

Abstract
Neisseria gonorrhoeae is an obligate pathogen that hijacks iron from the human iron transport protein, holo-transferrin (Fe2-Tf), by expressing TonB-dependent outer membrane receptor proteins, TbpA and TbpB. Homologous to other TonB-dependent outer membrane transporters, TbpA is thought to consist of a β-barrel with an N-terminal plug domain. Previous reports by our laboratories show that the sequence EIEYE in the plug domain is highly conserved among various bacterial species that express TbpA and plays a crucial role in iron utilization for gonococci. We hypothesize that this highly conserved EIEYE sequence in the TbpA plug, rich in hard oxygen donor groups, binds with Fe3+ through the transport process across the outer membrane through the β-barrel. Sequestration of Fe3+ by the TbpA-plug supports the paradigm that the ferric iron must always remain chelated and controlled throughout the transport process. In order to test this hypothesis here we describe the ability of both the recombinant wild-type plug, and three small peptides that encompass the sequence EIEYE of the plug, to bind Fe3+. This is the first report of the expression/isolation of the recombinant wild-type TbpA plug. Although CD and SUPREX spectroscopies suggest that a non-native structure is observed for the recombinant plug, fluorescence quenching titrations indicate that the wild-type recombinant TbpA plug binds Fe3+ with a conditional log Kd = 7 at pH 7.5, with no evidence of binding at pH 6.3. A recombinant TbpA plug with mutated sequence (NEIEYEN → NEIAAAN) shows no evidence of Fe3+ binding under our experimental set up. Interestingly, in silico modeling with the wild-type plug also predicts a flexible loop structure for the EIEYE sequence under native conditions which once again supports the Fe3+ binding hypothesis. These in vitro observations are consistent with the hypothesis that the EIEYE sequence in the wild-type TbpA plug binds Fe3+ during the outer membrane transport process in vivo.
Introduction
Iron is essential for survival of nearly all organisms, and once assimilated inside the living system it serves various biological functions.1–5 In spite of the high abundance of iron in the earth’s crust, the potential toxicity and extreme aqueous insolubility [Ksp for Fe3+ = 10−38] of free Fe3+ under biological conditions requires a delicate and efficient control mechanism during the iron uptake process.4,6 Living systems increase the bioavailability of this essential nutrient and lower its toxic effects by sequestering iron with biological ligands, such as proteins/peptides and organic chelators. Throughout the process of transport from the environment to the cell, iron remains sequestered.
Most bacterial species, like other living organisms, require iron for survival and they have developed specialized mechanisms to sequester iron from the environment. There are two major routes through which a bacterium can acquire iron from its environment: i) by secreting small Fe3+ binding molecules known as siderophores that ultimately are recognized at the transmembrane receptor proteins and/or ii) by stealing iron directly from host iron transport proteins (eg. transferrin and lactoferrin) with the help of membrane expressed receptors.7–12 The outer membrane expressed receptors found in Gram negative bacteria, which are responsible for iron acquisition, fall into a broader class of proteins known as TonB-dependent transporters. Although these receptors from different bacteria have very low sequence identity in most cases, they are structurally very similar to each other and consist of a β-barrel spanning the outer membrane that is occluded by an N-terminal plug domain.7–12 Once the cargo [siderophore-ferric complex or naked iron] is transported through these outer membrane receptors and reaches the periplasm, it is bound by a soluble periplasmic binding protein. An inner membrane ABC (ATP Binding Cassette) transporter system helps transport iron to the cytoplasm using ATP energy in the final step of iron transport.9–20
N. gonorrhoeae, the human pathogen responsible for the sexually transmitted infection gonorrhea, does not utilize siderophores for iron sequestration from its host.8,21–24 Instead, it expresses iron regulated surface exposed outer membrane receptor proteins, TbpA and TbpB, which recognize, bind and steal iron from holo-transferrin (Fe2-Tf) utilizing TonB derived energy.16,21,22,25–33 The crystal structure of the transferrin receptors for N. gonorrhoeae [TbpA/TbpB] are not yet available, but based on its sequence homology with other iron transporters [e.g. FepA and FhuA found in E. coli] a similar structure is proposed for TbpA. The proposed structure consists of 22 transmembrane β-strands arranged in the form of a barrel with surface exposed loops and an N-terminal plug domain occluding the barrel pore.19, 34–39 Earlier reports have shown that the gonococci cannot survive without TbpA in a medium where transferrin is the only iron source, whereas TbpB makes the iron acquisition more efficient as it can discriminate between holo and apo transferrin.40–44 The iron acquisition process in N. gonorrhoeae can conceptually be divided into the following steps (Fig. 1): recognition and binding of Fe2-Tf at the TbpA/TbpB surface; release of iron from Tf and removal of apo-Tf from TbpA surface using TonB derived energy; passage of “free iron” through the β-barrel of TbpA; binding of the iron at the periplasm by FbpA [which is a part of an ABC transport system]; and finally the passage of iron to the cytosol with the help of FbpB and FbpC (Fig. 1).27,45,46 Although the energy requirement of each step described above is not clearly established, it has been shown by our laboratories in earlier reports that iron removal from holo-Tf by TbpA/TbpB is not TonB energy dependent, whereas removal of apo-transferrin from TbpA requires TonB energy.27,45,46
Fig. 1.

Cartoon representation of the iron acquisition process of N. gonorrhoeae. Holo-transferrin is recognized and bound by TbpA/TbpB at the external surface of the outer membrane. Iron is released from transferrin and the apo-transferrin is released from the outer membrane receptor using TonB derived energy followed by transport of iron into the periplasm. In the periplasm apo-FbpA binds with TbpA and acquires the iron. FbpA then transports the iron across the periplasm and delivers it to the inner membrane receptors FbpB and FbpC which ultimately transport iron into the cytoplasm (the TbpA plug is not shown in the cartoon for clarity).
As mentioned earlier, the hollow pore of the TbpA barrel domain is occluded by the N-terminal plug domain, which probably prevents the passage of small molecules and antibiotics through the barrel. Interestingly, binding of Fe3+-siderophore complexes to similar plug domains in outer membrane transporters during iron internalization has been reported for E. coli receptors FepA and FhuA, which indicates an important role for these plug domains in iron transport rather than just acting as “gate keepers”.34–39, 47 There is considerable debate in the literature as to how the cargo enters the periplasm through a β-barrel transporter where the pore formed by β-barrel is occluded by a N-terminal plug domain. There are two major views regarding how cargo can pass through the barrel pore: i) the plug undergoes a conformational change forming a channel for the cargo to pass through and/or ii) the plug is dislocated completely or partially from the barrel pore making space for the passage of the cargo.10,36,48
Through mutagenesis and epitope (hemagglutinin motif) insertion studies done by us, it was shown that the plug domain of TbpA is surface exposed near the Ala110 residue.35 We have further shown that the sequence EIEYE (118–122) is highly conserved in the TbpA-plug domain from different bacteria that express TbpA and triple-alanine substitution of the Glu120. Tyr121 and Glu122 in a tbpA mutant (EIAAA 118–122) showed an 80% reduction in transferrin bound iron utilization (although the mutant binds transferrin with wild type affinity).19 Thus, these experiments demonstrate the importance of the TbpA-plug sequence EIEYE in transferrin-iron utilization by gonococci. Due to the high insolubility and toxicity of Fe3+, biological systems evolve so as to scavenge and coordinate available free Fe3+. The EIEYE motif is conserved and has an abundance of hard donor atoms suitable for coordinate-covalent bonding to Fe3+. Therefore we hypothesize that iron released from Tf at the TbpA surface interacts and binds with this plug sequence (EIEYE) and is subsequently transported through the transporter β-barrel.
