

Abstract
Heterochromatin comprises tightly compacted repetitive regions of eukaryotic chromosomes. The inheritance of heterochromatin through mitosis requires RNA interference (RNAi), which guides histone modification 1 during the DNA replication phase of the cell cycle2. Here, we show that the alternating arrangement of origins of replication and non-coding RNA in pericentromeric heterochromatin results in competition between transcription and replication. Co-transcriptional RNAi releases RNA polymerase II (PolII), allowing completion of DNA replication by the leading strand DNA polymerase, and associated histone modifying enzymes3 which spread heterochromatin with the replication fork. In the absence of RNAi, stalled forks are repaired by homologous recombination without histone modification.
In fission yeast, the Rik1/CLRC (Recombination in K, Cryptic Locus Regulator) complex silences heterochromatin via Clr4 and Lid2, which methylate histone H3 lysine 9 (H3K9) and demethylate histone H3 lysine 4 (H3K4), respectively2. This complex is recruited in part by RNA interference, which processes non-coding transcripts found in the pericentromeric heterochromatin1,4. Interactions between the RITS (RNAi transcriptional silencing) complex and CLRC have recently been found5,6, but spreading of the Rik1 complex into reporter genes depends on the catalytic activity of RNAi, and the mechanism remains unknown7. Recently, we found that Cdc20 and Mms19 interact with Rik1 and are required for histone modification3. Cdc20 is the catalytic subunit of the leading strand DNA polymerase Polε, while Mms19 is a regulatory subunit of the PolII transcription factor TFIIH. Both proteins participate in transcription coupled nucleotide excision repair (TC-NER) which depends on damage-stalled PolII to detect structural lesions in the DNA which are repaired by the Polε after PolII release8.
The pericentromeric heterochromatin of fission yeast comprises outermost (otr) repeats called dg (5kb) and dh (1-6kb), flanked by innermost (imr) repeats (~6kb) containing clusters of tRNA genes (Fig. 1a). Histone H3 lysine-9 methylation is associated with dg and dh repeats (Fig. 1b), but ends abruptly at the tRNA clusters, and so is confined to heterochromatin9. The dg and dh repeats are transcribed by RNA polymerase II 10, and processed into siRNA clusters up to 4.5kb in length (Fig. 1b). To investigate the extent of siRNA precursor transcripts, we first cloned and sequenced dh and dg repeat complementary DNA from dcr1Δ mutants (Fig. 1c). Polyadenylation sites were then identified using RACE-PCR (Methods), and sequencing revealed they were located within the clusters of siRNA (Fig. 1c). In previous studies of dcr1Δ mutants, PolII enrichment was detected by ChIP11, while transcriptional run-on (TRO) analysis indicated over-accumulation of forward (but not reverse) transcripts1. We found that these PolII ChIP (cen-dg) and TRO probes lie downstream of “forward” polyA sites (Fig. 1d), indicating inefficient termination and PolII readthrough in the absence of RNAi. To confirm readthrough, Northern blots of polyadenylated and total RNA from dcr1Δ, ago1Δ and rdp1Δ mutants were probed with strand specific probes. Transcripts corresponding to full length dh (1.3kb) and dg (1.3-2.3kb) cDNA clones were enriched in polyA+ RNA, as expected, but much longer readthrough transcripts up to 4.5kb could also be detected (Supplementary Fig. 1) indicating that polyadenylation was highly inefficient at these internal sites.
Figure 1. Transcription and replication of pericentromeric heterochromatin in fission yeast.
