

Abstract
Gene duplication provides an essential source of novel genetic material to facilitate rapid morphological evolution. Traits involved in reproduction and sexual dimorphism represent some of the fastest evolving traits in nature, and gene duplication is intricately involved in the origin and evolution of these traits. Here, we review genomic research on stalk-eyed flies (Diopsidae) that has been used to examine the extent of gene duplication and its role in the genetic architecture of sexual dimorphism. Stalk-eyed flies are remarkable because of the elongation of the head into long stalks, with the eyes and antenna laterally displaced at the ends of these stalks. Many species are strongly sexually dimorphic for eyespan, and these flies have become a model system for studying sexual selection. Using both expressed sequence tag and next-generation sequencing, we have established an extensive database of gene expression in the developing eye-antennal imaginal disc, the adult head and testes. Duplicated genes exhibit narrower expression patterns than non-duplicated genes, and the testes, in particular, provide an abundant source of gene duplication. Within somatic tissue, duplicated genes are more likely to be differentially expressed between the sexes, suggesting gene duplication may provide a mechanism for resolving sexual conflict.
Keywords: stalk-eyed flies, gene duplication, sexual conflict, spermatogenesis, tubulin genes
1. Introduction
Sexual dimorphism is a characteristic of many of the most bizarre and conspicuous morphological and behavioural traits found in nature. The evolution of these characters is driven by differences in the selection pressures affecting the sexes, and they often exhibit rapid rates of change [1–4]. Understanding the genetic basis of these traits and how they contribute to the diversification of animals represents a fundamental goal of evolutionary biology. One central feature when examining the genetic architecture of sexual dimorphism is that males and females essentially share the same genetic material. There are few genes that reside exclusively in one sex and this is expected to lead to substantial conflict between the sexes as to how the genome evolves because males and females will often have different selective optima for any given trait or gene [5–7]. Recently, there has been considerable interest concerning the prevalence and evolutionary significance of intralocus sexual conflict, and this research suggests that sexual conflict has had a profound impact on genomic structure and variation [5,6]. Here, we summarize recent research on the genetic architecture of sexual dimorphism in stalk-eyed flies and highlight how new genomic techniques can be applied to the study of sexual selection and sexual conflict in a non-model organism. In the first section, we review the recent literature on the genomics of sexual dimorphism and sexual conflict, focusing primarily on research in Drosophila because of its relatively close evolutionary proximity to stalk-eyed flies. The second section provides an introduction to sexual selection in stalk-eyed flies. In the third section, we summarize our genomic data on chromosomal gene content, gene duplication and sex-biased gene expression focusing in particular on its relevance to sexual conflict and reproductive investment in stalk-eyed flies.
2. Genetic architecture of sexual dimorphism and sexual conflict
Seminal research conducted by Rice and co-workers [7–10] provided the first extensive demonstration of the widespread nature of sexually antagonistic loci. In a series of experiments on Drosophila melanogaster, they manipulated the chromosomal composition and breeding regime of flies in the laboratory to relax or eliminate selection in one of the sexes while the other sex was allowed to accumulate sexually antagonistic variation. Consistent with theoretical predictions, loci that were harmful to one sex or beneficial to the other developed as a result of sex-limited evolution [8,10]. Additional research in Drosophila has demonstrated substantial negative fitness correlations between the sexes [9–12], although this relationship is present only in adults and not in larvae, presumably because there is little divergence in their adaptive interests at this stage. As sexually antagonistic variation builds up in the genome, the average fitness of the sexes is reduced, a phenomenon called gender load [13], and selection is expected to break or reduce the genetic correlation between the sexes that is the basis for the sexual conflict. This can be accomplished by a few different mechanisms. First, sex-specific regulatory DNA can evolve to allow a given gene to be expressed at a higher level in one sex [14–16]. Second, sex-specific splicing mechanisms can evolve to create the formation of alternative transcripts, as in many sex determination pathways [17], in a single sex. Finally, gene duplication can occur to produce an additional copy of a gene that is free from the pleiotropic constraints operating on the original copy and facilitate the evolution of sex-specific expression [18,19].
Sex chromosomes are predicted to be a hotspot for sexual conflict because each sex has an advantage with respect to the evolutionary outcome of sexually antagonistic variation on these chromosomes, depending on the dominance of a given allele [20,21]. Because sex-linked genes spend twice as much time in the homogametic sex (e.g. XX in flies and mammals) than in the heterogametic sex, the homogametic sex is expected to control the outcome of sexual conflict whenever alleles have dominant effects. Alternatively, alleles that are recessive and beneficial to males are predicted to increase in frequency when the gene is on the X chromosome because they are exposed to selection in males but hidden from selection in females. Theory predicts that little sexually antagonistic variation will reside on autosomes, and recent work in Drosophila estimated that nearly 97 per cent of all sexually antagonistic variation was located on the X chromosome [12]. A recent paper [22], however, argued that there are several conditions under which the autosomes, rather than the X chromosome, are more likely to accumulate sexually antagonistic variation.
The development of microarray and next-generation sequencing technologies has resulted in a flourish of genomic research comparing the pattern of sex-biased genetic effects between the sex chromosomes and autosomes. Initial microarray experiments comparing gene expression between the sexes in Drosophila revealed that a large portion of the genome is differentially expressed between the sexes, and that there is a significant underrepresentation of male-biased genes (i.e. genes with higher levels of expression in males) on the X chromosome [14–16,23]. A similar relationship between sex-biased genes and chromosomal location has been found in other Drosophila species [16,23], including the neo-X chromosome in Drosophila pseudoobscura [16], as well as in mosquitoes [24] and flour beetles [25]. This ‘demasculinization’ of the X involves extensive gene movement off of the X chromosome on to an autosome, and, therefore, is consistent with the resolution of sexual conflict resulting from genes with dominant effects. Alternative explanations, however, have also been proposed to explain this pattern. In Drosophila, the X chromosome has been thought to undergo inactivation during meiotic stages of spermatogenesis, a process called meiotic sex chromosome inactivation (MSCI) [26–29]. This transcriptional silencing would make the X chromosome a disfavoured location for genes that are important to sperm development. Many male-biased genes are expressed at high levels in the male germline and, therefore, these genes are under strong selective pressures to avoid the X chromosome [30–32]. While genes expressed in testes represent the vast majority of male-biased genes in these microarray studies, the demasculinization pattern also held for male-biased genes in somatic tissue, suggesting MSCI alone was not sufficient to explain the gene movement [14]. Furthermore, sexual antagonism and MSCI are not mutually exclusive explanations for biased sex chromosome composition because one process may drive the other [33]. Increased dosage requirements have also been hypothesized as a mechanism driving the lack of male-biased genes on the X chromosome [25,34].
The relocation of genes off of the X chromosome onto an autosome is driven largely by gene duplication. The movement or creation of autosomal male-biased genes accounts for nearly all of the biased chromosomal gene content in Drosophila, whereas evolutionary shifts in the expression patterns of genes have little impact on these patterns [16]. Retroposition events, in particular, enable biased gene movement across chromosomes. Retroposition creates new genes when reverse-transcribed mRNAs are inserted into the genome at different locations from their parental copy and survive to form functioning genes. In Drosophila, the distribution of retrogenes is characterized by excess movement off of the X chromosome, with the derived copy exhibiting elevated expression levels in the testes in the majority of cases [28,30,32,35]. Analyses of retrogene polymorphism suggest that this movement is driven primarily by selection and not by mutational bias [36]. While this pattern is consistent with avoidance of MSCI, other studies in Drosophila have demonstrated that the testes-specific expression of derived retrogenes also applies to gene movement between two different autosomes, indicating that there are processes unrelated to MSCI affecting gene distribution [32]. In addition, a few recent studies have questioned the existence of MCSI in Drosophila [37–39]. They found only minor reductions in the level of gene expression for X-linked genes during the transition to the meiotic phase of spermatogenesis and suggested that previous estimates of MSCI may have resulted from a lack of dosage compensation in the male germline.
Regardless of the mechanism driving chromosomal movement, it is clear that gene duplication is intrinsically linked to sex-biased gene expression and possibly to the resolution of sexual conflict [18,19]. The vast majority of sex-biased genes are expressed in the gonads. Spermatogenesis in Drosophila involves a unique transcriptional apparatus involving thousands of genes that are expressed exclusively or primarily in the testes and many of these testes-specific genes derive from recent duplication events [40–43]. For instance, the basal transcription factor complex TFIID contains several testes-specific paralogues that are critical to sperm production [44,45]. This complex is part of the general machinery responsible for initiating transcription at core promoters in eukaryotic cells. The complex includes 12 TATA binding protein-associated factors (TAFs), five of which have been duplicated in Drosophila and are expressed exclusively in the testes. These genes are required for progression of the meiotic cell cycle, and mutations in the genes result in meiotic arrest and sterility [44,45]. Similarly, the proteasome, a complex that controls protein degradation, also contains numerous testes-specific components that have originated recently, via gene duplication, within Drosophila [46,47]. Not only is much of spermatogenesis regulated by testes-specific duplicates, but there are several cases of recurrent duplication in which a given gene has undergone multiple independent duplications and acquired testes-specific expression in different lineages [46–49].
In a series of recent articles, Gallach et al. [19,50,51] have explored in detail the connection between gene duplication, testes-specific gene expression and the resolution of sexual conflict. They present a model in which sexual conflict arises in a ubiquitously expressed parental gene that contains an allele that increases fitness when expressed in the testes but is harmful relative to other alleles when expressed in other tissues. In this situation, selection will favour the duplication of the gene and relocation of a copy elsewhere in the genome to avoid homogenizing processes such as gene conversion. The allelic divergence that arises from the sexual conflict may even promote the duplication event [52]. Released from pleiotropic constraints, the derived copy is free to evolve testes-specific gene expression, potentially resulting in rapid rates of protein evolution. Gallach et al. [51] stress that gene duplication will often be the favoured mechanism to resolve sexual conflict because most genes have important functions in both sexes; so it is not possible to simply downregulate a gene in one sex when conflict arises. Gene duplication allows the sexual conflict to be resolved without disrupting the function of the parental copy in other tissues and in the opposite sex. They argue that this scenario represents the primary mechanism driving the pattern of biased chromosomal gene content and sex-biased gene expression. The testes are under intense selective pressures generated by sperm competition, meiotic drive and parasite infections that are not likely to be applicable to females or other tissues in males. This tissue then becomes the primary generator of sexual conflict and, consequently, the overwhelming cause for gene duplication in the genome.
3. Sexual selection in stalk-eyed flies
Sexual dimorphism in head morphology is common in adult flies. Modification of the male head into elaborate and exaggerated structures, such as eye-stalks or antlers, that play a critical role in their mating system has evolved independently in several dipteran families. Wilkinson & Dodson [53] list over 40 groups, in eight different families, in which head projections have evolved. All of these flies belong to the Acalyptratae, a group that comprises approximately 20 per cent of all described fly species [54]. Within the Acalyptratae, sexually dimorphic head modification is significantly overrepresented in certain families. For instance, in the Platystomatidae, there are 11 different genera that contain species with eye-stalks or broad heads (collectively termed hypercephaly) and seven other genera with species that possess cheek processes. This clustered phylogenetic distribution suggests that these flies may possess some physiological or developmental mechanism that predisposes them to evolve this extreme phenotype.
Stalk-eyed flies in the family Diopsidae provide some of most spectacular examples of head elongation within Diptera [53,55]. They are the most species-rich group of hypercephalic flies, as all species within the family possess eye-stalks to some degree. Diopsids are found primarily in the old-world tropics although flies in the basal genus Sphyracephala have a cosmopolitan distribution. While currently the subject of taxonomic revisions [56], there are approximately 200–300 described species in 10–14 different genera within the family [57]. There is substantial variation in the family in terms of the size of the eye-stalks as well as the extent of the dimorphism [55,58]. For instance, flies in the genus Sphyracephala have eye-stalks that are approximately one-third of their body length, and most species are monomorphic or slightly dimorphic, while some Teleopsis species have eye-stalks that are nearly two times their body length. Phylogenetic analysis of 35 species sampled from each of the major genera demonstrated that eye-stalk sexual dimorphism evolved independently at least four times within the family, and there have been several substantial reductions in eyespan and dimorphism [59].
