

Abstract
The current construct of schizophrenia as a unitary disease is far from satisfactory, and is in need of reconceptualization. The first five papers in our “facts” series reviewed what is known about schizophrenia to date, and a limited number of key facts appear to stand out. Schizophrenia is characterized by persistent cognitive deficits, positive and negative symptoms typically beginning in youth, substantive heritability, and brain structural, functional and neurochemical alterations including dopaminergic dysregulation. Several pathophysiological models have been proposed with differing interpretations of the illness, like the fabled six blind Indian men groping different parts of an elephant coming up with different conclusions. However, accumulating knowledge is integrating the several extant models of schizophrenia etiopathogenesis into unifying constructs; we discuss an example, involving a neurodevelopmental imbalance in excitatory/inhibitory neural systems leading to impaired neural plasticity. This imbalance, which may be proximal to clinical manifestations, could result from a variety of genetic, epigenetic and environmental causes, as well as pathophysiological processes such as inflammation and oxidative stress. Such efforts to “connect the dots” (and visualizing the elephant) are still limited by the substantial clinical, pathological, and etiological heterogeneity of schizophrenia and its blurred boundaries with several other psychiatric disorders leading to a “fuzzy cluster” of overlapping syndromes, thereby reducing the content, discriminant and predictive validity of a unitary construct of this illness. The way ahead involves several key directions: a) choosing valid phenotype definitions increasingly derived from translational neuroscience; b) addressing clinical heterogeneity by a cross-diagnostic dimensional and a staging approach to psychopathology; c) addressing pathophysiological heterogeneity by elucidating independent families of “extended” intermediate phenotypes and pathophysiological processes (e.g. altered excitatory/inhibitory, salience or executive circuitries, oxidative stress systems) that traverse structural, functional, neurochemical and molecular domains; d) resolving etiologic heterogeneity by mapping genomic and environmental factors and their interactions to syndromal and specific pathophysiological signatures; e) separating causal factors from consequences and compensatory phenomena; and f) formulating or reformulating hypotheses that can be refuted/tested, perhaps in the mouse or other experimental models. These steps will likely lead to the current entity of schizophrenia being usefully deconstructed and reconfigured into phenotypically overlapping, but etiopathologically unique and empirically testable component entities (similar to mental retardation, epilepsy or cancer syndromes). The mouse may be the way to rescue the trapped elephant!
Keywords: schizophrenia, models, heterogeneity, etiology, pathophysiology, phenotype, treatment, biology
Introduction
The last several decades have witnessed a steady accrual of a substantive body of knowledge in schizophrenia. However, the concept of schizophrenia as a disease entity, which has survived for over a century, is still mired in controversy and remains unsatisfactory to researchers and clinicians. The central roadblock to progress in this field is the lack of a cohesive, integrated model that incorporates the known facts of the illness.
Progress in a body of scientific knowledge requires a set of established facts based on which models are built that generate falsifiable hypotheses, which in turn refine the model and expand knowledge (Platt, 1964). Building upon a seminal paper two decades earlier (Wyatt et al., 1988, Tandon, 1999), the authors compiled an extensive set of “facts” for the purpose of evaluating conceptual models or theoretical constructs developed to understand the nature of schizophrenia. Major findings in the extant literature were identified and evaluated in terms of their relevance to schizophrenia, reproducibility, and durability over time (Tandon et al., 2008c). In the subsequent four papers of this series [parts II–V], the etiological, pathophysiological, phenotypic and treatment related facts of schizophrenia were reviewed in detail (Tandon et al., 2008b, Keshavan et al., 2008, Tandon et al., 2009, Tandon et al., 2010). In this article that concludes the series, we weave together key strands from the afore-mentioned systematic reviews, propose an integrative model of what we know and outline the critical next steps to refine our understanding of this illness.
The core “facts” of schizophrenia
A traditional approach to defining a disease is to extract the established individual facts of the illness and, at the end of an inductive process, identify the necessary and sufficient conditions which make that disease unique. The first five papers in this series (see Table 1 in Tandon) (Tandon et al., 2008c) compiled 77 facts of schizophrenia with this goal in mind. Somewhat overlapping sets of facts were outlined by another similar recent review (MacDonald and Schulz, 2009). These facts illustrate what we currently know about this illness: Etiology of schizophrenia definitely involves genetic factors but the precise genes and gene environmental interactions are yet to be clarified. Pathology of schizophrenia involves widespread but variable brain structural, functional and neurochemical alterations that appear to involve dopamine dysregulation. Phenotype of schizophrenia is characterized by persistent cognitive deficits and negative symptoms episodically punctuated by the hallucinations and delusions of psychosis, typically beginning in youth. Treatment does not address etiology, and involves a combination of dopamine D2 receptor blocking agents and psychotherapies tailored to the individual patient’s specific psychosocial deficits.
Table 1 further distills these observations to a core set of eight facts. The first seven “facts” stand out as a) highly replicable, with many independent studies, consistent replication, and no contradictory findings (reproducibility), b) highly durable, having been known for over 30 years, and being listed in both 1988 (Wyatt et al., 1988)and 1999 (Tandon, 1999)(durability), and c) rated as being not explainable by some other confounding variable and likely (though not definitely) related to schizophrenia (primacy). The eighth fact met the first two, but not the last, criteria. These eight facts in essence summarize what we know about the etiology, pathology, clinical features, and treatment response in this illness.
Table 1.
The “facts” of schizophrenia that are rated as having high reproducibility, durability and primacy to the illness. For details of the list of 77 facts of schizophrenia see the first paper in this series (Tandon et al., 2008c).
|
It is clear from Table 1 that while certain key findings in schizophrenia represent replicability, durability, and primacy to schizophrenia as we currently know it, none of them can be considered to be a necessary and sufficient feature of this illness. Similar symptoms, brain structural alterations, onset and course characteristics, genetic liability, and response to dopamine-blocking antipsychotics are seen across a variety of neuropsychiatric states. This sobering realization brings up the key question: just what is schizophrenia and what may be the best model(s) that can define the essence of this disease given the limitations of our current knowledge?
Extant models
Models of a disease are working hypotheses about its nature around which data about the disease are organized; thus, they have both an explanatory and predictive function. There may be few other diseases in medicine with a larger number of conceptual models proposed than in schizophrenia. These models are designed to explain some aspect/s of the body of etiological, pathological, clinical, and treatment facts of schizophrenia. Some of these models are modular: i.e., they try to explain relationships between different facts within a single domain. Other models are broader and hypothesize relationships between facts across multiple domains. These broader models can be grouped into etio-pathogenetic and pathophysiologic models, with the former models designed to explain the relationships between etiological and pathological facts and the latter models designed to explain relationships between pathological facts and clinical + treatment facts.
In this paper, we briefly list the representative models and attempt to evaluate a) their value in explaining what we know about the etiology, pathology, clinical features and treatment of schizophrenia as outlined earlier and b) their ability to generate testable predictions, including the availability of animal models (Table 2). Early and primarily psychodynamic theories of schizophrenia such as the schizophrenogenic mother and/or covert parental discord (Lidz, 1985), double bind communication (Bateson, 1956)and repressed homosexuality (Freud, 1911) have not survived as viable models, and will not be discussed here. Extant models may be organized by the primary domain of inquiry from which they were derived, i.e. clinical expression, pathophysiology, and etiology. This list is not exhaustive, but seeks to outline the widely prevalent models. Detailed descriptions of the models are beyond the scope of this paper, and the reader is referred to the original articles as well as previous papers in this series (Tandon et al., 2008c, Tandon et al., 2008b, Keshavan et al., 2008, Tandon et al., 2009, Tandon et al., 2010). We outline both what contributions these models have made toward our understanding of schizophrenia as also what they do not explain.
Table 2.
Extant Models of schizophrenia. This list is not meant to be exhaustive, but illustrates examples of the main lines of thinking about schizophrenia on the domains of clinical disease expression, pathophysiology, and etiology. The extent to which each of these models explains the known etiologic, pathophysiologic, and/or clinical facts of the entity of schizophrenia are rated in successive columns, and discussed further in the text.
| Model | Main refs | Etiology | Patho-physiology | Clinical features | Treatment | Testability (animal models) |
|---|---|---|---|---|---|---|
| Models of disease expression | ||||||
| Self-monitoring dysfunction | (Frith and Done, 1988, Feinberg, 1978) | NC | + | + | NC | NA |
| Deficit syndrome | (Carpenter et al., 1988) | NC | + | + | ± | NA |
| Impaired Information processing | (Hemsley and Zawada, 1976) | NC | + | + | NC | |
| Aberrant salience | (Kapur, 2003) (Howes and Kapur, 2009) | NC | + | + | + | A |
| Language development and speciation | (Crow, 1995) | ± | + | + | NC | NA |
| Models of pathophysiology | ||||||
| Heteromodal association cortex | (Ross and Pearlson, 1996) | NC | + | + | NC | NA |
| Cortico-cerebellar- thalamic-cortical circuit [CCTCC]). | (Andreasen, 1999) | NC | + | + | NC | NA |
| Aberrant default state networks | (Williamson, 2007) | NC | + | + | NC | NA |
| Disturbances of Inhibitory Neural Circuits and Gamma Oscillations | (Kwon et al., 1999) | NC | + | + | NC | A |
| Dopamine imbalance hypothesis | (Weinberger, 1987); (Grace, 1991) | ± | + | ++ | + | A |
| Amphetamine sensitization | (Lieberman et al., 1990) | NC | + | + | + | A |
| Altered Glutamatergic neurotransmission | (Kim et al., 1980); (Javitt and Zukin, 1991); (Olney and Farber, 1995); (Coyle, 2006) | ± | + | + | + | A |
| Altered GABA neurotransmission | (Lewis and Hashimoto, 2007); (Benes and Berretta, 2001) | ± | + | + | − | A |
| Altered membrane integrity | (Hitzemann et al., 1984, Horrobin et al., 1994) | ± | + | + | + | NA |
| Increased oxidative | (Reddy and stress Yao, 1996) | + | + | + | + | NA |
| Inflammatory response | (Smith, 1991) | + | + | + | + | A |
| Models of pathogenesis | ||||||
| “early” developmental models | (Murray, 2002); (Weinberger, 1987); (Akbarian et al., 1993) | + | + | + | NC | A |
| “Late” developmental models | (Feinberg, 1982); (Hoffman and McGlashan, 1994); (Keshavan et al., 1994, DeLisi, 2008) | ± | + | + | NC | NA |
| Progressive/developmental models (“2-hit” or “3-hit” models | (DeLisi, 1997); (Weinberger, 1987); (Woods et al., 2009) | ± | + | + | NC | NA |
| Models of etiology | ||||||
| Genetic models | (Gottesman and Gould, 2003) | ++ | + | + | NC | A |
| Environmental models: early (intra/perinatal infections, nutrition) and later environmental factors (cannabis exposure, psychosocial stress) | (van Os et al., 2005) | ++ | + | + | NC | A |
| Epigenetic and gene-environmental interaction models | (Petronis et al., 1999); (Costa et al., 2001) | ++ | + | + | NC | A |
NC= not considered in this theory; ++ = most of the facts in this domain are explained by the theory; + some but not all facts are explained or are consistent with this theory; ±= some data but not conclusive; − and no evidence at this time for treatment relevance despite available studies. A= animal models available (briefly mentioned in text); NA= not available.
