

Abstract
Ideal tests of the effects of therapeutic interventions measure the desired outcomes; however, the desired outcomes are not always easily measured or may be long-term objectives. Biomarkers and surrogate end-points are often cheaper and easier to measure and can be measured over a shorter time span. They can be used in screening, diagnosing, staging, and monitoring diseases, in monitoring responses to interventions, and in various aspects of drug discovery and development. They can be extrinsic to the body or intrinsic, and can relate to any point in the pharmacological chain, at the molecular, cellular, tissue, or organ level. Problems arise when the relation between the pathophysiology of the disease and the mechanism of action of the intervention is not properly understood; when adverse effects obviate therapeutic effects; when confounding factors, such as other drugs, alter the surrogate independently of the final end-point; when a biomarker persists after resolution of the disease; and when the concentration–effect curves for the effects of an intervention on the primary outcome and the surrogate are different. Use of biomarkers may also be hindered by poor reproducibility of measurement techniques. Challenges for clinical pharmacologists are to devise biomarker tests that are reliable, reproducible, sensitive, and specific, and surrogate end-points that are associated with the clinical outcomes of concern and useful. A robust taxonomy is needed of the relations that link the pathophysiology of disease, the mechanisms of action of interventions and their adverse effects, the desired clinical outcomes, and the surrogate end-points that predict them.
Introduction
Ideal tests of the effects of therapeutic interventions measure the desired outcomes. For example, the desired outcome in the management of pneumonia is resolution of its signs and symptoms (such as fever, breathlessness, chest pain, and auscultatory signs), which can be monitored during treatment. Other measures related to effects of the infection can also be assessed, such as the chest X-ray, inflammatory markers in the blood, and the presence of the organism in the sputum or antibodies to it in the blood, but as they are not the clinically relevant end-points they are regarded as surrogate end-points. Although in this case the surrogate end-points are very close in the pathophysiological chain to the clinically relevant end-points, there are important differences. For example, the time course of changes in a chest X-ray is not the same as the time course of changes in the clinical features of pneumonia; the former can take longer to show evidence of pneumonia and may take longer to resolve [1]. In some infections (for example, typhoid), an individual can continue to carry the organism long after recovery from the infection [2]; after recovery from infection with Clostridium difficile the toxin can be detected in the stools [3], and after recovery from viral diseases the serum antibodies can persist for years. These differences reduce the value of such markers in monitoring the disease and its response to treatment.
Definitions
A biomarker has been defined as ‘a characteristic that is objectively measured and evaluated as an indication of normal biologic processes, pathogenic processes, or pharmacologic responses to a therapeutic intervention’[4]. A surrogate end-point has been defined as ‘a biomarker intended to substitute for a clinical end-point’, the latter being ‘a characteristic or variable that reflects how a patient feels, functions, or survives’[5]. Thus, all surrogate end-points are biomarkers, but not all biomarkers are surrogate end-points, because biomarkers can substitute for end-points that are not clinical.
A surrogate marker can be defined as a surrogate that substitutes not for an end-point but for some other measure. For example, the plasma concentration of a drug is a surrogate marker for the concentration of the drug at its site of action.
Uses and advantages of surrogate end-points
Surrogate end-points can be used for different purposes, in screening, diagnosing, staging, and monitoring diseases, or in monitoring responses to interventions. They can also be used in various aspects of drug discovery and development, as follows:
as targets for drug actions in drug discovery (for example, cyclo-oxygenase-2 activity as a target for anti-inflammatory drugs);
as end-points for pharmacodynamic studies of drug action (for example, serum cholesterol as a marker for the action of a drug that is intended to be used to prevent cardiovascular disease) and in pharmacokinetic/pharmacodynamic studies;
in studying concentration–effect (dose–response) relations;
in clinical trials;
for studying adverse drug effects and adverse drug reactions.
Although surrogates can be used to study the risks of harms, it is preferable to study the harms themselves; here, I shall deal only with surrogates of benefits.