The evolution of eye-stalk sexual dimorphism is mediated by a breeding system that involves both male–male competition and female choice. In many species, females aggregate at dusk on leaves or root hairs, and males fight for control of these mating sites. In a typical confrontation, males align themselves face-to-face and grapple with their forelegs until one male flies away. Males with larger eye-stalks are more likely to win these battles and displace other males from the aggregations [60–62]. In the morning, the males controlling the sites mate numerous times with several females. Mating rate and ejaculate size vary considerably among species, but in the most well-studied, and highly dimorphic, species (Teleopsis dalmanni and Teleopsis whitei), matings last for approximately 60 s and the ejaculate size is small [63,64]. Both field and laboratory experiments have shown that females in sexually dimorphic species prefer to roost and mate with males with larger eye-stalks, but females in monomorphic species exhibit little female choice [65–68]. Consistent with genetic models of sexual selection [69], female preference behaviour is genetically linked with male eyespan. After 13 generations of bidirectional selection on male eyespan in T. dalmanni, females from the long-eyespan lines and the control lines exhibited preference for males with larger eyespan, while females from the short-eyespan lines preferred males with shorter eyespan [70].
Despite the strong selection pressures operating on eyespan and the rapid evolution of this trait, there is still abundant genetic variation for male eyespan in sexually dimorphic species [71,72]. Elevated levels of genetic variation are common for sexually dimorphic traits [73]. Genic capture models explain this result by proposing that there is substantial variation for general condition within a population, and male ornamental traits accurately capture and advertise this variation because they are costly to produce [74,75]. Females, therefore, can assess the genetic quality of a male through the expression of these exaggerated traits and evolve mating preference for males that possess them. While the evolutionary significance of ‘good genes’ models of sexual selection remains controversial [76–78], there is evidence that some male ornamental traits exhibit heightened condition-dependence [79–81]. Several studies on stalk-eyed flies have measured condition-dependence for sexual and non-sexual traits in both monomorphic and dimorphic species, and also found results consistent with the genic capture models [82–84]. Relative eyespan (i.e. body size effects removed) in sexually dimorphic males has higher condition dependence than eyespan in females, eyespan in monomorphic males and non-ornamental traits such as wingspan in sexually dimorphic males. Furthermore, this condition-dependence appears to have a genetic basis as certain genotypes produce males with larger eyespan across a range of stressful environments, while other genotypes consistently produce males with smaller eye-stalks [85].
Another evolutionary pressure driving the evolution of diopsid eye-stalks is the presence of X-linked meiotic drive in many species [86,87]. Meiotic drive is a selfish genetic element that segregates non-randomly into gametes, promoting itself at the expense of gametes that do not carry the drive element. In males that carry the drive element, Y-bearing sperm either fail to develop properly or are destroyed during spermatogenesis, resulting in the transfer to females of only X-bearing sperm and, consequently, the production of a highly female-biased sex ratio [87,88]. As a population becomes skewed towards females, the reproductive success of individual males increases but females that mate with drive males will predominately produce daughters and therefore have lower reproductive success than females that mate with non-drive males. Therefore, mating with drive males is costly to females and they are expected to evolve mechanisms to counter the effects of drive [89]. One mechanism that females use is to evaluate the presence of meiotic drive based on male eyespan. In T. dalmanni, males without a drive element have larger eyespan than drive carrying males; so eyespan serves, to some degree, as an indicator of the presence of the drive element in males [90]. Therefore, females that choose to mate with males with longer eyespan are more likely to avoid the drive genotype, and this provides an explanation for the possible genetic benefits that females derive from preferring to mate with males with larger eyespan [90].
The relationship between meiotic drive and male eyespan results from genetic linkage [90,91]. In T. dalmanni, the genetic region with the largest effect on eyespan is located on the X chromosome, explaining over 30 per cent of the variation in male eyespan [92]. Quantitative trait locus analysis has demonstrated that loci influencing drive and eyespan are tightly linked on this chromosome and a lack of recombination between drive and non-drive individuals along much of the X chromosome suggests that these genes are located in one or more inversions [91]. Meiotic drive has been found in other dimorphic Teleopsis species, but not in monomorphic ones, suggesting that it may consistently influence sexual selection and the evolution of eyespan across the clade [86]. In addition, recent breeding experiments involving two T. dalmanni populations have revealed numerous cases of cryptic drive, indicating that drive elements have evolved and been suppressed numerous times in the genus (G. S. Wilkinson 2012, unpublished data). The inversion complex on the drive X chromosome can influence other traits as well as eyespan. For example, drive males have shorter sperm than non-drive males, and drive males suffer in post-copulatory sperm competition [93,94]. The presence of drive and the superior competitive ability of non-drive sperm may also influence the evolution of female mating rate in stalk-eyed flies. Females that mate often, thereby mixing the sperm of drive and non-drive males, may reduce the deleterious effects associated with mating with a drive male, and a comparative analysis has revealed a correlation across populations between female mating rate and the prevalence of drive [86]. Additional female traits exhibit a relationship with male eyespan and the X chromosome. Selection on male eyespan produced a correlated response in female eyespan, female internal reproductive morphology (ventral receptacle size) and egg size, all of which were due, in part, to X-linked genes that are in linkage disequilibrium or exhibit pleiotropy [95].
Overall, stalk-eyed flies provide an excellent model system for studying the evolution of sexual dimorphism. Numerous aspects of diopsid biology are affected by sexual selection and many of the genetic and phenotypic relationships among these components have been described. These interactions often involve male and female phenotypic traits and therefore provide opportunities for sexual conflict. The rapid change in eye-stalk morphology and repeated independent evolution of sexual dimorphism within the family enable comparative analysis. Furthermore, the close evolutionary proximity of diopsids to Drosophila facilitates functional annotation of the genome. In developing genomic resources for species within this family, we hope to encourage additional genetic and developmental studies of these fascinating animals.
4. Stalk-eyed fly genomics
Technological advances—such as expressed sequence tag (EST) libraries, microarrays and next-gen sequencing—that allow users to screen the expression and nucleotide sequence of thousands of genes at a time have enhanced the exploration of the genetic basis of sexual dimorphism in non-model organisms. In the past few years, we have conducted genomic research on stalk-eyed flies to assess the gene content of the X chromosome, quantify levels of sex-biased gene expression and annotate various transcriptomes. In the following sections, we summarize much of this work, focusing in particular on the prevalence of gene duplication and tissue-enriched gene expression and how these biological phenomena relate to sexual conflict.
(a). Sex-linkage
As summarized earlier, selective forces acting on genes located on the sex chromosomes are distinct from those on the autosomes, and consequently sex chromosomes are expected to have important influence on the evolution of sexual dimorphism [20,21]. In stalk-eyed flies, the significance of the X chromosome to the evolution of sexual dimorphism is well established [90–92,95,96] but, other than a few microsatellite markers, the genetic composition of this chromosome has been entirely unknown. Recently, we undertook a project to identify the sex-linkage of genes that are particularly relevant to eye-stalk development and evolution in T. dalmanni. The first step was to establish a database of genes expressed in the developing eye-antennal imaginal disc, the tissue that forms the adult head and eye-stalks. We sequenced over 40 000 clones from eye-antennal imaginal disc EST libraries made at three developmental stages and, after assembly and annotation, identified approximately 3500 genes that had significant homology to a known protein in D. melanogaster [97]. On the basis of this gene database, we then developed a microarray platform to identify sex-linkage.
Microarrays are best known as a method for comparing levels of gene expression between two different tissues, but they can also be used to compare DNA levels between tissues. This technique is called comparative genomic hybridization (CGH) and it is commonly used to identify duplicated chromosomal regions associated with disease [98,99]. In XY systems, because females have two X chromosomes and males have a single X chromosome, there should be a hybridization intensity difference for X-linked genes versus autosomal genes when male DNA is compared with female DNA. The expectation is that X-linked genes will have twice the signal for females than males, whereas autosomal genes will have the same signal intensity between the sexes. For all the genes in the EST database, we designed 60 bp probes and constructed microarray slides that contained 6–10 probes per gene. We labelled male DNA with a green dye and female DNA with a red dye, and hybridized the two to the slide.
Results from the CGH experiment clearly distinguished sex-linked genes from autosomal genes [100]. A histogram of the relative intensities for each gene on the slide shows a bimodal distribution, with the major peak reflecting no difference in DNA quantity for males and females and the smaller peak reflecting greater DNA abundance for females than males (figure 1a). Therefore, autosomal genes are represented by the major peak and X-linked genes by the minor peak. Approximately, 15 per cent of the genes on the microarray slides were scored as X-linked, which corresponds to the approximate size of the X chromosome based on karyotype images [92]. This experiment also identified a single gene that had a female/male log ratio of −5. We suspected that this gene was located on the Y chromosome and subsequent polymerase chain reaction (PCR) of the gene for over 40 males and females confirmed Y-linkage.
Figure 1.

Identifying sex-linkage in T. dalmanni. (a) Distribution of T. dalmanni CGH log2 ratio values. Male and female genomic DNA was hybridized to microarray slides containing probes designed from EST sequence data. A total of 3444 genes are represented. The large peak indicates autosomal genes while the smaller peak are genes on the X chromosome. (b) T. dalmanni chromosomal synteny with D. melanogaster indicates the diopsid X chromosome is homologous to chromosome 2L in Drosophila. The autosomal category includes 2891 T. dalmanni genes and the X chromosome comprises 533 T. dalmanni genes.
Examination of the chromosomal location of the homologues of the T. dalmanni genes in Drosophila reveals a strong syntenic relationship [100]. Over 90 per cent of the X-linked genes in T. dalmanni are located on chromosome 2L in Drosophila (Muller element B), suggesting that the T. dalmanni X chromosome is derived directly from an autosome (figure 1b). The formation of new X chromosomes, called neo-X chromosomes, has been found in several Drosophila species, but the neo-X found in T. dalmanni represents one of the largest wholesale reconstitutions of the X chromosome, at least in flies, and possibly in insects. In order to verify the chromosomal designations indicated by the CGH experiments, we mapped the location of a handful of genes that had probes on the chip. We examined a total of 28 genes, all of which had polyglutamine repeat regions that varied between selection lines and therefore could be typed as markers in line crosses. In all cases, the CGH results were confirmed. The four genes that are located on 2L in Drosophila are all on the X chromosome in T. dalmanni and the remaining 24 non-2L genes are all autosomal in T. dalmanni. In addition, mapping the Drosophila location of the T. dalmanni autosomal genes suggests additional syntenic relationships between the species. Chromosome 1 in T. dalmanni appears to be homologous primarily to 3L and X in Drosophila (all the genes but one come from these two Drosophila arms), while chromosome 2 contains almost exclusively genes from 2R and 3R [100].
Syntenic relationships across Diptera suggest that the neo-X chromosome in T. dalmanni is a derived condition. Both Drosophila and the mosquito, Anopheles gambiae, have relatively large X chromosomes that share a majority of their genes, suggesting that this X is basal for the family [101,102]. Alternatively, several other calyptrate and acalyptrate species have a small X with very few genes [103]; so it is most parsimonious at this time to assume that the Drosophila–mosquito X was lost or greatly reduced and the ancestor to diopsids had a small X. If this is the case, then the neo-X in diopsids formed from either the fusion of an autosome to a small pre-existing X or the movement of sex-determining genes to an autosome that started the sex chromosome creation process anew [104].
CGH experiments were also conducted on three other Teleopsis species, one of which, Teleopsis quinqueguttata, is not sexually dimorphic. The results from these experiments also produced two distinct peaks with strong agreement across the species [100]. Nearly all of the X-linked genes in T. dalmanni are X-linked in the other three species. But some genes have differential sex-linkage within Teleopsis, and on the basis of the synteny both with Drosophila and among the Teleopsis species, we can tentatively map gene movement within the genus. This analysis reveals a biased pattern of gene movement with 23 genes moving off of the X chromosome onto an autosome and no genes moving onto the X chromosome on the branch leading to T. quinqueguttata. Furthermore, the branches leading to the most dimorphic species, T. dalmanni and Teleopsis thaii, exhibit movement primarily onto the X chromosome (20 out of 22 gene movements). Therefore, gene movement appears to be associated with sexual dimorphism, but not in the manner predicted by an X feminization hypothesis because the branches with the most male-biased phenotypes have genes moving primarily onto the X chromosome. Sex chromosome gene content in Drosophila, however, is also affected by the age of a gene with newly duplicated genes actually being overrepresented on the X [105], and the pattern in Teleopsis may be driven by a similar process.