Models of phenotypic expression
Several models have been derived from analyses of specific aspects of clinical expression of schizophrenia. The absence of a clear phenotypic distinction across diverse psychotic states such as schizophrenia, psychotic affective disorder and schizophrenia has led to one view that these disorders might represent a continuum (Crow, 1986). Alterations in attention and information processing in schizophrenia have been thought to result from a failure of filter mechanisms leading to schizophrenia related psychopathology (Hemsley and Zawada, 1976). Frith and Done (Frith and Done, 1988) originally proposed that symptoms of a lien control (i.e. first rank symptoms) in schizophrenia might result from problems with the central monitoring of responses. Crow (1998, 2008) has proposed that nuclear symptoms of schizophrenia may be related to a disintegration of language processes that is uniquely human and emerges in the context of speciation. Stephan and co-workers (Stephan et al., 2009) recently attempted an integration of this model with other aspects of schizophrenia such as disordered neural connectivity and abnormal synaptic plasticity. Carpenter and colleagues (Carpenter et al., 1988) proposed that enduring negative symptoms, or the deficit symptoms may be useful as a way toward resolving clinical heterogeneity in schizophrenia. Kapur (Kapur, 2003) proposed that psychosis may result from an aberrant assignment of salience to the elements of one’s experience as a result of dopaminergic dysregulation. However, while these models can elegantly explain some clinical aspects of schizophrenia and certain aspects of neurobiology, other central elements of the disorder are ignored. None of these models purports to explain the broad body of the “facts of schizophrenia”.
Pathophysiological models
Several conceptual models have stemmed from the observed pathophysiological alterations in schizophrenia, and have focused on one or other neurophysiological or neuronal network. One view is that abnormal transcallosal inter-hemispheric integration may underlie some aspects of schizophrenia such as delusions of alien control (Nasrallah, 1985). Observations of alterations in synchronous oscillations in the gamma range (30- 80 Hz) have led to the theory that psychopathology in schizophrenia may result from dysfunction of inhibitory circuits (Kwon et al., 1999). Feinberg (Feinberg, 1978) and Frith and Done (Frith and Done, 1988) proposed that defective corollary discharges may underlie failures of self-monitoring of thought processes leading to symptoms of psychosis. Another, more recent view is that some aspects of schizophrenia and its liability might stem from aberrant activity of default state network networks, which are normally anti-correlated with task-oriented networks (Williamson, 2007). Such connectivity changes may be related to white matter pathology (Konrad and Winterer, 2008), as evidenced by diffusion tensor imaging studies (Ardekani et al., 2010), and post-mortem findings of reduced oligodendrocytes which synthesize myelin (Segal et al., 2007). It should be noted that a white matter disease such as metachromatic leukodystrophy can mimic schizophrenia with its psychosis phenotype (Hyde et al., 1992).
The widely held neurochemical hypothesis, the classic hyperdopaminergic model (Carlsson, 1977, Randrup and Munkvad, 1967, Snyder, 1976) has in recent years given way to more complex and integrative views such as the prefrontal-limbic dopamine (DA) imbalance model (Weinberger, 1987), and the phasic-tonic DA imbalance model (Grace, 1991). Dopaminergic models are strongly supported by the fact that all currently available antipsychotics work by blocking dopaminergic receptors, and also by clinical observations of psychotic symptoms in individuals following repeated amphetamine use. DA blockade may be a necessary and sufficient explanation for antipsychotic effects (Kapur and Mamo, 2003), and by inference, to the positive symptom dimension of schizophrenia. DA imbalance models provide an integrative view of cognitive and negative symptoms with psychotic episodes, though not other aspects of the illness such as structural alterations and functional decline. Amphetamine-induced sensitized state has been used as an animal model for some domains such as psychotic symptoms, but not other domains such as negative symptoms (Featherstone et al., 2007). DA related genes do not appear to explain the substantive heritability of schizophrenia, and dopaminergic treatments have little effect of cognitive and negative symptoms. It is quite likely that DA alterations are involved downstream in the final common pathways that lead to psychosis (Seeman, 2010), though the primary causal events remain elusive.
Alteration in the excitatory neurotransmitter glutamate system, especially involving N-methyl D-aspartate (NMDA) receptor function (Olney and Farber, 1995, Javitt and Zukin, 1991, Kim et al., 1980) has emerged as a promising pathophysiological model. This model stemmed from early observations that phencyclidine (PCP) causes symptoms closely resembling schizophrenia (Lodge and Anis, 1982). Glutamatergic constructs such as the NMDA receptor hypofunction model have considerable explanatory power, and can integrate the early and late developmental deviations, the role of dopamine, as well as the post-illness onset deteriorative processes that occur in schizophrenia (Keshavan, 1999, Coyle, 2006). Many genes implicated in schizophrenia appear to converge on the glutamatergic system (Carter, 2006). However, this model does not clarify why, despite the ubiquity of glutamatergic neurons in the brain, schizophrenia is not associated with more generalized neurological impairment (Crow, 1995). Further, definitive evidence of alterations of brain glutamatergic neurotransmission in schizophrenia is still awaited. Medications that modulate the glycine site of the NMDA receptors have been reported to improve cognitive deficits in schizophrenia. However, interventions that therapeutically modify this system, while promising in preclinical trials, still await confirmation in double-blind, randomized controlled trials (Buchanan et al., 2007, Tandon et al., 2010).
Several lines of evidence indicate that gamma amino acid butyric acid (GABA) neurotransmission, the main inhibitory neurochemical system, is dysfunctional in schizophrenia (Lewis and Hashimoto, 2007, Benes and Berretta, 2001, Van Kammen, 1977). Post-mortem studies have consistently reported reductions in GABA neurons and/or key enzymes related to this pathway. It has been suggested that observations of altered neural synchrony and cognitive deficits in schizophrenia may be related to altered GABA neurotransmission. The genetic mouse model of reelin haploinsufficiency is associated with dendritic loss, presynaptic GABAergic as well as executive cognitive defects seen in schizophrenia (Costa et al., 2001, Brigman et al., 2006) The therapeutic value of GABAergic agents in schizophrenia remains to be established, and few GABA related genes have been definitively identified underlying the liability to schizophrenia. Other neurotransmitter systems such as the cholinergic system (Tandon and Greden, 1989) and serotonin pathway (Kapur and Remington, 1996) have been posited to account for the psychopathology of schizophrenia, but again, are yet to lead to specific therapeutic interventions.
Alternative neurochemical hypotheses have been proposed as well. The membrane hypothesis posits the role of membrane alterations in peripheral cells (Hitzemann et al., 1984) as well as the brain (Reddy and Keshavan, 2003). This model is appealing since membranes are critical for several aspects of neuronal function such as receptors, channels and signal transduction. The inflammation hypothesis of schizophrenia is based on observations that schizophrenia may be associated with an increased serum concentration of several pro-inflammatory cytokines that may reflect response by microglia to various pathological processes in the brain (Muller and Schwarz, 2008). Excessive free radicals from activated microglia may cause a decrease in neurogenesis as well as white matter abnormalities in the brains of patients with schizophrenia (Monji et al., 2009). The pathophysiology of schizophrenia has also been thought be related to increased oxidative stress (accumulation of reactive oxygen species that may damage neuronal function) in this illness, supported by observations of decreased antioxidant and increased pro-oxidant processes (Bitanihirwe and Woo, 2010, Yao et al., 2010).
In summary, several disparate pathophysiological models have been proposed for schizophrenia and may appear to reflect rather disparate views of this complex disease entity. However, the field is increasingly seeing efforts to “connect the dots”. For example, growing evidence suggests that the integrity of cortical and hippocampal NMDA receptors may be critical for maintaining normal GABAergic function. Belforte et al (2010) showed that a restricted deletion of NMDA receptors in corticolimbic interneurons in early postnatal but not post-adolescent mice induced features resembling human schizophrenia such as novelty-induced hyperlocomotion, deficits in pre-pulse inhibition, mating, and anhedonia-like and anxiety-like behaviors. Of note, reduced expression of glutamic acid decarboxylase and parvalbumin, disinhibition of cortical excitatory neurons, and reduced neuronal synchrony were also seen. NMDA receptor hypofunction early in development may therefore lead to reduced GABAergic inhibition of excitatory neuronal activity predisposing to psychotic symptoms later in development, i.e. adolescence. The glutamatergic and GABAergic alterations could lead to a functional imbalance between excitatory and inhibitory neural systems which may well be a fundamental deficit in at least a broad subgroup of schizophrenia; dopaminergic alterations could be secondary to this imbalance. NMDA receptor hypofunction itself may result from Kynurenic acid, an endogenous NMDA receptor antagonist, resulting from disturbed tryptophan/kynurenine metabolism which in turn might be secondary to inflammatory processes (Muller 2008).Clarifying the relationships between these alterations in excitatory and inhibitory neural systems may help develop a more integrative neurochemical model (Keshavan, 1999, Stone et al., 2007).
Models of pathogenesis
Disruptions in the normal maturation of brain structures have been widely held to underlie schizophrenia. The “early” developmental models suggest that disruptions in intrauterine or early postnatal life, i.e. neuronal proliferation, migration, differentiation and elimination, or neurogenesis processes, may lead to impaired neural infrastructure and abnormal brain maturation predisposing to premorbid dysfunction as well as psychopathology emerging later in adolescence or early adulthood (Murray, 2002, Weinberger, 1987, Akbarian et al., 1993). By contrast, the “late” developmental models, which account for the typical onset of schizophrenia during adolescence, propose deviations in later emerging processes such as synaptic/axonal pruning or neuronal apoptosis and/or myelination processes (Feinberg, 1982, Keshavan et al., 1994, Hoffman et al., 2007). Other developmental models argue for an interaction of early and late developmental processes leading to progressive pathology (Woods et al., 2009), or a cascade of pathogenetic events (Keshavan, 1999). Several models seek to explain why schizophrenic illness typically manifests in adolescence or early adulthood (Keshavan, 1999, Feinberg and Campbell, 2010) and how stress marked by hypothalamo-pituitary axis dysfunction (Tandon and Greden, 1991, McCormick et al., 2010, Tandon et al., 1991) contribute to its evolution. Models of neurodegeneration (or neuroprogression for lack of a better word) or negative neuroplasticity propose continuing atrophic processes even after illness onset (DeLisi, 1997, Garver et al., 1997). A recent view is that schizophrenia may be associated with acceleration of aging processes (Kirkpatrick, 2009). The hypothesized increase in corticolimbic glutamatergic activity (due to reduced inhibition by GABAergic interneurons), if it persists over time, may lead to excitotoxic neuronal loss and thereby account for progressive neuropathology and functional decline seen in at least some patients (Keshavan, 1999). These processes occur during adolescence and early adulthood, a critical phase of development characterized by pruning of excitatory synapses, proliferation of inhibitory synapses, increasing myelination (Paus et al., 2008) and dopaminergic innervation of the prefrontal cortex (Rosenberg and Lewis, 1995). These “late” developmental processes, designed to optimize the excitatory, inhibitory balance in the cortical and subcortical regions, may be impaired in those at risk for schizophrenia, leading to illness onset during adolescence of early adulthood.
Etiological models
It has been known for long that schizophrenia is highly heritable. The most promising etiological models therefore include the view that schizophrenia may be polygenic/multifactorial (Gottesman and Shields, 1967). Genetic models of schizophrenia have to account for its substantive heritability and a far greater degree of genetic heterogeneity than previously realized. Genetic models also have to account for the persistence and stable incidence of the disorder despite the disadvantages conferred by the genes, a central evolutionary paradox pertaining to this illness (Crow, 2000). Though recent linkage and genome-wide association studies have identified a large number of candidate genes and specific risk alleles for schizophrenia, replicated findings explain only a very small fraction of the heritability. The view that schizophrenia may be caused by multiple common genes each conferring a small effect has been supported by genome-wide association studies (GWAS). International consortia involving GWAS studies have generated evidence for a number of common polymorphisms showing association with schizophrenia (such as the major histocompatibility complex region at 6p22–p21; neuregulin on chromosome 8, transcription factor 4 on chromosome 18q21.2; and 2q32.1) (Stefansson et al., 2008, International Schizophrenia Consortium, 2008). These alleles present relatively modest odds ratios (the odds of a risk variant being present in cases vs. controls) and suggest causal roles of gene regulatory mechanisms in Schizophrenia. Considerable overlap is seen with autism and with bipolar disorder challenging conventional disease classifications. Much of heritability, however, remains unexplained (Duan et al., 2010).