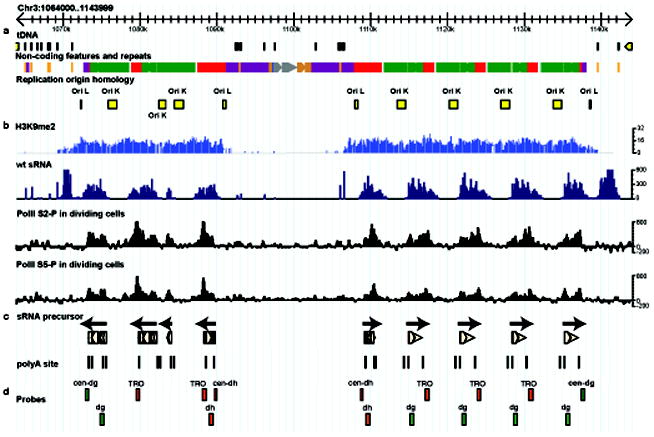
a. Pericentromeric heterochromatin on Centromere 3. dh (red), dg (green) and imr (magenta) repeats are shown, bordered by tRNA genes (brown). Replication origins (yellow) are found in each repeat. b. Tiling microarrays of K9me2 ChIP (light blue) and clusters of small RNA sequences (dark blue) from wild-type cells. ChIP-seq reads corresponding to poised (S5-P) and elongating (S2-P) RNA polymerase II enriched in dcr1Δ cells relative to WT cells are in black. c. cDNA clones (beige) from dcr1Δ cells. PolyA sites are indicated as vertical lines and correspond to peaks of PolII. Arrows indicate the direction of “Forward” transcription. d. Alignment of probes used in previous studies indicates that regions enriched for PolII11 (cen-dg) and transcriptional run-on probes1 (TRO) lie downstream of forward orientation polyA sites
Inefficient polyadenylation is a strong indication of failure to release RNA polymerase II12, and we hypothesized that slicing7 and dicing13 of nascent transcripts via RNAi promotes 3’-5’ degradation by the exosome7 and release of RNA Polymerase II from the 3’ end12. The exosome is required for silencing consistent with this idea14,15. To examine PolII release, we performed ChIP-seq with PolII antibodies, and found peaks of both poised (S5 phosphorylated) and elongating forms (S2 phosphorylated) of PolII in dcr1Δ mutants that corresponded to the polyadenylation sites on each strand (Fig. 1b,c). Peaks of siRNA accumulation mapped just downstream. Thus siRNA in WT cells accumulated where PolII was released (Fig. 1b).
siRNA accumulate during S phase2 and we found that siRNA clusters ended abruptly at the replication origin homology regions contained within each repeat16 (Fig. 1b). To assess the influence of DNA replication on PolII accumulation we blocked replication in high concentrations of hydroxyurea (HU) and performed ChIP-seq using PolII antibodies. In arrested dcr1Δ mutants, PolII accumulated throughout the otr repeats, but in dividing dcr1Δ cells, PolII accumulation was absent from replication origins (Fig. 2a and data not shown). To test if PolII was expelled by replication fork progression (Fig. 2b), HU-arrested dcr1Δ cells were released into the cell cycle (Fig. 2c). As predicted, accumulation at replication origins was quickly lost, and PolII was only found between origins, closer to promoters10, in each subsequent S phase.
Figure 2. RNA interference and DNA replication restrict RNA polymerase II accumulation and prevent DNA damage.

a. Small RNA (blue) and PolII ChIP-seq reads (black) and regions of significant enrichment (blue and red rectangles) from WT and dcr1Δ on the right arm of Centromere 1. b. A replication bubble is shown, initiated at one of the 3 origin homology regions at centromere 1 (yellow boxes). c. Chromatin immunoprecipitation for RNA PolII and Rad22Rad52 from HU-arrested and released wild-type (dashed lines) and dcr1Δ (solid lines). Cell cycle progression after release from HU block is monitored by septation index, which peaks coincident with S phase. d. Representative parental and non-parental di-type tetrads from crosses between rhp51Δ cells, defective in homologous recombination, and dcr1Δ or ago1Δ.
Failure to release RNA polymerase II during S phase is a strong and robust signal for DNA damage8. In order to monitor DNA repair, the HU-arrested cells contained a Rad22-fusion protein Rad22-YFP. Rad22 (Rad52 in budding yeast) is essential for homologous recombination (HR) and is associated with single stranded DNA ends early during DNA repair. Chromatin immunoprecipitation revealed that Rad22 Rad52 was weakly associated with heterochromatic origins in wild-type cells arrested with HU, but quickly declined following release (Fig. 2c). In dcr1Δ mutants, on the other hand, Rad22 Rad52 peaked early in each successive S phase, indicating engagement of the repair machinery during heterochromatin replication17. In order to exclude the impact of HU arrest on DNA damage, we also examined Rad22-YFP accumulation in untreated WT and dcr1Δ mutant cells by fluorescence microscopy (Supplementary Fig. 2). The results were consistent with chromatin IP, in that 6 times as many dcr1Δ than WT cells had Rad22 Rad52 foci during septation (early S phase). Therefore, Dcr1 activity prevents DNA damage and the engagement of HR at the centromere.