(b). Gene duplication
Much of the ‘off-of-the-X’ gene movement in Drosophila is driven by duplication events in which the derived copy, often through retroposition, moves off the X chromosome and subsequently acquires male-biased expression [28,30,32]. Therefore, we wanted to characterize the extent of gene duplication in Teleopsis and examine its relationship with chromosomal location and sex-biased gene expression. Comprehensive identification of gene duplication events from transcriptome data is problematic owing to incomplete assembly of genes and the assembly of alternative transcripts and divergent alleles into separate contigs. However, with substantial sequence coverage from several tissue sources and detailed sequence analysis of the resulting contigs, many of these difficulties can be mitigated. For this analysis, we collected four lanes of Illumina sequence data from testes, one lane from the adult male head, and one lane from the adult female head. These data comprised approximately 20 GB of short-read (60–80 bp) sequence information that was combined together with the EST data into a single grand transcriptome assembly.
In a two-step process, all the RNA-seq short reads were first assembled in ABySS [106,107] using five different k-mer sizes (33, 37, 41, 45 and 49). In the second step, the resulting contigs from each assembly were combined together, along with the contigs from the EST assembly, and assembled again in Geneious [108] with a minimum overlap of 30 bp and a minimum overlap identity of 96 per cent. This produced 55 792 contigs that were greater than 200 bp and these contigs were kept for annotation. All open-reading frames (ORFs) greater than 120 bp were extracted from these contigs and translated into their protein sequence using the ‘getorf’ module in Emboss [109]. The sequences were blasted, using blastp, against the D. melanogaster (Flybase r5.37) and A. gambiae (Ensembl P3.49) protein databases. Over 12 000 contigs contained ORFs with significant blast hits. In some cases, a given contig contained two or more ORFs that had significant hits to the same gene in D. melanogaster, which suggested the existence of a frame shift error in the contig sequence. These contigs were examined by eye and the frame shifts corrected by hand using the blastp alignment as a guide for identifying the source of the error. Gene expression values (expressed as reads per kilobase per million—RPKM) for each of the contigs were generated by mapping the reads back onto the contigs using Bowtie [110].
Within the grand transcriptome assembly, there were thousands of cases in which two or more contigs had their top blast hit to overlapping regions of the same gene in both Drosophila and Anopheles. In order to distinguish duplicated genes from allelic variants and alternative transcripts, we compared all the contigs that had significant homology to the same gene in D. melanogaster or A. gambiae to each other using a blastp search. Only contigs that contained greater that 50 per cent of the total protein sequence for the gene in Drosophila were included in the analysis. If any two contigs exhibited greater than 80 per cent protein similarity (based on blastp) across any portion of their sequence, these sequences were aligned together in Sequencher (v. 4.8; Gene Codes) and scored as paralogues, alternative transcripts or allelic variants. Paralogues can be distinguished from alternative transcripts based on the distribution of variation across the alignment. Sequence divergence between paralogues is typically spread across most of the alignment, whereas alternative transcripts are characterized by perfect identity for a subsection of the contigs (i.e. the regions of shared exons) and then a complete lack of homology across the remaining sequence. The difference between paralogues and allelic variants is simply a matter of protein divergence. We took a conservative approach and called two contigs as putative duplicates if they had greater than 10 per cent amino acid divergence across homologous regions of the protein. It is likely that some very recent duplication events are missed using this criterion. Overall, 685 genes appear to be single copy in D. melanogaster and A. gambiae but have multiple paralogues in T. dalmanni and comprise a total of 1787 paralogues, indicating that many genes have undergone multiple duplication events.
Gene duplication in Teleopsis is associated with the formation of the neo-X chromosome and gene movement on and off the sex chromosomes. Genes that reside on chromosome 2L in D. melanogaster, which is homologous to the neo-X chromosome in T. dalmanni, are significantly more likely to have duplicated in stalk-eyed flies than are genes located on other chromosomes (χ2 = 14.12, p = 0.002). The CGH experiments [100] have directly identified X-linkage in T. dalmanni for a subset of genes that have duplicated and these data confirm the association between the neo-X chromosome and elevated duplication rates (χ2 = 7.00, p = 0.008). Duplicated genes are also more likely to move between the autosomes and the sex chromosomes. Comparing D. melanogaster and T. dalmanni, 127 genes violate the 2L-X syntenic relationship between species and genes that have duplicated are over twice as likely to be represented in this subset of genes (χ2 = 19.56, p < 0.0001). Within Teleopsis, the relationship is even stronger, as genes that have duplicated are over three times more likely to have moved on or off the X chromosome of one of the four species examined in this genus than are genes that have not duplicated (χ2 = 17.05, p<0.0001). We are in the process of mapping the chromosomal location of all the genes identified in the testes transcriptome and will soon be able to provide a more detailed view of the pattern of duplication and gene movement on and off the neo-X chromosome.
Understanding how these duplication events are related to sex-biased gene expression requires mapping their expression relative to the tissue types used in the transcriptome assembly. For this analysis, we scored a gene as ‘tissue-enriched’ if the average level of gene expression was five times greater in one tissue than in the other tissue (e.g. head versus testes) and ‘tissue-specific’ if its expression level was 500 times greater in one of the tissues. It is important to point out that in this analysis all paralogues from a gene that has duplicated are counted as a ‘duplicate’ because we do not have the phylogenomic information to determine which of the copies is the original gene and which is the derived duplicated copy of a gene. Nevertheless, figure 2 clearly illustrates that duplicate paralogues are more likely to exhibit expression patterns that are enriched or specific to a given tissue. In both the testes and the head, genes that exhibit a more limited range of expression contain a higher percentage of duplicated genes. Over 70 per cent of all testes-specific genes and 26 per cent of all head-specific genes have been involved in a gene duplication event compared with only 11 per cent of gene that are expressed at similar levels between the tissues (‘ubiquitous’ in figure 2). Narrower expression patterns for duplicated genes are well documented [111–115] and occur either because the duplicate copy develops a new specialized function or because the regulatory role of the original gene is divided among the various paralogues created by the duplication events [116,117].
Figure 2.

The relationship between gene duplication and tissue specificity of gene expression in T. dalmanni. Columns indicate the percentage of genes for a given tissue expression pattern that are composed of genes that have undergone a gene duplication. The numbers within the columns provide the total number of duplicate paralogues that fall within that expression category. Tissue-‘enriched’ genes are defined as genes that are expressed five times more than in the other tissue and tissue-‘specific’ genes are expressed 500 times more than in the other tissue.
Genes expressed in the testes, in particular, are associated with duplication events in T. dalmanni (figure 2). There are over 20 times more testes-specific duplicate paralogues (590 paralogues) than head-specific duplicate paralogues (27 paralogues). In Drosophila, spermatogenesis uses a unique transcriptional programme that requires numerous genes that are expressed exclusively in the testes and male germline, many of which have originated from recent gene duplication events [40,42,43,47]. Stalk-eyed flies appear to have evolved a similar, or possibly greater, level of genetic diversity to function during this process. As summarized earlier, gene duplication and expression in the testes in Drosophila is the primary mechanism driving chromosomal relocation and biased sex chromosome composition. Similarly, in Teleopsis, duplicated testes genes also appear to be largely responsible for the association between gene duplication and the neo-X chromosome (figure 3). There is a significant over-representation of duplicated genes located on the neo-X (as inferred by their location on 2L in D. melanogaster) relative to the autosomes for testes-enriched genes (χ2 = 16.25, p<0.0001) but not for genes that are expressed at similar or greater levels in the head (head-enriched: χ2 = 0.88, p = 0.345; ubiquitous: χ2 = 0.00, p = 0.989).
Figure 3.
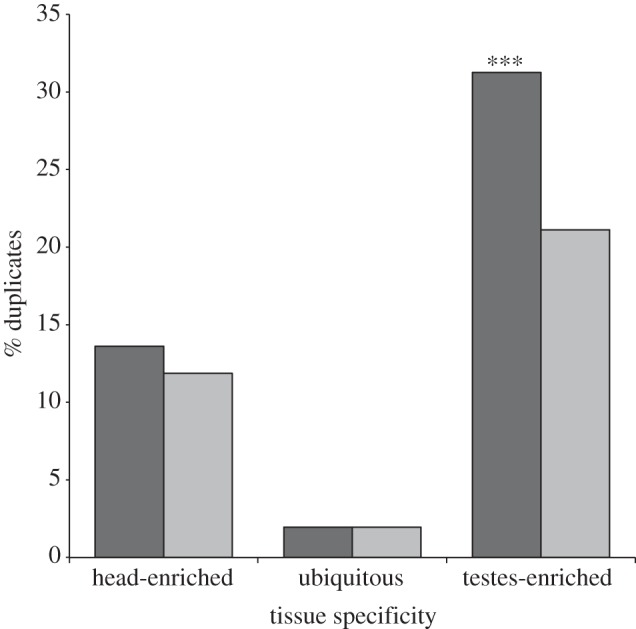
Association between chromosomal location and tissue specificity of gene expression. ‘2L’ genes in T. dalmanni have homologues on chromosome 2L in D. melanogaster, which is the source of the neo-X chromosome in T. dalmanni. ‘Non-2L’ genes in T. dalmanni have homologues on other chromosomes in D. melanogaster. Tissue-‘enriched’ genes are defined as genes that are expressed five times more than in the other tissue. A gene was scored as tissue-enriched if any of the paralogues associated with that gene was expressed five times more in one of the tissues, whereas ‘ubiquitous’ genes included only genes for which no paralogues were tissue-enriched. Dark grey bars, 2L; light grey bars, non-2L. p < 0.0001.
One prominent example of testes genes that have undergone abundant diversification in stalk-eyed flies involves the tubulin genes that are the central component of the axoneme, the structure in the sperm tail that provides motility. Two proteins, α-tubulin and β-tubulin, assemble into microtubules that are arranged in a stereotypic pattern within the axoneme—usually a ring of nine doublets surrounding two central pairs (termed 9 + 2 architecture) [118]—to form the basic motor structure of sperm. Mutations in these genes result in sterile males [119–121]. In Drosophila, the axonemal α-tubulin is also expressed in other tissues, including females, while the axonemal β-tubulin is specific to the male germline. Both genes are part of a gene family that diversified during insect evolution forming four primary clades for each gene family [49]. While reproductive genes often evolve rapidly [122,123], the tubulins are highly conserved at the protein level. In fact, there is not a single amino acid difference in the axonemal β-tubulin across all Drosophila species, a taxonomic range that spans approximately 60 million years [124].
Analysis of the testes transcriptome in T. dalmanni revealed numerous duplicate copies of both α-tubulin and β-tubulin. To examine the molecular evolution of this proliferation in more detail, we sequenced the duplicate paralogues for several T. dalmanni populations and closely related species [86,125] (sequence data have been deposited in NCBI Genbank under accession codes JQ866691–JQ866759). Stalk-eyed fly protein sequences were aligned with homologous proteins from other dipterans using the Muscle alignment function [126] in the Geneious analysis package [108] and maximum-likelihood trees were generated in PhyML [127] using a JTT + G model with 100 bootstrap replicates. The phylogenies for both α- and β-tubulins clearly show elevated rates of protein evolution for the duplicated copies in stalk-eyed flies relative to other flies (figures 4 and 5). It is noteworthy that a blast search of the Glossina morsitans supercontig database (provided by the G. morsitans group at the Wellcome Trust Sanger Institute and which can be searched at http://www.sanger.ac.uk/cgi-bin/blast/submitblast/g_morsitans) also identified six paralogous copies of β-tubulin that have arisen from recent duplication, but they have not differentiated to the extent of the stalk-eyed fly genes. Despite this rapid change relative to other dipteran homologues, tubulin evolution in stalk-eyed flies is still characterized by strong stabilizing selection. dN/dS ratios, which were calculated in HyPhY [128] using the SLAC method, for each duplicate paralogue range from 0.2 to 0.02.
Figure 4.

Phylogenetic analysis of α-tubulin genes in Diptera. Bootstrap values are provided for nodes defining and joining stalk-eyed fly paralogues. Dm, D. melanogaster, Dp, D. pseudoobscura, Sb, S. beccarri, Td (Cam), T. dalmanni (Cameron population), Td (Gom), T. dalmanni (Gombak population), Td (Lang), T. dalmanni (Langat population), Tq, T. quinqueguttata, Tt, T. thaii, Tw, T. whitei. Measurements of gene expression values (RPKM) are presented for each T. dalmanni (Gom) paralogue.
