

Primary human immunodeficiencies comprise a broad group of disorders ranging from severely impaired lymphocyte development to more subtle functional abnormalities (reviewed in ref. 1). The recent identification of the genetic basis of several of these diseases has enriched our understanding of the molecular events controlling the development and function of the immune system. These gene products fall into four major categories: enzymes regulating DNA recombination and nucleotide metabolism, cell cycle regulators, proteins involved in antigen presentation, and components of B and T cell signal transduction pathways (1). Nichols et al. (2) describe the most recent addition to the latter group in this issue of the Proceedings. DHSP encodes a single SH2 domain and is mutated in patients with X-linked lymphoproliferative syndrome (XLP). DHSP represents a new paradigm in signal transduction defects associated with immunodeficiencies. All signaling molecules previously shown to be involved in these diseases, including γc, JAK-3, and ZAP70 in severe combined immunodeficiency and Btk in X-linked agammaglobulinemia, are “hard wired” components essential for signal transmission (1). In contrast, DSHP is up-regulated late in the immune response and likely modulates signal duration or amplitude.
XLP is unique among immunodeficiencies in that it renders its victims particularly vulnerable to one pathogen, Epstein–Barr virus (EBV) (reviewed in refs. 3 and 4). The XLP phenotype is poorly penetrant before exposure to EBV. Although a small percentage of affected boys have either hyper IgM or hypogammaglobulinemia, they do not present with recurrent bacterial infections indicating that humoral immunity is largely intact in these patients. Upon EBV infection, however, the majority of patients succumb to fulminant infectious mononucleosis (IM). Those who survive the initial EBV infection suffer from hypogammaglobulinemia, malignant B or T cell lymphomas, aplastic anemia, and, rarely, granulomatous vaculitis. XLP carriers remain healthy despite random X-chromosome inactivation in B cells (5), demonstrating that XLP B cells do not have increased susceptibility to EBV infection or transformation. This suggests that vulnerability to EBV is due to an impaired immune response to the pathogen. Defects in both natural killer (NK) cell and cytotoxic T lymphocyte (CTL) activity have been reported in EBV-infected XLP patients (refs. 3 and 4 and refs. therein). Hypogammaglobulinemia and reduced titers of antibodies against EBV are common (refs. 3 and 4 and references therein). No effects on normal lymphocyte development have been observed. Normal numbers of B, T, and NK cells are present, although after EBV infection the ratio of CD4+ to CD8+ T cells in the periphery is inverted (ref. 3 and refs. therein).
The EBV life cycle has several aspects, which distinguish it from other viral pathogens (reviewed in ref. 6). EBV-infected B cells proliferate rapidly and grow indefinitely in vitro, but initial infection of immunocompetent hosts is usually only mildly symptomatic and nearly always results in a state of chronic latent infection of mature B cells. The EBV-encoded LMP1 gene is essential for B cell transformation and proliferation. It acts in part by constitutively activating the CD40-signaling pathway (7). B cell proliferation and survival also is enhanced by IL10 (8), which is secreted from EBV-infected cells in both endogenously (9) and virally (10) encoded forms. In addition to positively regulating the growth and survival of B cells, IL-10 impairs T and NK cell function. The majority of these inhibitory effects of IL-10 are mediated by macrophages (11–14). The specificity of XLP pathogenesis to EBV infection suggests that the normal immune response to these or other unique aspects of EBV are affected.
The genetic defect in XLP was identified by using a positional cloning approach (2). DSHP encodes a single SH2 domain, which is most homologous to the SH2 domain of SHIP, an inositol-phosphatase involved in negative regulatory signaling pathways in hematopoietic cells. DSHP is expressed in T cell lines, conA or anti-CD3 activated peripheral blood T cells, and in the T cell zone and germinal centers of reactive lymph nodes. It was not detected in most B cell lines, EBV-immortalized lymphoblasts from normal controls, or in anti-IgM-stimulated peripheral blood B cells. This expression pattern suggests that DSHP functions primarily in T (or possibly NK) cell-mediated immune responses.
The relatively mild immunodeficiency in XLP patients in the absence of EBV indicates that DSHP is not essential for most immune responses. While the signaling cascades that initiate cellular activation are triggered within seconds or minutes after stimulation, expression of DSHP is up-regulated much later, peaking at 48 hr. These observations imply that DSHP regulates the duration or amplitude of the immune response and suggest a mechanism by which it might be involved specifically in the defense against EBV. The immune response is normally limited by down-regulatory signals mediated by inhibitory receptors such as FcγRIIb, CD22, CTLA-4, and KIR (reviewed in refs. 15–17). The rapid proliferation of EBV-infected B cells may necessitate a prolonged immune response to provide time for EBV specific CTLs to amplify and eliminate their targets. DSHP might interfere with inhibitory signals, providing an extra “boost” at the end of the response. The regulated expression of DSHP would restrict its effect to a particular temporal window, thus preventing constitutive cellular activation and autoimmunity. Failure to induce DSHP may prevent this extended phase of the response, allowing EBV-infected cells to escape immune surveillance.
