

Abstract
Drug development has improved over recent decades, with refinements in analytical techniques, population pharmacokinetic–pharmacodynamic (PK–PD) modelling and simulation, and new biomarkers of efficacy and tolerability. Yet this progress has not yielded improvements in individualization of treatment and monitoring, owing to various obstacles: monitoring is complex and demanding, many monitoring procedures have been instituted without critical assessment of the underlying evidence and rationale, controlled clinical trials are sparse, monitoring procedures are poorly validated and both drug manufacturers and regulatory authorities take insufficient account of the importance of monitoring.
Drug concentration and effect data should be increasingly collected, analyzed, aggregated and disseminated in forms suitable for prescribers, along with efficient monitoring tools and evidence-based recommendations regarding their best use. PK–PD observations should be collected for both novel and established critical drugs and applied to observational data, in order to establish whether monitoring would be suitable. Methods for aggregating PK–PD data in systematic reviews should be devised.
Observational and intervention studies to evaluate monitoring procedures are needed. Miniaturized monitoring tests for delivery at the point of care should be developed and harnessed to closed-loop regulated drug delivery systems. Intelligent devices would enable unprecedented precision in the application of critical treatments, i.e. those with life-saving efficacy, narrow therapeutic margins and high interpatient variability.
Pharmaceutical companies, regulatory agencies and academic clinical pharmacologists share the responsibility of leading such developments, in order to ensure that patients obtain the greatest benefit and suffer the least harm from their medicines.
Introduction
Broadly speaking, clinical pharmacology can be designated as the capability to describe, explain and predict the fate and the effects of drugs in humans. This expertise is needed in pharmaceutical companies for the development of new therapeutic agents, in health services for provision of specialized advice to practitioners and patients, in regulatory authorities for appraisal of drug dossiers and in universities to teach health professionals the Effective, Rational, Adjusted, Safe and Monitored Use (ERASMUS) of medicines. We aim to show here how monitoring drug therapy typically fits among the core competencies to be fostered and developed by this discipline.
Monitoring drug therapy
Monitoring therapeutic interventions in the context of the management of chronic disease has been defined as repeated testing aimed at guiding and adjusting the management of a chronic or recurrent condition [1]. A major part of this activity is the monitoring of drug therapy, which can be defined as the measurement of a pharmacokinetic, pharmacodynamic or clinical outcome variable which, with appropriate interpretation, will directly influence the prescription of a medicine and management of the patient. For this to be the case, the monitored variable must either be the relevant clinical outcome itself or, more often, must represent a valid surrogate that can be used to predict treatment success, failure or toxicity. The circulating drug concentration is the classical pharmacokinetic (PK) surrogate in what has been traditionally called ‘therapeutic drug monitoring’ (TDM), while various response biomarkers are used as pharmacodynamic (PD) surrogates. The interpretation of such measurements thus requires prior knowledge of the pharmacokinetic–pharmacodynamic (PK–PD) relationships that characterize the drug and of their predictive value regarding clinically relevant outcomes.
Blood concentration measurement of drugs can probably be traced back to Widmark, who in 1932 introduced an equation for predicting the time course of blood alcohol concentrations [2], while lithium and digoxin were the first therapeutic agents recognized to be worth monitoring in this way [3], [4]. Among pharmacodynamic biomarkers, glycaemia and glycosuria were already by 1925 competing in monitoring insulin therapy [5], and prothrombin time has been used for about 60 years to monitor treatment with oral anticoagulants [6].
Since then, an increasing number of pharmaceutical agents have been recognized as deserving pharmacokinetic or pharmacodynamic monitoring. In the 1990s, practitioners were usually taught how to interpret blood (plasma or serum) concentration results of digoxin, phenytoin, lithium, theophylline, the aminoglycosides and ciclosporin [7]. Since then, further cardiac, antiepileptic, psychoactive, anti-infective and immunosuppressant agents have joined the list, as well as antiretroviral and anticancer drugs [8]. Criteria have been devised to identify candidate drugs for monitoring therapeutic concentrations or adverse effects [9]–[11].
