

Abstract
Human immunodeficiency virus Type-1 (HIV-1) protease is crucial for viral maturation and infectivity. Studies of protease dynamics suggest that the rearrangement of the hydrophobic core is essential for enzyme activity. Many mutations in the hydrophobic core are also associated with drug resistance and may modulate the core flexibility. To test the role of flexibility in protease activity, pairs of cysteines were introduced at the interfaces of flexible regions remote from the active site. Disulfide bond formation was confirmed by crystal structures and by alkylation of free cysteines and mass spectrometry. Oxidized and reduced crystal structures of these variants show the overall structure of the protease is retained. However, cross-linking the cysteines led to drastic loss in enzyme activity, which was regained upon reducing the disulfide cross-links. Molecular dynamics simulations showed that altered dynamics propagated throughout the enzyme from the engineered disulfide. Thus, altered flexibility within the hydrophobic core can modulate HIV-1 protease activity, supporting the hypothesis that drug resistant mutations distal from active site can alter the balance between substrate turnover and inhibitor binding by modulating enzyme activity.
1. INTRODUCTION
Human immunodeficiency virus type 1 (HIV-1) protease is a homodimeric, aspartyl protease with 99 residues in each monomer. Protease is essential for post-translational processing of the viral polyprotein Gag-Pro-Pol during the assembly and maturation of viral particles1,2. There is an internal twofold symmetry in the protease dimer. However, ligands are asymmetric and introduce asymmetry in the dimer upon binding. Under protease inhibitor (PI) therapy, drug resistance mutations arise within the active site of the enzyme. These active site mutations directly interfere with the inhibitor binding and are primary cause of drug resistance to PIs. Additional mutations occur throughout the enzyme, and although they have often been shown to contribute to drug ressistance3-5, the mechanism by which they do so is poorly understood.
Ligand binding in protease involves conformational changes in the protease flaps and the hydrophobic core residues. While the flaps of the unliganded protease are highly dynamic6-11 and exhibit large conformational changes during ligand binding, hydrophobic core regions also exhibit subtle conformational changes12. Of the 19 residues that comprise the hydrophobic core of HIV-1 protease in each monomer, 13 sites are associated with drug resistance12 (Figure. 1). The role of these core residues, which do not contact either substrates or inhibitors directly, in substrate binding or drug resistance is not obvious and remains largely unexplored. Molecular dynamic simulations on unliganded HIV protease suggested the conformational changes occurring during side chain repacking in the hydrophobic core or “Hydrophobic sliding” as possible mechanism affecting drug susceptibility12. The hydrophobic core domains of each protease monomer rearranges through correlated sliding motions facilitated by near-isoenergetic exchange of van der Waals contacts between hydrophobic side-chains such that the hydrogen bonding network within the core, primarily with the backbone, is conserved12. While these simulations indicate that the rearrangement of the core facilitates conformational changes in the protease, much remains unknown about the role of this rearrangement in catalytic function and drug resistance.
Figure 1.
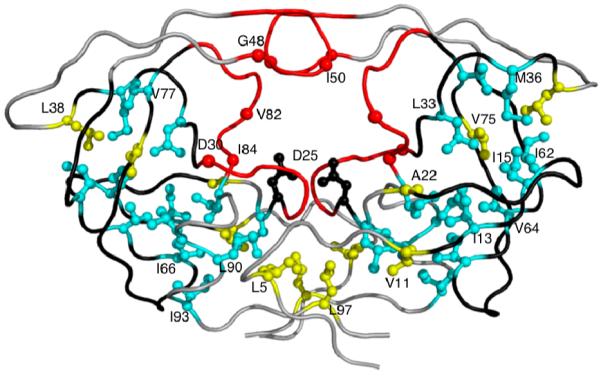
Drug resistance mutations in HIV-1 protease. Active site and the primary active site residues causing drug resistance (D30N, G48V,I50L/V, V82A/F/T, I84V) are colored in red. Hydrophobic core residues associated with drug resistance (I13V, I15V, L24I, L33F, M36I, I62V, I64V, I66F, V77I, I85V, L89M, L90M, I93L) are colored cyan. Remaining hydrophobic core residues (L5, V11, A22, L38, V75, L97) and rest of the protease are in yellow and gray, respectively. Catalytic Asps and the loops containing residues 11-22, 31-38 and 58-78 are displayed in black.