The most likely role for DSHP in signal transduction is to compete with other SH2-containing proteins for binding to phosphotyrosine. Based on its homology to the SHIP SH2 domain, Nichols et al. (2) suggest that DSHP sustains the immune response by blocking inhibitory signals mediated by SHIP. SHIP transmits FcγRIIb inhibitory signals in B and mast cells (18) and down-regulates cytokine receptor signaling in myeloid cells (19). SHIP dephosphorylates PIP3 (20, 21), the product of PI3, resulting in impaired activation of Tec family kinases and reducing sustained Ca2+ influx (22–24). Activation of Akt, which promotes cell survival and proliferation, also is dependent on PI3 kinase (25) and also is likely down-regulated by SHIP. SHIP also has been suggested to inhibit Ras signaling by competing with GRB2 for SHC binding (26).
Consistent with the proposed competition between DSHP and SHIP during the T cell response to EBV, SHIP is expressed in T cells and becomes tyrosine phosphorylated and associated with SHC in response to TCR stimulation (27). However, a direct negative regulatory role for SHIP has not been demonstrated in T cells. Further experiments are therefore required to confirm this model. This hypothesis suggests that PI3 kinase dependent signals such as IL-2-induced activation of Akt (25) and E2F (28) and TCR-stimulated activation of Itk and sustained Ca2+ influx (22–24) should be attenuated in T cells from XLP patients. Cells from mice lacking DSHP also should have heightened sensitivity to negative regulation, which would be alleviated by SHIP deficiency (19). Conversely, overexpression of DSHP should mimic SHIP deficiency and result in increased or prolonged activation of PI3 kinase- and Ras-dependent signaling pathways.
DSHP also may prolong T cell responses by blocking down-regulatory signals in a SHIP-independent manner (Fig. 1). The inhibitory receptors CTLA4 and KIR do not signal via SHIP but rather the protein tyrosine phosphatases SHP-2 (29) and SHP-1 (18, 30), respectively. Like DSHP, CTLA4 is expressed only in activated T cells (31). T cells from CTLA-4 deficient mice are hyperproliferative and have a constitutively activated phenotype (32, 33) associated with inappropriate activation of Fyn, Lck, Zap70, and the Ras pathway (29). The KIR family of inhibitory receptors are expressed constitutively in NK cells and inducibly in activated CD8+ T cells with a memory phenotype (refs. 15, 17, and 34 and refs. therein). Crosslinking of KIRs inhibits NK- or CTL-mediated target cell lysis and the production of cytokines such as IFN-γ (refs. 15, 17, and 34 and refs. therein). Interestingly, XLP T cells have defects in IFN-γ production in response to autologous EBV-infected B cell lines (refs. 3 and 4 and refs. therein). If DSHP is expressed in NK cells, increased sensitivity to KIR mediated down-regulation may explain both the NK and memory CTL defects in XLP patients.
Figure 1.
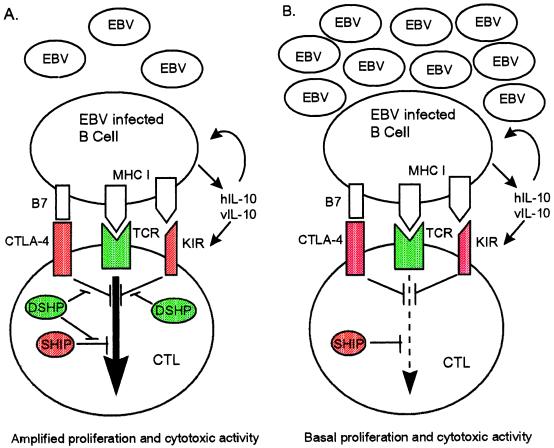
Model for the role of DSHP in the CTL response to EBV. (A) EBV-infected B cells secrete hIL-10 and vIL-10. These cytokines augment proliferation and survival of infected B cells, up-regulate the inhibitory receptor KIR on CTLs, and act on macrophages to inhibit T and NK cell function (not shown). During the CTL response to EBV in normal individuals, DSHP (green) attenuates inhibitory signals (red) from SHIP, CTLA-4, and/or KIR late in the immune response. This prolongs the response, allowing rapidly proliferating EBV-infected cells to be controlled. (B) In XLP patients, inhibitory signals (red) predominate in the absence of DSHP, and EBV-infected B cells escape immune surveillance.