To test this hypothesis, we carried out a series of in vitro and in silico analyses on the wild-type recombinant and mutated recombinant TbpA plug samples and small peptides (S1, S2 and S3) that mimic specific segments of the wild-type sequence of the TbpA plug domain (Fig. 2). The secondary structures of the recombinant plug samples (wild-type and mutated) and a probable change in the structures due to addition of Fe3+ were investigated using circular dichroism spectroscopy. The folding characteristics of the wild-type recombinant sample at different pH values in the absence and presence of Fe3+ (from different iron sources) were investigated by SUPREX (Stability of Unpurified Proteins from Rates of H/D Exchange), a MALDI-TOF method.49,50 Fluorescence emission titrations were performed to determine the conditonal binding constants of Fe3+ with the wild-type recombinant plug, the mutated recombinant plug sample and the model peptides. Finally, three protein models of the wild-type and triple alanine substituted (EIAAA) TbpA plugs were generated. These predict that EIEYE is part of a flexible loop that can act as an iron-binding site.
Fig. 2.

Cartoon representation of the wild-type recombinant TbpA plug, the mutated recombinant TbpA plug and the short peptides (S1, S2 and S3). The proposed iron binding sequence (EIEYE) is preserved in the wild-type recombinant plug and in the model peptides. However in the mutated recombinant plug this sequence is altered by alanine substitution of the three key potential iron binding residues making the sequence EIAAA. The three other tyrosine residues, Tyr58, 75 and 98, are also shown in the figure for both wild type and mutated recombinant plug proteins.
Materials and methods
Buffers used for circular dichroism, SUPREX and fluorescence studies were prepared in deionized water and chelexed (BioRad) over night to avoid iron contamination prior to use. Potassium dihydrogenphosphate, sodium perchlorate, EDTA, NTA, (Aldrich Chemical Company), Tris (Boehringer Mannheim Laboratory Reagents), Fmoc protected amino acids (CHEM IMPEX International) and the resin used for the solid state peptide synthesis (NOVA Biochem) were used as received. Buffers for SUPREX were made in D2O and the pD adjusted with NaOD or DCl (Aldrich Chemical Company). A stock solution of Fe(III)NTA was made in 50 mM MES, 200mM KCl, pH 6.5 and allowed to equilibrate overnight. Using 1:1 ligand to metal ratio, the speciation of the solution at this pH is 50% FeNTAH−1 and 50% FeNTAH−2.51 The material used for protein expression and purification are provided in the following sections.
Recombinant protein purification
Generation of recombinant E. coli strains over-expressing His-tagged plug proteins
The tbpA plug expression plasmids were constructed by PCR amplification of gonococcal chromosomal DNA with oVCU289 (CATATGGAAAATGTGCAAGCCGGACAAGC) and oVCU290 (CTCGAGCCTGCCTTCCCCGATAACATCGTCG). The resulting amplicons were initially cloned into pCR2.1TOPO and then subcloned into pET-22b(+) using restriction sites engineered into the oligonucleotide primers (sequences shown in bold). The resultant plasmids encoded 162 amino acids (E1 to R162) of the mature plug domain fused at the carboxy-terminus to the His6 tag. Plasmid pVCU263 was transformed into E. coli strain BL21(DE3), generating RIC264, which expressed the wild-type plug protein. Plasmid pVCU273 was transformed into E. coli strain BL21(DE3), generating RIC279, which expressed the triple-alanine substituted mutant of the plug protein. The DNA insert in pVCU273 was generated by PCR amplification from chomosomal DNA template isolated from the triple alanine mutant strain (MCV260) 19. The triple alanine mutant of N. gonorrhoeae was previously characterized as described.19
Protein expression and purification
Cultures of recombinant E. coli strains RIC264 and RIC279 were grown in LB media containing 100 μg/ml ampicillin. When cultures reached an optical density of between 0.4–0.8, protein expression was induced by the addition of 1mM IPTG (isopropyl-B-D-thiogalactopyranoside) (Sigma). The cultures were allowed to grow in the presence of IPTG for four hours at 37°C. After induction, cultures were centrifuged, washed and centrifuged again. The final cell pellets were resuspended in sterile IX PBS (pH 7.2) and stored at −20°C overnight. The bacterial pellets were thawed on ice and then resuspended in lysis buffer (50mM NaH2PO4, 300 mM NaCl, 10mM Imidazole, at pH 8.0). Lysozyme and His-tagged protease cocktail inhibitor (BioRad) were added at a concentration of 1mg/ml and 1ml/20gm wet weight, respectively. The suspension was incubated on ice for 30 min and then sonicated. The resulting lysate was centrifuged at 10,000 × g for 30 min to remove unlysed cells and debris. The supernatant was incubated with Ni-NTA agarose (Qiagen) overnight at 4°C. Subsequently, the Ni-NTA resin plus bound protein was added to a column and the flow-through fraction was collected in a fresh tube. The resin in the column was washed twice with wash buffer (50mM NaH2PO4, 300 mM NaCl, 20mM Imidazole, at pH 8.0). Finally, the recombinant proteins were eluted from the column in elution buffer (50mM NaH2PO4, 300 mM NaCl. 250mM Imidazole, at pH 8.0). Protein samples were solubilized in SDS (sodium dodecyl sulfate) containing loading dye and separated on 10% SDS-polyacrylamide gels along with molecular weight standards and concentration control (lysozyme).52 Gels were stained with Coomassie blue and subsequently destained in a solution containing 10% acetic acid and 40% methanol (Fig. 3). The purified proteins were dialyzed against IX PBS (pH 7.2) to remove the imidazole. The protein samples were aliquoted to avoid repeated freeze-thawing and each of the aliquots contained 2 mg/100 ml of protein.
Fig. 3.