Developing methods of monitoring therapy should be part of drug development
The modern clinical development of drugs has largely benefited from improvements in PK–PD modelling, in particular since the introduction of population approaches [12]. The ability to predict the average response profile to a given dosage is essential in elaborating appropriate standard therapeutic regimens. Assessment of the absorption and disposition of a drug defines its blood concentration vs. time curve (pharmacokinetics), while knowing how concentration exposure affects the evolution of relevant biomarkers (pharmacodynamics) helps in anticipating its therapeutic efficacy in terms of clinical outcomes, and often its adverse effects and consequent adverse reactions [13]. Assessing dose–response or concentration–effect relations is necessary in the evaluation of a drug's therapeutic margin, which indicates the range of exposure within which it is expected to be safe and effective.
In given conditions, several types of biomarkers are available, and they play different roles in clinical development programmes, according to the condition being treated and whether they are rapid reversible or slow cumulative response markers. For example, in the treatment of osteoporosis, collagen degradation products fall almost immediately after the administration of antiresorptive agents and are thus ideally suited for phase I trials; in contrast, bone mineral density takes months to change and there is some evidence that it correlates to some extent with the risk of fracture [14], [15] and hence is more useful during phase III. However, within-person variability and the limited extent to which bone mineral density predicts the risk of fractures vitiate its usefulness as a long term monitoring technique [16].
The second advantage that modern population PK–PD techniques confer is their ability to identify susceptibility factors that modify the therapeutic response or the adverse effects of drugs, namely genetic traits, physiological characteristics such as age, gender and pregnancy, pathophysiological conditions and drug–drug interactions [17]. Such knowledge is of prime importance in devising appropriate dosage adjustments in subgroups of patients in whom such susceptibility factors are identified.
The third type of information to be gained from PK–PD approaches in drug development is the quantification of variability in response. Besides the distinction between pharmacokinetic and pharmacodynamic components, total population variability can be split into a part that is explained by the above-mentioned susceptibility factors and a part that is determined by random population variability, which is not accounted for by defined factors. This unexplained variability can in turn be broken down into between-subject and within-subject (also called inter-occasion) components. Drug developers should pay attention to thorough estimation of all those sources of variability, as they determine the optimal treatment strategy.
Some agents can be prescribed based solely on standard dosage recommendations, when the therapeutic margin is larger than the total population variability. Others require dosage adjustment recommendations based on patient characteristics, when the therapeutic margin is still larger than the random population variability. Close monitoring becomes necessary when the therapeutic margin is narrower than the random population variability, but larger than the within-subject variability (otherwise, erratic fluctuations would preclude safe and effective dosing in any patient).
Depending on the relative importance of pharmacokinetic and pharmacodynamic components in response variability, monitoring drug therapy relies on measurement of blood concentrations or of appropriate biomarkers respectively.
Necessary improvements
Modern drug development has achieved a good degree of reliability in the elaboration of standard dosage recommendations. It has also become fairly good at providing prescribers with rational dosage recommendations in classical patient subgroups (for example, patients with renal or liver insufficiency). Conversely, its contributions to the practical management of disease and monitoring strategies are still unsatisfactory. Pharmaceutical manufacturers tend to leave these responsibilities largely to prescribers. Even though most physicians would not contest the clinical importance of monitoring drug therapy for some drugs, several arguments suggest that there is room for improvement in the way that it is currently performed and studied.
Most monitoring procedures have been devised and set up mainly, if not exclusively, on empirical grounds, without critical assessment of the underlying evidence and rationale [18]. This often tends to produce both over-sampling and under-interpretation of the results, and prescribers mostly implement therapeutic adjustments by trial and error. Data for formal dosage adjustments are often not recorded along with blood sampling [19]. Monitoring is insufficient in some therapeutic areas and exaggerated in others, and large numbers of tests requested by practitioners are of questionable usefulness. For example, regular cholesterol measurements in patients taking statins for secondary prevention, included by the NHS as an indicator in the Quality and Outcomes Framework, are not useful if performed more than once every 3–4 years [20] and may not be necessary at all in primary prevention [21].