The contributions of protein dynamics in both protein structure and function have been investigated in other systems. Studies on the multidrug efflux pump AcrB13 and caspase714 have shown the functional importance of flexible regions, but there has been little emphasis on studying the dynamics of the hydrophobic core of the proteins. The reasons are two fold: 1) difficulty in accessing the hydrophobic core in folded proteins and, 2) limited resolution of currently available experimental techniques to view protein dynamics. While NMR, Mass spec-trometry and other single molecule techniques have furthered our understanding of protein dynamics and conformational heterogeneity of proteins in bulk solution15-18, structural characterization of conformations other than those found in the crystal structures, remains challenging.
We experimentally investigated the role of hydrophobic core flexibility in HIV-1 protease function using site-directed cysteine cross-linking. Disulfide bonds are usually either structural and stabilize a protein or catalytic and modulate the function of proteins involved in cellular pathways19-22. Disulfide bonds which modulate activity are both useful in functional description of proteins and provide a method to evaluate the regulation of function in proteins13,23,24. In this study, site-directed cysteine cross-linking provided evidence for the requirement of conformational changes in the hydrophobic core of HIV-1 protease for efficient cleavage of its substrates. The reversibility of disulfide chemistry worked as a molecular switch to modulate protease activity confirming that cross-linking caused the reduced activity (Figure. 2). These results confirm the role of hydrophobic core flexibility in protease function.
Figure 2.
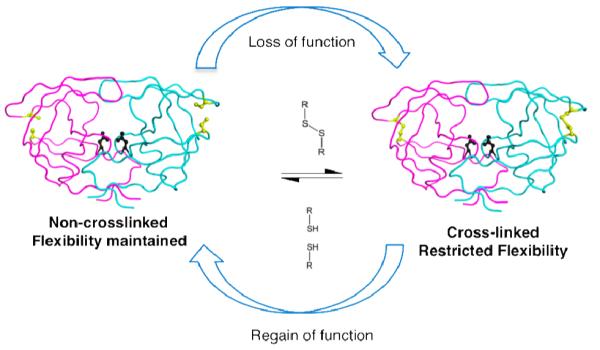
Schematic of cysteine redox chemistry used in this study. The cysteine substituted and non-cross-linked protease is flexible and as active as WT enzyme. Upon oxidation of cysteines, protease gets cross-linked via disulfide bond. Resulting loss of hydrophobic core flexibility is accompanied by loss of catalytic activity that is reversible upon reduction of the disulfide bond.
Since conformational flexibility in HIV-1 protease has also been implicated in drug resistance12,25,26, we hypothesize that the non-active site mutations may generate novel conformational heterogeneity in the protease, such that there is a preferential selection for conformers that process substrates than bind inhibitors. Therefore, intrinsic protein dynamics can mechanistically connect protein function, molecular recognition and drug resistance. More generally, hydrophobic core rearrangement may represent a mechanism by which proteins undergo the necessary conformational changes required for function.
2. RESULTS
2.1 Rationale for cysteine engineering and chosen cysteine pairs
Analysis of previous MD simulations12 revealed that 19 core residues in each protease monomer had limited solvent accessibility throughout the 14ns simulations (Figure 1). During the simulations these hydrophobic core residues undergo conformational changes concurrent with the opening of the active site cavity. We hypothesized that this hydrophobic sliding in HIV-1 protease is critical for proteolytic activity. To test this hypothesis, specific pairs of cysteines were introduced into the protease in a cysteine-free background. MD simulation trajectories12 were analyzed at various time points during the simulations and cysteine pairs were chosen at protease positions to restrict the movement of the hydrophobic domains using the Cβ-Cβ distance criterion for allowing disulfide bond formation27 (Table SI1). The selection of residue positions was based on their proximity to the rearranging hydrophobic core and was not limited to the 19 positions comprising the core. Three distinct regions containing residues (11-21), (31-38) and (58-78) were seen to be tethered to the core and exhibit extensive rearrangement. Specifically, G16, R14 and E65 residues, although not hydrophobic themselves, are contiguous with the hydrophobic core and along with L38 met the distance criteria. These solvent accessible sites were chosen as suitable to probe the hydrophobic core dynamics and are amenable to reduction-oxidation chemistry. Under appropriate oxidation conditions, the introduced cysteines should form disulfide cross-links, likely restricting hydrophobic core movement and potentially enzymatic activity.
