with the column affect solute progress unless a significantly high salt concentration is included to screen them. The salt sensitivity of E4-ORF3 prevents this. As an alternative, light scattering techniques provide a way to estimate the size of particles in solution and are less sensitive to ionic strength effects. To test the hypothesis that WT E4-ORF3 forms larger oligomers than L103A, we analyzed three fractions of WT E4-ORF3 (fractions A, B, and C; supplemental Fig. 1B) and the dimeric L103A sample (supplemental Fig. 1A) by dynamic light scattering. The quasielastic light scattering intensity has a direct dependence on particle size. As a consequence, larger particles can be studied at significantly lower concentrations than smaller particles. We found that it was necessary to concentrate dimeric L103A samples to achieve adequate light scattering intensity. The polydispersity increased slightly when the L103A mutant was concentrated but remained a small percentage of the total signal (confirmed by sedimentation equilibrium AUC, data not shown). The effective hydrodynamic diameter of L103A was found to be 4.4 nm. In contrast to L103A, fractions of WT E4-ORF3 could be characterized at low μm concentrations. Dynamic light scattering revealed that the WT E4-ORF3 protein formed particles significantly larger than those present in the L103A sample (Fig. 3B). Effective hydrodynamic diameters of 62.6, 18.2, and 5.9 nm were estimated for the three WT E4-ORF3 fractions. It is important to note that smaller hydrodynamic diameter was observed for the L103A protein even at a 40-fold molar excess over the WT protein. These data demonstrate that the L103A dimeric state cannot simply be overcome by concentration effects.
In addition, we used small angle x-ray scattering (SAXS) to examine the solution conformation of WT and L103A proteins. SAXS is a technique that is sensitive to the overall shape and size of a molecule. The scattering profiles collected for the two proteins differed significantly (Fig. 3C). Inspection of the small-angle region using the Guinier representation indicated molecular heterogeneity for the WT protein, as evidenced by data curvature. In contrast, the L103A protein produced a linear profile in the small-angle region, from which we extracted the radius of gyration (Rg) of 22.8 Å (2.2 nm) using the Guinier approximation. The determined Rg is within the range observed for other proteins of similar length. For example, the α-subunit tryptophan synthase is 268 residues long and has an Rg of 19.1 Å, whereas OspA has an Rg of 25.0 Å and is 257 residues long (44, 45). A homodimer of E4-ORF3 contains 272 amino acid residues, including the unstructured residues in the His6 tag. The predicted Rg for a 136-residue folded monomer is 14.2 Å, whereas the value predicted for a fully unfolded 136-residue monomer is 36.4 Å. The predicted values are based on correlations between the length of a protein chain in residues and the observed Rg (46, 47). The empirical relationship predicts an Rg of 18.5 Å for a dimer of 272 residues. The somewhat larger value observed for the L103A dimer likely reflects shape effects. The key point is that the experimental Rg is significantly larger than expected for a folded monomer and less than predicted for a fully unfolded monomer. A Kratky representation of the scattering data revealed the presence of a peak for both WT and L103A proteins, as is expected for a folded globular protein (Fig. 3D). Thus, the SAXS studies are fully consistent with the AUC data and the gel filtration experiments.
WT E4-ORF3 Is Well Folded and L103A Mutant Protein Adopts Same Structure
To confirm that the protein fold is not perturbed by the L103A substitution, we used CD spectroscopy to probe protein secondary structure. Far-UV spectra of the E4-ORF3 WT and L103A proteins revealed that the two have identical secondary structure, with pronounced spectral minima at 208 and 222 nm and a maximum at 195 nm (Fig. 4A); the spectra indicate significant helical content. These data demonstrate that the secondary structure is not perturbed by the L103A mutation. In addition, different gel filtration fractions of WT E4-ORF3 (supplemental Fig. 1B) possessed identical secondary structure (supplemental Fig. 3), suggesting that multimerization of E4-ORF3 is not accompanied by significant gain or loss of helical content.
FIGURE 4.