Figure 5.

Phylogenetic analysis of β-tubulin genes in Diptera. Bootstrap values are provided for nodes defining and joining stalk-eyed fly paralogues. Dm, D. melanogaster; Dp, D. pseudoobscura; Sb, S. beccarri; Td (Cam), T. dalmanni (Cameron population); Td (Gom), T. dalmanni (Gombak population); Td (Lang), T. dalmanni (Langat population); Tq, T. quinqueguttata; Tt, T. thaii; Tw, T. whitei. Measurements of gene expression values (RPKM) are presented for each T. dalmanni (Gom) paralogue, along with the sequence data for two important functional domains (see text for details). The axoneme motif in the C-terminal tail is underlined.
Quantification of expression values for the tubulin paralogues indicates that the duplicate copies are not minor isoforms but are expressed at high levels within the testes and therefore may play an important functional role in sperm production (figures 4 and 5). All the α-tubulin paralogues but one (α-tub5) are among the 40 most highly expressed genes in the testes. The more rapidly evolving copies (α-tub5, β-tub5 and β-tub6) are expressed at lower levels than the other tubulin genes, but even β-tub6 (RPKM = 85) is expressed at a substantially higher level than the median expression value for all testes genes (RPKM = 21.8). In order to determine the tissue specificity of the tubulin duplicates, we prepped RNA from adult females, gonadectomized males and mature testes and conducted PCR on the cDNA generated from these RNA samples. All the β-tubulin genes appear to be testes-specific, as no PCR bands were produced for the female or gonadectomized male samples (see electronic supplementary material, S1). The α-tub1 exhibits a ubiquitous expression pattern, while α-tub2, α-tub3 and α-tub4 (we have not yet succeeded in amplifying α-tub5) are all expressed in females but not gonadectomized males (see electronic supplementary material, S1). Given that none of these genes is expressed in the female head, we suspect that their expression is limited to reproductive tissues in the adult female. In the future, it will be essential to examine correlated patterns of gene expression across a range of spermatogenetic tissues and stages and to conduct functional assays (e.g. RNAi) that knockdown specific genes in order to identify whether specific pairs of α-tubulin and β-tubulin paralogues function together as a microtubule unit.
Mutagenesis assays in Drosophila have identified two sequence motifs in the β-tubulin protein that have strong functional significance. The first domain, termed the internal variable region (IVR), comprises three amino acids (TGA) at positions 55–57 and mediates physical interaction with the outer dynein arms in the axoneme [121]. Mutations in this region result in non-functioning sperm that do not maintain structural integrity along the length of the sperm tail [119]. The second domain, termed the axoneme motif, resides in the C-terminal tail of the protein and controls axoneme architecture [119,120]. Mutations in this region also produce non-functioning sperm, often lacking the central pair of microtubules [119,120]. These functional domains are highly conserved across insects [49,121,124] but the stalk-eyed fly β-tubulin paralogues exhibit substantial variation within these regions (figure 5). For instance, all axonemal β-tubulin genes examined thus far in animals possess a glycine at position 56 in the IVR [121], but four of the T. dalmanni paralogues possess an alternative amino acid at this position (figure 5). Understanding what factors are promoting this diversification and how this genetic diversity translates into phenotypic diversity requires additional studies on the developmental genetics of spermatogenesis in stalk-eyed flies. All the species in the family, except flies in the genus Diasemopsis, possess a heteromorphic sperm phenotype that is characterized by a long sperm type that is capable of fertilization and a short sperm type (approximately one-half to one-quarter the length of the long sperm) that probably cannot fertilize [129,130]. The diversity of the tubulin genes found within stalk-eyed flies suggests that there may be morphological complexity beyond this dimorphism.
The standard view of gamete investment is that egg production by females requires vast energetic demands, while sperm has relatively little cost and is produced in limitless quantity. While this dichotomy clearly applies to many species, there is an increased recognition that sperm production often entails non-trivial costs that affect the reproductive strategies of both sexes [131]. Several studies have shown that males modify their ejaculate size based on various aspects of female quality [132–134]. Flies, in particular, are noteworthy for the investment that many species make in individual sperm. Drosophila contains numerous species that produce giant sperm, including Drosophila bifurca, a species that has the longest sperm recorded for any animal [135]. The large sperm production entails a physiological cost in terms of delayed sexual maturity [136] and males modulate sperm production based on the availability of females [137].
Stalk-eyed flies exhibit a similar investment strategy in sperm production. While not approaching the extremes found within Drosophila, many diopsid species, particularly in the genus Diasemopsis, produce large sperm [129] and numerous aspects of their reproductive biology suggest that there is a substantial physiological cost associated with sperm production. First, males transfer very few sperm during a single copulation. In T. dalmanni, estimates range from between 65 and 140 sperm per mating [95,138] and, while males can mate several times in a morning, sperm transfer become limited after several sequential copulations [139]. Second, females in many species are highly sperm-limited owing, largely, to extensive infertile or unsuccessful copulations. In a laboratory survey of T. dalmanni, approximately 12 per cent of all matings of a standard duration failed to transfer any sperm [140]. Measurements in the wild indicate that between 20 and 65 per cent of all eggs laid by T. dalmanni females are not fertile [141,142]. Finally, diopsids take an extremely long time to reach sexual maturity, and this time period is characterized by substantial reproductive gland growth. In T. dalmanni, males reach sexual maturity about 25 days after eclosion, which is even longer than in D. bifurca, and experimental manipulation of their diet reduces testes and accessory gland growth and extends their time to sexual maturity [143]. Overall, these results suggest that the fitness effects associated with sperm production in male diopsids are not simply a function of sperm quantity but are likely to involve several aspects of sperm quality. Future research will need to examine how the high degree of genetic diversity reflected in the tubulin genes and the numerous other testes-specific duplicates relate to sperm fitness and investment, and the extent to which this diversification is driven by sperm competition and male–female coevolution.
(c). Sexual conflict
The abundance of gene duplication associated with reproductive tissues suggests that it may play a central role in the evolution of sexual dimorphism and the resolution of sexual conflict [18,19,51]. The process is particularly powerful because it allows differentiation between the sexes at multiple levels of transcriptional complexity. Not only can duplicate copies evolve sex-biased gene expression when freed from the pleiotropic constraints operating on the original gene, but also they can diverge at the protein level to favour the sex in which the duplicate is primarily expressed. The majority of studies examining the relationship between sex-biased gene expression and gene duplication have focused on expression in the germline [16,30,32]. Our catalogue of duplicated genes expressed in the adult head of T. dalmanni allows us to examine this relationship in somatic tissues. Male and female T. dalmanni exhibit numerous behavioural differences related to mating and reproduction [61,62,65,144,145], so it is reasonable to expect expression differences between the sexes in this tissue. For this analysis, we excluded all genes that had a testes-enriched or testes-specific pattern of gene expression to eliminate genes whose expression patterns were driven largely by selective forces related to sperm production. For the remaining 7018 genes, 174 exhibit twofold expression differences between the sexes, and genes that have duplicated were significantly over-represented in this group (figure 6). Genes that have been involved in a duplication event are over twice as likely to exhibit a twofold sex bias (χ2 = 24.70, p < 0.0001) and seven times as likely to exhibit a fourfold sex bias (χ2 = 22.40, p < 0.0001). Currently, we do not know whether these differentially expressed genes have been subject to different selective pressures in males and females, but if sex-biased gene expression reflects previous sexual conflict, then this relationship between duplication and sex-biased expression suggests that gene duplication provides a prominent mechanism for resolving this conflict.
Figure 6.
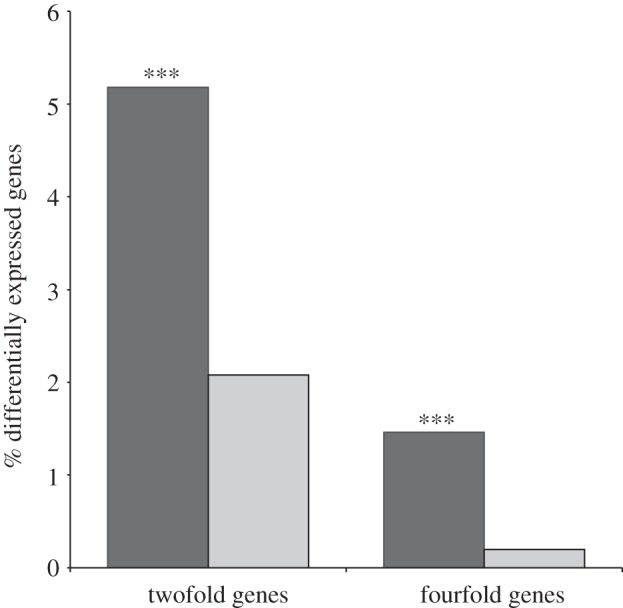
Association between sex-biased gene expression and gene duplication. Black bars, duplicates; grey bars, non-duplicates. ‘Duplicates’ include all paralogues for any gene that has duplicated in T. dalmanni; ‘non-duplicates’ have not been involved in a gene duplication event. Bars indicate the percentage of genes in each category that exhibit twofold or fourfold expression differences between males and females in the adult head. Black bars, duplicates; grey bars, non-duplicates. ***p < 0.0001.
To illustrate the evolution of tissue- and sex-specific gene expression that can result from gene duplication, figure 7 presents the phylogenetic relationships and expression values for two gene families that have both testes-specific and head-enriched sex-biased gene copies. Both genes, ferritin 1 heavy chain homologue (Fer1HCH) and Ance-4, have four paralogues in T. dalmanni, two of which are expressed primarily in the head and two of which are highly specific to the testes. Sphyracephala beccarri, a monomorphic species with relatively short eye-stalks that belongs to the basal genus in the family [59], has at least one copy of the testes-specific paralogues for each gene, indicating that the initial duplication is ancestral to the diversification of diopsids. It is possible this duplication event is related to the formation of the neo-X chromosome in the family. For the paralogues expressed in the head, the duplication of each gene is relatively recent, occurring after the split with the congeneric species, T. quinqueguttata, and involves the evolution of sex-biased gene expression (male-biased for Fer1HCH and female-biased for Ance-4) for one of the duplicates. Fer1HCH forms part of a complex with Fer2LCH that mediates iron-binding and storage and affects numerous aspects of behaviour in Drosophila [146] but is also expressed ubiquitously throughout the body [147]. The second member of the complex, Fer2LCH, also has a male-biased duplicate copy in T. dalmanni, suggesting this complex may have been influenced by selective pressures specific to male behaviour. If these pressures favoured changes at the protein level that were harmful to females, then the generation of a duplicate complex allows each sex to approach their respective fitness optima.
Figure 7.

Phylogenetic analysis and gene expression values for two genes—Fer1HCH (a) and Ance-4 (b)—that have duplicated and exhibit sex-biased gene expression in the adult head of T. dalmanni. In the gene expression boxes, M, male head; F, female head; T, testes. Dm, D. melanogaster; Dp, D. pseudoobscura; Sb, S. beccarri; Tq, T. quinqueguttata.
The phylogenies in figure 7 also reveal accelerated protein evolution for the duplicate copies that have acquired sex-biased gene expression. Rapid evolution of genes involved in reproduction has been well documented in Drosophila [122,123,148], so we wanted to examine whether duplication and the evolution of sex-biased gene expression were associated with faster protein evolution for genes expressed primarily in somatic tissue. Accurate measurements of protein divergence on a genomic scale are not available within diopsids; so we used D. melanogaster as the reference taxon to assess protein evolution. On the basis of the percent similarity scores from a blastp search against the D. melanogaster protein database, protein divergence in T. dalmanni is greater for both duplicated and sex-biased genes (figure 8). There is also a significant interaction term, based on a least-squares model, between these variables indicating that the difference between unbiased and sex-biased genes is greater for duplicated genes than genes that have not duplicated (Model effects: Duplicate—F1,6986 = 89.34, p < 0.0001, Sex-bias—F1,6986 = 39.19, p < 0.0001, Duplicate × Sex-bias—F1,6986 = 12.13, p = 0.0005). This result supports the hypothesis that the resolution of sexual conflict from gene duplication may also involve extensive protein evolution [18].
Figure 8.