Two protease variants, each with a unique cysteine pair, were constructed (Figure 3a). The first pair (G16C/L38C) would establish an intra-monomeric cross-link that should severely restrict hydrophobic core rearrangements. An alanine control variant G16A/L38A was generated to ensure that these sites could tolerate substitutions without compromising protease activity. The second cysteine pair (R14C/E65C) was designed as a control to form cross-links at sites that should not restrict hydrophobic core rearrangements, as the relative distance between these residues did not significantly change during the simulations12 (Table SI1). Cross-linking in both these variants should occur under oxidizing conditions, but loss of protease function was predicted, based on our hypothesis, in only the G16C/L38C variant. Cross-linking in G16C/L38C should restrict the hydrophobic core dynamics and hinder activity. In R14C/E65C variant, protease function should be unaffected under both oxidizing and reducing (cross-linked (oo) and non-cross-linked (rr)) conditions because core rearrangements should be unaffected (Figure 2).
Figure 3.
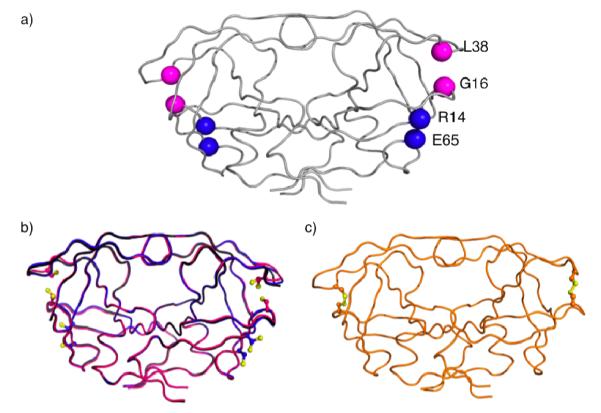
Ribbon diagrams of crystal structures of protease with engineered cysteines (a) Sites for engineering cysteines in HIV-1 protease are shown with backbone Cα (G16C/L38C) in red and (R14C/E65C) in blue. (b) The backbone structural superposition of DRV complexes of (G16C/L38C)rr and (R14C/E65C)rr on WT in black. Under reducing conditions, no disulfide bonds were observed. Alternate conformations for cysteine side chains were, however, seen for 50% of substituted cysteines in each pair analyzed. (c) apo(G16C/L38C)oo with disulfide bonds on both sides of the dimer shown in orange color.
2.2 Modulation of protease activity
By exploiting the reversibility of disulfide bonding, the effect of restricted flexibility on protease function was evaluated. Protease mediated cleavage of a fluorogenic peptide substrate derived from the native CA-p2 cleavage site on Gag polyprotein was measured in the presence and absence of reducing agent. The WT, (G16C/L38C)rr, (G16A/L38A)ctrl, and (R14C/E65C)rr proteases all showed similar catalytic activity (Table 1). In contrast, the cross-linked protease (G16C/L38C)oo showed 146-fold lower activity than the non-cross-linked (G16C/L38C)rr, demonstrating dramatic loss of function. 70% of this function was recovered upon addition of the reducing agent TCEP, demonstrating that the loss of activity was due to the G16C/L38C disulfide bond. The R14C/E65C variant exhibited comparable catalytic activity under both oxidizing and reducing conditions, as expected. These results confirm that the activity of disulfide-crosslinked variants depends on the location of the cross-link. Protease activity was only sensitive to the cross-link that restricted the flexibility of HIV-1 protease hydrophobic core.
Table 1.