Clinical researchers have not yet devoted sufficient attention to the question of monitoring drug therapy and controlled clinical trials remain scarce in this area. Moreover, we still lack a widely accepted conceptual framework for the evidence-based appraisal of monitoring procedures. Parts of such a framework have been elaborated [22], but without having yet reached the level of recognition of theoretical constructions developed for diagnostic tests. In particular, such elaborations still rely insufficiently on PK–PD modelling and disease evolution modelling, despite noteworthy appeals from early pioneers [23], [24].
Poor use of controlled clinical trials is a problem for monitoring procedures, even more than for therapeutic or diagnostic innovations, in which the need for clinical research has progressively become established. In our experience with antiretroviral drugs and new targeted anticancer agents, denial of access to a monitoring test for a control group is often felt to be unethical by both practitioners and patients, as soon as a test becomes available, bringing the promise of improved efficacy and safety of life-saving treatments. This should stimulate the search for original alternative study designs that can prevent frustration among participants and their caregivers.
The elaboration and clinical validation of monitoring procedures are generally disregarded by both private and public sponsors. In the competitive economic framework that currently characterizes drug development, pharmaceutical companies are understandably reluctant to encourage approaches that could complicate to any extent prescription of their drugs. Nor are public funders interested in supporting trials, which would be mainly aimed at improving the use of profit-generating medicines. There are therefore few incentives to optimize long- term therapies [25].
There is insufficient awareness among regulatory authorities of the importance of monitoring. While the role of biomarkers as surrogates for clinical outcomes during drug development has been widely promoted by the USA's Food and Drug Administration (FDA) during the past decade, it is still uncertain whether practical management of patients has much benefited from such developments [26]. Monitoring tests are considered by the authorities as a business mainly related to diagnostic tests and their approval is disconnected from drug innovation [27]. Pharmacogenetic tests today represent a noteworthy exception. For example, the FDA encourages their development and evaluation jointly with the development of new drugs [28]. By contrast, there is no indication that regulatory authorities are interested in treatment individualization sufficiently to encourage drug developers to assess monitoring using drug concentrations or biomarkers. This is surprising, as monitoring addresses phenotypic traits that integrate both genetic and non-genetic influences, thus representing a further relevant step towards individualized medicine.
The last but probably most important obstacle to the development of monitoring is that it is complex and demanding. Drug concentration monitoring usually requires blood sampling, pre-analytical precautions, thorough clinical data recording (current dosage, last time of dosing, patient characteristics), liaison with a distant laboratory, analysis using sophisticated and costly apparatus, delivery of the results after a significant delay, and interpretation according to complex algorithms that are generally not mastered by practitioners, thus requiring the intervention of a specialist. This is far too complicated. The situation is similar for pharmacodynamic biomarkers that are characterized by rapid reversible changes (for example, glycaemia), while slow cumulative response markers are definitely easier to use for monitoring in this type of setting (for example HbA1c in diabetes), although they are often poorly established as useful tools. In a few cases it has been possible to simplify the monitoring loop, bringing it nearer to cases that merely require clinical monitoring (such as the treatment of pain or hypertension). For example, INR monitoring of oral anticoagulation or HbA1c determination during oral antidiabetic treatment can now be satisfactorily performed in general practice using desktop devices at the point of care [29], without loss of efficacy [30]. A further step forward, self-monitoring and self-management of anticoagulation by patients themselves, when capable, has brought definite improvements in treatment efficacy and safety [31].
The future
Progress in pharmacotherapy continues to make life-threatening diseases amenable to long term stabilization during continuous therapy, using pharmaceutical agents that have been called ‘critical drugs’. During the recent past, this has typically been the case for HIV infection, systemic fungal infections and an increasing number of cancers (imatinib therapy of chronic myeloid leukaemia being a paradigmatic example). Many of these critical drugs meet the classical criteria for monitoring [7], [8]: long term use, measurability, consistent concentration–response relations, a narrow or intermediate therapeutic margin, high inter-individual but restricted intra-individual variability, limited pharmacokinetic predictability, the problem of drug–drug interactions and reversibility of effect. However, most of them have been commercialized in fixed dosage regimens. In the absence of suitable rapid reversible markers of efficacy and toxicity, such drugs undoubtedly represent candidates for concentration monitoring. Several older drugs used in neglected tropical or orphan diseases are similar (including antituberculosis drugs). We therefore consider it both probable and suitable that monitoring should expand, despite the barriers outlined above, so that patients with serious diseases will benefit optimally from the promises brought by novel therapeutic agents. The following developments could ensure progress in monitoring.