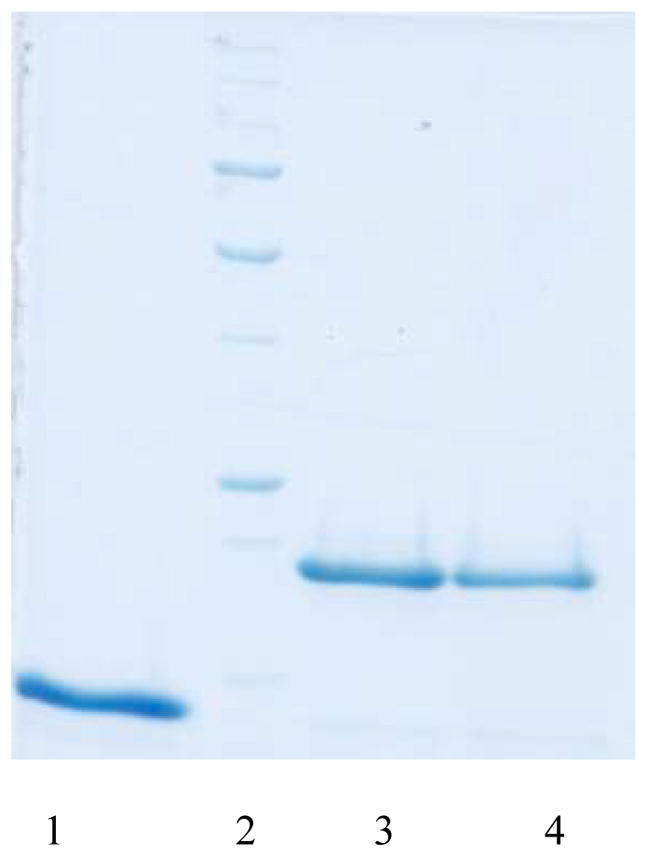
Characterization of purified wild-type and mutated recombinant TbpA plug proteins. Plug proteins were separated by SDS-PAGE and subsequently visualized with Coomassie blue. The lanes contain the following samples: Lane 1, 20 μg lysozyme; Lane 2, molecular weight markers; Lane 3, 20 μg wild-type plug protein; Lane 4, 20 μg EYE mutant plug protein.
Peptide synthesis
The small peptides with sequences NEIEYEN (S1), SSGAINEIEYENVKAVEISK (S2) and NEIEYENVKAVEISKGSNSV (S3) were synthesized in 0.1 mmol scale on PAL-PEG-PS resin (Applied Biosystems). Standard Fmoc (9-fluorenylmethoxy-carbonyl)-protected amino acids (Chem-Impex and Novabiochem) were coupled in 20 min cycles with HBTU (O-benzotriazole-N,N,N′,N′-tetramethyluronium hexafluorophosphate) (Novabiochem) and N-methylmorpholine (NMM) (Acros) in N,N′-dimethylformamide (DMF) (Caledon). Fmoc protecting groups were removed by using 20% piperidine in DMF. The N-termini of peptides were acetylated using acetic anhydride and NMM. Cleavage from the resin and removal of side chain protecting groups was accomplished by treating the resin with a 10 mL mixture of 95% trifluoroacetic acid (TFA) and 2.5% triisopropylsilane (TIS) under nitrogen while shaking for 4 h. Peptide was precipitated from solution by evaporating off TFA with a nitrogen stream, followed by three washes with diethyl ether (Caledon). Purification was accomplished by semi-preparative reversed-phase HPLC on a YMC C18 column with a linear 40 min gradient from 7 to 93% acetonitrile in water with 0.1% TFA. Purity was validated to be greater than 95% by analytical HPLC. The mass of each peptide was determined by ESI-MS.
Circular dichroism (CD)
CD spectra of the wild-type and mutated recombinant TbpA-plug samples were recorded on an Aviv model 202 CD spectropolarimeter in 50 mM NaClO4 chelexed buffer at pH 7.5. Typically 5 μL of the wild type/mutated plug (10/20 μM respectively) was diluted in 3 ml of the above buffer in the absence or presence of increasing concentrations of Fe(ClO4)3 (0–10 μM). Spectra were acquired between 190–300 nm at 25°C. The raw data (in millidegrees), after correction for buffer and cuvette contributions, were converted to mean residue ellipticity (in degrees squared cm per decimole) using Equation (1), where
| (1) |
C = mM concentration of the plug samples, n = number of residues in the plugs (162 amino acids for both wild type and mutated recombinant plugs) and 1 = path length in cm.53
Stability of Unpurified Proteins from rates of H/D Exchange (SUPREX)
In order to study the effect of pH on the folding and Fe3+ sequestering ability of the wild-type recombinant TbpA-plug sample, SUPREX experiments were performed in 50 mM MES with 200 mM KCl (pH = 6.3), 50 mM Tris (pH = 7) and 20 mM phosphate (pH = 7.2) using a high sensitivity SUPREX protocol.49,50,54 The wild-type recombinant plug (200 μM) was incubated with FeNTA (2 mM) for 15 minutes prior to SUPREX analysis. The guanidinium chloride (GdmCl) concentrations varied from 0–6 M and an exchange time of 5 minutes was maintained. For the other set of SUPREX experiments 50 mM Tris buffer at pH 7 was used and wild-type recombinant TbpA-plug was incubated with Fe(ClO4)3. The GdCl concentration was varied from 0–3 M using a 5 min exchange time. In all experiments, concentrated proteins were diluted ten times in SUPREX buffers in absence or presence of iron (> 10 fold excess). The exchange reactions were quenched by 0.1% TFA followed by the addition of a saturated solution of sinapinic acid in 1:1 acetonitrile-water mixture. Data analyses were performed as described previously.49,50,54
Fluorescence titration
Fluorescence spectroscopy was used to calculate the binding affinity of the wild-type and mutated recombinant TbpA plugs, and model peptides S1, S2 and S3 (Fig. 2) with Fe3+. Fluorescence spectra were recorded in 100 mM Tris at pH 7.5 on JOBIN-YVON-SPEX Fluorolog3 fluorimeter in right angle mode.
The wild-type recombinant TbpA plug and the peptides S1, S2 and S3 all have at least one tyrosine residue (Fig. 2) and thus are capable of giving an emission band at 310 nm when excited at 285 nm. When a free tyrosine is complexed with Fe3+ this emission is quenched and by following this tyrosine quenching at 310 nm as a function of increasing Fe3+ concentration we calculated the dissociation constant for the equilibrium shown in Equation (2), where P represents protein or peptide and Fe represents Fe3+.
| (2) |
As stated in the Introduction, the solubility of Fe3+ at physiological pH is very low due to hydrolysis and this imposes a limit to the titrations described in this work. Moreover as we anticipated a weak binding between the plug/peptide and Fe3+ the concentration of Fe3+ was kept low throughout the titration to avoid Fe3+ precipitation from the protein/peptide solution. Under this limiting condition the dissociation constant that we calculate for Equation (2) is a conditional Kd (Equation (3)) and is not a “true” thermodynamic parameter.55,56
| (3) |
[P]eq. and [PFe]eq. in Equation (3) represent the equilibrium concentration of free protein/peptide and protein/peptide-Fe3+ complex respectively and [Fe]T is the total concentration of bound and free Fe3+. The concentration of the wild-type protein used for the emission study was 20 μM and for the peptides was kept at either 20 or 50 μM.