We performed genetic tests to determine the role of RNAi in preventing DNA damage during S phase. DNA damage during replication can be rescued by HR repair, and we found that double mutants in the RecA homolog rhp51 rad51 and dcr1Δ or ago1Δ were inviable or formed microcolonies (Fig. 2d). A similar requirement for Rhp51 Rad51 has been demonstrated for convergent stalled replication forks18, which are protected from collapse in fission yeast by a stable replication-pausing complex comprising Swi1/Swi3 and Mrc1 (Mediator of replication checkpoint 1)19. Low concentrations of HU stalls replication forks, and we found that while dcr1Δ, ago1Δ and rdp1Δ cells were insensitive, double mutants with swi3Δ or mrc1Δ were very sensitive to low concentrations of HU (Supplementary Fig. 3). Similar results were obtained with Camptothecin (CPT) which causes arrest during S phase when the replication fork encounters the CPT-topoisomerase I complex. In genome-wide epistasis tests, mutants in more than 30 genes, mostly encoding proteins involved in DNA repair and histone modification interacted significantly with both mrc1 and dcr1, forming a striking genetic network (Supplementary Table 1). This indicates that loss of Dcr1 activity engages replication fork protection.
In order to assess fork integrity, we examined replication of the repeats by 2D gel electrophoresis using probes from the ura4 transgene, which was inserted into a passively replicated dg repeat on chromosome 1 (Fig. 2b). In WT cells, we detected strong X intermediates, indicative of joint molecules, as well as the expected fork or Y molecules (Fig. 3a). Similar X-DNA sister chromatid junctions arise at origins20 but also at stalled replication forks21. These X-molecules were unaffected in dcr1Δ (Fig. 3b) but reduced in mms19Δ, in swi6Δ and especially in clr4Δ cells (Fig. 3c-e). Both Mms19 and Clr4 interact with Rik1, and Mms19 participates in transcription initiation3. Swi6 on the other hand is required to initiate replication within heterochromatic repeats17, and recruitment depends on Clr4. Thus simultaneous replication and transcription of heterochromatic repeats promote local replication fork stalling.
Figure 3. Replication fork stalling during heterochromatin replication.
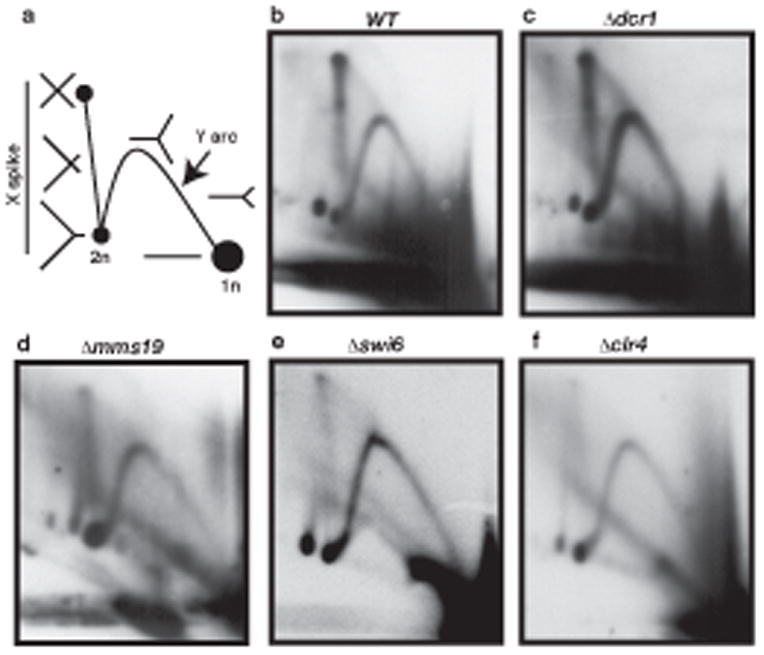
Replication intermediates in wild-type and mutant cells resolved by 2D gel electrophoresis and probed with the unique DS/E probe from the ura4 transgene within the dg repeat on chromosome 1 (Fig. 2a). (a) A schematic of replication intermediates in 2D gels indicates joint molecules (X), and forks (Y). Junction molecules indicate fork stalling in (b) WT and (c) dcr1Δ mutant cells, and are reduced in (d) mms19Δ, (e) swi6Δ and (f) clr4Δ.