Large scale collection of PK–PD observations should be pursued and encouraged for both novel and established critical drugs. Pharmaceutical companies should be pressurized to release and publish their data in timely fashion and clinical laboratories in the public health service should complement this effort through multicentre research collaborations. Children and old people, often neglected during drug development, deserve such observational PK–PD studies.
State of the art PK–PD analyses should be further applied to observational data, with the intention of establishing whether monitoring would be suitable, and to estimate the clinically important parameters that are necessary to support the development of rational monitoring strategies. Redirecting PK–PD modelling towards monitoring questions might well change the way that it is performed, compared with its application in pre-marketing drug development.
Methods for aggregating PK–PD data into systematic PK–PD reviews should be devised, very much like meta-analyses are used to aggregate results from interventional or diagnostic trials. Central coordination and repositories for such data would facilitate their dissemination and clinical use. This task should ideally be devolved to a consortium working under strict rules of independence and transparency, which might bring together people from academies, medical associations, regulatory authorities and pharmaceutical companies.
A consistent set of conceptual criteria for the elaboration and assessment of monitoring procedures, relying on the best available evidence, remains to be elaborated and validated. Facing a drug concentration or biomarker measurement, the prescriber essentially has three questions to answer: (i) Is this result expected in this patient, considering his/her treatment and personal characteristics? (ii) Is this result suitable with regards to appropriate therapeutic target ranges? (iii) How does this result enable prediction of the effect of dosage adjustment decisions aiming to optimize therapy? The provision of quantitative answers to those three questions, and of methods for their critical appraisal, would represent the global aim of this process. These concepts will have to be disseminated widely into the medical culture, so that practitioners adopt them in every day health care. Clinical pharmacologists, even with the assistance of clinical pharmacists, will be insufficient in number to do the job by themselves; their role will be mainly to produce and teach about efficient tools, ensuring wide access of primary care practitioners and patients to appropriate monitoring resources.
Technological efforts towards the miniaturization of monitoring tests and their delivery at the point of care are under way. Recent lab-on-chip methods carry a promise in the near future that drug concentrations or relevant biomarkers may be measurable by desktop devices, of sufficient performance to rival standard laboratory techniques, such as immunoassays or chromatography. However, the mere production of accurate and precise concentration results will not enable practitioners to transform measurements into sound dosage adjustment decisions. Robust and user-friendly computer tools connected to the measurement devices, and implementing the conceptual approaches and PK–PD tools mentioned above, will be necessary to provide seamless and efficient monitoring services in the clinics. One may also consider proposing self-monitoring of drug concentrations for some patients, particularly when there is high inter-occasion variability.
Consequently, observational studies and intervention trials that evaluate monitoring procedures need to be designed and performed, so that monitoring develops according to the standards of evidence-based medicine. As stated above, methodological progress is still expected in this area (for example, substituting randomized monitoring trials with target concentration interventions [32]). The connection of point-of-care monitoring devices to central databases can be envisioned, given the widespread availability of Internet and mobile communications networks, and will facilitate large scale surveys (once significant privacy and security issues have been dealt with), and even the performance of intervention trials.
A further step might lead to closed-loop regulated drug delivery systems, such as have already been proposed, for example, in the treatment of diabetes [33]. This extreme degree of control engineering would enable prescribers to abandon formal interpretation and decision algorithms almost completely. A few drugs outside intensive care may be candidates for such a ‘pharmacostat’ approach in the near future, at least before more basic needs in monitoring are satisfactorily covered.
The possibility of implementing monitoring techniques of this sort is illustrated in Figure 1, in which potential monitoring methods are mapped by analogy on to modern methods of navigation.
Figure 1.