Kinetic values for the WT, cross-linked, non-cross-linked variants of G16C/L38C and R14C/E65C and G16A/L38A variants of HIV-1 protease.
| Protease Variants | Catalytic Activity, Kcat/Km (μm−1s−1) |
Normalized |
|---|---|---|
| Wild-type | 6.8×10−2 ± 0.002 | 1 |
| G16C_L38C (rr) | 7.6 ×10−2 ± 0.004 | 1.1 |
| G16C_L38C (oo) | 5.2 ×10−4 ± 2.5 ×10−6 | 7.6 ×10−3 |
| G16C_L38C(oo) + 5mM TCEP (regain of function) |
4.7 ×10−2 ± 0.006 | 0.69 |
| G16A_G38A | 6.2 ×10−2 ± 0.002 | 0.91 |
| R14C_E65C (rr) | 6.5 ×10−2 ± 0.003 | 0.96 |
| R14C_E65C (oo) | 7.1 ×10−2 ± 0.004 | 1.04 |
2.3 Structural analysis
Six crystal structures of the engineered protease variants were determined (Table SI2). Five structures were in complex with DRV, a 4 pM inhibitor of WT28, including the oxidized and reduced forms of G16C/L38C, reduced R14C/E65C, WT and G16A/L38A. The final structure was an apo structure of the oxidized G16C/L38C variant. In the oxidized G16C/L38C variant in complex with DRV, perhaps due to the high affinity of DRV and/or the asymmetry of the crystal packing, although contiguous electron density is observed, one of the disulfide bridges is in a highly strained geometry. However, in the apo form of G16C/L38C variant both disulfides are clearly observed, (Figure SI1) although this structure was excluded from all comparative structural analyses because of the relatively low resolution.
The overall structural fold of all cysteine substituted protease complexes was well preserved when compared to the WT, with RMSD ranging from 0.06 to 0.09 (Figure. 3b and c). Hydrogen bond analysis revealed that all structurally important hydrogen bonds were conserved for all DRV complexes confirming that the known mode of ligand binding was intact in these proteases. The disulfide bonds observed in DRV(G16C/L38C)or and apo(G16C/L38C)oo structures fall into geometric categories (-RHHook and –LHHook respectively) commonly observed in natural proteins19.
2.4 Molecular dynamic simulation analysis of cross-linked and non-crosslinked structures
To evaluate the ability of engineered cross-links to restrict hydrophobic core dynamics, MD simulations were carried out for 20 ns on the cross-linked and non-crossliked protease. DRV was removed from the starting structures, DRV(G16C/L38C)rr and a model of DRV(G16C/L38C)oo. Extensive structural differences were observed in the protease backbone for the (oo) compared to the (rr) structures of G16C/L38C protease between 0 ns and 20 ns (Figure. 4a and 4b). These differences map to backbone shifts of greater than 1Å and are most pronounced for the protease flaps (residues 45-55) as indicated by the double difference plots. The presence of disulfide bonds affected the dynamics in (G16C/L38C)oo protease simulations leading the flaps to rearrange and begin to open after 20 ns. In comparison, for the (G16C/L38C)rr simulation, the unrestricted hydrophobic core region showed subtle motions, with no significant motion elsewhere in the protease dimer on the time scale of these simulations. The RMSF (root mean square fluctuation) for each protease residue over the 20ns MD simulations for (G16C/L38C)oo and (G16C/L38C)rr structures further confirmed these results (Figure 4c). This suggests that flap movements can be modulated in response to the restricted hydrophobic core dynamics as caused by the formation of a disulfide between G16C and L38C.
Figure 4.

Molecular dynamics simulation analyses. (a) Backbone superposition of (G16C/L38C)oo from 0ns (cyan) and at 20ns (pale cyan) and (G16C/L38C)rr from 0ns (light pink) to 20ns (magenta) in MD simulations. The side chains of active site aspartic acids and the engineered cysteines are displayed. (b) Differences in internal Cα-Cα distances between the 0ns and 20ns snapshots of the cross-linked (oo) and non-crosslinked (rr) forms of (G16C/L38C) variant are shown in the double difference plots. Each contour line represents a deviation by 0.5Å. Black, green, blue and red distinguish the contour ranges −1.0 Å and below, −1.0 to −0.5 Å, 0.5-1.0 Å and 1 Å and above, respectively. (c) Average RMSF of protease residues in (G16C/L38C)oo and (G16C/L38C)rr proteases from 20ns MD simulation trajectories. Protease molecules from 5, 10, 15 and 20ns simulations were superposed on to the 0ns crystal structure using the most invariant residues, 24 to 26 and 85 to 95. The average Cα RMSFs were calculated and mapped on to a representative protease molecule with the most variable regions depicted in red and the most invariant regions depicted in blue.