In recent years, observations have emerged of rare and unique mutations and copy number variations (CNV) conferring possibly severe risks seen in a significant proportion of patients, though no single locus explains more than approximately 1% of cases. Genes involved in cell signaling, brain development and glutamate appear to be differentially affected, and seem to cut across diagnostic boundaries, being seen in other developmental disorders such as autism (Walsh et al., 2008, Stefansson et al., 2008, McClellan and King, 2010, International Schizophrenia Consortium, 2008). However, CNVs have been criticized on methodological grounds (Craddock et al., 2010); most variants are not specific to schizophrenia, and some (e.g., the DISC translocation) occur only in a single family. Finally, the relationships between genes (e.g. DISC1, neuregulin) and their neuropathological consequences (such as dendritic morphology) are not straightforward, and are likely to be influenced by profound gene-gene and gene-protein interactions, leading to incredibly complex “interactomes” (Camargo et al., 2007, Hayashi-Takagi et al., 2010, Jaaro-Peled et al., 2010).
Any etiological model of schizophrenia needs to integrate genetic risk with environmental factors associated with the disorder (van Winkel et al., 2010, Tandon et al., 2008b). Several well-known environmental factors such as prenatal infection, maternal malnutrition, obstetric trauma, trauma, urbanicity and cannabis exposure, either individually or collectively, can account for a variable proportion of the etiology of schizophrenia (Tandon et al., 2008b). Some of these environmental factors have relatively large odds ratios, ranging from 2.4 (urbanicity) to immigrant status (4.4) (Boydell, 2004). Differential sensitivity to environment may be determined by genetic susceptibility; translational studies, using novel paradigms examining stress sensitivity, are needed to identify underlying mechanisms and point the way to possible interventions (Van Os, 2010). Developmental risk to schizophrenia may stem from either direct influence of these environmental factors or via indirect effects (e.g. maternal inflammatory responses such as cytokines) to intrauterine viral exposure (Ellman et al., 2010).
Gene-environmental models suggest that interactive effects of genes and environment on biological pathways may have larger effects than either genes or environment (Meyer and Feldon, 2010). A large fraction of the heritability of schizophrenia may be accounted for by gene-environment interaction (van Os et al., 2008). An integrative approach is the “two-hit hypothesis” (Bayer et al., 1999, Huttunen et al., 1994, Maynard et al., 2001) which proposes that genetic risk and early developmental alterations (first hit) may prime the person to react to a second hit in the form of an environmental factor later during development leading to the illness. Treating genetically altered mice with putative mutations with environmental perturbations can help model such integrative models for this complex mental illness (Robertson et al., 2006). Even beyond elucidating models of disease pathogenesis, animal models are critical to identifying treatment targets (Javitt et al., 2008).
An epigenetic model, which is increasingly gaining recognition, posits that environmental factors impact on gene expression via changes in DNA methylation and chromatin structure which, in turn, may play a role in the etiology of schizophrenia (Petronis et al., 1999). This research is in its infancy and few definitive findings have emerged to support this model in schizophrenia yet. However, since it is possible by pharmacological approaches to modify epigenetic processes (Gavin and Sharma, 2010), this model has a prominent potential to pave a path to therapeutic advances.
In summary, each of the listed models is derived from one or other well-known features of the schizophrenic illness, and therefore represents a valuable step towards explaining what we know about that aspect of the illness. However, none of the models, in and of itself, explains a substantive set of facts of schizophrenia, all have inconsistencies and discrepant facts, which they cannot account for, and not all are equally testable. Many models do not lend themselves to generate animal models, which are particularly useful in evaluating testable predictions (Feifel and Shilling, 2010). It is therefore instructive to review the potential roadblocks that have stymied efforts to develop a unitary model of schizophrenia.
Roadblocks to the unitary models of schizophrenia
Several constraints underlie the current state of impasse in our conceptualization of schizophrenia. While reliability of diagnoses has improved with the recurrent revisions of the Diagnostic and Statistical Manual of mental disorders (DSM), validity of the disorder remains in serious question. First, a key roadblock to discriminant validity of schizophrenia is the blurred boundary between schizophrenia and normalcy (van Os, 2003) as well as between schizophrenia and other major psychiatric disorders such as bipolar disorder, a challenge to the century-old Kraepelinian view that these are distinct illnesses (Craddock and Owen, 2007). Central to this debate is the concept of schizoaffective disorder as an entity that straddles both illnesses. Is this disorder a valid entity, circa 2010? Because psychiatric disorders generally do not respect the time-honored expectations (Sydenham, 1966) that symptom constellations (syndromes) would result from specific pathology which maps to specific etiology, Robins and Guze (Robins and Guze, 1970) proposed four tenets of a valid psychiatric diagnosis. These include the need for a distinct signature in phenomenology, course, family history, and biology. Schizoaffective disorder fails to meet these criteria, being characterized by having overlaps with schizophrenia and bipolar disorder in each of these domains (Heckers, 2009, Tandon and Maj, 2008). The interface between schizophrenia and the continuum of “health” is also fuzzy, leading to the intermediate syndromes of schizotypal and brief psychotic disorders. The predictive validity of schizophrenia as a construct is limited by the large variability in longitudinal course of this illness (Deister and Marneros, 1993, Harrison et al., 2001, Kendler et al., 1985), as is the response to different treatments (Tandon et al., 2008a). The content validity of the schizophrenia construct is also seriously limited by the substantive heterogeneity of schizophrenia in cross-sectional symptomatology, as well as the etiopathological factors implicated (Tsuang and Faraone, 1995). In the schizophrenia literature authors often invoke heterogeneity to “explain” inconsistent findings. As previously argued in our series (Keshavan et al., 2008), heterogeneity is the problem rather than a solution; heterogeneity may be central to revisiting the inadequate conceptualization of this disease entity.
Heterogeneity is not unique to schizophrenia. Small pox is an example of a phenomenological heterogeneity, being caused by a virus, leading to purulent pustules, resulting in diverse manifestations based on location (e.g. blindness and facial disfiguration). Sickle cell disease illustrates pathophysiological heterogeneity; this classic Mendelian disease is caused by a single point mutation leading to altered oxygen affinity of the hemoglobin molecule and in turn the deformed (sickle shaped) erythrocyte. Clinical manifestations range from mild anemia, to hemolytic crises and acute vascular events. The diverse phenotypes result from an interaction between the genetic alteration and other disease modifying genes as well as environmental factors such as dehydration and ambient oxygen concentrations (Kato et al., 2007). Etiological heterogeneity characterizes neurofibromatosis, which may present either as multiple tumors, i.e. neurofibromas and/or cafe au lait spots, and may be caused by multiple genes. Some syndromes such as heart failure are characterized by etiological heterogeneity (viral cardiomyopathy, coronary disease etc) and phenotypic heterogeneity (edema, breathlessness, liver enlargement), but with some readily discernible pathophysiological underpinnings. Cancer syndromes are another example of such complex diseases. Discovering cause in such syndromes, while complicated by the multiple etiological factors involved, is still facilitated by the fact that pathology is tractable and laboratory tests and biomarkers are available. Schizophrenia, however, presents an even more complex problem, since it is associated with phenotypic, pathophysiological, as well as etiological heterogeneity, and with no specific marker or pathology defined yet. A syndromal entity that comes close to schizophrenia is chronic fatigue syndrome (CFS), a syndrome diagnosed by the presence of a constellation of symptoms, a duration of at least six months, and requiring ruling out multiple possible medical or psychiatric causes that may explain these symptoms. As in schizophrenia, no specific laboratory tests are available, and CFS has been considered a heterogeneous entity. Originally a vague constellation of symptoms of weaknesses, aches and pains, CFS is being deconstructed into a number of syndromes with simple etiology (Lyme disease, EBV infection) as well as heterogeneous psychiatric conditions (somatoform disorders and depression) with outwardly similar manifestations. Schizophrenia is likely not much different.
Inconsistencies in observed relationships between genetic variation, phenotypic syndromes, and putative biomarkers pose another significant hurdle in the path of progress. An example is the lack of consistent relationships between symptoms and brain structural alterations (Andreasen, 1999). It has been suggested that distinct aspects of the negative syndrome such as the deficit syndrome may be related to specific neuroanatomical changes (Carpenter et al., 1993), but this has not been consistently borne out (Galderisi and Maj, 2009). Similarly, the relations between syndromal dimensions and biomarkers appear to cut across traditional diagnostic categories such as schizophrenia (Baumann and Bogerts, 1999). Neural circuits appear to correspond better to cognitive or behavioral functions (e.g. dorsal frontostriatal systems with attention, ventral prefrontal/amygdala with affect regulation, etc) but not as well with symptom constellations defined by diagnoses (Beardenet al., 2007). Clearly, the key to surpass the logjam of schizophrenia, a term famously articulated by Zubin and Steinhauer (Zubin and Steinhauer, 1981), lies in unraveling a) the blurred boundaries, b) heterogeneity within the entity, and c) the unclear syndromal and biological relationships. However, how do we embark on such a complex task?
Redefining schizophrenia: leveraging old facts into new models
Defining complex psychiatric disorders based on overt externally observable phenotypes (such as behaviors) is not optimal for determining etiology of these entities. As we stated earlier in this series (Tandon et al., 2008b, Keshavan et al., 2008), the logical next step is to deconstruct schizophrenia into its multiple component parts and reconfigure these components in a more meaningful and testable manner. What strategies can be employed towards this end? We propose ten key steps that can help move forward in this path.
1. Better phenotype definitions
Poor phenotype definition is one of the major obstacles for etiologic research in schizophrenia. Freely available informatics tools such as those developed by the Consortium for Neuropsychiatric Phenomics (CNP) offer a framework for defining and refining latent constructs used in cognitive neuroscience research (Sabb et al., 2009). Such approaches can help define phenotypes traversing multiple levels of expression from proteome to syndrome. Considerations for choosing optimum phenotypes for studies will include high heritability, relation to known functional genetic variation, reliability of measurement, validity to known endophenotypes (or better termed intermediate phenotypes), relevance to and clinical outcomes; translational relevance is also important, as evidenced by relationships between known neural systems and treatment targets, as well as homologies of expression across species enabling both basic and clinical investigation (Bilder et al., 2009). Rapid advances in transgenic modeling allow phenotypes to be studied across species; such phenotypes, derived by bottom-up evaluation, may offer the best traction.
2. Thinking in categories vs. dimensions (Tandon et al., 2009)
From the foregoing discussion it is clear that unequivocal, necessary, and sufficient characteristics of schizophrenia are not easily identifiable; so schizophrenia resembles more a constellation with blurred borders. Such constellations, elsewhere in medicine, have been called “fuzzy sets” (Vineis, 2008). Artificial intelligence-derived neural network approaches are now being use to address psychiatric diagnostic questions such as prediction of suicide attempts (Modai et al., 2004) and autism (Arthi and Tamilarasi, 2008).
This approach to thinking about diagnosis is not a fuzziness of logic but is instead a logic of fuzziness. In fuzzy logic, as contrasted with binary (either-or) information, fuzzy variable sets are proposed to have a truth value that ranges between 0 and 1 and are thus not constrained to the two truth values of classic propositional logic (Zadeh, 1965). This concept helps reducing loss of information when complexity of a disease increases.