The principal advantages of surrogate end-points are that they are often cheaper and easier to measure than clinical end-points and can be measured over a shorter time span. For example, it is easier to measure a patient's blood pressure than to use echocardiography to measure left ventricular function, and it is much easier to carry out echocardiography than to measure long-term morbidity and mortality from hypertension. Blood pressure can be measured today, whereas it takes several years to collect mortality data. In clinical trials, the use of surrogates leads to smaller sample sizes. For example, to determine the effect of a new drug on blood pressure a relatively small sample size of say 100–200 patients would be needed and the trial would be relatively quick (1–2 years). To study the prevention of deaths from strokes, a much larger study group would be needed and the trial would take many years. There may also be ethical problems associated with measuring final end-points. For example, in paracetamol overdose it is unethical to wait for evidence of liver damage before deciding whether or not to treat a patient; instead, a pharmacological surrogate, the plasma paracetamol concentration, is used to predict whether treatment is required [6]; the challenges for clinical pharmacologists in that case are outlined in another paper in this issue of British Journal of Clinical Pharmacology[7].
Particular problems arise when monitoring intermittent disorders and the effects of preventive therapies.
Monitoring intermittent disorders
Intermittent disorders can be very difficult to monitor. For example, while a patient with epilepsy can keep a diary of the number of seizures, absence of seizures over a period of time during therapy cannot necessarily be attributed to the treatment but might simply be a reflection of periodicity. Measuring the plasma concentration of an antiepileptic drug will reveal whether there is enough there to have a putative effect, but not that it is the drug that is having the apparent beneficial effect.
Monitoring preventive therapies
When an intervention is aimed at preventing the end-point, the end-point is not itself suitable for monitoring. The best that one can do is to find a surrogate event that can be monitored in advance of the end-point and that predicts the efficacy of the preventive intervention. In hypertension, the blood pressure is a surrogate end-point, changes in which predict the success or failure of antihypertensive therapy in preventing heart attacks and strokes [8].
Classifying surrogate end-points
There are different ways of classifying surrogate end-points [9].
By the pathophysiology of the disorder or illness
Surrogate end-points can be classified in terms of the pathophysiology of a disorder or illness at different levels in the pharmacological chain from molecular to clinical. Figures 1 and 2 illustrate the example of asthma.
Figure 1.
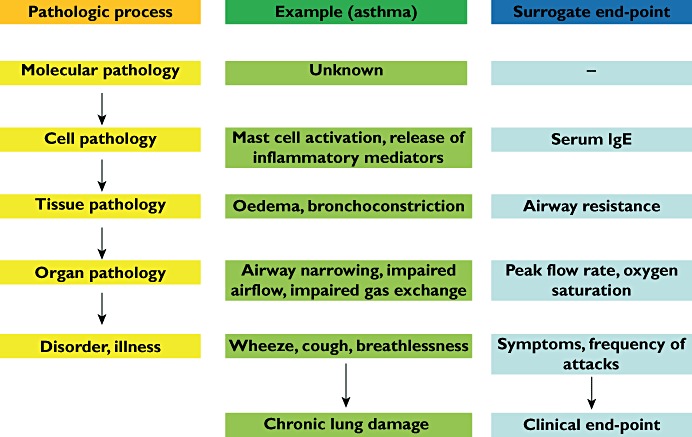
Surrogate end-points in the pathophysiology of asthma
Figure 2.
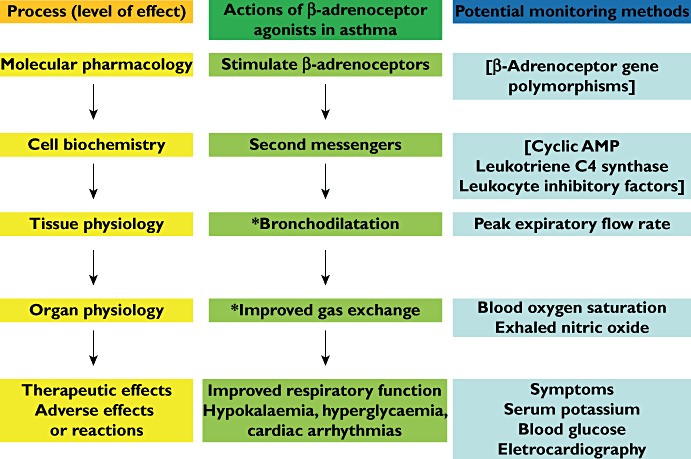
The chain in the pharmacodynamic process for β-adrenoceptor agonists in the treatment of acute severe asthma and the potential monitoring measurements that might be made at each pharmacodynamic level. * Relevant to the pathophysiology of the condition. [Items in brackets are not used in monitoring]
By the mechanism of action of the intervention (targets)
Classification of surrogate end-points according to the level at which they occur in the pharmacological chain is illustrated in Figure 3 for different drugs according to their targets. The nearer the therapeutic or adverse effect a surrogate end-point is, the better a measure of the clinically relevant end-point it is likely to be.