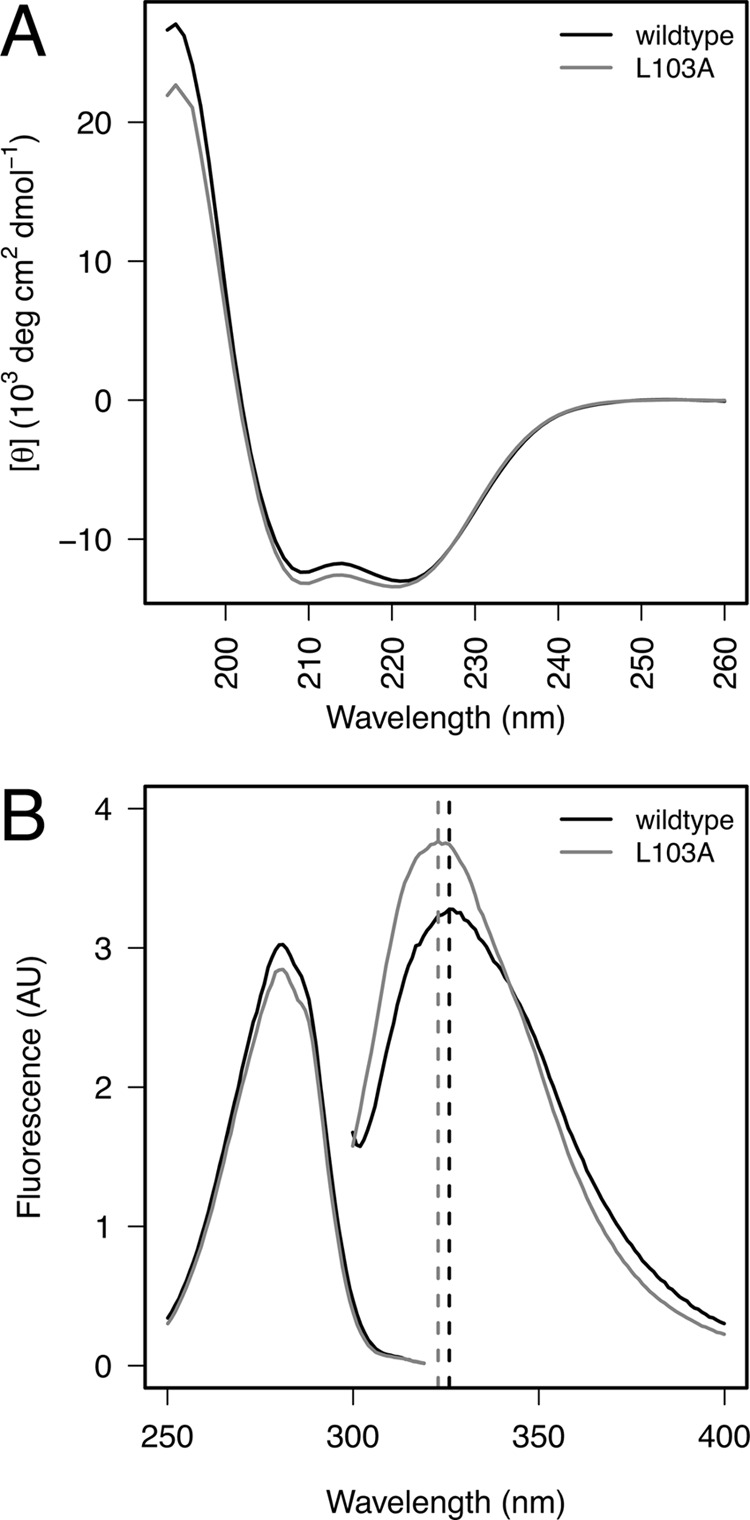
Structural characterization of E4-ORF3 WT and L103A proteins. A, far-ultraviolet CD spectra of the E4-ORF3 WT and L103A proteins contain a nearly identical secondary structure. Mean residue ellipticity is presented as a function of wavelength. Spectral minima at 208 and 222 nm as well as the maximum at 195 nm are consistent with significant helical content of the proteins, which is predicted from their sequence. B, fluorescence intensity shown as a function of wavelength. Fluorescence excitation (left) and emission (right) spectra of WT and L103A suggest that Trp-35, the unique Trp in the E4-ORF3 sequence, is partially sequestered from solvent and is in a similar environment in both proteins. Wavelength of maximal emission intensity of both samples is indicated with a dashed vertical line. AU, arbitrary units.
E4-ORF3 contains a single Trp residue (Trp-35), which we hypothesized may serve as a probe of tertiary structure in the protein. Near-UV CD spectra of WT and L103A proteins (supplemental Fig. 4A) revealed a number of narrow concordant bands arising from the residues, which absorb in this region (Trp, Tyr, Phe). A near-UV CD spectrum is a hallmark of a compact cooperatively folded protein, and many partially folded proteins give no near-UV CD signal even when the far-UV CD shows significant secondary structure. The spectral bands seen for WT and L103A were not identical in intensity but displayed remarkable agreement in their position and line width. We further probed the environment of Trp-35 using fluorescence spectroscopy. The fluorescence emission and excitation properties of Trp residues are sensitive to solvent accessibility and proximity to quenching groups. Fig. 4B displays the fluorescence emission and excitation spectra of the E4-ORF3 WT and L103A proteins. The excitation spectra are superimposeable, indicating that the fluorophore is present at the same concentration in both samples and that the local fluorophore environments are similar. When Trp-35 was selectively excited, the emission spectra of the two proteins indicate that the molecular environment is similar in both proteins. The emission maxima of the E4-ORF3 WT and L103A proteins were 326 and 323 nm, respectively; both were blue-shifted from a solvent-exposed maximum of ∼350 nm. The quantum yield of L103A was ∼10% greater than that of WT E4-ORF3. We used acrylamide quenching as an independent confirmation of solvent accessibility of Trp-35. Fluorescence quenching of Trp by acrylamide requires the two to come into physical contact; the ease of quenching thus reports on the solvent accessibility of the indole side chain of Trp. We found that Trp35 in WT and L103A proteins was similarly shielded from solvent, with a Stern-Volmer constant of ∼1 m−1 (supplemental Fig. 4B). Therefore, the results of three independent techniques agree and indicate that Trp-35 is sequestered from solvent in both the WT and L103A E4-ORF3 proteins; the small change in the quantum yield could arise from subtle changes in the environment, but the overall environments are similar.