How do we proceed to disentangle a fuzzy set? This situation is not unique to schizophrenia; for example, there is no single criterion that allows us to define unequivocally the concept of cancer: neither cellular morphology (there are borderline situations between benign and malignant), nor clinical features (many cancers have diverse manifestations). While few sufficient and necessary characteristics of schizophrenia have been identified, they offer a useful starting point to define the constellation of syndromes to investigate. If one were to visualize the challenge as one of disentangling a fuzzy ball made of multiple colored threads, a logical first step is to begin thread by thread, and even better, by focusing on bundles of connected threads (extended intermediate phenotypes (Keshavan et al., 2008). In order to allow this, the concept of disease has to move away from monothetic definitions and must embrace a “polythetic” approach that is “agnostic” to current diagnostic categories. An example of such an approach to trans-diagnostic evaluation of uniformly collected datasets is the ongoing Bipolar- Schizophrenia Network for Intermediate Phenotypes (B-SNIP) (http://b-snip.org/).
Efforts to elucidate the etiopathology of schizophrenia have been impeded by its imprecise definition and continuing transformations in its categorical conceptualization (Tandon and Maj, 2008). Dimensional approaches may be more powerful in addressing issues of validity (Esterberg and Compton, 2009) and may help address clinical heterogeneity of schizophrenia better than categorical approaches (Peralta and Cuesta, 2008). While there are good scientific reasons for considering dimensional approaches to psychiatric classification (better construct validity and greater power to characterize latent structure of psychopathology), categorical diagnoses may be preferable for communication, clinical decision-making, and to distinguish between individuals with and without a mental disorder (Kamphuis and Noordhof, 2009). The combined goals of validity and utility may be better served by using a combination of categorical and dimensional approaches as we move toward the next iterations of psychiatric nosology.
3. Staging the disease to chart the illness trajectory
An important way ahead in resolving clinical heterogeneity of schizophrenia is to “stage the longitudinal trajectories of this illness in a developmentally meaningful manner, into asymptomatic, early and late prodromal, psychotic and persistent stages of the illness (McGorry et al., 2006). This approach which characterizes disease in terms of severity and progression, and may guide optimization of preventive and/or therapeutic steps, has been underutilized in psychiatry compared to the rest of medicine (e.g. hypertension, cancer). This approach has utility and predictive validity. The main challenge lies in the rather indistinct boundaries between stages as clinically defined, necessitating development of clear definitions including biomarkers (Keshavan et al., 2009).
4. “Extended” intermediate phenotypes and cross-cutting pathophysiological processes
The inherent imprecision of phenotypic boundaries which underlies the failure to discover the biological factors in schizophrenia points to the need to identify biomarkers that cut across syndromal categories. An important paradigm shift in the field in recent years is the concept of intermediate phenotypes, which provide footholds in the path from the phenome to the genome (Gottesman and Gould, 2003). Several neurophysiological, biochemical, and neuroanatomical biomarkers have been identified in schizophrenia (Keshavan et al., 2008, Ivleva et al., 2010) that can potentially serve as intermediate phenotypes. An important step ahead is to delineate the relationships amongst these measures, so that independent “families” (or extended) intermediate phenotypes can be identified, that link etiology, pathophysiology and clinical expression (e.g. abnormal gamma synchrony, inefficient task-related prefrontal activation and impaired working memory). This approach offers the promise of unscrambling and/or deconstructing the current concept of schizophrenia, thereby allowing a more straightforward and successful analysis of this etiologically heterogeneous entity. Doing so will move the field from arbitrary and imprecise definitions of phenotypic categories toward well-delineated natural entities that are defined by multi-level molecular, physiological and behavioral data (Figure 1). Such categorization is more likely to carve nature at its joints, define entities which are more likely to show predictable treatment responses, and to offer promising and productive targets for further research. Several examples exist in medical literature of such an approach leading to pathophysiologically defined subsets of syndromally defined disease (e.g. breast cancer, chronic fatigue syndrome). In an effort to reformulate psychiatric diagnosis, the National Institute of Mental Health has recently initiated the Research Domain Criteria (RDoC) project, which seeks to define mental disorders based on emerging knowledge on pathophysiological processes (e.g. fear circuitry) rather than the symptom-based nosology, which is based on clinical consensus (Insel and Cuthbert, 2009). Future editions of the Diagnostic and Statistical Manual will probably heed those ideas and suggestions (Hyman, 2010). Reverting to the example of the cancers, progress in disentangling their nature was not made by moving phenotypic boundaries between metaplasia and dysplasia to get it “just right”, but by delineating distinct pathophysiolocal processes (e.g. angiogenesis, apoptosis, growth factors), which in turn allowed development of specific treatments (Hanahan and Weinberg, 2000). Similar strands of treatment-relevant pathophysiological processes, such as alterations excitatory/inhibitory, executive function and salience circuitries, oxidative stress, inflammation and membrane dysfunction may become discernible across psychotic and related disorders in the not too distant future.
Figure 1.
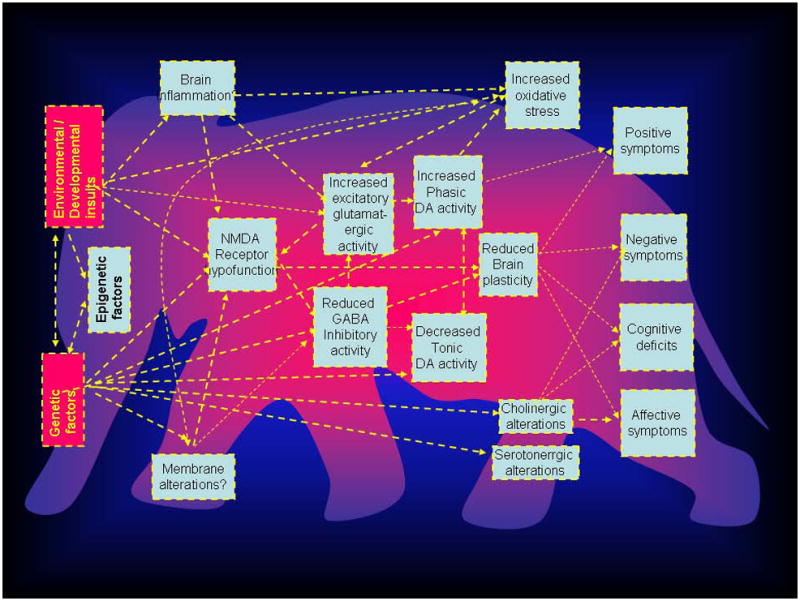
Schematic diagram showing extant models of schizophrenia that encompass the known etiological, pathophysiological facts of schizophrenia. Arrows illustrate possible connections between these models, which need to be investigated to develop a coherent, overarching concept of schizophrenia. The proverbial elephant (see text) in the background is presented to illustrate the fact that several theorists may see one or other aspect of the disease as an explanation of the entire concept. Note that not all theories may be accommodated within the traditional concept of schizophrenia as currently defined, and may cut across other disease boundaries.
5. Identification of latent structures in cross-diagnostic multi-domain datasets
Recent advances in biometrics make it possible to more clearly delineate illness boundaries using a data-centric approach thus integrating information across phenomic, endophenomic and genomic/enviromic domains (Barabasi, 2007, Loscalzo et al., 2007). The sequencing of the human genome and the growing body of transcriptomic, proteomic, and metabolomic data sets in health and disease make it now possible to characterize human disease more precisely at clinical as well as preclinical levels. Sophisticated statistical modeling approaches are now available to investigate genetic overlap and identify “disease networks between multiple seemingly disparate phenotypes such as schizophrenia, bipolar disorder and autism (Rzhetsky et al., 2007). Integration of high throughput data (“omics”) from heterogeneous datasets across multiple domains allows identification of emergent, hitherto unknown relationships between disease related variables, and thereby helps redefine complex disease entities. One such approach is “redescription mining” that helps identify latent significant relationships in multi-omics data; as an example, using such an approach, the heterogeneous chronic fatigue syndrome has been resolved into etiological meaningful subtypes (Waltman et al., 2006). These approaches will elucidate the relations between disease susceptibility, environmental influence; and phenotypic manifestations of the disease to more lucidly define disease prognosis and to eventually individualize treatment for optimal therapeutic efficacy.
6. Connecting the dots: toward integrative models
One approach to “reconstruct” schizophrenia is to build all-encompassing integrative models. Current theorists might be viewing different aspects of the disease, like the proverbial six blind men of Hindustan (India) articulated in John Saxe’s famous poem groping an elephant in a dark room and coming up with the varying descriptions (Saxe, 1849). It is quite possible that such an elephant exists; it may be seen from Figure 2 it may be possible that several extant models may be conceptually linked. Genetic, and/or environmental factors (such as hypoxia and brain inflammation) may lead to NMDA receptor hypofunction. NMDA receptor hypofunction, by inducing a state of hypofunctioning GABAergic interneurons, might lead to increased glutamatergic excitatory activity; this would in turn cause cognitive impairments via altered gamma synchrony and gating processes, and cause psychosis via secondary dopaminergic alterations. The glutamatergic alterations may affect neuroplasticity leading to cognitive impairments as well as increased oxidative stress leading to further neuronal damage. Such formulations which traverse molecular, cellular and systems levels of explanation, have only rarely been done before (Insel, 2010); to paraphrase David Lewis, doing so would in essence allow the field to get a better picture of the fabled elephant, or at least for the “blind men to begin talking to each other” (Dobbs, 2010, Lewis and Hashimoto, 2007). It is also possible, however, that there may be no elephant, more than one elephant, or many different animals in the room.
7. Towards a personalized and preemptive approach
The ground seems to be shifting underneath the traditional and aging unitary concept of schizophrenia. It is puzzling why we continue to debate whether schizophrenia is one or many diseases, or drawing an arbitrary boundary between schizophrenia and normality. We argue that such binary thinking has not advanced our field. Classification mainly on phenotypic features, i.e. psychological symptoms and signs tends to perpetuate Cartesian dichotomy that lingers in our field. One way ahead is to replace categorical thinking by a continuum model. In such a model, each patient may be placed on a unique location in the multi-dimensional “disease space” along the tripartite coordinates of the patient’s genomic and environmental risk/resilience factors and disease expression (at molecular, physiological and behavioral levels). Doing so is the central tenet of personalized medicine; charting person-specific hallmarks of a complex disorder will not only offer novel targets for future pathophysiological and therapeutic research, but will also help optimize diagnosis and treatment for each individual patient.
8. Clarifying causes vs. consequences and compensatory phenomena
A major roadblock to unraveling biomarkers of psychiatric disorders such as schizophrenia is the difficulty in disentangling causal factors from those that are consequences or maladaptive or adaptive responses to the pathophysiologic process. As may be seen in Figure 1, the relationships between purported pathophysiologic processes may be bidirectional. For example, NMDA receptor hypofunction proposed to underlie pathophysiology of schizophrenia may lead to increased oxidative stress (Forder and Tymianski, 2009); on the other hand, it has also been suggested that alterations in the antioxidant system such as glutathione may induce a state of NMDA receptor hypofunction leading to schizophrenia (Do et al., 2009). Animal models are critical for teasing apart such cause-effect relationships (Nestler and Hyman, 2010). In a sense, models of mice may be a step forward from those that seek to define the elephant; this would be consistent with another Indian fable ascribed to Vishnu Sharma (~200 BC), that of a grateful mouse nibbling away at the threads, and rescuing an elephant trapped in a hunter’s net (Govindan, 2007).
9. Refining and developing testable hypotheses
The field of schizophrenia is replete with examples of theories that have remained unproven, but still held with tenacity by their proponents. At least in part, this situation prevails because the theories in question have not been so specifically stated or precisely defined so as to lead to explicit predictions that can then be formally tested. Models are necessary to make explicit our efforts at making sense of a body of information (“facts”) about an illness concept such as schizophrenia. For them to be useful, however, they need to precisely address the following: a) exactly which aspect/s of schizophrenia does the model pertain to and/or connect? (etiology – pathophysiology – clinical expression – treatment); b) what specific “facts of schizophrenia” does the model rest upon? c) explanatory value of model: how does the model comport with other “facts of schizophrenia”? What known findings in schizophrenia can be explained by the model, and what “facts” is the model not consistent with? d) Does the model have specific testable predictions, and have they been tested? And finally, e) is the model of continuing relevance?