Figure 3.
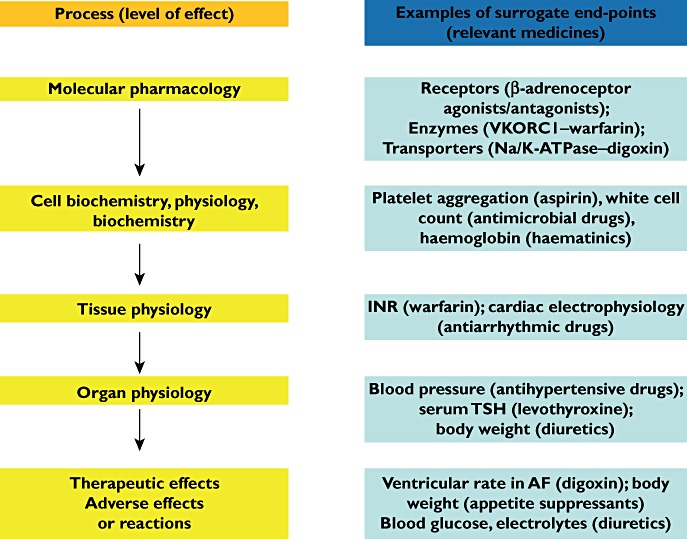
Examples of surrogate end-points at different pharmacodynamic levels (from molecular to whole body) in the management of diseases
By the nature of the measurement
A surrogate can be extrinsic to the individual, for example cigarette smoking as a surrogate end-point for lung cancer, or intrinsic. Intrinsic end-points can be physical (signs and symptoms), psychological, or laboratory measurements. Examples are shown in Table 1. These categories could be further subdivided according to whether the markers are being used for diagnosis, staging, or monitoring of disease, or for determining its response to an intervention. They could also be divided according to the level at which they occur (molecular, cellular, etc.) and according to whether they relate to susceptibility factors, primary or secondary pathology, or complications of the disease.
Table 1.
Classifying biomarkers by the type of measurement
| Types of biomarker | Examples of surrogate end-points | The relevant clinical end-points |
|---|---|---|
| A. Extrinsic markers | Cigarette consumption | Lung cancer |
| Daily defined dose | Drug consumption | |
| B. Intrinsic markers | ||
| 1. Physical evaluation | ||
| a. Symptoms | Breathlessness | Heart failure |
| b. Signs | Lid lag | Hyperthyroidism |
| 2. Psychological evaluation | Likert scales | Pain |
| Questionnaires | Self-harm | |
| 3. Laboratory evaluation | ||
| a. Physiological | Blood pressure | Heart attacks and strokes |
| b. Pharmacological | ||
| i. Exogenous | Inhibition of CYP enzymes | Routes of drug metabolism |
| ii. Endogenous | Docetaxel clearance | Febrile neutropenia |
| c. Biochemical | Blood glucose concentration | Complications of diabetes |
| d. Haematological | INR with warfarin | Pulmonary embolism |
| e. Immunological | Autoantibodies | Autoimmune diseases |
| f. Microbiological | Clostridium difficile toxin | Pseudomembranous colitis |
| g. Histological | Jejunal biopsy | Gluten-sensitive enteropathy |
| h. Radiographic | White dots on MRI scan | Lesions of multiple sclerosis |
| i. Genetic | CYP2C9 isoenzymes/VKORC1 genotype | Warfarin dosage |
INR, international normalized ratio.