How might DSHP act to prevent CTLA-4 or KIR signaling? Because SHIP plays no role in these pathways (18, 29, 30), DSHP would have to compete with another SH2 containing protein. SH2 domains have been grouped according to their preference for sequences adjacent to the phosphotyrosine to which they bind (35, 36). Much of this binding specificity is dictated by the amino acid residue at the βD5 position of the SH2 domain (37). Based on overall homology and the βD5 residue, both SHIP and DSHP are group I SH2 domains (38). DSHP should thus share preferences for phosphotyrosine context with other group I SH2-containing proteins in addition to SHIP. These include Abl, Itk, Syk, Zap 70, GRB2, and GAP (35, 36), many of which are important for T cell signaling. The major phosphorylation site of SHP-1 is contained within a sequence, pYXNX, preferred by several group I SH2 domains including GRB2 (39). DSHP therefore may prevent the binding of tyrosine phosphorylated SHP-1 or SHP-2 to SH2 domains of their substrates. Because the SH2 domains of SHP-1 and SHP-2 belong to group III (35, 36), DSHP is unlikely to affect their binding to tyrosine-phosphorylated immunoreceptor tyrosine-based inhibitory motifs (ITIMs) on the inhibitory receptors.
In addition to inducing rapid proliferation of infected cells, EBV attempts to evade the immune response by stimulating secretion of both hIL-10 (9) and vIL-10 (10) by B cells. Interestingly, IL-10 can induce KIR expression on CTLs (34). IL-10 mediated up-regulation of KIR in CTLs has been proposed as a mechanism by which EBV escapes CTL control (34). XLP CTLs, lacking DSHP, might be overwhelmed by the inhibitory effect of EBV/IL-10 induced KIR. Many of the other inhibitory effects of IL-10 on T and NK cells are mediated by macrophages (11–14). DSHP expression was not detected in the myeloid progenitor cell lines HL-60 and U937; so it is unlikely that IL-10 signaling is affected in XLP macrophages. However, expression of DSHP in more mature myeloid cells cannot be ruled out. It also is possible that T or NK cells lacking DSHP are particularly sensitive to IL-10-induced changes in cytokine secretion or antigen presentation by macrophages.
Whether one or more of these potential signaling pathways are affected, the concept that DSHP acts to enhance or prolong T- and/or NK-mediated immunity has important therapeutic implications. EBV-associated lymphoproliferative disorders occur commonly in other immunosuppressed patients such as those receiving solid organ and bone marrow transplants as well as in some HIV-infected individuals (40). Therapies aimed at replacement of the defective DSHP gene in XLP or enhancement of DSHP expression in T cells from other immunosuppressed patients might be effective in preventing the expansion of EBV-infected cells. This may be a feasible approach since a major current therapy for these lymphoproliferative disorders is the adoptive transfer of autologous T cells. Interventions that specifically increase transcription of the DSHP gene also might be possible in non-XLP, EBV-induced malignancies. Inappropriate expression of DSHP might result in the inhibition of multiple signaling pathways, so such therapeutic approaches must be undertaken with caution and only when there is a better understanding of DSHP function.
Mutations in DSHP were identified in only 60% of XLP patients. Nichols et al. (2) suggest that they might have missed subtle regulatory mutations in DSHP in the remaining patients or that some of these patients might have been misdiagnosed. More interestingly, they hypothesize that these patients might carry mutations in other components of signaling pathways, which regulate response to EBV infection. Identification of such mutations would shed light on which DSHP-mediated signals are critical for this response and how these signals are transmitted.
Acknowledgments
A.B.S. is a Special Fellow of the Leukemia Society of America. D.J.R. is a recipient of an National Institutes of Health Physician Scientist Award and a McDonnell Scholar Award. O.N.W. is an Investigator of the Howard Hughes Medical Institute.
ABBREVIATIONS
- XLP
X-linked lymphoproliferative syndrome
- EBV
Epstein–Barr virus
- NK
natural killer
- CTL
cytotoxic T lymphocyte
Footnotes
The companion to this Commentary begins on page 13765.