In WT cells (Fig. 4a), modified histones recruit Swi6 and the Rik1 complex via chromo- and other domains. Swi6 promotes early replication, and the Rik1 complex interacts with DNA Polε, which allows spreading of histone modification along with fork progression3. Flanking tRNA genes (Fig. 1a) pause replication22, preventing further spreading into neighboring euchromatin9,23. Transcription during S phase stalls the replication fork, accounting for interactions between the replication and transcription machineries3, but RNAi releases PolII allowing replication to proceed. In the absence of RNAi (Fig. 4b), PolII remains stalled at replication forks and signals DNA repair by homologous recombination, which restarts blocked forks24. The Rik1 complex is lost along with the replisome, preventing spreading of heterochromatin into reporter genes, which lose H3K9 methylation entirely. Recombination also removes modified histones from at least one of the two daughter chromatids25 reducing, but not eliminating, methylation of the repeats as previously observed7.
Figure 4. Replication-coupled transcriptional silencing through histone modification and RNAi.
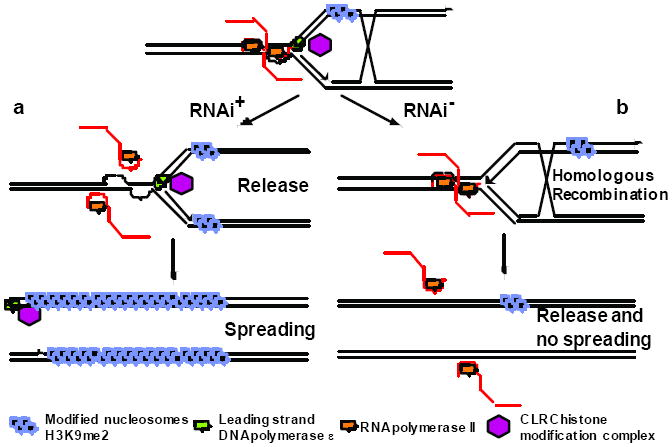
a. The Rik1 complex (red octagon) is recruited to heterochromatic replication forks by interactions with methylated histone H3K9me2 and with the leading strand DNA polymerase (Pol ε, green). Swi6 induces origin firing, but collision with RNA polymerase II (orange) stalls replication forks. RNAi releases PolII by processing of pre-siRNA transcripts (red lines) allowing leading strand DNA polymerase to complete DNA replication and the associated Rik1 histone modification complex (red hexagon) to spread histone modification (black circles).
b. In the absence of RNAi, origins fire but PolII is not released, stalling replication forks. Stalled PolII signals repair via homologous recombination instead. Recombination could in principle occur with sister chromatids (shown here) or with other copies of the same repeat (not shown). DNA polymerase ε and the associated Rik1 complex are lost along with the replisome, and fail to spread histone modification into neighboring reporter genes.
We tested this model in several ways. First, we predicted that the interaction between the Rik1 complex and Polε should depend on RNAi, and we found that co-immunoprecipitation of Cdc20/ Polε with Dos2/Clr7 was reduced in dcr1Δ cells, along with H3K9me2 (Supplemental Fig. 4). Second, we observed that mutants in the cyclin-dependent PolII CTD kinase Cdk9 display slow growth and loss of pericentromeric silencing and sRNA (Supplemental Fig. 5). cdk9 is a central regulator of transcription elongation that links cell-cycle regulated pre-mRNA processing, co-transcriptional histone methylation and DNA damage26. Finally, Clr4 has recently been found to have additional roles in recruiting the RNA-induced transcriptional silencing (RITS) complex to accessory PolII factors27, providing a potential mechanism for PolII release by RNAi. We found long transcripts indicative of strong transcriptional readthrough in clr4Δ mutant cells consistent with this model (Supplementary Fig. 1).
In the budding yeast S. cerevisiae, the Dicer-related RNAse III Rnt1 releases PolII during transcription termination28 while in E.coli, failure of transcription termination stalls replication forks and triggers recombination29, providing a precedent for the mechanism we propose. According to this mechanism, transcription during S phase triggers histone modification, so long as RNA polymerase is released by RNAi, and not by homologous recombination repair. In plants, fungi and invertebrates, heterochromatic silencing may involve similar mechanisms (Supplemental Table 1), while in mammals, both X inactivation and imprinting require transcription of non-coding RNA in dividing cells30. In each case, release of PolII during S phase, by RNAi or by other means, could allow fork restart and spreading of histone modification in a similar way.
Methods summary
Non-coding transcripts were cloned from a cDNA phage library by hybridization to dh and dg consensus probes. Cloning and high throughput sequencing of sRNA was performed using the Illumina genome analyzer according to manufacturer’s instructions. Two dimensional gel electrophoresis of replication intermediates from steady state cultures was performed with probes to the otr1∷ura4+ insertion. For ChIP experiments, cultures were arrested in 15mM HU for 4.5 hours, released and harvested at indicated times, to be crosslinked and processed for Chromatin immunoprecipitation.