The structural regions containing residues (11-21), (31-38) and (58-78) that were previously implicated in the hydrophobic core sliding12 (Figure 1) were further analyzed for the effect of physical 16C/38C cross-linking on the side chain conformational rearrangement in the core during the MD simulations. While quantifying the restricted sliding is challenging, structural analysis of MD trajectories at various time points revealed that side chain repacking within the hydrophobic core was restricted in the (G16C/L38C)oo simulations compared to the (G16C/L38C)rr simulations. In (G16C/L38C)rr simulations, the hydrophobic core side chains sampled a variety of conformations, exchanging van der Waals contacts with neighboring residues. However in the (G16C/L38C)oo simulations, most of the hydrophobic core interactions were maintained. These results suggest that the restricted hydrophobic core dynamics observed in (G16C/L38C)oo simulations is a direct consequence of disulfide cross-linking and likely impair catalysis.
2.5 Conclusions
In this study site-directed cysteine engineering was used to reversibly restrict the internal dynamics in protease and evaluate the effects on function. The double-cysteine mutant (G16C/L38C) was selected as a pivotal pair of residues that varied significantly in separation distance depending on the conformational state of the protein. Thus cross linking them would severely restrict the internal core dynamics. The double-cysteine mutant (R14C/E65C) was used as internal controls, as these residues, though on separate loops, were not seen to change their separation distance significantly. Reduction-oxidation of the disulfide bridges was used as a switch to regulate hydrophobic core dynamics and eventually protease function (Figure. 2). The formation of disulfide (G16C/L38C) cross-links in fact exerted conformational restraints on the hydrophobic core, compromising protease activity. This mechanism of core rearrangement explains the results in the observed inactivation of (G16C/L38C)oo that was largely restored upon reversing the disulfide bonds to (G16C/L38C)rr (Table 1). Activity was lost and regained only in the (G16C/L38C) variant, where hydrophobic core movements were expected to be crucial for protease function. In the control case of (R14C/E65C) variant crosslinking did not alter enzyme activity. These results strongly support the mechanistic role of hydrophobic core rearrangement in protease function.
3. DISCUSSION
HIV-1 protease undergoes large conformational changes to bind ligands. We show that conformational changes are essential to protease activity. Flap motion and hydrophobic core repacking have been previously implicated in modulating pro-tease activity. However, the inherent challenges of manipulating protein dynamics, make testing these motions experimentally difficult. Our results provide strong support that hydro-phobic core flexibility contributes to the efficiency of substrate cleavage by HIV-1 protease.
When HIV protease processes substrates, the enzyme undergoes large conformational changes upon substrate recognition, cleavage and product release. Competitive active site inhibitors lock down these conformational changes by tightly binding to the active site of the enzyme, mimicking a transition state. Drug resistance occurs when the balance between sub-strate recognition and turnover and inhibitor binding is perturbed. In the case of the active site, resistance-conferring mutations occur in such a manner that directly preserves substrate recognition and turnover while compromising inhibitor binding29. However many mutations associated with resistance occur outside the active site within the protease hydro-phobic core3,5,12,30 (Figure 1). Mutations that modulate the dynamics of the hydrophobic core of HIV-1 could modulate the functional properties of the active site, and thus the relative binding affinities for substrates and inhibitors. We hypothesize that distal mutations outside the active site cause drug resistance when the balance between substrate recognition and turnover, a dynamic process, and inhibitor binding, a locking down of the enzyme, is altered in a manner to favor substrate turnover by modulating the flexibility of the core of HIV-1 protease.
This study demonstrates the essential role of the hydrophobic core in modulating the activity of HIV-1 protease. The use of specific cross-links to probe the impact of dynamics in pro-tease activity provides a new tool to investigate this enzyme. Such engineered cross-links may be useful to probe our hypothesis of the mechanism of action of mutations outside the active site modulating drug resistance in HIV-1 protease.