In certain cases, some proteins can adopt partially folded structures, which are compact and contain significant secondary structure, but lack the specific sidechain-sidechain tertiary interactions; these states are known as “molten globules.” Although fluorescence and CD spectroscopies can report on tertiary packing of fluorophores or aromatic groups, respectively, NMR spectroscopy provides a more detailed view. We collected a proton NMR spectrum of the E4-ORF3 L103A protein (supplemental Fig. 4). The spectrum revealed the presence of methyl resonances, which were ring current-shifted below 0 ppm; these resonances belong to protein side chains whose environment is influenced by close packing against aromatic side chains, such as those of Phe, Tyr, or Trp. The presence of ring current-shifted resonances is an indication of close packing, unequivocally demonstrating that the L103A mutation does not induce a molten globule state in E4-ORF3 and that the protein is well folded. The heterogeneous, higher-order oligomerization of WT E4-ORF3 prevents detailed NMR studies.
E4-ORF3 L103A Mutant Inhibits WT E4-ORF3 Protein in Vivo
If the dimer formed by the L103A mutant protein represents a trapped intermediate normally involved in higher order E4-ORF3 protein assembly, then one would anticipate that L103A could interfere with higher assembly of the WT E4-ORF3 protein by sequestering WT E4-ORF3 into nonproductive dimers. To test this hypothesis, recombinant Ad5 vectors were used that express WT and L103A E4-ORF3 proteins with separate epitope tags. The WT E4-ORF3 protein was tagged with a FLAG epitope, and the L103A mutant protein was tagged with an HA epitope to allow the visualization of each protein within the same infected cell. HeLa cells were infected individually with Ad5 vectors that express each E4-ORF3 protein at low multiplicities of infection (MOI; 200 physical virus particles/cell corresponding to ∼10 infectious viruses/cell). Typical nuclear tracks were found in cells that express WT E4-ORF3 (Fig. 5A), whereas diffuse nuclear localization of the L103A mutant protein was observed (Fig. 5B), as reported previously (17). Coinfections were performed with a low MOI (200 virus particles/cell) of the FLAG-tagged WT E4-ORF3 expression vector and a high MOI (1250 virus particles/cell) of the HA-tagged E4-ORF3 L103A mutant protein expression vector. Our rationale was that overexpression of the L103A mutant protein relative to the WT E4-ORF3 protein may drive the WT protein to largely form dimers with the L103A mutant protein. A significant reduction in the ability of the WT E4-ORF3 protein to form nuclear tracks was observed in coinfected cells. Representative examples are shown in Fig. 5 (C–H) where two patterns of WT E4-ORF3 expression were detected. In some cells, small nuclear punctae of WT E4-ORF3 were evident that colocalized with the L103A mutant protein (Fig. 5, F–H). Of note, such punctae were never observed with the L103A mutant protein alone at any MOI; therefore, the WT E4-ORF3 protein contributes to this process. In other cells, few nuclear punctae of WT E4-ORF3 were observed; instead, a diffuse nuclear staining pattern of both WT and the L103A mutant protein was detected (Fig. 5, C–E). These results were specific to the coexpression of the E4-ORF3 WT and L103A proteins because an augmentation of nuclear track formation was observed when cells were coinfected with a low MOI of FLAG-tagged WT E4-ORF3 vector and a high MOI of HA-tagged WT E4-ORF3 vector (Fig. 5, I–K).
FIGURE 5.

L103A inhibits WT E4-ORF3 protein activity in vivo. A, FLAG-tagged WT E4-ORF3 localization following infection of HeLa cells at a low MOI was analyzed by IF microscopy using TRITC-labeled secondary antibody. The image is of one cell with E4-ORF3 tracks evident within the nucleus as filamentous structures. B, HA-tagged L103A E4-ORF3 localization following infection of HeLa cells at high MOI was analyzed by IF microscopy using FITC-labeled secondary antibody as in A. C–H, coinfection of HeLa cells with a low MOI of FLAG-E4-ORF3-WT plus a high MOI of HA-L103A. E4-ORF3 localization was analyzed by IF microscopy using FITC-labeled (anti-HA, C and F) and TRITC-labeled (anti-FLAG, D and G) secondary antibodies as in A. Merged FITC and TRITC images are shown in E and H. I–K, coinfection of HeLa cells with a low MOI of FLAG-E4-ORF3-WT plus a high MOI of HA-E4-ORF3-WT. E4-ORF3 localization was analyzed by IF microscopy using FITC-labeled (anti-HA, I) and TRITC-labeled (anti-FLAG, J) secondary antibodies as in A. A merged FITC and TRITC image is shown in K. L, PML-NB evident in uninfected HeLa cells represented as discrete punctae. M–O, PML-NB reorganization following infection of HeLa cells at low MOI with Ad-CMV-FLAG-WT E4-ORF3. P–R, PML-NB formation following infection of HeLa cells at high MOI with Ad-CMV-HA-L103A. S–U, PML-NB formation following coinfection of HeLa cells at low MOI with Ad-CMV-FLAG WT E4-ORF3 and high MOI Ad-CMV-HA-L103A. PML detected in M, P, and S using rabbit anti-PML antibody and FITC-conjugated secondary antibody. E4-ORF3 proteins were detected in panels N, Q, and T using anti-E4-ORF3 monoclonal antibody and TRITC-conjugated secondary antibody. Merged FITC and TRITC images are shown in O, R, and U. Arrows in S–U indicate E4-ORF3 and PML colocalization.