10. Rethinking the current terminology
While we will still need schizophrenia as a useful starting point for identifying populations for treatment and research studies, the term has a stigmatizing effect (Van Os, 2010), denotes a false sense of certain comprehension and perpetuates “in the box” thinking (Hyman, 2010). Alternative names have been suggested such as “integration disorder” in Japan (Sato, 2006); this name change actually reduced negative perceptions to this illness (Takahashi et al., 2009). The term integrative disorder, however, is too vague and fails to capture the central features of the disorder. Van Os (van Os, 2009)has suggested the term “salience disorder” arguing that aberrant salience is a central feature of this illness. However, this name again does not do much justice to key aspects of the illness such as negative and cognitive symptoms. We may use a term that calls this entity what it actually is, e.g., youth onset conative, cognitive and reality distortion (CONCORD) syndromes (conative, a time-honored but not-so frequently used term for motivational behavior (Reitan and Wolfson, 2000). While this has the advantage of being less stigmatizing, not all patients who we currently define as having schizophrenia currently (e.g. those who do not show all these features) may belong to such a category. At the very least, we should consistently use the term “schizophrenias” to avoid the illusion this entity being a single disease entity.
Conclusions
With a view towards compiling a defined set of facts about the nature of schizophrenia, we began this series with the tentative assumption that schizophrenia is a unitary disease entity (Tandonet al., 2008c). The varying nature of certainty about this body of facts (Tandon et al., 2008b, Keshavan et al., 2008, Tandon et al., 2009, Tandon et al., 2010) and the unsatisfactory nature of extant models seeking to explain this body of facts leads us to conclude that the current world of schizophrenia likely includes multiple phenotypically overlapping syndromes and diseases, and that the unitary concept of schizophrenia may have outlived its usefulness. The current construct of schizophrenia, as defined by clinical characteristics, seems awkwardly juxtaposed against the emerging findings from the vibrantly evolving worlds of neuroscience and genetics (Insel and Cuthbert, 2009). As our review indicates, a more fruitful approach is to move beyond simply rearranging symptom constellations, and to configure how known facts across the genomic, enviromic, endophenomic and phenomic domains may be reassembled to identify clusters of etiopathologically meaningful and empirically testable entities while remaining agnostic to traditional, phenotypic boundaries. If we are to make real progress in unraveling the nature of the many disorders that are all currently called schizophrenia, the field must boldly step outside the schizophrenia box. The conceptual trappings of the elephant-like unitary constructs may be rescued by the mouse and like models!
Acknowledgments
We thank Ryan Mears PhD, John Sweeney PhD, Jai Shah MD, and Anthony Ahmed PhD for their helpful comments on this paper.
Role of Funding Source Role of Funding Source
This work was supported in part by NIMH grants MH64023, 60902 and 78113 (MSK).
Footnotes
Conflict of Interest
The authors report no conflicts relevant to this paper.
Contributors
Matcheri Keshavan wrote the first draft of the manuscript. All authors contributed to and have approved the final manuscript.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Akbarian S, Bunney WE, Jr, Potkin SG, Wigal SB, Hagman JO, Sandman CA, Jones EG. Altered distribution of nicotinamide-adenine dinucleotide phosphate-diaphorase cells in frontal lobe of schizophrenics implies disturbances of cortical development. Arch Gen Psychiatry. 1993;50:169–77. doi: 10.1001/archpsyc.1993.01820150007001. [DOI] [PubMed] [Google Scholar]
- Andreasen NC. A unitary model of schizophrenia: Bleuler’s “fragmented phrene” as schizencephaly. Arch Gen Psychiatry. 1999;56:781–7. doi: 10.1001/archpsyc.56.9.781. [DOI] [PubMed] [Google Scholar]
- Ardekani BA, Tabesh A, Sevy S, Robinson DG, Bilder RM, Szeszko PR. Diffusion tensor imaging reliably differentiates patients with schizophrenia from healthy volunteers. Hum Brain Mapp. 2010 doi: 10.1002/hbm.20995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arthi K, Tamilarasi A. Prediction of autistic disorder using neuro fuzzy system by applying ANN technique. Int J Dev Neurosci. 2008;26:699–704. doi: 10.1016/j.ijdevneu.2008.07.013. [DOI] [PubMed] [Google Scholar]
- Barabasi AL. Network medicine--from obesity to the “diseasome”. N Engl J Med. 2007;357:404–7. doi: 10.1056/NEJMe078114. [DOI] [PubMed] [Google Scholar]
- Bateson GJ, DD, Haley J, Weakland J. Toward a theory of schizophrenia. Behavioral Science. 1956;1:251–264. [Google Scholar]
- Baumann B, Bogerts B. The pathomorphology of schizophrenia and mood disorders: similarities and differences. Schizophr Res. 1999;39:141–8. doi: 10.1016/s0920-9964(99)00113-9. discussion 162. [DOI] [PubMed] [Google Scholar]
- Bayer TA, Falkai P, Maier W. Genetic and non-genetic vulnerability factors in schizophrenia: the basis of the “two hit hypothesis”. J Psychiatr Res. 1999;33:543–8. doi: 10.1016/s0022-3956(99)00039-4. [DOI] [PubMed] [Google Scholar]
- Bearden CE, Van Erp TG, Thompson PM, Toga AW, Cannon TD. Cortical mapping of genotype-phenotype relationships in schizophrenia. Hum Brain Mapp. 2007;28:519–32. doi: 10.1002/hbm.20404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benes FM, Berretta S. GABAergic interneurons: implications for understanding schizophrenia and bipolar disorder. Neuropsychopharmacology. 2001;25:1–27. doi: 10.1016/S0893-133X(01)00225-1. [DOI] [PubMed] [Google Scholar]
- Bilder RM, Sabb FW, Cannon TD, London ED, Jentsch JD, Parker DS, Poldrack RA, Evans C, Freimer NB. Phenomics: the systematic study of phenotypes on a genome-wide scale. Neuroscience. 2009;164:30–42. doi: 10.1016/j.neuroscience.2009.01.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bitanihirwe BK, Woo TU. Oxidative stress in schizophrenia: An integrated approach. Neurosci Biobehav Rev. 2010 doi: 10.1016/j.neubiorev.2010.10.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boydell J. In: Neurodevelopment and Schizophrenia. MS MK, KENNEDY J, MURRAY R, editors. London/New York, NY: Cambridge University Press; 2004. [Google Scholar]
- Brigman JL, Padukiewicz KE, Sutherland ML, Rothblat LA. Executive functions in the heterozygous reeler mouse model of schizophrenia. Behav Neurosci. 2006;120:984–8. doi: 10.1037/0735-7044.120.4.984. [DOI] [PubMed] [Google Scholar]
- Buchanan RW, Javitt DC, Marder SR, Schooler NR, Gold JM, Mcmahon RP, Heresco-Levy U, Carpenter WT. The Cognitive and Negative Symptoms in Schizophrenia Trial (CONSIST): the efficacy of glutamatergic agents for negative symptoms and cognitive impairments. Am J Psychiatry. 2007;164:1593–602. doi: 10.1176/appi.ajp.2007.06081358. [DOI] [PubMed] [Google Scholar]
- Camargo LM, Collura V, Rain JC, Mizuguchi K, Hermjakob H, Kerrien S, Bonnert TP, Whiting PJ, Brandon NJ. Disrupted in Schizophrenia 1 Interactome: evidence for the close connectivity of risk genes and a potential synaptic basis for schizophrenia. Mol Psychiatry. 2007;12:74–86. doi: 10.1038/sj.mp.4001880. [DOI] [PubMed] [Google Scholar]
- Carlsson A. Does dopamine play a role in schizophrenia? Psychol Med. 1977;7:583–97. doi: 10.1017/s003329170000622x. [DOI] [PubMed] [Google Scholar]
- Carpenter WT, Jr, Buchanan RW, Kirkpatrick B, Tamminga C, Wood F. Strong inference, theory testing, and the neuroanatomy of schizophrenia. Arch Gen Psychiatry. 1993;50:825–31. doi: 10.1001/archpsyc.1993.01820220081009. [DOI] [PubMed] [Google Scholar]
- Carpenter WT, Jr, Heinrichs DW, Wagman AM. Deficit and non deficit forms of schizophrenia: the concept. Am J Psychiatry. 1988;145:578–83. doi: 10.1176/ajp.145.5.578. [DOI] [PubMed] [Google Scholar]
- Carter CJ. Schizophrenia susceptibility genes converge on interlinked pathways related to glutamatergic transmission and long-term potentiation, oxidative stress and oligodendrocyte viability. Schizophr Res. 2006;86:1–14. doi: 10.1016/j.schres.2006.05.023. [DOI] [PubMed] [Google Scholar]
- Costa E, Davis J, Grayson DR, Guidotti A, Pappas GD, Pesold C. Dendritic spine hypoplasticity and downregulation of reelin and GABAergic tone in schizophrenia vulnerability. Neurobiol Dis. 2001;8:723–42. doi: 10.1006/nbdi.2001.0436. [DOI] [PubMed] [Google Scholar]
- Coyle JT. Glutamate and schizophrenia: beyond the dopamine hypothesis. Cell Mol Neurobiol. 2006;26:365–84. doi: 10.1007/s10571-006-9062-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Craddock N, Hurles ME, Cardin N, Pearson RD, Plagnol V, Robson S, Vukcevic D, Barnes C, Conrad DF, Giannoulatou E, Holmes C, Marchini JL, Stirrups K, Tobin MD, Wain LV, Yau C, Aerts J, Ahmad T, Andrews TD, Arbury H, Attwood A, Auton A, Ball SG, Balmforth AJ, Barrett JC, Barroso I, Barton A, Bennett AJ, Bhaskar S, Blaszczyk K, Bowes J, Brand OJ, Braund PS, Bredin F, Breen G, Brown MJ, Bruce IN, Bull J, Burren OS, Burton J, Byrnes J, Caesar S, Clee CM, Coffey AJ, Connell JM, Cooper JD, Dominiczak AF, Downes K, Drummond HE, Dudakia D, Dunham A, Ebbs B, Eccles D, Edkins S, Edwards C, Elliot A, Emery P, Evans DM, Evans G, Eyre S, Farmer A, Ferrier IN, Feuk L, Fitzgerald T, Flynn E, Forbes A, Forty L, Franklyn JA, Freathy RM, Gibbs P, Gilbert P, Gokumen O, Gordon-Smith K, Gray E, Green E, Groves CJ, Grozeva D, Gwilliam R, Hall A, Hammond N, Hardy M, Harrison P, Hassanali N, Hebaishi H, Hines S, Hinks A, Hitman GA, Hocking L, Howard E, Howard P, Howson JM, Hughes D, Hunt S, Isaacs JD, Jain M, Jewell DP, Johnson T, Jolley JD, Jones IR, Jones LA, et al. Genome-wide association study of CNVs in 16,000 cases of eight common diseases and 3,000 shared controls. Nature. 2010;464:713–20. doi: 10.1038/nature08979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Craddock N, Owen MJ. Rethinking psychosis: the disadvantages of a dichotomous classification now outweigh the advantages. World Psychiatry. 2007;6:84–91. [PMC free article] [PubMed] [Google Scholar]