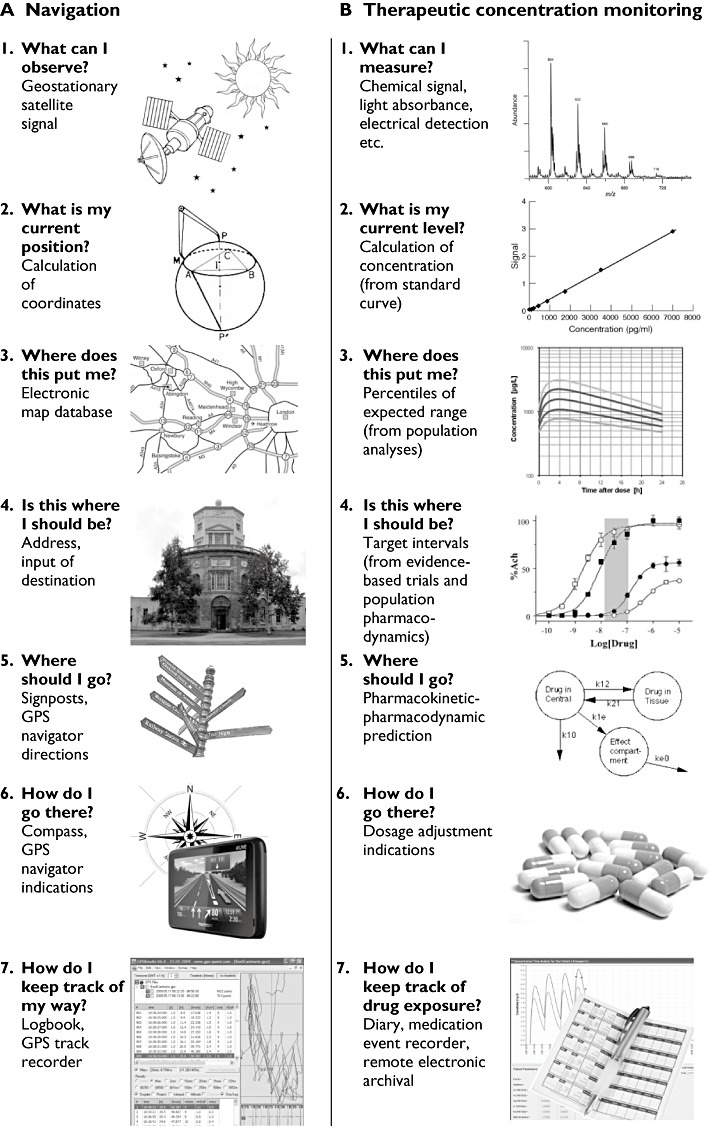
A) Sequential steps of modern navigation, as currently implemented in GPS-based satellite navigation devices. B: Potential modern methods of monitoring drug therapy, mapped onto the methods of navigation
Conclusions
Progress in various aspects of monitoring drug therapy is needed and possible. It represents the natural extension of improvements that have characterized clinical drug development during the past few decades. This constitutes an important duty for today's clinical pharmacologists, whatever their involvement in universities and hospitals, pharmaceutical companies or regulatory authorities. Patients are entitled to expect the greatest benefits and the least harm from medicines that they receive, which is the very purpose of dosage individualization based on monitoring.
Acknowledgments
The authors' research on monitoring has benefited from the support of the Nano-Tera programme from the Swiss National Science Foundation (ISyPeM project).
Competing Interests
There are no competing interests to declare.