These results suggest that the L103A mutant protein may act as a dominant-negative effector to antagonize the activity of WT E4-ORF3. To analyze this possibility, we examined whether the L103A mutant protein interferes with the ability of WT E4-ORF3 to reorganize PML-NB. HeLa cells were infected individually with Ad vectors that express FLAG-tagged WT E4-ORF3 (low MOI), HA-tagged L103A (high MOI), or coinfected with both viruses (low and high MOIs, respectively). Subsequently, PML-NB relocalization was analyzed by IF microscopy (Fig. 5). In uninfected cells, typical PML-NB were evident (Fig. 5L); PML was relocalized into nuclear tracks following expression of WT E4-ORF3 (M–O) but not L103A (P–R). Interestingly, the L103A mutant protein interfered with the ability of WT E4-ORF3 to relocalize PML (Fig. 5, S–U). In coinfected cells, colocalization with PML-NB was observed with some (arrows) but not all of the E4-ORF3 punctae that formed.
To probe the interactions between WT and L103A E4-ORF3 further, in vitro binding studies were performed. We analyzed whether the WT E4-ORF3 and L103A proteins directly interact by coexpressing the two in E. coli and in mammalian cells. In E. coli, untagged WT E4-ORF3 and His6-tagged L103A were purified on a Ni2+-NTA affinity column, followed by gel filtration. The salting out step was omitted to avoid coprecipitating the two proteins; we wanted to visualize protein complexes formed between the two during protein expression, rather than those that might arise as a consequence of salt precipitation. The gel filtration profile of the Ni2+-NTA-purified proteins differed significantly from those of WT E4-ORF3 or L103A alone (supplemental Fig. 5A) and contained a broad distribution of E4-ORF3 oligomers. Remarkably, we found that both proteins were present in each gel filtration fraction. SDS-PAGE revealed that WT E4-ORF3 preferentially populated larger oligomers than the L103A protein and that fractions containing the highest abundance of L103A eluted at the same time as dimeric L103A characterized previously. The identity of the two proteins was confirmed using MALDI/TOF mass spectrometry, and their abundance was estimated using SDS-PAGE and analytical HPLC (supplemental Fig. 5, B and C). To probe interactions between the two E4-ORF3 proteins in mammalian cells, HeLa cells were infected individually with Ad vectors that express FLAG-tagged WT E4-ORF3 (low MOI), HA-tagged L103A (high MOI), or coinfected with both viruses (low and high MOIs, respectively). Subsequently, whole cell extracts were prepared, and anti-FLAG antibody was used to immunoprecipitate WT E4-ORF3. Coprecipitation of the L103A mutant protein with WT E4-ORF3 was analyzed by Western blot (Fig. 6). These results demonstrated that the L103A protein coimmunoprecipitated with WT E4-ORF3 (IP: FLAG).
FIGURE 6.
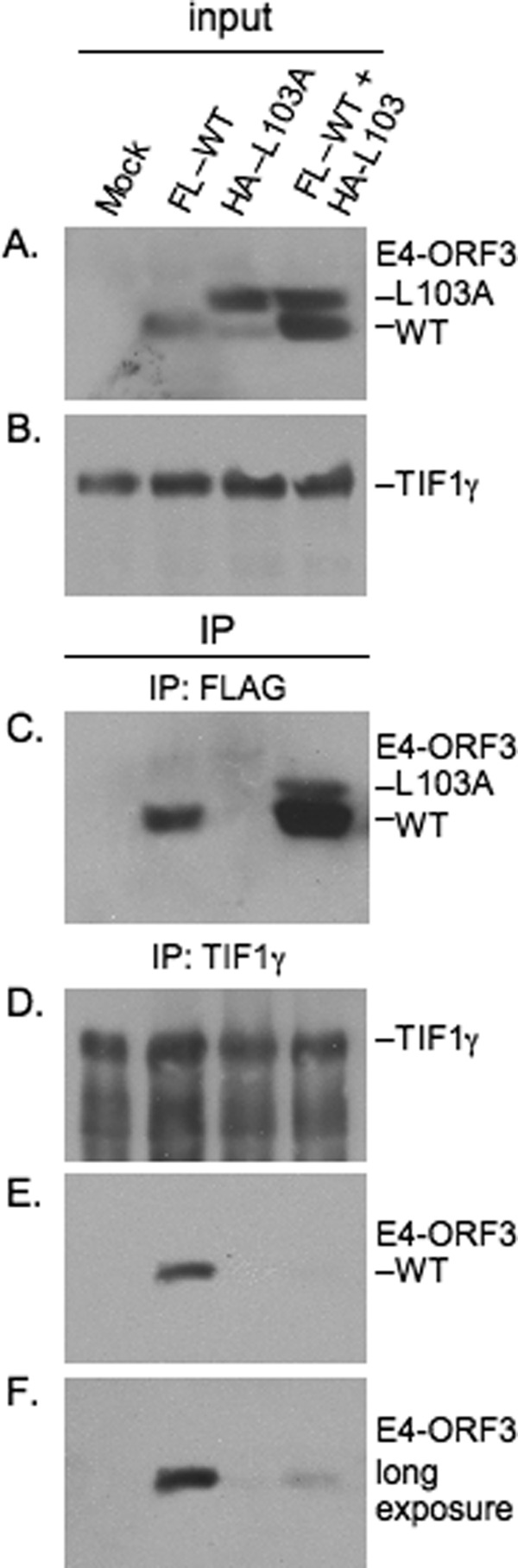
WT E4-ORF3 and L103A coimmunoprecipitate and L103A inhibits binding of WT-ORF3 to TIF1γ. HeLa cells were mock-infected (Mock), individually infected with low MOI and high MOI of Ad-CMV vectors that express WT E4-ORF3 and HA-L103A, or coinfected with low MOI FLAG-WT-ORF3 and high MOI HA-L103A. E4-ORF3 (A) and TIF1γ (B) in cells extracts (input) were detected by Western blot. FLAG-WT-ORF3 migrated faster than L103A in the gel due to the presence of a smaller epitope tag; both proteins were detected using an anti-E4-ORF3 monoclonal antibody. The major L103A species corresponds to the epitope-tagged protein recognized using an anti-HA antibody; the weaker, faster migrating L103A was not recognized by an anti-HA antibody an may reflect translation initiation at an internal methionine residue or proteolytic cleavage of the epitope tag. E4-ORF3 (C, E, and F) and TIF1γ (D) proteins present in anti-FLAG (C) and anti-TIF1γ (D, E, and F) immunoprecipitates (IP) were detected by Western blot. F is a longer exposure of the same gel shown in E.