For the fluorescence experiments described here the emission intensity at 310 nm before starting the titration experiment is a measure of total protein concentration and at each stage of the titration the emission intensity at 310 nm is due to the available free protein in solution. Thus we assume the quenching in fluorescence by adding Fe3+ is proportional to the fraction of protein bound (Fb).55,56
| (4) |
Replacing the value of [PFe]eq from Equation (4) into Equation (3) and on rearrangement we obtain:55,56
| (5) |
If F0 is the emission intensity of the protein/peptide before addition of Fe3+ and Fi is the emission intensity at each stage of the titration, then percentage quenching (Q%) can be defined as:55,56
| (6) |
Since Q% is directly proportional to Fb under the experimental conditions used,55 a plot Q% vs [Fe]T is a plot of Equation (7), which can yield the conditional Kd.
| (6) |
Qmax represents the maximum quenching that can be obtained at the end of the titration. Kd and Qmax values were obtained from plots of Equation (7) using SigmaPlot version 9.0.55,56 The Kd values reported here are the result of four independent determinations for the wild-type recombinant TbpA plug and model peptide S1.
Protein model creation
The plug domain from Neisseria gonorrhoeae TbpA (GenBank: YP_208545) includes 162 aa (including the predicted TonB box, but not including the 24 aa signal peptide). This sequence was submitted to Swiss-Model,57 Phyre2 58 and I-TASSER,59 and the resulting models were evaluated with Erratv2 60 and ProCheck 61 using the SAVE server at UCLA (http://nihserver.mbi.ucla.edu/SAVES/). The best model. generated by I-TASSER, had an ERRAT quality score of 98.7. The top templates chosen by I-TASSER for model creation were 3FHH:A (ShuA heme transporter from Shigella 62) and 2HDI:A and 2GSK:A (Cir colicin receptor and BtuA cobalamin transporter from Escherichia coli). I-TASSER was also used to create a model of the TbpA plug with three amino acids (120–122) changed from EYE to AAA. The mutated TbpA model had an ERRAT score of 82.6 and used the same templates as the original model. PDBsum (http://www.ebi.ac.uk/pdbsum/) was used to create topology plots of the two models.63
Results and discussion
CD spectroscopy: Wild-type recombinant and mutated recombinant TbpA-plugs are predominantly unfolded
We examined the secondary structural content of the wild-type recombinant and mutated recombinant plug domain of TbpA expressed in E. coli without the barrel. The CD spectra of the wild-type and mutated recombinant TbpA-plug samples are presented in Fig. 4. In the absence of any Fe3+ both protein samples showed strong negative CD signals centered at 200 nm indicating mostly unfolded structures (solid lines in Fig. 4).47 Usher et al has previously reported similar CD behavior for recombinant wild type FepA plug.47 An unfolded structure for the recombinant TbpA plugs, expressed without the barrel, is not unexpected given that when inside the barrel the plug is expected to form H-bonds. which will stabilize and induce native secondary structure. Thus our CD result is consistent with previous observations for the expressed FepA plug.47
Fig. 4.

CD spectra of A) wild-type recombinant and B) mutated recombinant TbpA plug in 50 mM NaClO4 buffer at pH 7.5, [wild-type recombinant TbpA plug] = 10 μM, [mutated recombinant TbpA plug] = 20 μM and [Fe(ClO4)3] = 0–10 μM. The solid line spectra shown in A) and B) are for wild-type or mutated recombinant TbpA plug proteins in the absence of Fe3+, and the dashed and dotted lines are after addition of Fe3+. Both spectra show strong negative bands centered at 200 nm indicating a mostly unfolded structure. Fe3+ addition did not induce any significant structural change for either, which is reflected by very small change in CD absorption signals.
In homologous β-barrel iron transport proteins from E. coli, the respective cargo (in those cases an iron-siderophore complex) has been shown to interact with the recombinant plug domain of that system.34–39,47 To investigate if a similar interaction between Fe3+ and the wild-type or mutated recombinant TbpA plug proteins can give rise to a change in the conformation of the plug, we carried out CD titration experiments with Fe3+. As shown in Fig. 4 these titration experiments did not exhibit any significant change (either in intensity or position) in the CD signal (dashed and dotted lines in Fig. 4) for either of the protein samples, which suggests no or very little structural/conformational change in both wild-type and mutated recombinant TbpA plug proteins subsequent to Fe3+ addition. This observation does not discount a binding event, but rather suggests that even if a binding event does occur it does not produce a structural change in the plug that can be monitored by CD.
SUPREX: The folding behavior of wild-type recombinant TbpA-plug does not change with pH and the presence of iron
SUPREX provides information regarding the folding thermodynamics of proteins.49,50,54,64–66 Here we used SUPREX as a complement to our CD studies to further examine the folding behavior of the wild-type recombinant TbpA plug as a function of pH and in the presence of Fe3+. This represents the first characterization of the mass and the folding energetics of the wild-type recombinant TbpA plug domain.
The mass of the wild-type recombinant TbpA-plug, measured using MALDI-MS with trypsin inhibitor protein as an internal standard, was calculated to be 18282 ±3 Da. A set of SUPREX experiments at pH 6.3 and 7.2 were carried out to see if the wild-type recombinant plug shows any observable difference in folding and unfolding energetics due to a change in pH; none were observed. The ΔMass values shown in Fig. 5 are all around 100 Da. Considering that ~30–40% of the amide protons exchanged for deuterons during the H/D exchange reaction in SUPREX are typically back-exchanged to protons during the MALDI analysis step, the data in Fig. 5 suggest that nearly all of the amide protons in the 162 amino acid wild-type recombinant TbpA-plug were exchanged for deuterons in the SUPREX experiment. This and the absence of clear denaturant dependence of the ΔMass values indicates there are no globally protected protons in the wild-type recombinant TbpA plug samples at pH 6.3 or 7.2. These data are consistent with a predominantly unfolded protein, or a protein that does not contain a detectable amount of secondary or tertiary structure at these pH values. This is not surprising given that the plug was expressed without the β-barrel, and thus was never in an in vivo like environment to promote folding.
Fig. 5.

SUPREX behavior of wild-type recombinant TbpA plug at pD 7.4 (dark squares) and pD 6.5 (open circles). Change in observed protein mass due to H/D exchange as a function of denaturant concentration is plotted to obtain the SUPREX curve. Conditions: exchange time 5 min, trypsin inhibitor internal standard, 50 mM MES buffer/200 mM KCl pD 6.5 or 20 mM phosphate buffer/pD 7.4. Error bars indicate the variation between the 10 mass spectra collected for each denaturant concentration. When error bars are not visible the mass variation is smaller than the symbol designating the datum point.