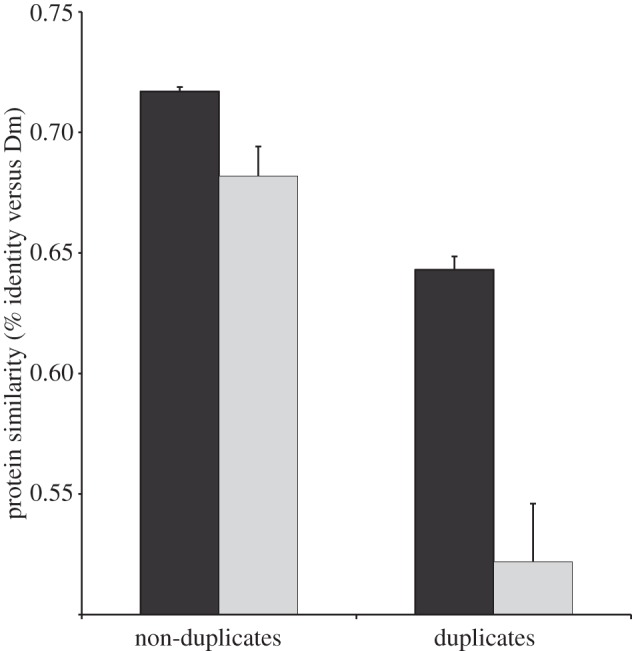
Rate of protein evolution for sex-biased and duplicated genes in the adult head. All genes that are expressed five times more in the testes were excluded from the analysis. ‘Sex-biased’ indicates genes with twofold expression differences between the sexes. Dark grey bars, unbiased; light grey bars, sex-biased. Dm, D. melanogaster.
Much of the data from the stalk-eyed fly transcriptomes is consistent with the proposal by Gallach and co-workers [19,50,51] that gene duplication resolves sexual conflict. The duplication rate is much higher for genes that function in the testes than for genes that are expressed in the head or in both tissues. However, it is important to note that we do not yet have data to exclude other factors, such as MSCI, that might be influencing the testes-specific duplication rate. Furthermore, two patterns suggest that a large proportion of the sex-biased duplications in T. dalmanni are not driven by sexual conflict within the testes. First, many of the duplications of testes-expressed genes involve multiple copies that are all testes-enriched or testes-specific. If the original copy of a gene duplication is already expressed in a male-limited pattern in the testes, then it is unlikely that sexual conflict is relevant to the origin and maintenance of the duplicate copy. This pattern is illustrated by the β-tubulin genes, which are all testes-specific, but also includes several other gene families. For instance, there are 31 genes that have at least four testes-enriched or testes-specific duplicates and no more than one other paralogue expressed at a similar level in the head. These genes comprised a total of 221 paralogues of which 210 are testes-enriched (40) or testes-specific (170). Many of these duplicates may have resulted from selection pressures associated with sperm competition rather than sexual conflict. The rate of testes-specific duplication may also be enhanced by certain properties of transcription within this tissue, such as open chromatic structure and elevated levels of the core transcriptional machinery [149]. Second, the analysis of the head duplicates indicates that tissues other than the testes may be a rich source of sexual conflict that is resolved through gene duplication. It will be essential for future experiments on stalk-eyed flies to collect data from additional tissues and species in order to more accurately identify the phylogenetic origin of duplication events and the extent of tissue- and sex-specific gene expression [19].
5. Conclusions and future directions
Stalk-eyed flies have become a valuable model system for studying the ecological and evolutionary aspects of sexual dimorphism, as they contain numerous classic features of a sexually selected system including ornamental traits, female preference behaviour, aggressive male interactions, sperm competition, condition-dependence and meiotic drive. Recent advances in sequencing technologies and other genomic methodologies now allow researchers to explore, on a wide scale, the genetic basis of these various components and the associations among them. Using many of these techniques, our research has focused on identifying chromosomal gene content, gene duplication and sex-biased gene expression in both sexually dimorphic and monomorphic species. In this review, we show that stalk-eyed flies possess a neo-X chromosome that, similar to Drosophila, contains a biased distribution of sex-biased genes. We have identified an extensive list of gene duplications that have arisen in the lineage leading to stalk-eyed flies and are associated with numerous aspects of their reproductive biology, including being preferentially associated with the neo-X chromosome, chromosomal gene movement, testes-expression and sex-biased gene expression in the head. Future studies will need to identify chromosomal gene content, gene presence/absence and tissue- and sex-specific gene expression across a wide-range of stalk-eyed fly species in order to provide a more fine-scaled picture of the evolutionary relationships among these variables and various sexual traits such as eyespan, female preference and sperm morphology. We also hope that the genomic resources we have developed will encourage more developmental research on these remarkable insects.
Acknowledgements
We thank Dustin Rubenstein and Mike Levandowsky for organizing a stimulating symposium and Rob DeSalle for his generous support of this research. Grace Chan, Samantha Eng, Yoko Higuchi and Fadwa Saber, high school students in the Science Research Mentoring Programme at the American Museum of Natural History, collected the sequence data for the tubulin genes. Bard College undergraduates Deven Connelly and Prabarna Ganguly assisted in the preparation of the adult head RNA-seq libraries through the Bard Summer Research Institute and Cara Brand assisted in the preparation of the testes RNA-seq libraries at the University of Maryland. We thank the Cullman Program in Molecular Systematics at AMNH for support. Funding for the research was provided by National Science Foundation grant nos DEB-0951816 (RHB) and DEB-0952260 (GSW).
References
- 1.Emlen D. J., Corley Lavine L., Ewen-Campen B. 2007. On the origin and evolutionary diversification of beetle horns. Proc. Natl Acad. Sci. USA 104, 8661–8668 10.1073/pnas.0701209104 (doi:10.1073/pnas.0701209104) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Hegyi G., Török J., Tóth L., Garamszegi L. Z., Rosivall B. 2006. Rapid temporal change in the expression and age-related information content of a sexually selected trait. J. Evol. Biol. 19, 228–238 10.1111/j.1420-9101.2005.00970.x (doi:10.1111/j.1420-9101.2005.00970.x) [DOI] [PubMed] [Google Scholar]
- 3.Meyer A. 1997. The evolution of sexually selected traits in male swordtail fishes (Xiphophorus: Poeciliidae). Heredity 79, 329–337 10.1038/hdy.1997.161 (doi:10.1038/hdy.1997.161) [DOI] [Google Scholar]
- 4.Omland K. E., Lanyon S. M. 2000. Reconstructing plumage evolution in orioles (Icterus): repeated convergence and reversal in patterns. Evolution 54, 2119–2133 10.1111/j.0014-3820.2000.tb01254.x (doi:10.1111/j.0014-3820.2000.tb01254.x) [DOI] [PubMed] [Google Scholar]
- 5.van Doorn G. S. 2009. Intralocus sexual conflict. Ann. NY Acad. Sci. 1168, 52–71 10.1111/j.1749-6632.2009.04573.x (doi:10.1111/j.1749-6632.2009.04573.x) [DOI] [PubMed] [Google Scholar]
- 6.Bonduriansky R., Chenoweth S. F. 2009. Intralocus sexual conflict. Trends Ecol. Evol. 24, 280–288 10.1016/j.tree.2008.12.005 (doi:10.1016/j.tree.2008.12.005) [DOI] [PubMed] [Google Scholar]
- 7.Rice W. R. 1992. Sexually antagonistic genes: experimental evidence. Science 256, 1436–1439 10.1126/science.1604317 (doi:10.1126/science.1604317) [DOI] [PubMed] [Google Scholar]
- 8.Rice W. R. 1996. Sexually antagonistic male adaptation triggered by experimental arrest of female evolution. Nature 381, 232–235 10.1038/381232a0 (doi:10.1038/381232a0) [DOI] [PubMed] [Google Scholar]
- 9.Chippindale A. K., Gibson J. R., Rice W. R. 2001. Negative genetic correlation for adult fitness between sexes reveals ontogenetic conflict in Drosophila. Proc. Natl Acad. Sci. USA 98, 1671–1675 10.1073/pnas.041378098 (doi:10.1073/pnas.041378098) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Prasad N. G., Bedhomme S., Day T., Chippendale A. K. 2007. An evolutionary cost of separate genders revealed by male-limited evolution. Am. Nat. 169, 29–37 10.1086/509941 (doi:10.1086/509941) [DOI] [PubMed] [Google Scholar]
- 11.Morrow E. H., Stewart A. D., Rice W. R. 2008. Assessing the extent of genome-wide intralocus sexual conflict via experimentally enforced gender-limited selection. J. Evol. Biol. 21, 1046–1054 10.1111/j.1420-9101.2008.01542.x (doi:10.1111/j.1420-9101.2008.01542.x) [DOI] [PubMed] [Google Scholar]
- 12.Gibson J. R., Chippindale A. K., Rice W. R. 2002. The X chromosome is a hot spot for sexually antagonistic fitness variation. Proc. R. Soc. Lond. B 269, 499–505 10.1098/rspb.2001.1863 (doi:10.1098/rspb.2001.1863) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Rice W. R., Chippindale A. K. 2002. The evolution of hybrid infertility: perpetual coevolution between gender-specific and sexually antagonistic genes. Genetica 116, 179–188 10.1023/A:1021205130926 (doi:10.1023/A:1021205130926) [DOI] [PubMed] [Google Scholar]
- 14.Parisi M., Nuttall R., Naiman D., Bouffard G., Malley J., Andrews J., Eastman, S., Oliver B. 2003. Paucity of genes on the Drosophila X chromosome showing male-biased expression. Science 299, 697–700 10.1126/science.1079190 (doi:10.1126/science.1079190) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Ranz J. M., Castillo-Davis C. I., Meiklejohn C. D., Hartl D. L. 2003. Sex-dependent gene expression and evolution of the Drosophila transcriptome. Science 300, 1742–1745 10.1126/science.1085881 (doi:10.1126/science.1085881) [DOI] [PubMed] [Google Scholar]
- 16.Sturgill D., Zhang Y., Parisi M., Oliver B. 2007. Demasculinization of X chromosomes in the Drosophila genus. Nature 450, 238–241 10.1038/nature06330 (doi:10.1038/nature06330) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Schutt C., Nothiger R. 2000. Structure, function and evolution of sex-determining systems in Dipteran insects. Development 127, 667–677 [DOI] [PubMed] [Google Scholar]
- 18.Connallon T., Clark A. G. 2011. The resolution of sexual antagonism by gene duplication. Genetics 187, 919–937 10.1534/genetics.110.123729 (doi:10.1534/genetics.110.123729) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Gallach M., Betran E. 2011. Intralocus sexual conflict resolved through gene duplication. Trends Ecol. Evol. 26, 222–228 (doi:10.1016/j.tree.2011.02.004) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Vicoso B., Charlesworth B. 2006. Evolution on the X chromosome: unusual patterns and processes. Nat. Rev. Genet. 7, 645–653 10.1038/nrg1914 (doi:10.1038/nrg1914) [DOI] [PubMed] [Google Scholar]
- 21.Rice W. R. 1984. Sex chromosomes and the evolution of sexual dimorphism. Evolution 38, 735–742 10.2307/2408385 (doi:10.2307/2408385) [DOI] [PubMed] [Google Scholar]