Supplementary Material
Acknowledgments
We thank Donna Roh and Tom Volpe for isolating cDNA clones. DI was supported by a NHMRC CJ Martin Postdoctoral Research Fellowship. MZ was supported by a fellowship from the Spanish Ministry of Science. This work was supported by grants BFU2008-01919 and Consolider-Ingenio CSD2007-00015 from the Spanish Ministry of Science and Innovation to FA, and NIH R01 GM076396 to ZC and RM.
Footnotes
Author contributions. SC, DVI, AK, JR contributed equally to this work and are listed in alphabetical order. MZ, SC, DVI, AK, JR, FL, EdC, LM, AC and DG performed experiments, and SC analyzed the data. WZC, FA, BA and RM designed experiments and RM and MZ wrote the manuscript.
Supplementary Information is linked to the online version of the paper at www.nature.com/nature.
Author Information. Genomics data and analysis are available from the Gene Expression Omnibus (http://www.ncbi.nlm.nih.gov/geo/) accession number GSE30837. Individual cDNA sequences are available from GenBank (http://www.ncbi.nlm.nih.gov/genbank/) with accession numbers JN388396 to JN388565. Reprints and permissions information is available at www.nature.com/reprints.
The authors declare no competing financial interests.
References
- 1.Volpe TA, et al. Regulation of heterochromatic silencing and histone H3 lysine-9 methylation by RNAi. Science. 2002;297:1833–1837. doi: 10.1126/science.1074973. [DOI] [PubMed] [Google Scholar]
- 2.Kloc A, Zaratiegui M, Nora E, Martienssen R. RNA Interference Guides Histone Modification during the S Phase of Chromosomal Replication. Curr Biol. 2008;18:490–495. doi: 10.1016/j.cub.2008.03.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Li F, Martienssen R, Cande WZ. Coordination of DNA replication and histone modification by the Rik1-Dos2 complex. Nature. 2011;475:244–248. doi: 10.1038/nature10161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Verdel A, et al. RNAi-mediated targeting of heterochromatin by the RITS complex. Science. 2004;303:672–676. doi: 10.1126/science.1093686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Motamedi MR, et al. HP1 proteins form distinct complexes and mediate heterochromatic gene silencing by nonoverlapping mechanisms. Mol Cell. 2008;32:778–790. doi: 10.1016/j.molcel.2008.10.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Bayne EH, et al. Stc1: a critical link between RNAi and chromatin modification required for heterochromatin integrity. Cell. 2010;140:666–677. doi: 10.1016/j.cell.2010.01.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Irvine DV, et al. Argonaute slicing is required for heterochromatic silencing and spreading. Science. 2006;313:1134–1137. doi: 10.1126/science.1128813. [DOI] [PubMed] [Google Scholar]
- 8.Svejstrup JQ. The interface between transcription and mechanisms maintaining genome integrity. Trends Biochem Sci. 2010;35:333–338. doi: 10.1016/j.tibs.2010.02.001. [DOI] [PubMed] [Google Scholar]
- 9.Cam HP, et al. Comprehensive analysis of heterochromatin- and RNAi-mediated epigenetic control of the fission yeast genome. Nat Genet. 2005;37:809–819. doi: 10.1038/ng1602. [DOI] [PubMed] [Google Scholar]
- 10.Djupedal I, et al. RNA Pol II subunit Rpb7 promotes centromeric transcription and RNAi-directed chromatin silencing. Genes Dev. 2005;19:2301–2306. doi: 10.1101/gad.344205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Buhler M, Verdel A, Moazed D. Tethering RITS to a nascent transcript initiates RNAi- and heterochromatin-dependent gene silencing. Cell. 2006;125:873–886. doi: 10.1016/j.cell.2006.04.025. [DOI] [PubMed] [Google Scholar]
- 12.Rosonina E, Kaneko S, Manley JL. Terminating the transcript: breaking up is hard to do. Genes Dev. 2006;20:1050–1056. doi: 10.1101/gad.1431606. [DOI] [PubMed] [Google Scholar]