Criteria for useful surrogate end-points – a taxonomy
Surrogates can be used at any point in the pharmacological chain, at the molecular, cellular, tissue, or organ levels (Figures 1–3). Likewise, a therapy might be developed to attack any one of the links in the chain, in order to try to manipulate the disease, symptomatically or therapeutically. Any measurement short of the actual outcome could be regarded as a biomarker.
However, there are different scenarios that link the biomarker or surrogate end-point to the disease and its outcome, as illustrated in Figure 4. These examples are not exhaustive.
Figure 4.
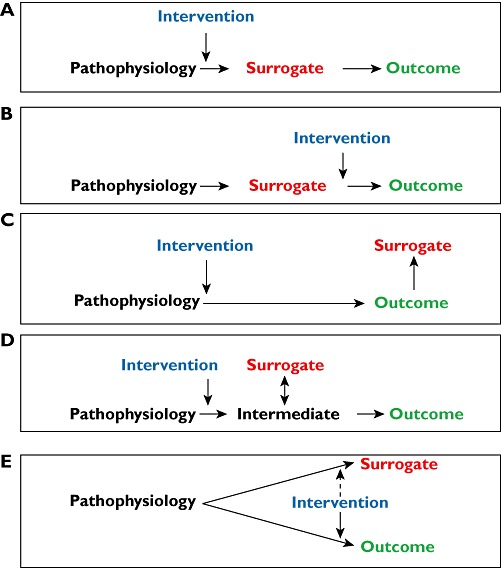
Five different scenarios relating pathophysiology, surrogate end-points and primary outcomes (see main text for discussion)
Scenario A
The ideal surrogate is one through which the disease comes about or through which an intervention alters the disease. For example, the serum cholesterol concentration should be an excellent diagnostic marker for cardiovascular disease; however, there is no clear cut-off point, and only about 10% of those who are going to have a stroke or heart attack have a serum cholesterol concentration above the reference range [10]. But even if cholesterol is not a good diagnostic marker, it can still be used as a biomarker of the therapeutic response to cholesterol-lowering drugs.
Scenario B
Even if a surrogate is in the pathway leading from the pathophysiology of the disease to its final outcomes, the intervention may not alter it. For example, most antihypertensive drugs lower the blood pressure by mechanisms that are probably not directed specifically at the prime cause of hypertension, whatever that is. Any surrogate marker earlier in the pathway than the raised blood pressure itself is therefore unlikely to be a good surrogate.
Scenario C
In some cases, the surrogate comes after the outcome rather than before it. For example, carcinoembryonic antigen, which is produced by cancer cells, is not useful in diagnosing ovarian carcinoma, because it is nonspecific, but can be used to monitor its response to treatment [11].
Scenario D
In some cases, a surrogate is closely related to an intermediary that mediates the effect of the pathophysiology but is not itself suitable as a biomarker. In this case, the surrogate is a kind of meta-marker; it is a marker of a marker. For example, in Gram-negative septicaemia the release of cytokines can cause a major primary outcome, such as hypotension. Cytokines are not suitable as biomarkers, but fever, another effect of cytokines, can be used as a biomarker of the response to antimicrobial drug therapy.
Scenario E
An important pitfall to avoid is to assume that an epiphenomenon or secondary outcome is a good surrogate marker. If the pathophysiology produces an effect by a different mechanism than that by which it produces the disease outcome, that effect (the epiphenomenon or secondary outcome) will not be a useful surrogate unless it is affected in the same way by the intervention as the primary outcome. There are many examples of epiphenomena.
Each of these scenarios is susceptible to modification by other factors (see the example of amiodarone in thyroid disease, in the Pitfralls and Problems section). In order to understand the value of a biomarker in monitoring therapy it is necessary to know which type of model fits the disease best.
Identifying biomarkers
The first step in identifying suitable biomarkers is to understand the pathophysiology of the disease and to find factors that determine it. For example (Figure 1), understanding the pathophysiology of asthma allows one to identify factors that might be useful as biomarkers. In a study of the use of biomarkers in heart failure, biomarkers that were linked to mechanisms involved in the aetiology seemed to be best suited to serve as early markers to predict and diagnose disease, select therapy, or assess progression [12].
The next step is to identify potential biomarkers based on the mechanism of action of the intervention related to the pathophysiology of the disease.
Finally, one must determine the extent to which the putative marker correlates with the process and how useful it is in predicting the final outcome.