- Crow TJ. The continuum of psychosis and its implication for the structure of the gene. Br J Psychiatry. 1986;149:419–29. doi: 10.1192/bjp.149.4.419. [DOI] [PubMed] [Google Scholar]
- Crow TJ. Constraints on concepts of pathogenesis. Language and the speciation process as the key to the etiology of schizophrenia. Arch Gen Psychiatry. 1995;52:1011–4. doi: 10.1001/archpsyc.1995.03950240029006. discussion 1019–24. [DOI] [PubMed] [Google Scholar]
- Crow TJ. Schizophrenia as the price that homo sapiens pays for language: a resolution of the central paradoxin the origin of the species. Brain Res Brain Res Rev. 2000;31:118–29. doi: 10.1016/s0165-0173(99)00029-6. [DOI] [PubMed] [Google Scholar]
- Deister A, Marneros A. Long-term stability of subtypes in schizophrenic disorders: a comparison of four diagnostic systems. Eur Arch Psychiatry Clin Neurosci. 1993;242:184–90. doi: 10.1007/BF02189961. [DOI] [PubMed] [Google Scholar]
- Delisi LE. Is schizophrenia a lifetime disorder of brain plasticity, growth and aging? Schizophr Res. 1997;23:119–29. doi: 10.1016/S0920-9964(96)00079-5. [DOI] [PubMed] [Google Scholar]
- Delisi LE. The concept of progressive brain change in schizophrenia: implications for understanding schizophrenia. Schizophr Bull. 2008;34:312–21. doi: 10.1093/schbul/sbm164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Do KQ, Cabungcal JH, Frank A, Steullet P, Cuenod M. Redox dysregulation, neurodevelopment, and schizophrenia. Curr Opin Neurobiol. 2009;19:220–30. doi: 10.1016/j.conb.2009.05.001. [DOI] [PubMed] [Google Scholar]
- Dobbs D. Schizophrenia: The making of a troubled mind. Nature. 2010;468:154–6. doi: 10.1038/468154a. [DOI] [PubMed] [Google Scholar]
- Duan J, Sanders AR, Gejman PV. Genome-wide approaches to schizophrenia. Brain Res Bull. 2010;83:93–102. doi: 10.1016/j.brainresbull.2010.04.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ellman LM, Deicken RF, Vinogradov S, Kremen WS, Poole JH, Kern DM, Tsai WY, Schaefer CA, Brown AS. Structural brain alterations in schizophrenia following fetal exposure to the inflammatory cytokine interleukin-8. Schizophr Res. 2010;121:46–54. doi: 10.1016/j.schres.2010.05.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Esterberg ML, Compton MT. The psychosis continuum and categorical versus dimensional diagnostic approaches. Curr Psychiatry Rep. 2009;11:179–84. doi: 10.1007/s11920-009-0028-7. [DOI] [PubMed] [Google Scholar]
- Featherstone RE, Kapur S, Fletcher PJ. The amphetamine-induced sensitized state as a model of schizophrenia. Prog Neuropsychopharmacol Biol Psychiatry. 2007;31:1556–71. doi: 10.1016/j.pnpbp.2007.08.025. [DOI] [PubMed] [Google Scholar]
- Feifel D, Shilling PD. Promise and pitfalls of animal models of schizophrenia. Curr Psychiatry Rep. 2010;12:327–34. doi: 10.1007/s11920-010-0122-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feinberg I. Efference copy and corollary discharge: implications for thinking and its disorders. Schizophr Bull. 1978;4:636–40. doi: 10.1093/schbul/4.4.636. [DOI] [PubMed] [Google Scholar]
- Feinberg I. Schizophrenia and late maturational brain changes in man. Psychomarmacology Bulletin. 1982;18:29–31. [Google Scholar]
- Feinberg I, Campbell IG. Sleep EEG changes during adolescence: an index of a fundamental brain reorganization. Brain Cogn. 2010;72:56–65. doi: 10.1016/j.bandc.2009.09.008. [DOI] [PubMed] [Google Scholar]
- Forder JP, Tymianski M. Postsynaptic mechanisms of excitotoxicity: Involvement of postsynaptic density proteins, radicals, and oxidant molecules. Neuroscience. 2009;158:293–300. doi: 10.1016/j.neuroscience.2008.10.021. [DOI] [PubMed] [Google Scholar]
- Freud S. The Schreber Case. Vol. 2003 New York: Penguin Classics Psychology; 1911. [Google Scholar]
- Frith CD, Done DJ. Towards a neuropsychology of schizophrenia. Br J Psychiatry. 1988;153:437–43. doi: 10.1192/bjp.153.4.437. [DOI] [PubMed] [Google Scholar]
- Galderisi S, Maj M. Deficit schizophrenia: an overview of clinical, biological and treatment aspects. Eur Psychiatry. 2009;24:493–500. doi: 10.1016/j.eurpsy.2009.03.001. [DOI] [PubMed] [Google Scholar]
- Garver DL, Nair TR, Christensen JD. ‘Schizophrenia as a chronic active brain process ...’: perhaps, but only in part. Psychiatry Res. 1997;76:131–8. doi: 10.1016/s0925-4927(97)00069-3. [DOI] [PubMed] [Google Scholar]
- Gavin DP, Sharma RP. Histone modifications, DNA methylation, and schizophrenia. Neurosci Biobehav Rev. 2010;34:882–8. doi: 10.1016/j.neubiorev.2009.10.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gottesman Ii, Gould TD. The endophenotype concept in psychiatry: etymology and strategic intentions. Am J Psychiatry. 2003;160:636–45. doi: 10.1176/appi.ajp.160.4.636. [DOI] [PubMed] [Google Scholar]
- Gottesman Ii, Shields J. A polygenic theory of schizophrenia. Proc Natl Acad Sci U S A. 1967;58:199–205. doi: 10.1073/pnas.58.1.199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Govindan S. 71 Golden Tales of Panchatantra (ascribed to Vishnu Sharma, ~ 200 BC) Unicorn Books; 2007. [Google Scholar]
- Grace AA. Phasic versus tonic dopamine release and the modulation of dopamine system responsivity: a hypothesis for the etiology of schizophrenia. Neuroscience. 1991;41:1–24. doi: 10.1016/0306-4522(91)90196-u. [DOI] [PubMed] [Google Scholar]
- Hanahan D, Weinberg RA. The hallmarks of cancer. Cell. 2000;100:57–70. doi: 10.1016/s0092-8674(00)81683-9. [DOI] [PubMed] [Google Scholar]
- Harrison G, Hopper K, Craig T, Laska E, Siegel C, Wanderling J, Dube KC, Ganev K, Giel R, An Der Heiden W, Holmberg SK, Janca A, Lee PW, Leon CA, Malhotra S, Marsella AJ, Nakane Y, Sartorius N, Shen Y, Skoda C, Thara R, Tsirkin SJ, Varma VK, Walsh D, Wiersma D. Recovery from psychotic illness: a 15-and 25 -year international follow-up study. Br J Psychiatry. 2001;178:506–17. doi: 10.1192/bjp.178.6.506. [DOI] [PubMed] [Google Scholar]
- Hayashi-Takagi A, Takaki M, Graziane N, Seshadri S, Murdoch H, Dunlop AJ, Makino Y, Seshadri AJ, Ishizuka K, Srivastava DP, Xie Z, Baraban JM, Houslay MD, Tomoda T, Brandon NJ, Kamiya A, Yan Z, Penzes P, Sawa A. Disrupted-in-Schizophrenia 1 (DISC1) regulates spines of the glutamate synapse via Rac1. Nat Neurosci. 2010;13:327–32. doi: 10.1038/nn.2487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heckers S. Is schizoaffective disorder a useful diagnosis? Curr Psychiatry Rep. 2009;11:332–7. doi: 10.1007/s11920-009-0048-3. [DOI] [PubMed] [Google Scholar]
- Hemsley DR, Zawada SL. ‘Filtering’ and the cognitive deficit in schizophrenia. Br J Psychiatry. 1976;128:456–61. doi: 10.1192/bjp.128.5.456. [DOI] [PubMed] [Google Scholar]
- Hitzemann R, Hirschowitz J, Garver D. Membrane abnormalities in the psychoses and affective disorders. J Psychiatr Res. 1984;18:319–26. doi: 10.1016/0022-3956(84)90022-0. [DOI] [PubMed] [Google Scholar]
- Hoffman RE, Mcglashan TH. Corticocortical connectivity, autonomous networks, and schizophrenia. Schizophr Bull. 1994;20:257–61. doi: 10.1093/schbul/20.2.257. [DOI] [PubMed] [Google Scholar]
- Hoffman RE, Woods SW, Hawkins KA, Pittman B, Tohen M, Preda A, Breier A, Glist J, Addington J, Perkins DO, Mcglashan TH. Extracting spurious messages from noise and risk of schizophrenia-spectrum disorders in a prodromal population. Br J Psychiatry. 2007;191:355–6. doi: 10.1192/bjp.bp.106.031195. [DOI] [PubMed] [Google Scholar]
- Horrobin DF, Glen AI, Vaddadi K. The membrane hypothesis of schizophrenia. Schizophr Res. 1994;13:195–207. doi: 10.1016/0920-9964(94)90043-4. [DOI] [PubMed] [Google Scholar]
- Howes OD, Kapur S. The dopamine hypothesis of schizophrenia: version III--the final common pathway. Schizophr Bull. 2009;35:549–62. doi: 10.1093/schbul/sbp006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huttunen MO, Machon RA, Mednick SA. Prenatal factors in the pathogenesis of schizophrenia. Br J Psychiatry Suppl. 1994:15–9. [PubMed] [Google Scholar]
- Hyde TM, Ziegler JC, Weinberger DR. Psychiatric disturbances in metachromatic leukodystrophy. Insights into the neurobiology of psychosis. Arch Neurol. 1992;49:401–6. doi: 10.1001/archneur.1992.00530280095028. [DOI] [PubMed] [Google Scholar]
- Hyman SE. The diagnosis of mental disorders: the problem of reification. Annu Rev Clin Psychol. 2010;6:155–79. doi: 10.1146/annurev.clinpsy.3.022806.091532. [DOI] [PubMed] [Google Scholar]
- Insel TR. Rethinking schizophrenia. Nature. 2010;468:187–93. doi: 10.1038/nature09552. [DOI] [PubMed] [Google Scholar]
- Insel TR, Cuthbert BN. Endophenotypes: bridging genomic complexity and disorder heterogeneity. Biol Psychiatry. 2009;66:988–9. doi: 10.1016/j.biopsych.2009.10.008. [DOI] [PubMed] [Google Scholar]
- International Schizophrenia Consortium. Rare chromosomal deletions and duplications increase risk of schizophrenia. Nature. 2008;455:237–41. doi: 10.1038/nature07239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ivleva EI, Morris DW, Moates AF, Suppes T, Thaker GK, Tamminga CA. Genetics and intermediate phenotypes of the schizophrenia--bipolar disorder boundary. Neurosci Biobehav Rev. 2010;34:897–921. doi: 10.1016/j.neubiorev.2009.11.022. [DOI] [PubMed] [Google Scholar]
- Jaaro-Peled H, Ayhan Y, Pletnikov MV, Saw A. Review of pathological hallmarks of schizophrenia: comparison of genetic models with patients and nongenetic models. Schizophr Bull. 2010;36:301–13. doi: 10.1093/schbul/sbp133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Javitt DC, Spencer KM, Thaker GK, Winterer G, Hajos M. Neurophysiological biomarkers for drug development in schizophrenia. Nat Rev Drug Discov. 2008;7:68–83. doi: 10.1038/nrd2463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Javitt DC, Zukin SR. Recent advances in the phencyclidine model of schizophrenia. Am J Psychiatry. 1991;148:1301–8. doi: 10.1176/ajp.148.10.1301. [DOI] [PubMed] [Google Scholar]