The interaction between the wild-type recombinant TbpA-plug and FeNTA at pH 6.3 and 7.2 was also monitored using SUPREX (similar SUPREX plot as Fig. 5; data not shown) and no change of SUPREX behavior was observed. Apparently either NTA is too strong an iron chelator, thus precluding any competitive binding by the wild-type recombinant plug with a lower binding affinity, under our experimental conditions, or Fe3+ binding does not appreciably alter the folding stability of the recombinant plug protein. An analogous SUPREX experiment was performed using Fe(ClO4)3 as the iron source instead of FeNTA in 50 mM Tris buffer at pH 7 with very similar results; that is, the presence of Fe(ClO4)3 does not cause any detectable change in the SUPREX behavior of the wild-type recombinant plug as well (data not shown).
These SUPREX results neither confirm nor disprove the hypothesis that the wild-type TbpA plug binds iron temporarily during the transport mechanism. Rather these data suggest that Fe3+ binding is either relatively weak and/or not significant, or if binding does occur, it does not change the global unfolding/refolding properties of the wild-type recombinant TbpA plug. These results are consistent with the CD data. Both CD and SUPREX suggest a largely unfolded structure both in the presence and absence of Fe3+ for the recombinant plug protein, although smaller and more local structural changes induced by Fe3+ can not be ruled out.
Fluorescence titration: The wild-type recombinant TbpA plug and peptide models exhibit weak binding to Fe3+
In general, tryptophan and tyrosine residues are responsible for fluorescence emission properties of proteins. The surface exposed tyrosines emit at 310 nm when excited at 285 nm around neutral pH (whereas tyrosinate will emit around 334 nm).67 TbpA wild-type recombinant plug contains four tyrosines. one of which is part of the EIEYE sequence (Fig. 2). On the other hand, in the mutated recombinant plug, the tyrosine is replaced by an alanine (EIAAA), leaving the remaining three tyrosine residues intact (Fig. 2). The model peptides S1, S2 and S3 all have one tyrosine each (Fig. 2). Binding between these tyrosine(s) and Fe3+ would lead to a decrease in free tyrosine(s) in solution and a subsequent quenching of this 310 nm band. Consequently, fluorescence emission experiments provide an opportunity to quantitatively investigate binding interactions between the TbpA plugs/model peptides and Fe3+ using the 310 nm band as a probe.
The inset of Fig. 6A shows the fluorescence emission titration spectra for a wild-type recombinant TbpA plug sample in 100 mM Tris buffer at pH 7.5 when increasing aliquots of Fe3+ were added to the protein solution. Care was taken while adding Fe3+ to a solution of the protein/peptide to avoid precipitation. The 310 nm band shows quenching upon addition of Fe3+ solution, indicating a binding event. (An emission band was also observed around 405 nm the intensity of which fluctuated with time. This peak can be attributed to an excited state complex formation between two tyrosine residues).67 Fig. 6A is a plot of % quenching (Q%) at 310 nm as a function of added Fe3+ ([Fe]T) according to Equation (7). The conditional Kd for the wild-type recombinant TbpA plug with Fe3+ is 10−7 M (Equation (2)). The model peptides S1, S2 and S3 (Fig. 2) were similarly titrated with Fe3+. The fluorescence emission titration spectra for S1 is presented in the inset of Fig. 6B and shows similar quenching behavior as the wild-type recombinant plug (representative fluorescence emission spectra for model peptide S3 is presented in Supplementary Material as Fig. SP1). Titration data analyses for the model peptides according to Equation (7) are shown in Fig 6B and in Fig. SP2, with conditional Kd values calculated for S1, S2 and S3 as 1 × 10−4, 5 × 10−4 and 1 × 10−6 M respectively.
Fig. 6.

Repesentative plots of % quenching of the 310 nm fluorescence emission band (Q%) vs total Fe3+ added ([Fe]T) according to Equation (7) for A) wild-type recombinant TbpA plug and B) S1 peptide in 100 mM Tris buffer at pH 7.5, [wild type recombinant plug] = 20 μM; [S1] = 20 μM. The dots represent actual data points and the smooth lines are the best fit for the data according to Equation (7) where Qmax = 40% and Kd = 1 × 10−7 M for A) and Qmax = 100% and Kd = 1 × 10−4 M for B). The insets show the titration spectra of A) recombinant wild-type TbpA plug (20 μM) and B) S1 peptide (20 μM), in the absence (highest intensity band) and in presence of increasing aliquots of Fe(ClO4)3. Conditions: 100 mM Tris at pH 7.5, excitation at 285 nm, using a 1 cm path length cuvette at room temperature the 310 nm band is for surface exposed tyrosine(s); fluorescence data collected in a quartz cuvette of 1 cm path length at room temperature; the final concentration for the wild-type plug titration is [Protein]:[Fe3+] = 20:1 and that of for S1 is [S1]:[Fe3+] = 1:50.
The fact that we observe different conditional Kd values for different peptides all containing at least one tyrosine residue is an indication that the conditional Kd obtained using our fluorescence assay method is not just a characteristic of any surface exposed tyrosine in a protein/peptide. The higher binding affinity observed for the wild-type TbpA plug protein compared to the smallest peptide S1 may be due to participation of other amino acids in stabilizing the protein/peptide-Fe3+ complex. And lastly, S1 being a very small peptide, is not expected to have any secondary structure and hence the emission band at 310 nm for solvent exposed tyrosine is reasonable. The fact that we observed an emission band at the same position for the wild-type TbpA plug protein is an indication that at least one of the total of four tyrosines in the wild-type recombinant plug is solvent exposed as well.
In the plots of Equation 7 for the wild-type recombinant TbpA plug protein the Qmax value is ~ 40%, whereas for the model peptides Qmax ~100% (Fig. 6B and Fig. SP2). The wild-type recombinant TbpA plug protein has four tyrosine residues which can potentially compete for the added Fe3+ whereas each peptide has just one tyrosine which is part of the EIEYE sequence. The fact that even at the theoretical end point of the titration the Qmax for the recombinant TbpA plug protein does not reach 100% indicates that not all of the tyrosines present in the protein show binding and hence quenching upon Fe3+ addition. On the other hand as the peptides show a Qmax ~100% this indicates that at the theoretical end point all of the available tyrosines are bound to added Fe3+. This suggests that in the wild-type recombinant TbpA plug there are at least two different types of tyrosine residues giving rise to the 310 nm band present under our experimental conditions. The first type interacts and binds with Fe3+ and gives rise to the quenching of the 310 nm band where as the second type does not interact with this ligand and does not show quenching. Further, as the model peptides S1, S2 and S3 all contain this putative Fe3+ binding site (EIEYE) and show a 100% quenching of the 310 nm band upon ligand binding, we suggest that this sequence (EIEYE) is actually responsible for showing quenching in the wild-type recombinant plug and binds with Fe3+. This is further confirmed by experiments using the mutant TbpA plug described below.