Supplementary Material
ACKNOWLEDGMENT
We thank Dr. William E Royer for assistance with initial data processing and refinement; Dr. Jill A Zitzewitz and Dr. Sagar V Kathuria for assistance with circular dichroism experiments, Dr. William R Kobertz and Dr. Keith P Romano for helpful scientific discussions; Ms. Claire Baldwin and Dr. Nese Kurt Yilmaz for editorial assistance; Dr. Scott Shaffer, Karin M Green and Stephanie Maniatis at the “UMMS Proteomics and Mass Spec-trometry Facility’ for collecting and analyzing mass spec data. We also thank the beamline staff at BioCARS Sector 14 at Argonne National Laboratory for their help during data collection. Argonne is operated by the University of Chicago Argonne, LLC, for the U.S. Department of Energy, Office of Biological and Environmental Research, under contract DE-AC02-06CH11357. Use of the BioCARS Sector 14 was supported by the National Institutes of Health, National Center for Research Resources, under grant No. RR007707
Funding Sources This work was supported by grants from the National Institutes of Health P01 GM66524.
ABBREVIATIONS
- HIV-1
Human Immunodeficiency virus type-1
- TCEP
tris 2-carboxyethyl phosphine
- WT
wild type
- RMSD
root mean square deviation
- MD
molecular dynamics
- DRV
darunavir
Footnotes
Author Contributions The manuscript was written through contributions of all authors. All authors have given approval to the final version of the manuscript.
SUPPORTING INFORMATION AVAILABLE Supporting Information. Further experimental details, protein expression, crystallographic statistics and anlaysis, enzyme kinet-ics and detection of disulfide bonds are available in the supporting information. This information is available free of charge via the Internet at http://pubs.acs.org/.
REFERENCES
- (1).Kohl NE, Emini EA, Schleif WA, Davis LJ, Heimbach JC, Dixon RA, Scolnick EM, Sigal IS. Proc Natl Acad Sci U S A. 1988;85:4686–4690. doi: 10.1073/pnas.85.13.4686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (2).Kramer RA, Schaber MD, Skalka AM, Ganguly K, Wong-Staal F, Reddy EP. Science. 1986;231:1580–4. doi: 10.1126/science.2420008. [DOI] [PubMed] [Google Scholar]
- (3).Muzammil S, Ross P, Freire E. Biochemistry. 2003;42:631–8. doi: 10.1021/bi027019u. [DOI] [PubMed] [Google Scholar]
- (4).Ohtaka H, Schon A, Freire E. Biochemistry. 2003;42:13659–66. doi: 10.1021/bi0350405. [DOI] [PubMed] [Google Scholar]
- (5).Clemente JC, Moose RE, Hemrajani R, Whitford LR, Govindasamy L, Reutzel R, McKenna R, Agbandje-McKenna M, Goodenow MM, Dunn BM. Biochemistry. 2004;43:12141–12151. doi: 10.1021/bi049459m. [DOI] [PubMed] [Google Scholar]
- (6).Ishima R, Freedberg DI, Wang YX, Louis JM, Torchia DA. Structure. 1999;7:1047–55. doi: 10.1016/s0969-2126(99)80172-5. [DOI] [PubMed] [Google Scholar]
- (7).Freedberg DI, Ishima R, Jacob J, Wang YX, Kustanovich I, Louis JM, Torchia DA. Protein Sci. 2002;11:221–32. doi: 10.1110/ps.33202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (8).Ishima R, Louis JM. Proteins. 2008;70:1408–15. doi: 10.1002/prot.21632. [DOI] [PubMed] [Google Scholar]
- (9).Perryman AL, Lin JH, McCammon JA. Protein Sci. 2004;13:1108–23. doi: 10.1110/ps.03468904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (10).Perryman AL, Lin JH, McCammon JA. Biopolymers. 2006;82:272–84. doi: 10.1002/bip.20497. [DOI] [PubMed] [Google Scholar]