Finally, we examined whether the L103A mutant interferes with the ability of WT E4-ORF3 to bind to the known cellular binding partner TIF1γ. Single virus infections and coinfections were performed as described above, TIF1γ was immunoprecipitated from whole cell extracts, and E4-ORF3 binding analyzed by Western blot (Fig. 6). These results demonstrated that the L103A mutant protein interfered with the binding of WT E4-ORF3 to TIF1γ (IP: TIF1γ). Collectively, these results are consistent with the hypothesis that the L103A mutant protein is able to sequester the WT E4-ORF3 protein within the nucleus and inhibit higher-order protein assembly. These results also support the conclusion that the stable dimer observed with the L103A mutant protein represents a bona fide intermediate in E4-ORF3 nuclear track formation.
DISCUSSION
Our biophysical analyses show that WT E4-ORF3 and the L103A mutant have very similar overall secondary structure content and both form compact globular structures with spectroscopic properties expected for native proteins. The major difference between the two proteins is in their ability to oligomerize. Dynamic light scattering, analytical ultracentrifugation, gel filtration, and SAXS all reveal the WT forms as heterogeneous mixtures of higher-order oligomers, whereas self-assembly of the L103A mutant is largely stopped at the dimer stage. Spectroscopic studies show that the WT oligomers contain native-like secondary structure and are folded. Thus, WT E4-ORF3 oligomerization fundamentally differs from nonspecific aggregation of unfolded proteins.
Given the small size of the E4-ORF3 protein (116 amino acids) and the large number of known cellular and viral interaction partners, we believe that higher-order multimerization is necessary for E4-ORF3 activity. Higher-order assembly will lead to the presentation of multiple binding sites. Our results are consistent with the hypothesis that multimeric WT E4-ORF3 has a higher affinity than the L103A mutant protein for cellular binding partners such as TIF1γ due to the presence of multiple binding interfaces on the WT protein. We propose that higher-order multimerization of the WT E4-ORF3 protein forms a polyvalent scaffold, which is required for the activity of E4-ORF3. Each individual scaffold building block (a dimer) has low affinity for E4-ORF3 binding partners. However, this low affinity can be overcome through avidity effects; higher-order multimerization of E4-ORF3 in the nucleus presents a polyvalent surface that greatly reduces the entropic costs of binding. The fact that the dimeric L103A mutant protein is incapable of binding both the E1B-55K and TIF1γ proteins supports this hypothesis. The L103A mutant protein may contain the binding regions for E1B-55K and TIF1γ, but its inability to multimerize prevents it from raising the binding affinity of the interaction to sufficient levels for efficient binding partner interactions. This model also is supported by the results of the IF experiments, which demonstrated that coexpression of the L103A mutant protein with WT E4-ORF3 interfered with the ability of the WT protein to form nuclear tracks and reorganize PML-NB. Additionally, the L103A mutant protein interfered with the binding of WT E4-ORF3 to TIF1γ in coimmunoprecipitation experiments. We hypothesize that upon coexpression, the L103A mutant protein functions as a dominant-negative effector by forming trapped heterodimers, or short multimers, with WT E4-ORF3. This hypothesis is supported by the IF analyses, which demonstrated the recruitment of the L103A mutant into small punctae only upon coexpression with the WT protein. Also supporting this hypothesis are the observations that WT E4-ORF3 and the L103A mutant protein coimmunoprecipitate from cellular extracts when expressed in vivo and cofractionate from cellular extracts when coexpressed in E. coli. Although we favor the possibility that WT E4-ORF3 and the L103A mutant protein heterodimerize, resulting in interference of WT activity by L103A, it is also possible that the region near Leu-103 may identify a critical binding surface for cellular binding partners, which is perturbed upon mutation, and that such interactions are required for higher-order E4-ORF3 protein oligomerization.