References
- 1.Fischer A, Cavazzana-Calvo M, De Saint Basile G, DeVillartay J P, Di Santo J P, Hivroz C, Rieux-Laucat F, Le Deist F. Annu Rev Immunol. 1997;15:93–124. doi: 10.1146/annurev.immunol.15.1.93. [DOI] [PubMed] [Google Scholar]
- 2.Nichols K E, Harkin D P, Levitz S, Krainer M, Kolquist K A, Genovese C, Bernard A, Ferguson M, Zuo L, Snyder E, et al. Proc Natl Acad Sci USA. 1998;95:13765–13770. doi: 10.1073/pnas.95.23.13765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Purtilo D T. Springer Semin Immunopathol. 1991;13:181–197. doi: 10.1007/BF00201468. [DOI] [PubMed] [Google Scholar]
- 4.Seemayer T A, Gross T G, Egeler R M, Pirruccello S J, Davis J R, Kelly C M, Okano M, Lanyi A, Sumegi J. Pediatr Res. 1995;38:471–478. doi: 10.1203/00006450-199510000-00001. [DOI] [PubMed] [Google Scholar]
- 5.Conley M E, Sullivan J L, Neidich J A, Puck J M. Clin Immunol Immunopathol. 1990;55:486–491. doi: 10.1016/0090-1229(90)90133-b. [DOI] [PubMed] [Google Scholar]
- 6.Cohen J I. J Am Med Assoc. 1997;278:510–513. [Google Scholar]
- 7.Kilger E, Kieser A, Baumann M, Hammerschmidt W. EMBO J. 1998;17:1700–1709. doi: 10.1093/emboj/17.6.1700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Rousset F, Garcia E, Defrance T, Peronne C, Vezzio N, Hsu D H, Kastelein R, Moore K W, Banchereau J. Proc Natl Acad Sci USA. 1992;89:1890–1893. doi: 10.1073/pnas.89.5.1890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Burdin N, Peronne C, Banchereau J, Rousset F. J Exp Med. 1993;177:295–304. doi: 10.1084/jem.177.2.295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Hsu D H, de Waal Malefyt R, Fiorentino D F, Dang M N, Vieira P, de Vries J, Spits H, Mosmann T R, Moore K W. Science. 1990;250:830–832. doi: 10.1126/science.2173142. [DOI] [PubMed] [Google Scholar]
- 11.Fiorentino D F, Zlotnik A, Vieira P, Mosmann T R, Howard M, Moore K W, O’Garra A. J Immunol. 1991;146:3444–3451. [PubMed] [Google Scholar]
- 12.De Waal Malefyt R, Haanen J, Spits H, Roncarolo M G, te Velde A, Figdor C, Johnson K, Kastelein R, Yssel H, de Vries J E. J Exp Med. 1991;174:915–924. doi: 10.1084/jem.174.4.915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Bejarano M T, de Waal Malefyt R, Abrams J S, Bigler M, Bacchetta R, de Vries J E, Roncarolo M G. Int Immunol. 1992;4:1389–1397. doi: 10.1093/intimm/4.12.1389. [DOI] [PubMed] [Google Scholar]
- 14.D’Andrea A, Aste-Amezaga M, Viliante N M, Ma X, Kubin M, Trinchieri G. J Exp Med. 1993;178:1041–1048. doi: 10.1084/jem.178.3.1041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Unkeless J C, Jin J. Curr Opin Immunol. 1997;9:338–343. doi: 10.1016/s0952-7915(97)80079-9. [DOI] [PubMed] [Google Scholar]
- 16.Healy J L, Goodnow C C. Annu Rev Immunol. 1998;16:645–670. doi: 10.1146/annurev.immunol.16.1.645. [DOI] [PubMed] [Google Scholar]
- 17.Scharenberg A M, Kinet J-P. Cell. 1996;87:961–964. doi: 10.1016/s0092-8674(00)81790-0. [DOI] [PubMed] [Google Scholar]
- 18.Ono M, Okada H, Bolland S, Yanagi S, Kurosaki T, Ravetch J V. Cell. 1997;90:293–301. doi: 10.1016/s0092-8674(00)80337-2. [DOI] [PubMed] [Google Scholar]
- 19.Helgason C D, Damen J E, Rosten P, Grewal R, Sorensen P, Chappel S M, Borowski A, Jirik F, Krystal G, Humphries R K. Genes Dev. 1998;12:1610–1620. doi: 10.1101/gad.12.11.1610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Damen J E, Liu L, Rosten P, Humphries R K, Jefferson A B, Majerus P W, Krystal G. Proc Natl Acad Sci USA. 1996;93:1689–1693. doi: 10.1073/pnas.93.4.1689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Lioubin M N, Algate P A, Tsai S, Carlberg K, Aebersold A, Rohrschneider L R. Genes Dev. 1996;10:1084–1095. doi: 10.1101/gad.10.9.1084. [DOI] [PubMed] [Google Scholar]