Fluorescence emission experiments were performed with the mutant TbpA plug (EIAAA; Fig. 2) to further establish the Fe3+ binding site in the wild-type plug. The fluorescence emission spectra for the mutated recombinant plug (EIEYE → EIAAA) did not show an emission maximum at 310 nm (Fig. SP3). Instead, a new band is observed that is centered at 334 nm which showed very large fluctuations in intensity with time. From the literature it is known that an emission band around 334 nm for proteins that contain only tyrosines as the fluorophore arise due to formation of free tyrosinate.67 Tyrosine in the ground state has a pKa ~ 11 and at the pHa of our experiments (7.5) it should not deprotonate. However, the excited state pKa value for tyrosine is ~ 4, and therefore in the presence of nearby proton acceptors tyrosinate can be formed at our experimental pH.67 The fluctuations observed in the intensity of the 334 nm band is another good indication of excited state deprotonation phenomena. Stabilization of the 334 nm band for the mutated recombinant TbpA plug may be interpreted as due to subtle and local changes in protein structure, which were not detectable by CD or SUPREX, and bringing in close proximity amino acid residues which will stabilize the tyrosinate residue at the excited state. Although the wild-type and mutated recombinant TbpA plug emission properties were not comparable, we propose, based on the lack of a 310 nm band or quenching in the presence of Fe3+ for the wild-type recombinant plug, that Fe3+ does not bind specifically to the mutated recombinant plug. This supports the EIEYE sequence as the Fe3+ binding site in the wild-type TbpA plug. This is further supported by the computer modeling discussed below.
Fluorescence emission spectra of the wild-type TbpA plug at pH 6.4 were very similar to that observed at physiological pH (spectra not shown). Upon addition of increasing amounts of Fe3+ solution to the TbpA plug protein the 310 nm band did not show any significant quenching. This can be interpreted as at this pH the tyrosines in the wild-type plug do not bind Fe3+ and hence show no quenching. Consequently, we observe a difference in iron binding to the TbpA plug depending on the pH. This is consistent with protonation or environmental pH possibly playing a role in iron release from the plug.
The reversibility of Fe3+ binding to the wild-type recombinant TbpA plug and model peptides was established by exposing an Fe3+ equilibrated protein or peptide sample to EDTA in 1:1, 1:5 and 1:10 protein/peptide:EDTA ratios. Emission spectra were recorded after 45 mins with recovery of emission intensity. This observation is consistent with reversible binding (necessary for in vivo periplasmic hand-off of Fe3+ to FbpA) and indicates that the quenching observed by addition of Fe3+ to the protein/peptide solutions is not due to paramagnetic quenching. This also confirms that the protein/peptide has a relatively low Fe3+ binding constant and cannot effectively compete for Fe3+ with a strong chelator like EDTA or FbpA.
TbpA wild-type and mutant plug models: EIEYE is part of a flexible loop near a surface exposed “top hat” region of the plug
The wild-type and mutant TbpA plug models under native conditions predicted by I-TASSER are shown in Fig. 7. In both models the core of the plug domain includes a series of beta strands connected by loops of varying lengths. These are also found in the 3FHH ShuA template structure, which shares a 28% sequence identity with the query sequences. The RMSD (Root Mean Square Deviation) value for the wild-type model/template comparison was 1.02.
Fig. 7.

Predicted structure of Neisseria gonorrhoeae TbpA plug domain. (A) Wild-type; (B) Mutant (EIEYE → EIAAA). The EIEYE sequence is postulated to be important for iron transport functions. Tyrosine side chains are shown as wire-frames. The possible importance of the “Top Hat” region (red circle) for interaction with transferrin is discussed in the text. The model was created with I-TASSER based on amino acids 25–187 from NCBI sequence YP_208545.
The EIEYE motif is in a long loop, anchored by beta strands. Just prior to this motif is another sequence, strongly conserved among the Neisseria species (Fig. 8), which forms a loop at the top of the molecule (‘top hat’ region). It has been shown previously that a portion of the TbpA plug is surface-exposed, specifically Ala110,35 and this residue lies within this top region along with one of the four tyrosines (Tyr98) found in the TbpA plug (Fig. 9). The predicted TonB box sequence (DTITV) lies within the N-terminal beta strand, as found in other plug domain structures.68 For the mutated TbpA structure, the EIAAA motif is still part of a loop. However, the change of just three amino acids caused a predicted overall shape change in the model (Fig. 7 and 9) particularly noticeable in the ‘top hat’ region.
Fig. 8.

Conservation of the NEIEYEN sequence and ‘Top Hat’ regions among gonococci. Dark shading represents conserved sequence positions; the orange block highlights the sequence conserved in Neisseria sequences. Stars indicated the positions of Tyr98 and Ala110.
Fig. 9.

Comparison of topology of wild type and mutant plug domain structures. Pink arrows represent beta strands; red tubes represent alpha helices. While the beta strand backbones of the TbpA model are nearly identical to the ShuA protein structure used as a template (data not shown), a change of just three amino acids in the TbpA sequence had dramatic effects on the overall predicted structure. Yellow spots indicate position of tyrosines. Images created with PDBsum.
Prior to binding Fe3+, the region of the plug containing the sequence EIEYE needs to reorganize its donor atoms to create a maximum binding interaction. This reorganization would require more energy if this sequence was part of a rigid secondary structure; however, as predicted by the present model, this sequence is in a flexible loop, making this reorganization more facile and supporting the hypothesis of ligand binding. These data also support our CD and SUPREX experiments on wild-type and mutated recombinant TbpA plugs where we did not observe any change in structure/folding stability of the proteins in the presence of Fe3+. There are three more tyrosines (Tyr 58, 75 and 98) in the TbpA plug that can act as potential donors for Tf released Fe3+. However, since the sequence EIEYE is very near the surface exposed top hat region and contains more than one potential donor group, this suggests that Fe3+ will have a preference for this sequence over the isolated tyrosines. Finally, by mutating this sequence (EIEYE) to EIAAA, the most dramatic change in the structure of the plug was observed in the surface exposed top hat region. Our previous report demonstrated that this mutant binds Tf with wild-type affinity, but shows an 80% reduction in iron uptake (TbpB− background) in an environment where Tf is the sole iron source.19 These two facts taken together indicate the importance of this region of the TbpA plug in Tf-iron internalization.