The results from coexpression of WT E4-ORF3 and the L103A mutant protein in E. coli indicate that the L103A mutant protein is able to limit the ability of the WT E4-ORF3 to multimerize. When the His6-tagged L103A and untagged WT proteins were coexpressed in E. coli, the L103A protein clearly reduced the multimeric state of the WT protein. When expressed individually, the WT and L103A proteins had distinct gel filtration profiles that did not overlap to any significant extent. The L103A mutant protein also interfered with the ability of WT E4-ORF3 to reorganize PML-NB in vivo. Some of the nuclear punctae that form when WT E4-ORF3 and the L103A mutant protein were coexpressed in vivo colocalized with PML-NB, but PML-NB integrity was not disrupted. This result suggests that the ability of E4-ORF3 to interact with one or more components of PML-NB may be uncoupled from the activity of E4-ORF3 to relocalize PML-NB proteins into nuclear tracks. Because the L103A mutant protein is not able to colocalize with PML-NB, we infer that WT E4-ORF3 in nuclear punctae with the L103A mutant protein directs PML-NB colocalization and that higher-order E4-ORF3 multimerization may be required for PML-NB disruption.
Although the isoelectric point of E4-ORF3 at pH 5.5 would suggest a highly charged, hydrophilic protein at physiological pH, the protein possesses a large amount of hydrophobic character (supplemental Fig. 6). The presence of a run of five leucine residues that are spaced between two and four residues apart near the C terminus of E4-ORF3 may contribute to its poor solubility. Mutation of these residues to alanine has severe phenotypic consequences on viral growth (17).6 The roughly even spacing of these residues suggests that they may play a role in generating a short amphipathic α-helix. This could mediate the self-associative properties of E4-ORF3, as suggested by the analytical ultracentrifugation data of the L103A mutant protein whose mutation maps to this region. Based on spectroscopic evidence, we demonstrated that the L103A mutant protein contains essentially identical secondary and tertiary structure compared with WT E4-ORF3. The L103A mutant protein largely forms dimers in solution and is deficient in the self-associative properties of WT E4-ORF3. Furthermore, the secondary structure of WT E4-ORF3 is invariant to multimerization, suggesting that the mechanism of E4-ORF3 polymerization involves the association of well folded proteins rather than major conformational rearrangement. These results provide a basis for understanding the self-associative properties of E4-ORF3 as they pertain to the activity of the protein. It will be interesting to explore the consequences of other critical E4-ORF3 mutations on the oligomeric state and functions of the protein.
Supplementary Material
Acknowledgments
We thank Dr. Thomas Dobner for antibody against E4-ORF3, Dr. Daiqing Liao for antibody against E1B-55K, and Dr. Nicole S. Sampson for use of the dynamic light scattering instrument. Use of the National Synchrotron Light Source, Brookhaven National Laboratory, was supported by the U. S. Department of Energy, Office of Science, Office of Basic Energy Sciences under Contract DE-AC02–98CH10886.
This work was supported, in whole or in part, by National Institutes of Health Grants CA122677 (to P. H.), GM078114 (to D. P. R.), and GM086199 (to D. F. G.) and by Training Grants CA009176 and GM007518 (to M. A. Y.).

This article contains supplemental “Experimental Procedures” and Figs. S1–S6.
M. A. Yondola, E. Zheng, and P. Hearing, unpublished results.
- PML-NB
- promyelocytic leukemia protein nuclear bodies
- Ad
- adenovirus
- TIF1
- transcription intermediary factor 1
- DDR
- DNA damage response
- MRN
- Mre11·Rad50·Nbs1
- TRITC
- tetramethyl rhodamine isothiocyanate
- IF
- immunofluorescence
- Ni2+-NTA
- nickel-nitrilotriacetic acid
- AUC
- analytical ultracentrifugation
- MOI
- multiplicity of infection.