- 13.Djupedal I, et al. Analysis of small RNA in fission yeast; centromeric siRNAs are potentially generated through a structured RNA. EMBO J. 2009;28:3832–3844. doi: 10.1038/emboj.2009.351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Buhler M, Haas W, Gygi SP, Moazed D. RNAi-dependent and -independent RNA turnover mechanisms contribute to heterochromatic gene silencing. Cell. 2007;129:707–721. doi: 10.1016/j.cell.2007.03.038. [DOI] [PubMed] [Google Scholar]
- 15.Murakami H, et al. Ribonuclease activity of Dis3 is required for mitotic progression and provides a possible link between heterochromatin and kinetochore function. PLoS One. 2007;2:e317. doi: 10.1371/journal.pone.0000317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Smith JG, et al. Replication of centromere II of Schizosaccharomyces pombe. Mol Cell Biol. 1995;15:5165–5172. doi: 10.1128/mcb.15.9.5165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Hayashi MT, Takahashi TS, Nakagawa T, Nakayama J, Masukata H. The heterochromatin protein Swi6/HP1 activates replication origins at the pericentromeric region and silent mating-type locus. Nat Cell Biol. 2009;11:357–362. doi: 10.1038/ncb1845. [DOI] [PubMed] [Google Scholar]
- 18.Lambert S, Watson A, Sheedy DM, Martin B, Carr AM. Gross chromosomal rearrangements and elevated recombination at an inducible site-specific replication fork barrier. Cell. 2005;121:689–702. doi: 10.1016/j.cell.2005.03.022. [DOI] [PubMed] [Google Scholar]
- 19.Shimmoto M, et al. Interactions between Swi1-Swi3, Mrc1 and S phase kinase, Hsk1 may regulate cellular responses to stalled replication forks in fission yeast. Genes Cells. 2009;14:669–682. doi: 10.1111/j.1365-2443.2009.01300.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Segurado M, Gomez M, Antequera F. Increased recombination intermediates and homologous integration hot spots at DNA replication origins. Mol Cell. 2002;10:907–916. doi: 10.1016/s1097-2765(02)00684-6. [DOI] [PubMed] [Google Scholar]
- 21.Minca EC, Kowalski D. Multiple Rad5 activities mediate sister chromatid recombination to bypass DNA damage at stalled replication forks. Mol Cell. 2010;38:649–661. doi: 10.1016/j.molcel.2010.03.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Deshpande AM, Newlon CS. DNA replication fork pause sites dependent on transcription. Science. 1996;272:1030–1033. doi: 10.1126/science.272.5264.1030. [DOI] [PubMed] [Google Scholar]
- 23.Scott KC, Merrett SL, Willard HF. A heterochromatin barrier partitions the fission yeast centromere into discrete chromatin domains. Curr Biol. 2006;16:119–129. doi: 10.1016/j.cub.2005.11.065. [DOI] [PubMed] [Google Scholar]
- 24.Lambert S, et al. Homologous recombination restarts blocked replication forks at the expense of genome rearrangements by template exchange. Mol Cell. 2010;39:346–359. doi: 10.1016/j.molcel.2010.07.015. [DOI] [PubMed] [Google Scholar]
- 25.Groth A, Rocha W, Verreault A, Almouzni G. Chromatin challenges during DNA replication and repair. Cell. 2007;128:721–733. doi: 10.1016/j.cell.2007.01.030. [DOI] [PubMed] [Google Scholar]
- 26.Pirngruber J, Shchebet A, Johnsen SA. Insights into the function of the human P-TEFb component CDK9 in the regulation of chromatin modifications and co-transcriptional mRNA processing. Cell Cycle. 2009;8:3636–3642. doi: 10.4161/cc.8.22.9890. [DOI] [PubMed] [Google Scholar]
- 27.Zhang K, et al. Clr4/Suv39 and RNA quality control factors cooperate to trigger RNAi and suppress antisense RNA. Science. 2011;331:1624–1627. doi: 10.1126/science.1198712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Ghazal G, et al. Yeast RNase III triggers polyadenylation-independent transcription termination. Mol Cell. 2009;36:99–109. doi: 10.1016/j.molcel.2009.07.029. [DOI] [PubMed] [Google Scholar]
- 29.Washburn RS, Gottesman ME. Transcription termination maintains chromosome integrity. Proc Natl Acad Sci U S A. 2011;108:792–797. doi: 10.1073/pnas.1009564108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Pauler FM, Koerner MV, Barlow DP. Silencing by imprinted noncoding RNAs: is transcription the answer? Trends Genet. 2007;23:284–292. doi: 10.1016/j.tig.2007.03.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.