Conclusions
The previous in vivo experiments reported by our group and in vitro and in silico results reported here strongly support our hypothesis that the EIEYE conserved sequence has the potential to bind Fe3+ as it is released from Tf at the TbpA/TbpB receptor. In the experiments reported here we used wild-type and triple alanine mutated recombinant TbpA plugs (without the barrel) purified from E. coli. In addition we synthesized the small peptides S1, S2 and S3 which encompass the EIEYE sequence of the plug that is hypothesized to bind iron.
Both CD and SUPREX yielded results that suggest a predominantly unfolded structure for the recombinant plug samples. We interpret this as a result of the fact that the plugs were expressed outside of the barrel in a heterologous bacterial expression system and consequently did not fold into a native conformation. Using fluorescence quenching spectroscopy we calculate conditional Kd values for Fe3+ with the wild type recombinant plug of TbpA (10−7M) and model peptides S1, S2 and S3 (10−4 to 10−6 M) at pH 7.5. The wild-type plug did not show any quenching of its tyrosine band upon addition of Fe3+ at pH 6.4, indicating that although the plug can bind Fe3+ at physiological pH, it loses this property at slightly acidic pH, suggesting protons or environmental pH could play a role in iron release from the plug in the periplasm. We did not detect Fe3+ binding for the mutated plug (EIEYE → EIAAA), further supporting the hypothesis that the EIEYE sequence of the plug is involved in Fe3+ sequestration and transport through the outer membrane. This is consistent with in vivo studies where an 80% reduction in transferrin bound iron utilization was observed for the mutant relative to the wild type plug.19
The binding event between the sequence EIEYE and Fe3+ is also supported by in silico modeling, as this sequence is part of a flexible loop that can reorganize more easily around the cargo. Moreover, the models predict another important conserved sequence, which is surface exposed and shows that a considerable conformational change between the wild type and recombinant plug is an important region for Tf-iron utilization. We hypothesize that this “top hat” region has the potential to act as a secondary recognition site for Tf during the iron internalization process. This hypothesis suggests further biological and biophysical experiments.
As mentioned the wild-type and mutated recombinant plugs do not have the native structure that these will have when inside the barrel under native conditions. However, it is predicted by our modeling study that the EIEYE sequence in the wild-type TbpA plug under native conditions is part of an “unstructured” flexible loop. This gives us the opportunity to compare our recombinant plug (having an unfolded structure) results with plugs grown under native conditions, as the scope of this work concentrates on the sequence EIEYE of the TbpA plug. Our results suggest that although the recombinant wild type TbpA plug does not have its native conformation, it still has the ability to bind its presumed cargo, Fe3+, even without the barrel. A different binding interaction between the sequence EIEYE from TbpA plug and Fe3+ under in vivo conditions would not be surprising, given that the dielectric constant, hydrophobic environment, and a change in protein structure may facilitate this binding. Nevertheless the low binding constant (Kd ~10−7 M) of the recombinant plug with Fe3+ in absence of the barrel is very reasonable as ultimately the Fe3+ cargo has to bind with the periplasmic iron binding and transporter protein FbpA. So to avoid competition and to keep the iron trafficking unidirectional, a low binding constant is biologically relevant.
A proposed, stepwise process for Fe3+ transport in Neisseria gonnorhoeae is illustrated in Fig. 10 which shows a cascade of events whereby Fe3+ is handed off from the cell exterior to the periplasm such that it is never present as “naked” or unchelated iron. The strong affinity of Tf for Fe3+ is diminished by docking at the exterior surface of TbpA, presumably via a conformational change,45 releasing the Fe3+ to the β-barrel interior. The Fe3+ then moves through the barrel weakly bound to the EIEYE sequence of the plug. Weak binding to the plug facilitates the exchange of Fe3+ to FbpA at the interior lip of the TbpA receptor. Fe3+ is strongly bound to FbpA, which is a part of the periplasmic ABC transport system that delivers iron to the cytosol.45
Fig. 10.

A) Cartoon representation of the proposed stepwise movement of Fe3+ from transferrin across the outer membrane and insertion into periplasmic iron transport protein FbpA: (i) Diffusion of Fe loaded Tf (Fe3+ tightly bound with conditional Kd ~10−20 M) to the outer surface of TbpA/TbpB receptor; (ii) Fe2Tf docking at TbpA (Fe2Tf Kd ≫ 10−20 M) where Fe3+ is released;45 (iii) TbpA plug “catches” released Fe3+ by binding at the EIEYE sequence and moves through the β-barrel of TbpA; (iv) Fe3+ bound to periplasm exposed plug (conditional Kd = 10−7 M) is handed off to apo-FbpA bound to inner lib of TbpA; (v) FeFbpA (conditional Kd ~10−18 M) diffuses through the periplasm ultimately releasing the iron at the inner membrane receptor TbpB/TbpC.45 Kd values shown are conditional not true thermodynamic constants. Relative values, however, illustrate the feasibility of unidirectional Fe3+ exchange across the outer membrane and into the periplasm.
Supplementary Material
Acknowledgments
ALC thanks the National Science Foundation (CHE 0809466) for funding this work and the laboratory of Prof. K. J. Franz, especially Drew Folk, Duke University for helping in the solid state peptide synthesis. CNC thanks the National Institute of Allergy and Infectious Diseases (AI147141) for funding these and previous studies of the gonococcal TbpA plug domain and MCF thanks the National Institutes of Health (GM084174) for funding the SUPREX experiments.