REFERENCES
- 1. Geoffroy M. C., Chelbi-Alix M. K. (2011) Role of promyelocytic leukemia protein in host antiviral defense. J. Interferon Cytokine Res. 31, 145–158 [DOI] [PubMed] [Google Scholar]
- 2. Everett R. D., Chelbi-Alix M. K. (2007) PML and PML nuclear bodies: Implications in antiviral defense. Biochimie 89, 819–830 [DOI] [PubMed] [Google Scholar]
- 3. Doucas V., Ishov A. M., Romo A., Juguilon H., Weitzman M. D., Evans R. M., Maul G. G. (1996) Adenovirus replication is coupled with the dynamic properties of the PML nuclear structure. Genes Dev. 10, 196–207 [DOI] [PubMed] [Google Scholar]
- 4. Ullman A. J., Hearing P. (2008) Cellular proteins PML and Daxx mediate an innate antiviral defense antagonized by the adenovirus E4 ORF3 protein. J. Virol. 82, 7325–7335 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Ullman A. J., Reich N. C., Hearing P. (2007) Adenovirus E4 ORF3 protein inhibits the interferon-mediated antiviral response. J. Virol. 81, 4744–4752 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Forrester N. A., Patel R. N., Speiseder T., Groitl P., Sedgwick G. G., Shimwell N. J., Seed R. I., Catnaigh P. Ó., McCabe C. J., Stewart G. S., Dobner T., Grand R. J., Martin A., Turnell A. S. (2012) Adenovirus E4orf3 targets transcriptional intermediary factor 1γ for proteasome-dependent degradation during infection. J. Virol. 86, 3167–3179 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Vink E. I., Yondola M. A., Wu K., Hearing P. (2012) Adenovirus E4-ORF3-dependent relocalization of TIF1α and TIF1γ relies on access to the coiled-coil motif. Virology 422, 317–325 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Yondola M. A., Hearing P. (2007) The adenovirus E4 ORF3 protein binds and reorganizes the TRIM family member transcriptional intermediary factor 1 α. J. Virol. 81, 4264–4271 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Carvalho T., Seeler J. S., Ohman K., Jordan P., Pettersson U., Akusjärvi G., Carmo-Fonseca M., Dejean A. (1995) Targeting of adenovirus E1A and E4-ORF3 proteins to nuclear matrix-associated PML bodies. J. Cell Biol. 131, 45–56 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Leppard K. N., Everett R. D. (1999) The adenovirus type 5 E1b 55K and E4 Orf3 proteins associate in infected cells and affect ND10 components. J. Gen. Virol. 80, 997–1008 [DOI] [PubMed] [Google Scholar]
- 11. Lethbridge K. J., Scott G. E., Leppard K. N. (2003) Nuclear matrix localization and SUMO-1 modification of adenovirus type 5 E1b 55K protein are controlled by E4 Orf6 protein. J. Gen. Virol. 84, 259–268 [DOI] [PubMed] [Google Scholar]
- 12. Weitzman M. D., Lilley C. E., Chaurushiya M. S. (2010) Genomes in conflict: Maintaining genome integrity during virus infection. Annu. Rev. Microbiol. 64, 61–81 [DOI] [PubMed] [Google Scholar]
- 13. Weitzman M. D., Ornelles D. A. (2005) Inactivating intracellular antiviral responses during adenovirus infection. Oncogene 24, 7686–7696 [DOI] [PubMed] [Google Scholar]
- 14. Karen K. A., Hoey P. J., Young C. S., Hearing P. (2009) Temporal regulation of the Mre11-Rad50-Nbs1 complex during adenovirus infection. J. Virol. 83, 4565–4573 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Weiden M. D., Ginsberg H. S. (1994) Deletion of the E4 region of the genome produces adenovirus DNA concatemers. Proc. Natl. Acad. Sci. U.S.A. 91, 153–157 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Barber G. N. (2011) Innate immune DNA sensing pathways: STING, AIMII, and the regulation of interferon production and inflammatory responses. Curr. Opin. Immunol. 23, 10–20 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Evans J. D., Hearing P. (2005) Relocalization of the Mre11-Rad50-Nbs1 complex by the adenovirus E4 ORF3 protein is required for viral replication. J. Virol. 79, 6207–6215 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Stracker T. H., Carson C. T., Weitzman M. D. (2002) Adenovirus oncoproteins inactivate the Mre11-Rad50-NBS1 DNA repair complex. Nature 418, 348–352 [DOI] [PubMed] [Google Scholar]
- 19. Lakdawala S. S., Schwartz R. A., Ferenchak K., Carson C. T., McSharry B. P., Wilkinson G. W., Weitzman M. D. (2008) Differential requirements of the C terminus of Nbs1 in suppressing adenovirus DNA replication and promoting concatemer formation. J. Virol. 82, 8362–8372 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Mathew S. S., Bridge E. (2007) The cellular Mre11 protein interferes with adenovirus E4 mutant DNA replication. Virology 365, 346–355 [DOI] [PubMed] [Google Scholar]
- 21. Mathew S. S., Bridge E. (2008) Nbs1-dependent binding of Mre11 to adenovirus E4 mutant viral DNA is important for inhibiting DNA replication. Virology 374, 11–22 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Harada J. N., Shevchenko A., Shevchenko A., Pallas D. C., Berk A. J. (2002) Analysis of the adenovirus E1B-55K-anchored proteome reveals its link to ubiquitination machinery. J. Virol. 76, 9194–9206 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Querido E., Blanchette P., Yan Q., Kamura T., Morrison M., Boivin D., Kaelin W. G., Conaway R. C., Conaway J. W., Branton P. E. (2001) Degradation of p53 by adenovirus E4orf6 and E1B55K proteins occurs via a novel mechanism involving a Cullin-containing complex. Genes Dev. 15, 3104–3117 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Baker A., Rohleder K. J., Hanakahi L. A., Ketner G. (2007) Adenovirus E4 34k and E1b 55k oncoproteins target host DNA ligase IV for proteasomal degradation. J. Virol. 