Pitfalls and problems
A major problem in the use of biomarkers is the failure to understand the relation between the pathophysiology of the problem and the mechanism of action of the intervention (see Figs 1 and 4). For example, smoking causes lung cancer, and a trial of the benefit of education in preventing lung cancer might use smoking as a surrogate end-point rather than the occurrence of the cancer itself. In contrast, if chemotherapy is used as a measure for treating lung cancer, smoking could not be used as a surrogate end-point. This is obvious (it is scenario A compared with scenario B in Figure 4), but it alerts us to the possibility of similar but less obvious examples, in which the mechanisms are not understood.
Ventricular arrhythmias cause sudden death, and antiarrhythmic drugs prevent ventricular arrhythmias. It was therefore expected that antiarrhythmic drugs would prevent sudden death. In fact, in the Cardiac Arrhythmia Suppression Trial [13] Class I antiarrhythmic drugs increased sudden death significantly in patients with asymptomatic ventricular arrhythmias after a myocardial infarction, and the trial was stopped prematurely. The mechanisms were not understood and the hypothesis was wrong [14].
Another good example is enalapril and vasodilators, such as hydralazine and isosorbide, whose haemodynamic effects and effects on mortality associated with heart failure are dissociated. Vasodilators improved exercise capacity and improved left ventricular function to a greater extent than enalapril; however, enalapril reduced mortality significantly more than vasodilators [15]. Thus, in this case the haemodynamic effects are not a good surrogate.
Confounding factors, particularly the use of drugs, can nullify the value of surrogate end-points. For example, serum free T3 is used as a marker of the tissue damage that thyroid hormone causes in patients with hyperthyroidism; however, its usefulness is blunted in patients taking amiodarone, which interferes with the peripheral conversion of T4 to T3 without necessarily altering thyroid function. This is a modification of scenario A. In a patient with gastrointestinal bleeding, the heart rate may not increase if the patient is also taking a β-blocker, leading the clinician to underestimate the severity of the condition. Likewise, corticosteroids can mask the signs of an infection or inflammation.
When a biomarker persists after resolution of the disease, its subsequent use is vitiated, as in the examples given in the Introduction.
As a general principle, if the concentration–effect curves for the effects of an intervention on the primary outcome and the surrogate are different, a change in the surrogate may not truly reflect the degree of change in the outcome (Figure 5). This is potentially true for any biomarker that does not lie on the same line as the pathophysiology and the outcome.
Figure 5.
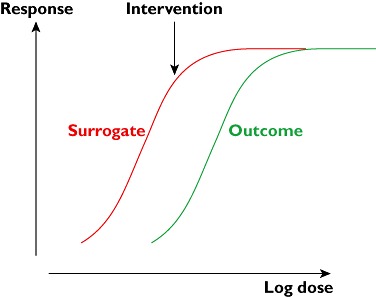
If the surrogate and the final outcome have different concentration–effect (dose–response) curves, an intervention may produce a large change in the surrogate and no change in the outcome (as shown) or vice versa (if the curves are switched)
Proper application of useful biomarkers may be hindered by lack of reproducibility of the methods used to measure them. For example, there are differences between ciclosporin concentrations measured in serum and blood and between blood ciclosporin concentrations measured using radioimmunoassay and high-performance liquid chromatography [16]. Despite the fact that the association between thiopurine methyltransferase (TPMT) activity and the risk of adverse effects from mercaptopurine was described several years ago [17], methods for measuring the enzyme are not standardized [18] and optimal treatment is often not achieved [19]. Another problem with TMPT is that in someone who has had a recent transfusion the activity of the enzyme in the recipient's erythrocytes may be contaminated by that in the donor's [20].
It is unusual for a single biomarker to provide all the information one needs in monitoring interventions. For example, patients with asthma feel breathless if they have a low peak expiratory flow rate (PEFR). In one study, however, different drugs produced different relations between PEFR and breathlessness [21]. Patients taking beclomethasone did not feel as breathless as those taking theophylline for a given PEFR. So what should the surrogate marker be; the ‘hard’ end-point of peak flow or the ‘soft’ end-point of how the patients felt? Probably both should be used. This stresses the potential usefulness of combinations of surrogates.