- Kamphuis JH, Noordhof A. On categorical diagnoses in DSM-V: cutting dimensions at useful points? Psychol Assess. 2009;21:294–301. doi: 10.1037/a0016697. [DOI] [PubMed] [Google Scholar]
- Kapur S. Psychosis as a state of aberrant salience: a framework linking biology, phenomenology, and pharmacology in schizophrenia. Am J Psychiatry. 2003;160:13–23. doi: 10.1176/appi.ajp.160.1.13. [DOI] [PubMed] [Google Scholar]
- Kapur S, Mamo D. Half a century of antipsychotics and still a central role for dopamine D2 receptors. Prog Neuropsychopharmacol Biol Psychiatry. 2003;27:1081–90. doi: 10.1016/j.pnpbp.2003.09.004. [DOI] [PubMed] [Google Scholar]
- Kapur S, Remington G. Serotonin-dopamine interaction and its relevance to schizophrenia. Am J Psychiatry. 1996;153:466–76. doi: 10.1176/ajp.153.4.466. [DOI] [PubMed] [Google Scholar]
- Kato GJ, Gladwin MT, Steinberg MH. Deconstructing sickle cell disease: reappraisal of the role of hemolysis in the development of clinical subphenotypes. Blood Rev. 2007;21:37–47. doi: 10.1016/j.blre.2006.07.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kendler KS, Gruenberg AM, Tsuang MT. Subtype stability in schizophrenia. Am J Psychiatry. 1985;142:827–32. doi: 10.1176/ajp.142.7.827. [DOI] [PubMed] [Google Scholar]
- Keshavan MS. Development, disease and degeneration in schizophrenia: a unitary pathophysiological model. J Psychiatr Res. 1999;33:513–21. doi: 10.1016/s0022-3956(99)00033-3. [DOI] [PubMed] [Google Scholar]
- Keshavan MS, Anderson S, Pettegrew JW. Is schizophrenia due to excessive synaptic pruning in the prefrontal cortex? The Feinberg hypothesis revisited. J Psychiatr Res. 1994;28:239–65. doi: 10.1016/0022-3956(94)90009-4. [DOI] [PubMed] [Google Scholar]
- Keshavan MS, Dick RM, Diwadkar VA, Montrose DM, Prasad KM, Stanley JA. Striatal metabolic alterations in non -psychotic adolescent offspring at risk for schizophrenia: a (1)H spectroscopy study. Schizophr Res. 2009;115:88–93. doi: 10.1016/j.schres.2009.08.012. [DOI] [PubMed] [Google Scholar]
- Keshavan MS, Tandon R, Boutros NN, Nasrallah HA. Schizophrenia, “just the facts”: what we know in 2008 Part 3: neurobiology. Schizophr Res. 2008;106:89–107. doi: 10.1016/j.schres.2008.07.020. [DOI] [PubMed] [Google Scholar]
- Kim JS, Kornhuber HH, Schmid-Burgk W, Holzmuller B. Low cerebrospinal fluid glutamate in schizophrenic patients and a new hypothesis on schizophrenia. Neurosci Lett. 1980;20:379–82. doi: 10.1016/0304-3940(80)90178-0. [DOI] [PubMed] [Google Scholar]
- Kirkpatrick B. Schizophrenia as a systemic disease. Schizophr Bull. 2009;35:381–2. doi: 10.1093/schbul/sbn183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Konrad A, Winterer G. Disturbed structural connectivity in schizophrenia primary factor in pathology or epiphenomenon? Schizophr Bull. 2008;34:72–92. doi: 10.1093/schbul/sbm034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kwon JS, O’donnell BF, Wallenstein GV, Greene RW, Hirayasu Y, Nestor PG, Hasselmo ME, Potts GF, Shenton ME, Mccarley RW. Gamma frequency-range abnormalities to auditory stimulation in schizophrenia. Arch Gen Psychiatry. 1999;56:1001–5. doi: 10.1001/archpsyc.56.11.1001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lewis DA, Hashimoto T. Deciphering the disease process of schizophrenia: the contribution of cortical GABA neurons. Int Rev Neurobiol. 2007;78:109–31. doi: 10.1016/S0074-7742(06)78004-7. [DOI] [PubMed] [Google Scholar]
- Lidz TF. Schizophrenia and the family. 2. New York: International Universities; 1985. [Google Scholar]
- Lieberman JA, Kinon BJ, Loebel AD. Dopaminergic mechanisms in idiopathic and drug-induced psychoses. Schizophr Bull. 1990;16:97–110. doi: 10.1093/schbul/16.1.97. [DOI] [PubMed] [Google Scholar]
- Lodge D, Anis NA. Effects of phencyclidine on excitatory amino acid activation of spinal interneurones in the cat. Eur J Pharmacol. 1982;77:203–4. doi: 10.1016/0014-2999(82)90022-x. [DOI] [PubMed] [Google Scholar]
- Loscalzo J, Kohane I, Barabasi AL. Human disease classification in the postgenomic era: a complex systems approach to human pathobiology. Mol Syst Biol. 2007;3:124. doi: 10.1038/msb4100163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Macdonald AW, Schulz SC. What we know: findings that every theory of schizophrenia should explain. Schizophr Bull. 2009;35:493–508. doi: 10.1093/schbul/sbp017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maynard TM, Sikich L, Lieberman JA, Lamantia AS. Neural development, cell-cell signaling, and the “two-hit” hypothesis of schizophrenia. Schizophr Bull. 2001;27:457–76. doi: 10.1093/oxfordjournals.schbul.a006887. [DOI] [PubMed] [Google Scholar]
- Mcclellan J, King MC. Genetic heterogeneity in human disease. Cell. 2010;141:210–7. doi: 10.1016/j.cell.2010.03.032. [DOI] [PubMed] [Google Scholar]
- Mccormick CM, Mathews IZ, Thomas C, Waters P. Investigations of HPA function and the enduring consequences of stressors in adolescence in animal models. Brain Cogn. 2010;72:73–85. doi: 10.1016/j.bandc.2009.06.003. [DOI] [PubMed] [Google Scholar]
- Mcgorry PD, Hickie IB, Yung AR, Pantelis C, Jackson HJ. Clinical staging of psychiatric disorders: a heuristic framework for choosing earlier, safer and more effective interventions. Aust N Z J Psychiatry. 2006;40:616–22. doi: 10.1080/j.1440-1614.2006.01860.x. [DOI] [PubMed] [Google Scholar]
- Meyer U, Feldon J. Epidemiology-driven neurodevelopmental animal models of schizophrenia. Prog Neurobiol. 2010;90:285–326. doi: 10.1016/j.pneurobio.2009.10.018. [DOI] [PubMed] [Google Scholar]
- Modai I, Kuperman J, Goldberg I, Goldish M, Mendel S. Fuzzy logic detection of medically serious suicide attempt records in major psychiatric disorders. J Nerv Ment Dis. 2004;192:708–10. doi: 10.1097/01.nmd.0000142020.20038.dd. [DOI] [PubMed] [Google Scholar]
- Monji A, Kato T, Kanba S. Cytokines and schizophrenia: Microglia hypothesis of schizophrenia. Psychiatry Clin Neurosci. 2009;63:257–65. doi: 10.1111/j.1440-1819.2009.01945.x. [DOI] [PubMed] [Google Scholar]
- Muller N, Schwarz MJ. A psychoneuroimmunological perspective to Emil Kraepelins dichotomy: schizophrenia and major depression as inflammatory CNS disorders. Eur Arch Psychiatry Clin Neurosci. 2008;258(Suppl 2):97–106. doi: 10.1007/s00406-008-2012-3. [DOI] [PubMed] [Google Scholar]
- Murray JB. Phencyclidine (PCP): a dangerous drug, but useful in schizophrenia research. J Psychol. 2002;136:319–27. doi: 10.1080/00223980209604159. [DOI] [PubMed] [Google Scholar]
- Nasrallah HA. The unintegrated right cerebral hemispheric consciousness as alien intruder: a possible mechanism for Schneiderian delusions in schizophrenia. Compr Psychiatry. 1985;26:273–82. doi: 10.1016/0010-440x(85)90072-0. [DOI] [PubMed] [Google Scholar]
- Nestler EJ, Hyman SE. Animal models of neuropsychiatric disorders. Nat Neurosci. 2010;13:1161–9. doi: 10.1038/nn.2647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olney JW, Farber NB. Glutamate receptor dysfunction and schizophrenia. Arch Gen Psychiatry. 1995;52:998–1007. doi: 10.1001/archpsyc.1995.03950240016004. [DOI] [PubMed] [Google Scholar]
- Paus T, Keshavan M, Giedd JN. Why do many psychiatric disorders emerge during adolescence? Nat Rev Neurosci. 2008;9:947–57. doi: 10.1038/nrn2513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peralta V, Cuesta MJ. Exploring the borders of the schizoaffective spectrum: a categorical and dimensional approach. J Affect Disord. 2008;108:71–86. doi: 10.1016/j.jad.2007.09.009. [DOI] [PubMed] [Google Scholar]
- Petronis A, Paterson AD, Kennedy JL. Schizophrenia: an epigenetic puzzle? Schizophr Bull. 1999;25:639–55. doi: 10.1093/oxfordjournals.schbul.a033408. [DOI] [PubMed] [Google Scholar]
- Platt JR. Strong Inference: Certain systematic methods of scientific thinking may produce much more rapid progress than others. Science. 1964;146:347–53. doi: 10.1126/science.146.3642.347. [DOI] [PubMed] [Google Scholar]
- Randrup A, Munkvad I. Brain dopamine and amphetamine-induced stereotyped behaviour. Acta Pharmacol Toxicol (Copenh) 1967;25(Suppl 4):62. doi: 10.1111/j.1600-0773.1967.tb03049.x. [DOI] [PubMed] [Google Scholar]
- Reddy R, Keshavan MS. Phosphorus magnetic resonance spectroscopy: its utility in examining the membrane hypothesis of schizophrenia. Prostaglandins Leukot Essent Fatty Acids. 2003;69:401–5. doi: 10.1016/j.plefa.2003.08.011. [DOI] [PubMed] [Google Scholar]
- Reddy RD, Yao JK. Free radical pathology in schizophrenia: a review. Prostaglandins Leukot Essent Fatty Acids. 1996;55:33–43. doi: 10.1016/s0952-3278(96)90143-x. [DOI] [PubMed] [Google Scholar]
- Reitan RM, Wolfson D. Conation: a neglected aspect of neuropsychological functioning. Arch Clin Neuropsychol. 2000;15:443–53. [PubMed] [Google Scholar]
- Robertson GS, Hori SE, Powell KJ. Schizophrenia: an integrative approach to modelling a complex disorder. J Psychiatry Neurosci. 2006;31:157–67. [PMC free article] [PubMed] [Google Scholar]
- Robins E, Guze SB. Establishment of diagnostic validity in psychiatric illness: its application to schizophrenia. Am J Psychiatry. 1970;126:983–7. doi: 10.1176/ajp.126.7.983. [DOI] [PubMed] [Google Scholar]
- Rosenberg DR, Lewis DA. Postnatal maturation of the dopaminergic innervation of monkey prefrontal and motor cortices: a tyrosine hydroxylase immunohistochemical analysis. J Comp Neurol. 1995;358:383–400. doi: 10.1002/cne.903580306. [DOI] [PubMed] [Google Scholar]
- Ross CA, Pearlson GD. Schizophrenia, the heteromodal association neocortex and development: potential for a neurogenetic approach. Trends Neurosci. 1996;19:171–6. doi: 10.1016/s0166-2236(96)10022-9. [DOI] [PubMed] [Google Scholar]