REFERENCES
- 1.Glasziou P, Aronson JK. An introduction to monitoring therapeutic interventions in clinical practice. In: Glasziou P, Irwig L, Aronson JK, editors. Evidence-Based Medical Monitoring: From Principles to Practice. Oxford: Wiley-Blackwell Ltd; 2008. pp. 3–14. [Google Scholar]
- 2.Widmark EMP. Die theoreischen Grundlagen und praktische Verwendbarkeit der gerichtlich-medizinischen Alkoholbestimmung. Berlin: Urban & Schwarzenberg; 1932. [Google Scholar]
- 3.Van der Helm HJ, Andriesse D. The determination of lithium in serum during therapy with lithium salts. Clin Chim Acta. 1961;6:747–8. doi: 10.1016/0009-8981(61)90127-9. [DOI] [PubMed] [Google Scholar]
- 4.Löwenstein JM. A method for measuring plasma levels of digitalis glycosides. Circulation. 1965;31:228–33. doi: 10.1161/01.cir.31.2.228. [DOI] [PubMed] [Google Scholar]
- 5.Roe JH, Irish OJ. Sugar threshold in one hundred cases of diabetes. JAMA. 1925;84:1406–7. [Google Scholar]
- 6.Quick AJ, Hussey CV, Kaser M. The action of dicumarol in the human being on plasma prothrombin time and total prothrombin time. Am Heart J. 1952;44:119–23. doi: 10.1016/0002-8703(52)90179-8. [DOI] [PubMed] [Google Scholar]
- 7.Aronson JK, Hardman M, Reynolds DJM. ABC of Monitoring Drug Therapy. London: BMJ Publishing Group; 1993. [Google Scholar]
- 8.Burton M, Evans WE, Schentag JJ, Shaw L. Applied Pharmacokinetics and Pharmacodynamics: Principles of Therapeutic Drug Monitoring. 4th edn. Baltimore, MD: Williams & Wilkins; 2006. [Google Scholar]
- 9.Ensom MH, Davis GA, Cropp CD, Ensom RJ. Clinical pharmacokinetics in the 21st century; does the evidence support definitive outcomes? Clin Pharmacokinet. 1998;34:265–79. doi: 10.2165/00003088-199834040-00001. [DOI] [PubMed] [Google Scholar]
- 10.Pirmohamed M, Ferner RE. Monitoring drug treatment. BMJ. 2003;327:1179–81. doi: 10.1136/bmj.327.7425.1179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Gross AS. Best practice in therapeutic drug monitoring. Br J Clin Pharmacol. 2001;52(Suppl. 1):5S–10S. doi: 10.1046/j.1365-2125.2001.0520s1005.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Csajka C, Verotta D. Pharmacokinetic–pharmacodynamic modelling: history and perspectives. J Pharmacokinet Pharmacodyn. 2006;33:227–79. doi: 10.1007/s10928-005-9002-0. [DOI] [PubMed] [Google Scholar]
- 13.Lathia CD, Amakye D, Dai W, Girman C, Madani S, Mayne J, MacCarthy P, Pertel P, Seman L, Stoch A, Tarantino P, Webster C, Williams S, Wagner JA. The value, qualification, and regulatory use of surrogate end points in drug development. Clin Pharmacol Ther. 2009;86:32–43. doi: 10.1038/clpt.2009.69. [DOI] [PubMed] [Google Scholar]
- 14.Cummings SR, Karpf DB, Harris F, Genant HK, Ensrud K, LaCroix AZ, Black DM. Improvement in spine bone density and reduction in risk of vertebral fractures during treatment with antiresorptive drugs. Am J Med. 2002;112:281–9. doi: 10.1016/s0002-9343(01)01124-x. [DOI] [PubMed] [Google Scholar]
- 15.Wasnich RD, Miller PD. Antifracture efficacy of antiresorptive agents is related to changes in bone density. J Clin Endocrinol Metab. 2000;85:231–6. doi: 10.1210/jcem.85.1.6267. [DOI] [PubMed] [Google Scholar]