- 22.Fry J. D. 2010. The genomic location of sexually antagonistic variation: some cautionary comments. Evolution 64, 1510–1516 10.1111/j.1558-5646.2009.00898.x (doi:10.1111/j.1558-5646.2009.00898.x) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Zhang Y., Sturgill D., Parisi M., Kumar S., Oliver B. 2007. Constraint and turnover in sex-biased gene expression in the genus Drosophila. Nature 450, 233–238 10.1038/nature06323 (doi:10.1038/nature06323) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Baker D. A., Russell S. 2011. The role of testis-specific gene expression in sex chromosome evolution of Anopheles gambiae. Genetics 189, 1117–1120 10.1534/genetics.111.133157 (doi:10.1534/genetics.111.133157) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Prince E. G., Kirkland D., Demuth J. P. 2010. Hyperexpression of the X chromosome in both sexes results in extensive female bias of X-linked genes in the flour beetle. Genome Biol. Evol. 2, 336–346 10.1093/gbe/evq024 (doi:10.1093/gbe/evq024) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Hense W., Baines J. F., Parsch J. 2007. X chromosome inactivation during Drosophila spermatogenesis. PLoS Biol. 5, 2288–2295 10.1371/journal.pbio.0050273 (doi:10.1371/journal.pbio.0050273) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Lifschytz E., Lindsley D. L. 1972. The role of X-chromosome inactivation during spermatogenesis. Proc. Natl Acad. Sci. USA 69, 182–186 10.1073/pnas.69.1.182 (doi:10.1073/pnas.69.1.182) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Betran E., Thornton K., Long M. 2002. Retroposed new genes out of the X in Drosophila. Genome Res. 12, 1854–1859 10.1101/gr.6049 (doi:10.1101/gr.6049) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kemkemer C., Hense W., Parsch J. 2011. Fine-scale analysis of X chromosome inactivation in the male germ line of Drosophila melanogaster. Mol. Biol. Evol. 28, 1561–1563 10.1093/molbev/msq355 (doi:10.1093/molbev/msq355) [DOI] [PubMed] [Google Scholar]
- 30.Vibranovski M. D., Zhang Y., Long M. Y. 2009. General gene movement off the X chromosome in the Drosophila genus. Genome Res. 19, 897–903 10.1101/gr.088609.108 (doi:10.1101/gr.088609.108) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Vibranovski M. D., Lopes H. F., Karr T. L., Long M. 2009. Stage-specific expression profiling of Drosophila spermatogenesis suggests that meiotic sex chromosome inactivation drives genomic relocation of testis-expressed genes. PLoS Genet. 5, e1000731. 10.1371/journal.pgen.1000731 (doi:10.1371/journal.pgen.1000731) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Meisel R. P., Han M. V., Hahn M. W. 2009. A complex suite of forces drives gene traffic from Drosophila X chromosomes. Genome Biol. Evol. 2009, 176–188 10.1093/gbe/evp018 (doi:10.1093/gbe/evp018) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Wu C. I., Xu E. Y. 2003. Sexual antagonism and X inactivation—the SAXI hypothesis. Trends Genet. 19, 243–247 10.1016/S0168-9525(03)00058-1 (doi:10.1016/S0168-9525(03)00058-1) [DOI] [PubMed] [Google Scholar]
- 34.Vicoso B., Charlesworth B. 2009. The deficit of male-biased genes on the, D. melanogaster X chromosome is expression-dependent: a consequence of dosage compensation? J. Mol. Evol. 68, 576–583 10.1007/s00239-009-9235-4 (doi:10.1007/s00239-009-9235-4) [DOI] [PubMed] [Google Scholar]
- 35.Bai Y., Casola C., Feschotte C., Betran E. 2007. Comparative genomics reveals a constant rate of origination and convergent acquisition of functional retrogenes in Drosophila. Genome Biol. 8, R11. 10.1186/gb-2007-8-1-r11 (doi:10.1186/gb-2007-8-1-r11) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Schrider D. R., Stevens K., Cardeño C. M., Langley C. H., Hahn M. W. 2011. Genome-wide analysis of retrogene polymorphisms in Drosophila melanogaster. Genome Res. 21, 2087–2095 10.1101/gr.116434.110 (doi:10.1101/gr.116434.110) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Mikhaylova L., Nurminsky D. 2011. Lack of global meiotic sex chromosome inactivation, and paucity of tissue-specific gene expression on the Drosophila X chromosome. BMC Biol. 9, 29 (doi:10.1186/1741-7007-9-29) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Meiklejohn C. D., Landeen E. L., Cook J. M., Kingan S. B., Presgraves D. C. 2010. Sex chromosome-specific regulation in the Drosophila male germline but little evidence for chromosomal dosage compensation or meiotic inactivation. PLoS Biol. 9, e1001126. 10.1371/journal.pbio.1001126 (doi:10.1371/journal.pbio.1001126) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Metta M., Schlotterer C. 2010. Non-random genomic integration—an intrinsic property of retrogenes in Drosophila? BMC Evol. Biol. 10, 114. 10.1186/1471-2148-10-114 (doi:10.1186/1471-2148-10-114) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.White-Cooper H. 2010. Molecular mechanisms of gene regulation during Drosophila spermatogenesis. Reproduction 139, 11–21 10.1530/REP-09-0083 (doi:10.1530/REP-09-0083) [DOI] [PubMed] [Google Scholar]
- 41.Chintapalli V. R., Wang J., Dow J. A. 2007. Using FlyAtlas to identify better Drosophila melanogaster models of human disease. Nat. Genet. 39, 715–720 10.1038/ng2049 (doi:10.1038/ng2049) [DOI] [PubMed] [Google Scholar]
- 42.White-Cooper H., Bausek N. 2010. Evolution and spermatogenesis. Phil. Trans. R. Soc. B 365, 1465–1480 10.1098/rstb.2009.0323 (doi:10.1098/rstb.2009.0323) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Mikhaylova L. M., Nguyen K., Nurminsky D. I. 2008. Analysis of the Drosophila melanogaster testes transcriptome reveals coordinate regulation of paralogous genes. Genetics 179, 305–315 10.1534/genetics.107.080267 (doi:10.1534/genetics.107.080267) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Hiller M., et al. 2004. Testis-specific TAF homologs collaborate to control a tissue-specific transcription program. Development 131, 5297–5308 10.1242/dev.01314 (doi:10.1242/dev.01314) [DOI] [PubMed] [Google Scholar]
- 45.Hiller M. A., Lin T-Y., Wood C., Fuller M. T. 2001. Developmental regulation of transcription by a tissue-specific TAF homolog. Genes Dev. 15, 1021–1030 10.1101/gad.869101 (doi:10.1101/gad.869101) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Torgerson D. G., Singh R. S. 2004. Rapid evolution through gene duplication and subfunctionalization of the testes-specific α4 proteasome subunits in Drosophila. Genetics 168, 1421–1432 10.1534/genetics.104.027631 (doi:10.1534/genetics.104.027631) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Belote J. M., Zhong L. 2009. Duplicated proteasome subunit genes in Drosophila and their roles in spermatogenesis. Heredity 103, 23–31 10.1038/hdy.2009.23 (doi:10.1038/hdy.2009.23) [DOI] [PubMed] [Google Scholar]
- 48.Tracy C., Rio J., Motiwale M., Christensen S. M., Betran E. 2010. Convergently recruited nuclear transport retrogenes are male biased in expression and evolving under positive selection in Drosophila. Genetics 184, 1067–1076 10.1534/genetics.109.113522 (doi:10.1534/genetics.109.113522) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Nielsen M., Gadagkar S., Gutzwiller L. 2010. Tubulin evolution in insects: gene duplication and subfunctionalization provide specialized isoforms in a functionally constrained gene family. BMC Evol. Biol. 10, 113. 10.1186/1471-2148-10-113 (doi:10.1186/1471-2148-10-113) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Gallach M., Chandrasekaran C., Betran E. 2010. Analyses of nuclearly encoded mitochondrial genes suggest gene duplication as a mechanism for resolving intralocus sexually antagonistic conflict in Drosophila. Genome Biol. Evol. 2, 835–850 10.1093/gbe/evq069 (doi:10.1093/gbe/evq069) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Gallach M., Domingues S., Betran E. 2011. Gene duplication and the genome distribution of sex-biased genes. Int. J. Evol. Biol. 2011, 1–20 10.4061/2011/989438 (doi:10.4061/2011/989438) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Proulx S. R., Phillips P. C. 2006. Allelic divergence precedes and promotes gene duplication. Evolution 60, 881–892 10.1111/j.0014-3820.2006.tb01168.x (doi:10.1111/j.0014-3820.2006.tb01168.x) [DOI] [PubMed] [Google Scholar]
- 53.Wilkinson G. S., Dodson G. 1997. Function and evolution of antlers and eye stalks in flies. In The evolution of mating systems in insects and arachnids (eds Choe J., Crespi B.), pp. 310–328 Cambridge, UK: Cambridge University Press [Google Scholar]
- 54.Yeates D. K., Wiegmann B. M. 1999. Congruence and controversy: toward a higher-level phylogeny of Diptera. Annu. Rev. Entomol. 44, 397–428 10.1146/annurev.ento.44.1.397 (doi:10.1146/annurev.ento.44.1.397) [DOI] [PubMed] [Google Scholar]
- 55.Burkhardt D., de la Motte I. 1985. Selective pressures, variability, and sexual dimorphism in stalk-eyed flies (Diopsidae). Naturwissenschaften 72, 204–206 10.1007/BF01195763 (doi:10.1007/BF01195763) [DOI] [Google Scholar]
- 56.Feijen H. R. 2011. The genus Teleopsis Rondani (Diptera, Diopsidae): discussion of its taxonomic position and revision of the species occurring in the Philippines. Zool. Meded. 85, 79–140 [Google Scholar]
- 57.Feijen H. R. 1989. Diopsidae. In Flies of the nearctic region (ed. Griffiths G. C. D.), pp. 1–122 Stuttgart, Germany: E. Schweizerbart'sche Verlagsbuchhandlung [Google Scholar]
- 58.Baker R. H., Wilkinson G. S. 2001. Phylogenetic analysis of sexual dimorphism and eye-span allometry in stalk-eyed flies (Diopsidae). Evolution 55, 1373–1385 10.1111/j.0014-3820.2001.tb00659.x (doi:10.1111/j.0014-3820.2001.tb00659.x) [DOI] [PubMed] [Google Scholar]
- 59.Baker R. H., Wilkinson G. S., DeSalle R. 2001. The phylogenetic utility of different types of molecular data used to infer evolutionary relationships among stalk-eyed flies (Diopsidae). Syst. Biol. 50, 87–105 10.1080/106351501750107512 (doi:10.1080/106351501750107512) [DOI] [PubMed] [Google Scholar]
- 60.Burkhardt D., de la Motte I., Lunau K. 1994. Signalling fitness: larger males sire more offspring. Studies of the stalk-eyed fly Cyrtodiopsis whitei (Diopsidae, Diptera). J. Comp. Physiol. A 174, 61–64 10.1007/BF00192006 (doi:10.1007/BF00192006) [DOI] [Google Scholar]
- 61.Small J., Cotton S., Fowler K., Pomiankowski A. 2009. Male eyespan and resource ownership affect contest outcome in the stalk-eyed fly, Teleopsis dalmanni. Anim. Behav. 78, 1213–1220 10.1016/j.anbehav.2009.08.009 (doi:10.1016/j.anbehav.2009.08.009) [DOI] [Google Scholar]
- 62.Panhuis T. M., Wilkinson G. S. 1999. Exaggerated male eye span influences contest outcome in stalk-eyed flies. Behav. Ecol. Sociobiol. 46, 221–227 10.1007/s002650050613 (doi:10.1007/s002650050613) [DOI] [Google Scholar]
- 63.Lorch P. D., Wilkinson G. S., Reillo P. R. 1993. Copulation duration and sperm precedence in the stalk-eyed fly Cyrtodiopsis whitei (Diptera, Diopsidae). Behav. Ecol. Sociobiol. 32, 303–311 10.1007/BF00183785 (doi:10.1007/BF00183785) [DOI] [Google Scholar]
- 64.Kotrba M. 1996. Sperm transfer by spermatophore in Diptera: new results from the Diopsidae. Zool. J. Linn. Soc. 117, 305–323 10.1111/j.1096-3642.1996.tb02192.x (doi:10.1111/j.1096-3642.1996.tb02192.x) [DOI] [Google Scholar]
- 65.Wilkinson G. S., Kahler H., Baker R. H. 1998. Evolution of female mating preferences in stalk-eyed flies. Behav. Ecol. 9, 525–533 10.1093/beheco/9.5.525 (doi:10.1093/beheco/9.5.525) [DOI] [Google Scholar]