- 22.Scharenberg A M, El-Hillal O, Fruman D A, Beitz L O, Li Z, Lin S, Gout I, Cantley L C, Rawlings D J, Kinet J P. EMBO J. 1998;17:1961–1972. doi: 10.1093/emboj/17.7.1961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Fluckiger A-C, Li Z, Kato R M, Wahl M I, Ochs H D, Longnecker R, Kinet J-P, Witte O N, Scharenberg A M, Rawlings D J. EMBO J. 1998;17:1973–1985. doi: 10.1093/emboj/17.7.1973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Bolland S, Pearse R N, Kurosaki T, Ravetch J V. Immunity. 1998;8:509–516. doi: 10.1016/s1074-7613(00)80555-5. [DOI] [PubMed] [Google Scholar]
- 25.Ahmed N N, Grimes H L, Bellacomse A, Chan T O, Tsichlis P N. Proc Natl Acad Sci USA. 1997;94:3627–3632. doi: 10.1073/pnas.94.8.3627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Tridandapani S, Kelley T, Cooney D, Pradhan M, Coggeshall K M. Immunol Today. 1997;18:424–427. doi: 10.1016/s0167-5699(97)01112-2. [DOI] [PubMed] [Google Scholar]
- 27.Lamkin T D, Walk S F, Liu L, Damen J E, Krystal G, Ravichandran K S. J Biol Chem. 1997;272:10396–10401. doi: 10.1074/jbc.272.16.10396. [DOI] [PubMed] [Google Scholar]
- 28.Brennan P, Babbage J W, Burgering B M, Groner B, Reif K, Cantrell D A. Immunity. 1997;7:679–689. doi: 10.1016/s1074-7613(00)80388-x. [DOI] [PubMed] [Google Scholar]
- 29.Marengere L E M, Waterhouse P M, Duncan G S, Mittrucker H-W, Feng G-S, Mak T W. Science. 1996;272:1170–1173. doi: 10.1126/science.272.5265.1170. [DOI] [PubMed] [Google Scholar]
- 30.Gupta N, Scharenberg A M, Burshtyn D N, Wagtmann N, Lioubin M N, Rohrschneider L R, Kinet J P, Long E O. J Exp Med. 1997;186:473–478. doi: 10.1084/jem.186.3.473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Walunas T L, Lenschow D J, Bakker C Y, Linsley P S, Freeman G J, Green J M, Thompson C B, Bluestone J A. Immunity. 1994;1:405–413. [Google Scholar]
- 32.Tivol E A, Borriello F, Schweitzer A N, Lynch W P, Bluestone J A, Sharpe A H. Immunity. 1995;3:541–547. doi: 10.1016/1074-7613(95)90125-6. [DOI] [PubMed] [Google Scholar]
- 33.Waterhouse P, Penninger J M, Timms E, Wakeham A, Shahinian A, Lee K P, Thompson C B, Griesser H, Mak T W. Science. 1995;270:985–986. doi: 10.1126/science.270.5238.985. [DOI] [PubMed] [Google Scholar]
- 34.Mingari M C, Moretta A, Moretta L. Immunol Today. 1998;19:153–157. doi: 10.1016/s0167-5699(97)01236-x. [DOI] [PubMed] [Google Scholar]
- 35.Songyang Z, Shoelson S E, Chaudhrui M, Gish G, Pawson T, Haser W G, King F, Roberts T, Ratnofsky S, Lechleider R J, et al. Cell. 1993;72:767–778. doi: 10.1016/0092-8674(93)90404-e. [DOI] [PubMed] [Google Scholar]
- 36.Songyang Z, Shoelson S E, McGlade J, Olivier P, Pawson T, Bustelo X R, Barbacid M, Sabe H, Hanafusa H, Yi T, et al. Mol Cell Biol. 1994;14:2777–2785. doi: 10.1128/mcb.14.4.2777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Sonyang Z, Gish G, Mbamalu G, Pawson T, Cantley L C. J Biol Chem. 1995;270:26029–26032. doi: 10.1074/jbc.270.44.26029. [DOI] [PubMed] [Google Scholar]
- 38.Ware M D, Rosten P, Damen J E, Liu L, Humpries R K, Krystal G. Blood. 1996;88:2833–2840. [PubMed] [Google Scholar]
- 39.Bouchard P, Zhao Z, Banville D, Dumas F, Fischer E H, Shen S H. J Biol Chem. 1994;269:19585–19589. [PubMed] [Google Scholar]
- 40.List A F, Greco F A, Vogler L B. J Clin Oncol. 1987;5:1673–1689. doi: 10.1200/JCO.1987.5.10.1673. [DOI] [PubMed] [Google Scholar]