- (11).Galiano L, Bonora M, Fanucci GE. J Am Chem Soc. 2007;129:11004–5. doi: 10.1021/ja073684k. [DOI] [PubMed] [Google Scholar]
- (12).Foulkes-Murzycki JE, Scott WR, Schiffer CA. Structure. 2007;15:225–33. doi: 10.1016/j.str.2007.01.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (13).Seeger MA, von Ballmoos C, Eicher T, Brandstatter L, Verrey F, Diederichs K, Pos KM. Nat Struct Mol Biol. 2008;15:199–205. doi: 10.1038/nsmb.1379. [DOI] [PubMed] [Google Scholar]
- (14).Witkowski WA, Hardy JA. Protein Sci. 2009;18:1459–68. doi: 10.1002/pro.151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (15).Busenlehner LS, Armstrong RN. Arch Biochem Biophys. 2005;433:34–46. doi: 10.1016/j.abb.2004.09.002. [DOI] [PubMed] [Google Scholar]
- (16).Palmer AG. 3rd Annual Review of Biophysics & Bio-molecular Structure. 2001;30:129–55. doi: 10.1146/annurev.biophys.30.1.129. [DOI] [PubMed] [Google Scholar]
- (17).Parak FG. Curr Opin Struct Biol. 2003;13:552–7. doi: 10.1016/j.sbi.2003.09.004. [DOI] [PubMed] [Google Scholar]
- (18).Blackburn ME, Veloro AM, Fanucci GE. Biochemistry. 2009;48:8765–7. doi: 10.1021/bi901201q. [DOI] [PubMed] [Google Scholar]
- (19).Schmidt B, Ho L, Hogg PJ. Biochemistry. 2006;45:7429–33. doi: 10.1021/bi0603064. [DOI] [PubMed] [Google Scholar]
- (20).Brandes N, Schmitt S, Jakob U. Antioxid Redox Signal. 2009;11:997–1014. doi: 10.1089/ars.2008.2285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (21).Thornton JM. J Mol Biol. 1981;151:261–87. doi: 10.1016/0022-2836(81)90515-5. [DOI] [PubMed] [Google Scholar]
- (22).Spadaro D, Yun BW, Spoel SH, Chu C, Wang YQ, Loake GJ. Physiol Plant. 2010;138:360–71. doi: 10.1111/j.1399-3054.2009.01307.x. [DOI] [PubMed] [Google Scholar]
- (23).Lazear E, Carfi A, Whitbeck JC, Cairns TM, Krummenacher C, Cohen GH, Eisenberg RJ. Journal of Virology. 2008;82:700–9. doi: 10.1128/JVI.02192-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (24).Bass RB, Butler SL, Chervitz SA, Gloor SL, Falke JJ. Methods Enzymol. 2007;423:25–51. doi: 10.1016/S0076-6879(07)23002-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (25).Rose RB, Craik CS, Stroud RM. Biochemistry. 1998;37:2607–21. doi: 10.1021/bi9716074. [DOI] [PubMed] [Google Scholar]
- (26).Galiano L, Ding F, Veloro AM, Blackburn ME, Simmerling C, Fanucci GE. J Am Chem Soc. 2009;131:430–1. doi: 10.1021/ja807531v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (27).Hazes B, Dijkstra BW. Protein Eng. 1988;2:119–25. doi: 10.1093/protein/2.2.119. [DOI] [PubMed] [Google Scholar]
- (28).Surleraux DL, Tahri A, Verschueren WG, Pille GM, de Kock HA, Jonckers TH, Peeters A, De Meyer S, Azijn H, Pauwels R, de Bethune MP, King NM, Prabu-Jeyabalan M, Schiffer CA, Wigerinck PB. J. Med. Chem. 2005;48:1813–1822. doi: 10.1021/jm049560p. [DOI] [PubMed] [Google Scholar]
- (29).Prabu-Jeyabalan M, Nalivaika EA, Schiffer CA. Structure. 2002;10:369–381. doi: 10.1016/s0969-2126(02)00720-7. [DOI] [PubMed] [Google Scholar]
- (30).Rhee SY, Taylor J, Fessel WJ, Kaufman D, Towner W, Troia P, Ruane P, Hellinger J, Shirvani V, Zolopa A, Shafer RW. Antimicrob. Agents Chemother. 2010;54:4253–61. doi: 10.1128/AAC.00574-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.