- 66.Hingle A., Fowler K., Pomiankowski A. 2001. Size-dependent mate preference in the stalk-eyed fly Cyrtodiopsis dalmanni. Anim. Behav. 61, 589–595 10.1006/anbe.2000.1613 (doi:10.1006/anbe.2000.1613) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Burkhardt D., de la Motte I. 1988. Big ‘antlers’ are favoured: female choice in stalk-eyed flies (Diptera, Insecta), field collected harems and laboratory experiments. J. Comp. Physiol. A 162, 649–652 10.1007/BF01342640 (doi:10.1007/BF01342640) [DOI] [Google Scholar]
- 68.Cotton S., Rogers D. W., Small J., Pomiankowski A., Fowler K. 2006. Variation in preference for a male ornament is positively associated with female eyespan in the stalk-eyed fly Diasemopsis meigenii. Proc. R. Soc. B 273, 1287–1292 (doi:10.1098/rspb.2005.3449) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Lande R. 1981. Models of speciation by sexual selection on polygenic traits. Proc. Natl Acad. Sci. USA 78, 3721–3725 10.1073/pnas.78.6.3721 (doi:10.1073/pnas.78.6.3721) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Wilkinson G. S., Reillo P. R. 1994. Female choice response to artificial selection on an exaggerated male trait in a stalk-eyed fly. Proc. R. Soc. Lond. B 255, 1–6 10.1098/rspb.1994.0001 (doi:10.1098/rspb.1994.0001) [DOI] [Google Scholar]
- 71.Wilkinson G. S., Taper M. 1999. Evolution of genetic variation for condition dependent traits in stalk-eyed flies. Proc. R. Soc. Lond. B 266, 1685–1690 10.1098/rspb.1999.0832 (doi:10.1098/rspb.1999.0832) [DOI] [Google Scholar]
- 72.Wilkinson G. S. 1993. Artificial sexual selection alters allometry in the stalk-eyed fly Cyrtodiopsis dalmanni (Diptera: Diopsidae). Genet. Res. 62, 213–222 (doi:10.1017/S001667230003192X) [Google Scholar]
- 73.Pomiankowski A., Møller A. P. 1995. A resolution of the lek paradox. Proc. R. Soc. Lond. B 260, 21–29 10.1098/rspb.1995.0054 (doi:10.1098/rspb.1995.0054) [DOI] [Google Scholar]
- 74.Tomkins J. L., Radwan J., Kotiaho J. S., Tregenza T. 2004. Genic capture and resolving the lek paradox. Trends Ecol. Evol. 19, 323–328 10.1016/j.tree.2004.03.029 (doi:10.1016/j.tree.2004.03.029) [DOI] [PubMed] [Google Scholar]
- 75.Rowe L., Houle D. 1996. The lek paradox and the capture of genetic variance by condition-dependent traits. Proc. R. Soc. Lond. B 263, 1415–1421 10.1098/rspb.1996.0207 (doi:10.1098/rspb.1996.0207) [DOI] [Google Scholar]
- 76.Cotton S., Pomiankowski A. 2007. Sexual selection: does condition dependence fail to resolve the ‘lek paradox’? Curr. Biol. 17, R335–R337 (doi:10.1016/j.cub.2007.03.004) [DOI] [PubMed] [Google Scholar]
- 77.Van Homrigh A., Higgie M., McGuigan K., Blows M. W. 2007. The depletion of genetic variance by sexual selection. Curr. Biol. 17, 528–532 10.1016/j.cub.2007.01.055 (doi:10.1016/j.cub.2007.01.055) [DOI] [PubMed] [Google Scholar]
- 78.Pischedda A., Chippindale A. K. 2006. Intralocus sexual conflict diminishes the benefits of sexual selection. PLoS Biol. 4, e356. 10.1371/journal.pbio.0040356 (doi:10.1371/journal.pbio.0040356) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Johnstone R. A. 1995. Sexual selection, honest advertisement and the handicap principle: reviewing the evidence. Biol. Rev. 70, 11–65 10.1111/j.1469-185X.1995.tb01439.x (doi:10.1111/j.1469-185X.1995.tb01439.x) [DOI] [PubMed] [Google Scholar]
- 80.Cotton S., Fowler K., Pomiankowski A. 2004. Do sexual ornaments demonstrate heightened condition-dependent expression as predicted by the handicap hypothesis? Proc. R. Soc. Lond. B 271, 771–783 10.1098/rspb.2004.2688 (doi:10.1098/rspb.2004.2688) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Ahuja A., De Vito S., Singh R. 2011. Condition dependence and the nature of genetic variation for male sex comb bristle number in Drosophila melanogaster. Genetica 139, 505–510 10.1007/s10709-011-9572-2 (doi:10.1007/s10709-011-9572-2) [DOI] [PubMed] [Google Scholar]
- 82.David P., Hingle A., Greig D., Rutherford A., Pomiankowski A., Fowler K. 1998. Male sexual ornament size but not asymmetry reflects condition in stalk-eyed flies. Proc. R. Soc. Lond. B 265, 2211–2216 10.1098/rspb.1998.0561 (doi:10.1098/rspb.1998.0561) [DOI] [Google Scholar]
- 83.Cotton S., Fowler K., Pomiankowski A. 2004. Condition dependence of sexual ornament size and variation in the stalk-eyed fly Cyrtodiopsis dalmanni (Diptera:Diopsidae). Evolution 58, 1038–1046 10.1111/j.0014-3820.2004.tb00437.x (doi:10.1111/j.0014-3820.2004.tb00437.x) [DOI] [PubMed] [Google Scholar]
- 84.Cotton S., Fowler K., Pomiankowski A. 2004. Heightened condition dependence is not a general feature of male eyespan in stalk-eyed flies (Diptera:Diopsidae). J. Evol. Biol. 17, 1310–1316 10.1111/j.1420-9101.2004.00754.x (doi:10.1111/j.1420-9101.2004.00754.x) [DOI] [PubMed] [Google Scholar]
- 85.David P., Bjorksten T., Fowler K., Pomiankowski A. 2000. Condition-dependent signalling of genetic variation in stalk-eyes flies. Nature 406, 186–188 10.1038/35018079 (doi:10.1038/35018079) [DOI] [PubMed] [Google Scholar]
- 86.Wilkinson G. S., Swallow J. G., Christianson S. J., Madden K. 2003. Phylogeography of sex ratio and multiple mating in stalk-eyed flies from southeast Asia. Genetica 117, 37–46 10.1023/A:1022360531703 (doi:10.1023/A:1022360531703) [DOI] [PubMed] [Google Scholar]
- 87.Presgraves D. C., Severence E., Wilkinson G. S. 1997. Sex chromosome meiotic drive in stalk-eyed flies. Genetics 147, 1169–1180 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Wilkinson G. S., Sanchez M. I. 2001. Sperm development, age and sex chromosome meiotic drive in the stalk-eyed fly, Cyrtodiopsis whitei. Heredity 87, 17–24 10.1046/j.1365-2540.2001.00898.x (doi:10.1046/j.1365-2540.2001.00898.x) [DOI] [PubMed] [Google Scholar]
- 89.Lande R., Wilkinson G. S. 1999. Models of sex-ratio meiotic drive and sexual selection in stalk-eyed flies. Genet. Res. 74, 245–253 10.1017/S0016672399004218 (doi:10.1017/S0016672399004218) [DOI] [Google Scholar]
- 90.Wilkinson G. S., Presgraves D. C., Crymes L. 1998. Male eye span in stalk-eyed flies indicates genetic quality by meiotic drive suppression. Nature 391, 276–278 10.1038/34640 (doi:10.1038/34640) [DOI] [Google Scholar]
- 91.Johns P. M., Wolfenbarger L. L., Wilkinson G. S. 2005. Genetic linkage between a sexually selected trait and X chromosome meiotic drive. Proc. R. Soc. B 272, 2097–2103 10.1098/rspb.2005.3183 (doi:10.1098/rspb.2005.3183) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Wolfenbarger L. L., Wilkinson G. S. 2001. Sex-linked expression of a sexually selected trait in the stalk-eyed fly, Cyrtodiopsis dalmanni. Evolution 55, 103–110 10.1111/j.0014-3820.2001.tb01276.x (doi:10.1111/j.0014-3820.2001.tb01276.x) [DOI] [PubMed] [Google Scholar]
- 93.Wilkinson G. S., Fry C. L. 2001. Meiotic drive alters sperm competitive ability in stalk-eyed flies. Proc. R. Soc. Lond. B 268, 2559–2564 10.1098/rspb.2001.1831 (doi:10.1098/rspb.2001.1831) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Fry C. L., Wilkinson G. S. 2004. Sperm survival in female stalk-eyed flies depends on seminal fluid and meiotic drive. Evolution 58, 1622–1626 10.1111/j.0014-3820.2004.tb01743.x (doi:10.1111/j.0014-3820.2004.tb01743.x) [DOI] [PubMed] [Google Scholar]
- 95.Wilkinson G. S., Amitin E. G., Johns P. M. 2005. Sex-linked correlated responses in female reproductive traits to selection on male eye span in stalk-eyed flies. Int. Comp. Biol. 45, 500–510 10.1093/icb/45.3.500 (doi:10.1093/icb/45.3.500) [DOI] [PubMed] [Google Scholar]
- 96.Johns P. M., Wilkinson G. S. 2007. X chromosome influences sperm length in the stalk-eyed fly Cyrtodiopsis dalmanni. Heredity 99, 56–61 10.1038/sj.hdy.6800963 (doi:10.1038/sj.hdy.6800963) [DOI] [PubMed] [Google Scholar]
- 97.Baker R. H., Morgan J., Wang X., Boore J. L., Wilkinson G. S. 2009. Genomic analysis of a sexually-selected character: EST sequencing and microarray analysis of eye-antennal imaginal discs in the stalk-eyed fly Teleopsis dalmanni (Diopsidae). BMC Genomics 10, 1–20 10.1186/1471-2164-10-361 (doi:10.1186/1471-2164-10-361) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Stankiewicz P., Beaudet A. L. 2007. Use of array CGH in the evaluation of dysmorphology, malformations, developmental delay, and idiopathic mental retardation. Curr. Opin. Genet. Dev. 17, 182–192 10.1016/j.gde.2007.04.009 (doi:10.1016/j.gde.2007.04.009) [DOI] [PubMed] [Google Scholar]
- 99.Zhang F., Gu W., Hurles M. E., Lupski J. R. 2009. Copy number variation in human health, disease, and evolution. Annu. Rev. Genomics Hum. Genet. 10, 451–481 10.1146/annurev.genom.9.081307.164217 (doi:10.1146/annurev.genom.9.081307.164217) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Baker R. H., Wilkinson G. S. 2010. Comparative genomic hybridization (CGH) reveals a neo-X chromosome and biased gene movement in stalk-eyed flies (Genus Teleopsis). PLoS Genet. 6, e1001121. 10.1371/journal.pgen.1001121 (doi:10.1371/journal.pgen.1001121) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Zdobnov E. M., et al. 2002. Comparative genome and proteome analysis of Anopheles gambiae and Drosophila melanogaster. Science 298, 149–159 10.1126/science.1077061 (doi:10.1126/science.1077061) [DOI] [PubMed] [Google Scholar]
- 102.Severson D. W., deBruyn B., Lovin D. D., Brown S. E., Knudson D. L., Morlais I. 2004. Comparative genome analysis of the yellow fever mosquito Aedes aegypti with Drosophila melanogaster and the malaria vector mosquito Anopheles gambiae. J. Hered. 95, 103–113 10.1093/jhered/esh023 (doi:10.1093/jhered/esh023) [DOI] [PubMed] [Google Scholar]
- 103.Davies N., Roderick G. K. 2005. Dipteran sex chromosomes in evolutionary developmental biology. In The Evolutionary biology of flies (ed. Yeates D. K., Weigmann B. M.), pp. 196–213 New York, NY: Columbia University Press [Google Scholar]
- 104.Charlesworth D., Charlesworth B., Marais G. 2005. Steps in the evolution of heteromorphic sex chromosomes. Heredity 95, 118–128 10.1038/sj.hdy.6800697 (doi:10.1038/sj.hdy.6800697) [DOI] [PubMed] [Google Scholar]
- 105.Zhang Y. E., Vibranovski M. D., Krinsky B. H., Long M. 2010. Age-dependent chromosomal distribution of male-biased genes in Drosophila. Genome Res. 20, 1526–1533 10.1101/gr.107334.110 (doi:10.1101/gr.107334.110) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Birol I., et al. 2009. De novo transcriptome assembly with ABySS. Bioinformatics 25, 2872–2877 10.1093/bioinformatics/btp367 (doi:10.1093/bioinformatics/btp367) [DOI] [PubMed] [Google Scholar]
- 107.Simpson J. T., Wong K., Jackman S. D., Schein J. E., Jones S. J. M., Birol İ. 2009. ABySS: a parallel assembler for short read sequence data. Genome Res. 19, 1117–1123 10.1101/gr.089532.108 (doi:10.1101/gr.089532.108) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Drummond A. J., et al. 2010. Geneious v. 5.4. See http://www.geneious.com