81, 7034–7040 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Orazio N. I., Naeger C. M., Karlseder J., Weitzman M. D. (2011) The adenovirus E1b55K/E4orf6 complex induces degradation of the Bloom helicase during infection. J. Virol. 85, 1887–1892 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Araujo F. D., Stracker T. H., Carson C. T., Lee D. V., Weitzman M. D. (2005) Adenovirus type 5 E4orf3 protein targets the Mre11 complex to cytoplasmic aggresomes. J. Virol. 79, 11382–11391 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Liu Y., Shevchenko A., Shevchenko A., Berk A. J. (2005) Adenovirus exploits the cellular aggresome response to accelerate inactivation of the MRN complex. J. Virol. 79, 14004–14016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Soria C., Estermann F. E., Espantman K. C., O'Shea C. C. (2010) Heterochromatin silencing of p53 target genes by a small viral protein. Nature 466, 1076–1081 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Blanchette P., Kindsmüller K., Groitl P., Dallaire F., Speiseder T., Branton P. E., Dobner T. (2008) Control of mRNA export by adenovirus E4orf6 and E1B55K proteins during productive infection requires E4orf6 ubiquitin ligase activity. J. Virol. 82, 2642–2651 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Gonzalez R., Huang W., Finnen R., Bragg C., Flint S. J. (2006) Adenovirus E1B 55-kilodalton protein is required for both regulation of mRNA export and efficient entry into the late phase of infection in normal human fibroblasts. J. Virol. 80, 964–974 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Goodrum F. D., Ornelles D. A. (1999) Roles for the E4 orf6, orf3, and E1B 55-kilodalton proteins in cell cycle-independent adenovirus replication. J. Virol. 73, 7474–7488 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Nordqvist K., Ohman K., Akusjärvi G. (1994) Human adenovirus encodes two proteins, which have opposite effects on accumulation of alternatively spliced mRNAs. Mol. Cell. Biol. 14, 437–445 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Ohman K., Nordqvist K., Akusjärvi G. (1993) Two adenovirus proteins with redundant activities in virus growth facilitates tripartite leader mRNA accumulation. Virology 194, 50–58 [DOI] [PubMed] [Google Scholar]
- 34. Sandler A. B., Ketner G. (1989) Adenovirus early region 4 is essential for normal stability of late nuclear RNAs. J. Virol. 63, 624–630 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Shepard R. N., Ornelles D. A. (2003) E4orf3 is necessary for enhanced S-phase replication of cell cycle-restricted subgroup C adenoviruses. J. Virol. 77, 8593–8595 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Shepard R. N., Ornelles D. A. (2004) Diverse roles for E4orf3 at late times of infection revealed in an E1B 55-kilodalton protein mutant background. J. Virol. 78, 9924–9935 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Woo J. L., Berk A. J. (2007) Adenovirus ubiquitin-protein ligase stimulates viral late mRNA nuclear export. J. Virol. 81, 575–587 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Schiedner G., Hertel S., Kochanek S. (2000) Efficient transformation of primary human amniocytes by E1 functions of Ad5: Generation of new cell lines for adenoviral vector production. Hum. Gene Ther. 11, 2105–2116 [DOI] [PubMed] [Google Scholar]
- 39. Liao D., Yu A., Weiner A. M. (1999) Coexpression of the adenovirus 12 E1B 55 kDa oncoprotein and cellular tumor suppressor p53 is sufficient to induce metaphase fragility of the human RNU2 locus. Virology 254, 11–23 [DOI] [PubMed] [Google Scholar]
- 40. Nevels M., Täuber B., Kremmer E., Spruss T., Wolf H., Dobner T. (1999) Transforming potential of the adenovirus type 5 E4orf3 protein. J. Virol. 73, 1591–1600 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Evans J. D., Hearing P. (2003) Distinct roles of the adenovirus E4 ORF3 protein in viral DNA replication and inhibition of genome concatenation. J. Virol. 77, 5295–5304 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Konarev P. V., Volkov V. V., Sokolova A. V., Koch M. H. J., Svergun D. I. (2003) PRIMUS a Windows-PC based system for small-angle scattering data analysis. J. Appl. Crystallogr. 36, 1277–1282 [Google Scholar]
- 43. Guinier A., Fournet G. (1955) Small-angle X-ray Scattering, John Wiley and Sons, Inc., New York [Google Scholar]
- 44. Gualfetti P. J., Iwakura M., Lee J. C., Kihara H., Bilsel O., Zitzewitz J. A., Matthews C. R. (1999) Apparent radii of the native, stable intermediates and unfolded conformers of the α-subunit of tryptophan synthase from Escherichia coli, a TIM barrel protein. Biochemistry 38, 13367–13378 [DOI] [PubMed] [Google Scholar]
- 45. Koide S., Bu Z., Risal D., Pham T. N., Nakagawa T., Tamura A., Engelman D. M. (1999) Multistep denaturation of Borrelia burgdorferi OspA, a protein containing a single-layer β-sheet. Biochemistry 38, 4757–4767 [DOI] [PubMed] [Google Scholar]
- 46. Kohn J. E., Millett I. S., Jacob J., Zagrovic B., Dillon T. M., Cingel N., Dothager R. S., Seifert S., Thiyagarajan P., Sosnick T. R., Hasan M. Z., Pande V. S., Ruczinski I., Doniach S., Plaxco K. W. (2004) Random coil behavior and the dimensions of chemically unfolded proteins. Proc. Natl. Acad. Sci. U.S.A. 101, 12491–12496 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Skolnick J., Kolinski A., Ortiz A. R. (1997) MONSSTER: A method for folding globular proteins with a small number of distance restraints. J. Mol. Biol. 265, 217–241 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.