Statistical problems can arise with biomarkers and surrogate end-points. A surrogate end-point has been defined statistically as ‘a response variable for which a test of the null hypothesis of no relationship to the treatment groups under comparison is also a valid test of the corresponding null hypothesis based on the true end-point’[22]. Often the surrogate end-point is used as an entry criterion in clinical trials, and it is important to be aware that this can lead to statistical problems [4]. It introduces heterogeneous variance and the problem of regression to the mean. If someone is entered into a trial on the basis of an abnormal surrogate end-point and then receives no treatment, the surrogate end-point will still improve, simply because of the statistical variation in the measurement of variables. This reduces the power of a study. There is also a high likelihood of missing data when surrogate end-points are used. If the sample size when using a surrogate end-point is small, the study may not be big enough to detect adverse drug reactions.
Challenges for clinical pharmacology
There are clear potential benefits in using biomarkers. Information can be obtained earlier, more quickly, and more cheaply. However, the chain of events in a disease process linking molecular pathogenesis to clinical outcome is fragile, and there is a major challenge in improving our understanding of the nature of the paths that diseases take and the mechanisms of action of interventions that affect them. The better we understand these processes, the better biomarkers we shall be able to develop for diagnosing, staging, and monitoring disease and its response to therapy. There is also a challenge in extending the taxonomy shown in Figure 4 and in testing it in different cases to demonstrate how well the taxonomy correlates with the usefulness of the surrogate in different scenarios.
In its report on the evaluation of biomarkers and surrogate end-points in chronic disease [23], the US Institute of Medicine of the National Academies recommended that evaluation of biomarkers should consist of three steps.
Analytical validation, to ensure that biomarker tests are reliable, reproducible, and adequately sensitive and specific.
Qualification, to ensure the biomarker is associated with the clinical outcome of concern.
Utilization analysis, to determine that the biomarker is appropriate for the proposed use.
Clinical pharmacologists can contribute to all of these processes.
Acknowledgments
This paper is a revised and updated version of[24].
Competing Interests
There are no competing interests to declare.
REFERENCES
- 1.Kuru T, Lynch JP., III Nonresolving or slowly resolving pneumonia. Clin Chest Med. 1999;20:623–51. doi: 10.1016/s0272-5231(05)70241-0. [DOI] [PubMed] [Google Scholar]
- 2.Bourdain A. Typhoid Mary: An Urban Historical. New York: Bloomsbury; 2001. [Google Scholar]
- 3.Johnson S, Homann SR, Bettin KM, Quick JN, Clabots CR, Peterson LR, Gerding DN. Treatment of asymptomatic Clostridium difficile carriers (fecal excretors) with vancomycin or metronidazole. A randomized, placebo-controlled trial. Ann Intern Med. 1992;117:297–302. doi: 10.7326/0003-4819-117-4-297. [DOI] [PubMed] [Google Scholar]
- 4.Wittes J, Lakatos E, Probstfield J. Surrogate endpoints in clinical trials: cardiovascular diseases. Stat Med. 1989;8:415–25. doi: 10.1002/sim.4780080405. [DOI] [PubMed] [Google Scholar]
- 5.NIH Definitions Working Group. Biomarkers and surrogate endpoints in clinical research: definitions and conceptual model. In: Downing GJ, editor. Biomarkers and Surrogate Endpoints. Amsterdam: Elsevier; 2000. pp. 1–9. [Google Scholar]