- Rzhetsky A, Wajngurt D, Park N, Zheng T. Probing genetic overlap among complex human phenotypes. Proc Natl Acad Sci U S A. 2007;104:11694–9. doi: 10.1073/pnas.0704820104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sabb FW, Burggren AC, Higier RG, Fox J, He J, Parker DS, Poldrack RA, Chu W, Cannon TD, Freimer NB, Bilder RM. Challenges in phenotype definition in the whole-genome era: multivariate models of memory and intelligence. Neuroscience. 2009;164:88–107. doi: 10.1016/j.neuroscience.2009.05.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sato M. Renaming schizophrenia: a Japanese perspective. World Psychiatry. 2006;5:53–5. [PMC free article] [PubMed] [Google Scholar]
- Saxe JG. Poems (New enlarged edition), The blind men and the elephant. Ticknor, Reed and Fields 1849 [Google Scholar]
- Seeman P. All Roads to Schizophrenia Lead to Dopamine Supersensitivity and Elevated Dopamine D2 Receptors. CNS Neurosci Ther. 2010 doi: 10.1111/j.1755-5949.2010.00162.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Segal D, Koschnick JR, Slegers LH, Hof PR. Oligodendrocyte pathophysiology: a new view of schizophrenia. Int J Neuropsychopharmacol. 2007;10:503–11. doi: 10.1017/S146114570600722X. [DOI] [PubMed] [Google Scholar]
- Smith RS. Is schizophrenia caused by excessive production of interleukin-2 and interleukin-2 receptors by gastrointestinal lymphocytes? Med Hypotheses. 1991;34:225–9. doi: 10.1016/0306-9877(91)90215-k. [DOI] [PubMed] [Google Scholar]
- Snyder SH. The dopamine hypothesis of schizophrenia: focus on the dopamine receptor. Am J Psychiatry. 1976;133:197–202. doi: 10.1176/ajp.133.2.197. [DOI] [PubMed] [Google Scholar]
- Stefansson H, Rujescu D, Cichon S, Pietilainen OP, Ingason A, Steinberg S, Fossdal R, Sigurdsson E, Sigmundsson T, Buizer-Voskamp JE, Hansen T, Jakobsen KD, Muglia P, Francks C, Matthews PM, Gylfason A, Halldorsson BV, Gudbjartsson D, Thorgeirsson TE, Sigurdsson A, Jonasdottir A, Bjornsson A, Mattiasdottir S, Blondal T, Haraldsson M, Magnusdottir BB, Giegling I, Moller HJ, Hartmann A, Shianna KV, Ge D, Need AC, Crombie C, Fraser G, Walker N, Lonnqvist J, Suvisaari J, Tuulio-Henriksson A, Paunio T, Toulopoulou T, Bramon E, Di Forti M, Murray R, Ruggeri M, Vassos E, Tosato S, Walshe M, Li T, Vasilescu C, Muhleisen TW, Wang AG, Ullum H, Djurovic S, Melle I, Olesen J, Kiemeney LA, Franke B, Sabatti C, Freimer NB, Gulcher JR, Thorsteinsdottir U, Kong A, Andreassen OA, Ophoff RA, Georgi A, Rietschel M, Werge T, Petursson H, Goldstein DB, Nothen MM, Peltonen L, Collier DA, St Clair D, Stefansson K. Large recurrent microdeletions associated with schizophrenia. Nature. 2008;455:232–6. doi: 10.1038/nature07229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stephan KE, Friston KJ, Frith CD. Dysconnection in schizophrenia: from abnormal synaptic plasticity to failures of self-monitoring. Schizophr Bull. 2009;35:509–27. doi: 10.1093/schbul/sbn176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stone JM, Morrison PD, Pilowsky LS. Glutamate and dopamine dysregulation in schizophrenia--a synthesis and selective review. J Psychopharmacol. 2007;21:440–52. doi: 10.1177/0269881106073126. [DOI] [PubMed] [Google Scholar]
- Sydenham TA, editor. Dr. Thomas Sydenham (1624–1689): His life and original writings. Berkeley: University of California Press; 1966. [Google Scholar]
- Takahashi H, Ideno T, Okubo S, Matsui H, Takemura K, Matsuura M, Kato M, Okubo Y. Impact of changing the Japanese term for “schizophrenia” for reasons of stereotypical beliefs of schizophrenia in Japanese youth. Schizophr Res. 2009;112:149–52. doi: 10.1016/j.schres.2009.03.037. [DOI] [PubMed] [Google Scholar]
- Tandon R. Moving beyond findings: concepts and model-building in schizophrenia. J Psychiatr Res. 1999;33:467–71. doi: 10.1016/s0022-3956(99)00036-9. [DOI] [PubMed] [Google Scholar]
- Tandon R, Belmaker RH, Gattaz WF, Lopez-Ibor JJ, Jr, Okasha A, Singh B, Stein DJ, Olie JP, Fleischhacker WW, Moeller HJ. World Psychiatric Association Pharmacopsychiatry Section statement on comparative effectiveness of antipsychotics in the treatment of schizophrenia. Schizophr Res. 2008a;100:20–38. doi: 10.1016/j.schres.2007.11.033. [DOI] [PubMed] [Google Scholar]
- Tandon R, Greden JF. Cholinergic hyperactivity and negative schizophrenic symptoms. A model of cholinergic/dopaminergic interactions in schizophrenia. Arch Gen Psychiatry. 1989;46:745–53. doi: 10.1001/archpsyc.1989.01810080075010. [DOI] [PubMed] [Google Scholar]
- Tandon R, Greden JF. Negative symptoms of schizophrenia: the need for conceptual clarity. Biol Psychiatry. 1991;30:321–5. doi: 10.1016/0006-3223(91)90287-v. [DOI] [PubMed] [Google Scholar]
- Tandon R, Keshavan MS, Nasrallah HA. Schizophrenia, “just the facts” what we know in 2008. 2. Epidemiology and etiology. Schizophr Res. 2008b;102:1–18. doi: 10.1016/j.schres.2008.04.011. [DOI] [PubMed] [Google Scholar]
- Tandon R, Keshavan MS, Nasrallah HA. Schizophrenia, “Just the Facts”: what we know in 2008 part 1: overview. Schizophr Res. 2008c;100:4–19. doi: 10.1016/j.schres.2008.01.022. [DOI] [PubMed] [Google Scholar]
- Tandon R, Maj M. Nosological status and definition of schizophrenia: some considerations for DSM-V and ICD-11. Asian Journal of Psychiatry. 2008:1. doi: 10.1016/j.ajp.2008.10.002. [DOI] [PubMed] [Google Scholar]
- Tandon R, Mazzara C, Dequardo J, Craig KA, Meador-Woodruff JH, Goldman R, Greden JF. Dexamethasone suppression test in schizophrenia: relationship to symptomatology, ventricular enlargement, and outcome. Biol Psychiatry. 1991;29:953–64. doi: 10.1016/0006-3223(91)90353-n. [DOI] [PubMed] [Google Scholar]
- Tandon R, Nasrallah HA, Keshavan MS. Schizophrenia, “just the facts” 4. Clinical features and conceptualization. Schizophr Res. 2009;110:1–23. doi: 10.1016/j.schres.2009.03.005. [DOI] [PubMed] [Google Scholar]
- Tandon R, Nasrallah HA, Keshavan MS. Schizophrenia, “Just the Facts” 5. Treatment and prevention Past, present, and future. Schizophr Res. 2010 doi: 10.1016/j.schres.2010.05.025. [DOI] [PubMed] [Google Scholar]
- Tsuang MT, Faraone SV. The case for heterogeneity in the etiology of schizophrenia. Schizophr Res. 1995;17:161–75. doi: 10.1016/0920-9964(95)00057-s. [DOI] [PubMed] [Google Scholar]
- Van Kammen DP. gamma-Aminobutyric acid (Gaba) and the dopamine hypothesis of schizophrenia. Am J Psychiatry. 1977;134:138–43. doi: 10.1176/ajp.134.2.138. [DOI] [PubMed] [Google Scholar]
- Van Os J. Is there a continuum of psychotic experiences in the general population? Epidemiol Psichiatr Soc. 2003;12:242–52. doi: 10.1017/s1121189x00003067. [DOI] [PubMed] [Google Scholar]
- Van Os J. ‘Salience syndrome’ replaces ‘schizophrenia’ in DSM-V and ICD-11: psychiatry’s evidence-based entry into the 21st century? Acta Psychiatr Scand. 2009;120:363–72. doi: 10.1111/j.1600-0447.2009.01456.x. [DOI] [PubMed] [Google Scholar]
- Van Os J. Are psychiatric diagnoses of psychosis scientific and useful? The case of schizophrenia. J Ment Health. 2010;19:305–17. doi: 10.3109/09638237.2010.492417. [DOI] [PubMed] [Google Scholar]
- Van Os J, Krabbendam L, Myin-Germeys I, Delespaul P. The schizophrenia envirome. Curr Opin Psychiatry. 2005;18:141–5. doi: 10.1097/00001504-200503000-00006. [DOI] [PubMed] [Google Scholar]
- Van Os J, Rutten BP, Poulton R. Gene-environment interactions in schizophrenia: review of epidemiological findings and future directions. Schizophr Bull. 2008;34:1066–82. doi: 10.1093/schbul/sbn117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Winkel R, Esquivel G, Kenis G, Wichers M, Collip D, Peerbooms O, Rutten B, Myin-Germeys I, Van Os J. Genome-Wide Findings in Schizophrenia and the Role of Gene-Environment Interplay. CNS Neurosci Ther. 2010 doi: 10.1111/j.1755-5949.2010.00155.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vineis P. Methodological insights: fuzzy sets in medicine. J Epidemiol Community Health. 2008;62:273–8. doi: 10.1136/jech.2007.063644. [DOI] [PubMed] [Google Scholar]
- Walsh T, Mcclellan JM, Mccarthy SE, Addington AM, Pierce SB, Cooper GM, Nord AS, Kusenda M, Malhotra D, Bhandari A, Stray SM, Rippey CF, Roccanova P, Makarov V, Lakshmi B, Findling RL, Sikich L, Stromberg T, Merriman B, Gogtay N, Butler P, Eckstrand K, Noory L, Gochman P, Long R, Chen Z, Davis S, Baker C, Eichler EE, Meltzer PS, Nelson SF, Singleton AB, Lee MK, Rapoport JL, King MC, Sebat J. Rare structural variants disrupt multiple genes in neurodevelopmental pathways in schizophrenia. Science. 2008;320:539–43. doi: 10.1126/science.1155174. [DOI] [PubMed] [Google Scholar]
- Waltman P, Pearlman A, Mishra B. Interpreter of maladies: redescription mining applied to biomedical data analysis. Pharmacogenomics. 2006;7:503–9. doi: 10.2217/14622416.7.3.503. [DOI] [PubMed] [Google Scholar]
- Weinberger DR. Implications of normal brain development for the pathogenesis of schizophrenia. Arch Gen Psychiatry. 1987;44:660–9. doi: 10.1001/archpsyc.1987.01800190080012. [DOI] [PubMed] [Google Scholar]
- Williamson P. Are anticorrelated networks in the brain relevant to schizophrenia? Schizophr Bull. 2007;33:994–1003. doi: 10.1093/schbul/sbm043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Woods SW, Addington J, Cadenhead KS, Cannon TD, Cornblatt BA, Heinssen R, Perkins DO, Seidman LJ, Tsuang MT, Walker EF, Mcglashan TH. Validity of the prodromal risk syndrome for first psychosis: findings from the North American Prodrome Longitudinal Study. Schizophr Bull. 2009;35:894–908. doi: 10.1093/schbul/sbp027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wyatt RJ, Alexander RC, Egan MF, Kirch DG. Schizophrenia, just the facts. What do we know, how well do we know it? Schizophr Res. 1988;1:3–18. doi: 10.1016/0920-9964(88)90034-5. [DOI] [PubMed] [Google Scholar]
- Yao JK, Dougherty GG, Jr, Reddy RD, Keshavan MS, Montrose DM, Matson WR, Mcevoy J, Kaddurah-Daouk R. Homeostatic imbalance of purine catabolism in first-episode neuroleptic-naive patients with schizophrenia. PLoS One. 2010;5:e9508. doi: 10.1371/journal.pone.0009508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zadeh L. Fuzzy sets. Information and Control. 1965;8:338–353. [Google Scholar]
- Zubin J, Steinhauer S. How to break the logjam in schizophrenia. A look beyond genetics. J Nerv Ment Dis. 1981;169:477–92. doi: 10.1097/00005053-198108000-00002. [DOI] [PubMed] [Google Scholar]