- 109.Rice P., Longden I., Bleasby A. 2000. EMBOSS: the European molecular biology open software suite. Trends Genet. 16, 276–277 10.1016/S0168-9525(00)02024-2 (doi:10.1016/S0168-9525(00)02024-2) [DOI] [PubMed] [Google Scholar]
- 110.Langmead B., Trapnell C., Pop M., Salzberg S. 2009. Ultrafast and memory-efficient alignment of short DNA sequences to the human genome. Genome Biol. 10, R25. 10.1186/gb-2009-10-3-r25 (doi:10.1186/gb-2009-10-3-r25) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Aoyagi N., Wassarman D. A. 2000. Genes encoding Drosophila melanogaster RNA polymerase. II General transcription factors: diversity in TFIIA and TFIID components contributes to gene-specific transcriptional regulation. J. Cell Biol. 150, F45–F50 10.1083/jcb.150.2.F45 (doi:10.1083/jcb.150.2.F45) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Gu Z., Rifkin S. A., White K. P., Li W-H. 2004. Duplicate genes increase gene expression diversity within and between species. Nat. Genet. 36, 577–579 10.1038/ng1355 (doi:10.1038/ng1355) [DOI] [PubMed] [Google Scholar]
- 113.Huerta-Cepas J., Dopazo J., Huynen M. A., Gabaldón T. 2011. Evidence for short-time divergence and long-time conservation of tissue-specific expression after gene duplication. Brief. Bioinf. 12, 442–448 10.1093/bib/bbr022 (doi:10.1093/bib/bbr022) [DOI] [PubMed] [Google Scholar]
- 114.Iwabe N., Kuma K., Miyata T. 1996. Evolution of gene families and relationship with organismal evolution: rapid divergence of tissue-specific genes in the early evolution of chordates. Mol. Biol. Evol. 13, 483–493 10.1093/oxfordjournals.molbev.a025609 (doi:10.1093/oxfordjournals.molbev.a025609) [DOI] [PubMed] [Google Scholar]
- 115.Langille M. G. I., Clark D. V. 2007. Parent genes of retrotransposition-generated gene duplicates in Drosophila melanogaster have distinct expression profiles. Genomics 90, 334–343 10.1016/j.ygeno.2007.06.001 (doi:10.1016/j.ygeno.2007.06.001) [DOI] [PubMed] [Google Scholar]
- 116.Lynch M., Conery J. S. 2000. The evolutionary fate and consequences of duplicate genes. Science 290, 1151–1155 10.1126/science.290.5494.1151 (doi:10.1126/science.290.5494.1151) [DOI] [PubMed] [Google Scholar]
- 117.Zhang J. 2003. Evolution by gene duplication: an update. Trends Ecol. Evol. 18, 292–298 10.1016/S0169-5347(03)00033-8 (doi:10.1016/S0169-5347(03)00033-8) [DOI] [Google Scholar]
- 118.Mencarelli C., Lupetti P., Dallai R., Kwang W. J. 2008. New insights into the cell biology of insect axonemes. Int. Rev. Cell Mol. Biol. 268, 95–145 10.1016/S1937-6448(08)00804-6 (doi:10.1016/S1937-6448(08)00804-6) [DOI] [PubMed] [Google Scholar]
- 119.Nielsen M. G., Turner F. R., Hutchens J. A., Raff E. C. 2001. Axoneme-specific β-tubulin specialization: a conserved C-terminal motif specifies the central pair. Curr. Biol. 11, 529–533 10.1016/S0960-9822(01)00150-6 (doi:10.1016/S0960-9822(01)00150-6) [DOI] [PubMed] [Google Scholar]
- 120.Popodi E. M., Hoyle H. D., Turner F. R., Xu K., Kruse S., Raff E. C. 2008. Axoneme specialization embedded in a ‘Generalist’ β-tubulin. Cell Motil. Cytoskelet. 65, 216–237 10.1002/cm.20256 (doi:10.1002/cm.20256) [DOI] [PubMed] [Google Scholar]
- 121.Raff E. C., Hoyle H. D., Popodi E. M., Turner F. R. 2008. Axoneme β-tubulin sequence determines attachment of outer dynein arms. Curr. Biol. 18, 911–914 10.1016/j.cub.2008.05.031 (doi:10.1016/j.cub.2008.05.031) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Haerty W., et al. 2007. Evolution in the fast lane: rapidly evolving sex-related genes in Drosophila. Genetics 177, 1321–1335 10.1534/genetics.107.078865 (doi:10.1534/genetics.107.078865) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Swanson W. J., Vacquier V. D. 2002. The rapid evolution of reproductive protein. Nat. Rev. Genet. 3, 137–144 10.1038/nrg733 (doi:10.1038/nrg733) [DOI] [PubMed] [Google Scholar]
- 124.Nielsen M. G., Caserta J. M., Kidd S. J., Phillips C. M. 2006. Functional constraint underlies 60 million year stasis of Dipteran testis-specific β-tubulin. Evol. Dev. 8, 23–29 10.1111/j.1525-142X.2006.05072.x (doi:10.1111/j.1525-142X.2006.05072.x) [DOI] [PubMed] [Google Scholar]
- 125.Foldvari M., Pomiankowski A., Cotton S., Carr M. 2007. A morphological and molecular description of a new Teleopsis species (Diptera: Diopsidae) from Thailand. Zootaxa 1620, 37–51 [Google Scholar]
- 126.Edgar R. C. 2004. MUSCLE: multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res. 32, 1792–1797 10.1093/nar/gkh340 (doi:10.1093/nar/gkh340) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Guindon S., Gascuel O. 2003. A simple, fast, and accurate algorithm to estimate large phylogenies by maximum likelihood. Syst. Biol. 52, 696–704 10.1080/10635150390235520 (doi:10.1080/10635150390235520) [DOI] [PubMed] [Google Scholar]
- 128.Pond S. L. K., Frost S. D. W., Muse S. V. 2005. HyPhy: hypothesis testing using phylogenies. Bioinformatics 21, 676–679 10.1093/bioinformatics/bti079 (doi:10.1093/bioinformatics/bti079) [DOI] [PubMed] [Google Scholar]
- 129.Presgraves D. C., Baker R. H., Wilkinson G. S. 1999. Coevolution of sperm and female reproductive tract morphology in stalk-eyed flies. Proc. R. Soc. Lond. B 266, 1041–1047 10.1098/rspb.1999.0741 (doi:10.1098/rspb.1999.0741) [DOI] [Google Scholar]
- 130.Swallow J. G., Wilkinson G. S. 2002. The long and short of sperm polymorphisms in insects. Biol. Rev. 77, 153–182 10.1017/S1464793101005851 (doi:10.1017/S1464793101005851) [DOI] [PubMed] [Google Scholar]
- 131.Wedell N., Gage M. J. G., Parker G. A. 2002. Sperm competition, male prudence and sperm-limited females. Trends Ecol. Evol. 17, 313–320 10.1016/S0169-5347(02)02533-8 (doi:10.1016/S0169-5347(02)02533-8) [DOI] [Google Scholar]
- 132.Byrne P. G., Rice W. R. 2006. Evidence for adaptive male mate choice in the fruit fly Drosophila melanogaster. Proc. R. Soc. B 273, 917–922 10.1098/rspb.2005.3372 (doi:10.1098/rspb.2005.3372) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Edward D. A., Chapman T. 2011. The evolution and significance of male mate choice. Trends Ecol. Evol. 26, 647–654 10.1016/j.tree.2011.07.012 (doi:10.1016/j.tree.2011.07.012) [DOI] [PubMed] [Google Scholar]
- 134.Urban F. 2006. Male perception of female mating status: its effect on copulation duration, sperm defence and female fitness. Anim. Behav. 72, 1259–1268 10.1016/j.anbehav.2006.03.021 (doi:10.1016/j.anbehav.2006.03.021) [DOI] [Google Scholar]
- 135.Pitnick S., Hosken D. J., Birkhead T. R. 2009. Sperm morphological diversity. In Sperm biology: an evolutionary perspective (ed. Birkhead T. R., Hosken D. J., Pitnick S.), pp. 69–149 San Diego, CA: Academic Press [Google Scholar]
- 136.Pitnick S., Markow T. A., Spicer G. S. 1995. Delayed male maturity is a cost of producing large sperm in Drosophila. Proc. Natl Acad. Sci. USA 92, 10 614–10 618 10.1073/pnas.92.23.10614 (doi:10.1073/pnas.92.23.10614) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Bjork A., Dallai R., Pitnick S. 2007. Adaptive modulation of sperm production rate in Drosophila bifurca, a species with giant sperm. Biol. Lett. 3, 517–519 10.1098/rsbl.2007.0219 (doi:10.1098/rsbl.2007.0219) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Rogers D. W., Grant C. A., Chapman T., Pomiankowski A., Fowler K. 2006. The influence of male and female eyespan on fertility in the stalk-eyed fly, Cyrtodiopsis dalmanni. Anim. Behav. 72, 1363–1369 10.1016/j.anbehav.2006.03.027 (doi:10.1016/j.anbehav.2006.03.027) [DOI] [Google Scholar]
- 139.Rogers D. W., Chapman T., Fowler K., Pomiankowski A. 2005. Mating-induced reduction in accessory reproductive organ size in the stalk-eyed fly Cyrtodiopsis dalmanni. BMC Evol. Biol. 5, 37. 10.1186/1471-2148-5-37 (doi:10.1186/1471-2148-5-37) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Baker R. H., Ashwell R. I. S., Richards T. A., Fowler K., Chapman T., Pomiankowski A. 2001. Effects of multiple mating and male eye span on female reproductive output in the stalk-eyed fly, Cyrtodiopsis dalmanni. Behav. Ecol. 12, 732–739 10.1093/beheco/12.6.732 (doi:10.1093/beheco/12.6.732) [DOI] [Google Scholar]
- 141.Cotton S., Small J., Pomiankowski A. 2010. Eyespan reflects reproductive quality in wild stalk-eyed flies. Evol. Ecol. 24, 83–95 10.1007/s10682-009-9292-6 (doi:10.1007/s10682-009-9292-6) [DOI] [Google Scholar]
- 142.Harley E., Fowler K., Cotton S. 2010. No detectable fertility benefit from a single additional mating in wild stalk-eyed flies. PLoS ONE 5, e14309. 10.1371/journal.pone.0014309 (doi:10.1371/journal.pone.0014309) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Baker R. H., Denniff M., Futerman P., Fowler K., Pomiankowski A., Chapman T. 2003. Accessory gland size influences time to sexual maturity and mating frequency in the stalk-eyed fly, Cyrtodiopsis dalmanni. Behav. Ecol. 14, 607–611 10.1093/beheco/arg053 (doi:10.1093/beheco/arg053) [DOI] [Google Scholar]
- 144.Al-khairulla H., Warburton D., Knell R. J. 2003. Do the eyestalks of female diopsid flies have a function in intrasexual aggressive encounters? J. Insect Behav. 16, 679–686 10.1023/B:JOIR.0000007703.84691.ad (doi:10.1023/B:JOIR.0000007703.84691.ad) [DOI] [Google Scholar]
- 145.Worthington A. M., Swallow J. G. 2010. Gender differences in survival and antipredatory behavior in stalk-eyed flies. Behav. Ecol. 21, 759–766 10.1093/beheco/arq050 (doi:10.1093/beheco/arq050) [DOI] [Google Scholar]
- 146.Kosmidis S., Botella J. A., Mandilaras K., Schneuwly S., Skoulakis E. M. C., Rouault T. A., Missirlis F. 2011. Ferritin overexpression in Drosophila glia leads to iron deposition in the optic lobes and late-onset behavioral defects. Neurobiol. Dis. 43, 213–219 10.1016/j.nbd.2011.03.013 (doi:10.1016/j.nbd.2011.03.013) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Georgieva T., Dunkov B. C., Dimov S., Ralchev K., Law J. H. 2002. Drosophila melanogaster ferritin: cDNA encoding a light chain homologue, temporal and tissue specific expression of both subunit types. Insect Biochem. Mol. Biol. 32, 295–302 10.1016/S0965-1748(01)00090-X (doi:10.1016/S0965-1748(01)00090-X) [DOI] [PubMed] [Google Scholar]
- 148.Baines J. F., Sawyer S. A., Hartl D. L., Parsch J. 2008. Effects of X-linkage and sex-biased gene expression on the rate of adaptive protein evolution in Drosophila. Mol. Biol. Evol. 25, 1639–1650 10.1093/molbev/msn111 (doi:10.1093/molbev/msn111) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Kaessmann H. 2010. Origins, evolution, and phenotypic impact of new genes. Genome Res. 20, 1313–1326 (doi:10.1101/gr.101386.109) [DOI] [PMC free article] [PubMed] [Google Scholar]