- 6.Wallace CI, Dargan PI, Jones AL. Paracetamol overdose: an evidence based flowchart to guide management. Emerg Med J. 2002;19:202–5. doi: 10.1136/emj.19.3.202. [erratum: 376] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Thomas SHL. An agenda for UK clinical pharmacology: Developing and delivering clinical toxicology in the UK National Health Service. Br J Clin Pharmacol. 2012;73:878–83. doi: 10.1111/j.1365-2125.2012.04229.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Law MR, Morris JK, Wald NJ. Use of blood pressure lowering drugs in the prevention of cardiovascular disease: meta-analysis of 147 randomised trials in the context of expectations from prospective epidemiological studies. BMJ. 2009;338:b1665. doi: 10.1136/bmj.b1665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Aronson JK. Biomarkers and surrogate endpoints. Br J Clin Pharmacol. 2005;59:491–4. doi: 10.1111/j.1365-2125.2005.02435.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Wald NJ, Hacksaw AK, Frost CD. When can a risk factor be used as a worthwhile screening test? BMJ. 1999;319:1562–5. doi: 10.1136/bmj.319.7224.1562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Gargano G, Correale M, Abbate I, Falco G, De Frenza N, Lorusso V, De Lena M, De Leonardis A. The role of tumour markers in ovarian cancer. Clin Exp Obstet Gynecol. 1990;17:23–9. [PubMed] [Google Scholar]
- 12.Jortani SA, Prabhu SD, Valdes R., Jr Strategies for developing biomarkers of heart failure. Clin Chem. 2004;50:265–78. doi: 10.1373/clinchem.2003.027557. [DOI] [PubMed] [Google Scholar]
- 13.The Cardiac Arrhythmia Suppression Trial Investigators. Preliminary report: effect of encainide and flecainide on mortality in a randomised trial of arrhythmia suppression after myocardial infarction. N Engl J Med. 1989;321:406–12. doi: 10.1056/NEJM198908103210629. [DOI] [PubMed] [Google Scholar]
- 14.Howick J, Glasziou P, Aronson JK. Evidence-based mechanistic reasoning. J R Soc Med. 2010;103:433–41. doi: 10.1258/jrsm.2010.100146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Cohn J. Lessons from V-HeFT: questions for V-HeFT11 and the future therapy of heart failure. Herz. 1991;16:267–71. [PubMed] [Google Scholar]
- 16.Reynolds DJ, Aronson JK. ABC of monitoring drug therapy. Cyclosporin. BMJ. 1992;305:1491–4. doi: 10.1136/bmj.305.6867.1491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Lennard L, Rees CA, Lilleyman JS, Maddocks JL. Childhood leukaemia: a relationship between intracellular 6-mercaptopurine metabolites and neutropenia. Br J Clin Pharmacol. 2004;58:S867–71. doi: 10.1111/j.1365-2125.2004.02314.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Armstrong VW, Shipkova M, von Ahsen N, Oellerich M. Analytic aspects of monitoring therapy with thiopurine medications. Ther Drug Monit. 2004;26:220–6. doi: 10.1097/00007691-200404000-00024. [DOI] [PubMed] [Google Scholar]
- 19.Coulthard SA, Matheson EC, Hall AG, Hogarth LA. The clinical impact of thiopurine methyltransferase polymorphisms on thiopurine treatment. Nucleosides Nucleotides Nucleic Acids. 2004;23:1385–91. doi: 10.1081/NCN-200027637. [DOI] [PubMed] [Google Scholar]
- 20.Ford L, Prout C, Gaffney D, Berg J. Whose TPMT activity is it anyway? Ann Clin Biochem. 2004;41(Pt 6):498–500. doi: 10.1258/0004563042466866. [DOI] [PubMed] [Google Scholar]
- 21.Higgs CMB, Laszlo G. Influence of treatment with beclomethasone, cromoglycate and theophylline on perception of bronchoconstriction in patients with bronchial asthma. Clin Sci. 1996;90:227–34. doi: 10.1042/cs0900227. [DOI] [PubMed] [Google Scholar]
- 22.Prentice RL. Surrogate endpoints in clinical trials: definition and operational criteria. Stat Med. 1989;8:431–40. doi: 10.1002/sim.4780080407. [DOI] [PubMed] [Google Scholar]
- 23.Institute of Medicine of the National Academies. Evaluation of biomarkers and surrogate endpoints in chronic disease. 2010. Available at http://www.iom.edu/Reports/2010/Evaluation-of-Biomarkers-and-Surrogate-Endpoints-in-Chronic-Disease.aspx (last accessed 18 January 2012) [PubMed]
- 24.Aronson JK. Biomarkers and surrogate endpoints in monitoring therapeutic interventions. In: Glasziou P, Irwig L, Aronson JK, editors. Evidence-Based Medical Monitoring: From Principles to Practice. Oxford: Wiley-Blackwell Ltd; 2008. pp. 48–62. [Google Scholar]