- 16.Bell KJ, Hayen A, Macaskill P, Irwig L, Craig JC, Ensrud K, Bauer DC. Value of routine monitoring of bone mineral density after starting bisphosphonate treatment: secondary analysis of trial data. BMJ. 2009;338:b2266. doi: 10.1136/bmj.b2266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Aronson JK, Ferner RE. Joining the DoTS. New approach to classifying adverse drug reactions. BMJ. 2003;327:1222–5. doi: 10.1136/bmj.327.7425.1222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Glasziou P, Irwig L, Mant D. Monitoring in chronic disease: a rational approach. BMJ. 2005;330:644–8. doi: 10.1136/bmj.330.7492.644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ellington C, Grgurinovich N, Miners JO, Mangoni AA. Quality of requests for serum digoxin concentrations: experience from an Australian regional health service. Br J Clin Pharmacol. 2007;63:623–7. doi: 10.1111/j.1365-2125.2006.02802.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Glasziou PP, Irwig L, Heritier S, Simes RJ, Tonkin A LIPID Study Investigators. Monitoring cholesterol levels: measurement error or true change? Ann Intern Med. 2008;148:656–61. doi: 10.7326/0003-4819-148-9-200805060-00005. [DOI] [PubMed] [Google Scholar]
- 21.Wald NJ, Law MR. A strategy to reduce cardiovascular disease by more than 80% BMJ. 2003;326:1419. doi: 10.1136/bmj.326.7404.1419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Glasziou PP, Irwig L, Aronson J. Evidence-Based Medical Monitoring: Principles and Practice. Oxford: BMJ Books, Blackwell Publishing; 2008. [Google Scholar]
- 23.Sheiner LB, Beal S, Rosenberg B, Marathe VV. Forecasting individual pharmacokinetics. Clin Pharmacol Ther. 1979;26:294–305. doi: 10.1002/cpt1979263294. [DOI] [PubMed] [Google Scholar]
- 24.Holford NH, Sheiner LB. Understanding the dose–effect relationship: clinical application of pharmacokinetic–pharmacodynamic models. Clin Pharmacokinet. 1981;6:429–53. doi: 10.2165/00003088-198106060-00002. [DOI] [PubMed] [Google Scholar]
- 25.Buclin T, Widmer N, Biollaz J, Decosterd LA. Who is in charge of assessing therapeutic drug monitoring? The case of imatinib. Lancet Oncol. 2011;12:9–11. doi: 10.1016/S1470-2045(10)70258-8. [DOI] [PubMed] [Google Scholar]
- 26.Woodcock J. Assessing the clinical utility of diagnostics used in drug therapy. Clin Pharmacol Ther. 2010;88:765–73. doi: 10.1038/clpt.2010.230. [DOI] [PubMed] [Google Scholar]
- 27.Scherf U, Becker R, Chan M, Hojvat S. Approval of novel biomarkers: FDA's perspective and major requests. Scand J Clin Lab Invest Suppl. 2010;242:96–102. doi: 10.3109/00365513.2010.493415. [DOI] [PubMed] [Google Scholar]
- 28.Hamburg MA, Collins FS. The path to personalized medicine. N Engl J Med. 2010;363:301–4. doi: 10.1056/NEJMp1006304. [DOI] [PubMed] [Google Scholar]
- 29.Laurence CO, Gialamas A, Bubner T, Yelland L, Willson K, Ryan P, Beilby J Point of Care Testing in General Practice Trial Management Group. Patient satisfaction with point-of-care testing in general practice. Br J Gen Pract. 2010;60:e98–104. doi: 10.3399/bjgp10X483508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Gialamas A, St John A, Laurence CO, Bubner K PoC Management Committee. Point-of-care testing for patients with diabetes, hyperlipidaemia or coagulation disorders in the general practice setting: a systematic review. Fam Pract. 2010;27:17–24. doi: 10.1093/fampra/cmp084. [DOI] [PubMed] [Google Scholar]
- 31.Garcia-Alamino JM, Ward AM, Alonso-Coello P, Perera R, Bankhead C, Fitzmaurice D, Heneghan CJ. Self-monitoring and self-management of oral anticoagulation. Cochrane Database Syst Rev. 2010;(4) doi: 10.1002/14651858.CD003839.pub2. CD003839. [DOI] [PubMed] [Google Scholar]
- 32.Holford NH. Target concentration intervention: beyond Y2K. Br J Clin Pharmacol. 2001;52(Suppl. 1):55S–9S. doi: 10.1046/j.1365-2125.2001.0520s1055.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Hovorka R, Kumareswaran K, Harris J, Allen JM, Elleri D, Xing D, Kollman C, Nodale M, Murphy HR, Dunger DB, Amiel SA, Heller SR, Wilinska ME, Evans ML. Overnight closed loop insulin delivery (artificial pancreas) in adults with type 1 diabetes: crossover randomised controlled studies. BMJ. 2011;342:d1855. doi: 10.1136/bmj.d1855. [DOI] [PMC free article] [PubMed] [Google Scholar]