References
- 1.Briat JF. J Gen Microbiol. 1992;138:2475–2483. doi: 10.1099/00221287-138-12-2475. [DOI] [PubMed] [Google Scholar]
- 2.Griffiths E. Iron and Infection: molecular physiology and clinical aspects. :69–137. [Google Scholar]
- 3.Imbert M, Blondeau R. Curr Microbiol. 1998;37:64–66. doi: 10.1007/s002849900339. [DOI] [PubMed] [Google Scholar]
- 4.Ratledge C, Dover LG. Annu Rev Microbiol. 2000;54:881–941. doi: 10.1146/annurev.micro.54.1.881. [DOI] [PubMed] [Google Scholar]
- 5.Weinberg ED. Perspect Biol Med. 1997;40:578–583. doi: 10.1353/pbm.1997.0072. [DOI] [PubMed] [Google Scholar]
- 6.Bullen JJ, Rogers HJ, Griffiths E. Curr Top Microbiol Immunol. 1978;80:1–35. doi: 10.1007/978-3-642-66956-9_1. [DOI] [PubMed] [Google Scholar]
- 7.Krewulak KD, Vogel HJ. Biochim Biophys Acta-Biomembranes. 2008;1778:1781–1804. doi: 10.1016/j.bbamem.2007.07.026. [DOI] [PubMed] [Google Scholar]
- 8.Neilands JB. Can J Microbiol. 1992;38:728–733. doi: 10.1139/m92-119. [DOI] [PubMed] [Google Scholar]
- 9.Ferguson AD, Deisenhofer J. Biochim Biophys Acta. 2002;1565:318–332. doi: 10.1016/s0005-2736(02)00578-3. [DOI] [PubMed] [Google Scholar]
- 10.Chu BC, Garcia-Herrero A, Johanson TH, Krewualk KD, Lau CK, Peacock RS, Salvinskaya Z, Vogel HJ. Biomet. 2010;23:601–611. doi: 10.1007/s10534-010-9361-x. [DOI] [PubMed] [Google Scholar]
- 11.Mietzner TA, Toncza SB, Adhikari P, Vaughan KG, Nowalk AJ. Curr Top Microbiol. 1998;225:114–135. doi: 10.1007/978-3-642-80451-9_7. [DOI] [PubMed] [Google Scholar]
- 12.Clarke TE, Tari LW, Vogel HJ. Curr Top Med Chem. 2001;1:7–30. doi: 10.2174/1568026013395623. [DOI] [PubMed] [Google Scholar]
- 13.Chakraborty R, Storey E, van der Helm D. Biomet. 2007;20:263–274. doi: 10.1007/s10534-006-9060-9. [DOI] [PubMed] [Google Scholar]
- 14.Cornelissen CN. Front Biosci. 2003;8:D836–D847. doi: 10.2741/1076. [DOI] [PubMed] [Google Scholar]
- 15.Cornelissen CN, Anderson JE, Boulton IC, Sparling PF. Infect Immun. 2000;68:4725–4735. doi: 10.1128/iai.68.8.4725-4735.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Cornelissen CN, Biswas GD, Tsai J, Paruchuri DK, Thompson SA, Sparling PF. J Bacteriol. 1992;174:5788–5797. doi: 10.1128/jb.174.18.5788-5797.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Killmann H, Benz R, Braun V. Embo J. 1993;12:3007–3016. doi: 10.1002/j.1460-2075.1993.tb05969.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Killmann H, Benz R, Braun V. J Bacteriol. 1996;178:6913–6920. doi: 10.1128/jb.178.23.6913-6920.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Noto JM, Cornelissen CN. Infect Immun. 2008;76:1960–1969. doi: 10.1128/IAI.00020-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Rutz JM, Liu J, Lyons JA, Goranson J, Armstrong SK, McIntosh MA, Feix JB, Klebba PE. Science. 1992;258:471–475. doi: 10.1126/science.1411544. [DOI] [PubMed] [Google Scholar]
- 21.Archibald FS, Devoe IW. Infect Immun. 1980;27:322–334. doi: 10.1128/iai.27.2.322-334.1980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.West SEH, Sparling PF. Infect Immun. 1985;47:388–394. doi: 10.1128/iai.47.2.388-394.1985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.West SEH, Sparling PF. J Bacteriol. 1987;169:3414–3421. doi: 10.1128/jb.169.8.3414-3421.1987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Simonson C, Brener D, De Voe IW. Infect Immun. 1982;36:107–113. doi: 10.1128/iai.36.1.107-113.1982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Anderson JE, Sparling PF, Cornelissen CN. J Bacteriol. 1994;176:3162–3170. doi: 10.1128/jb.176.11.3162-3170.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Archibald FS, Devoe IW. Fems Microbiol Lett. 1979;6:159–162. [Google Scholar]
- 27.Cornelissen CN, Anderson JE, Sparling PF. Mol Microbiol. 1997;26:25–35. doi: 10.1046/j.1365-2958.1997.5381914.x. [DOI] [PubMed] [Google Scholar]
- 28.Cornelissen CN, Sparling PF. Mol Microbiol. 1994;14:843–850. doi: 10.1111/j.1365-2958.1994.tb01320.x. [DOI] [PubMed] [Google Scholar]
- 29.Dyer D, Carbonetti N, McKenna W, Adams J, West S, Sparling PF. J Cell Biochem. 1987;101-101 [Google Scholar]
- 30.McKenna WR, Mickelsen PA, Sparling PF, Dyer DW. Infect Immun. 1988;56:785–791. doi: 10.1128/iai.56.4.785-791.1988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Mickelsen PA, Sparling PF. Infect Immun. 1981;33:555–564. doi: 10.1128/iai.33.2.555-564.1981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Ronpirin C, Jerse AE, Cornelissen CN. Infect Immun. 2001;69:6336–6347. doi: 10.1128/IAI.69.10.6336-6347.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Schryvers AB, Morris LJ. Infect Immun. 1988;56:1144–1149. doi: 10.1128/iai.56.5.1144-1149.1988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Boulton IC, Yost MK, Anderson JE, Cornelissen CN. Infect Immun. 2000;68:6988–6996. doi: 10.1128/iai.68.12.6988-6996.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Yost-Daljev MK, Cornelissen CN. Infect Immun. 2004;72:1775–1785. doi: 10.1128/IAI.72.3.1775-1785.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Ferguson AD, Hofmann E, Coulton JW, Diederichs K, Welte W. Science. 1998;282:2215–2220. doi: 10.1126/science.282.5397.2215. [DOI] [PubMed] [Google Scholar]
- 37.Ferguson AD, Kodding J, Coulton JW, Diederichs K, Braun V, Welte W. Structure. 2001;9:707–716. doi: 10.1016/s0969-2126(01)00631-1. [DOI] [PubMed] [Google Scholar]
- 38.Ferguson AD, Welt W, Hoffmann E, Lindner B, Holst O, Coulton JW, Diederichs K. Structure. 2000;8:585–592. doi: 10.1016/s0969-2126(00)00143-x. [DOI] [PubMed] [Google Scholar]
- 39.Locher KP, Rees B, Koebnik R, Mitschler A, Moulinier L, Rosenbusch JP, Moras D. Cell. 1998;95:771–778. doi: 10.1016/s0092-8674(00)81700-6. [DOI] [PubMed] [Google Scholar]
- 40.Boulton IC, Gorringe AR, Allison N, Robinson A, Gorinsky B, Joannou CL, Evans RW. Biochem J. 1998;334:269–273. doi: 10.1042/bj3340269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Cornelissen CN, Sparling PF. J Bacteriol. 1996;178:1437–1444. doi: 10.1128/jb.178.5.1437-1444.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Renauld-Mongenie G, Latour M, Poncet D, Naville S, Quentin-Millet MJ. Fems Microbiol Lett. 1998;169:171–177. doi: 10.1111/j.1574-6968.1998.tb13314.x. [DOI] [PubMed] [Google Scholar]
- 43.Retzer MD, Yu R, Zhang YP, Gonzalez GC, Schryvers AB. Microb Pathog. 1998;25:175–180. doi: 10.1006/mpat.1998.0226. [DOI] [PubMed] [Google Scholar]
- 44.DeRocco AJ, Yost-daljev MK, Kenney CD, Cornelissen CN. Biomet. 2010;22:493–451. doi: 10.1007/s10534-008-9179-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Siburt CJP, Roulhac PL, Weaver KD, Noto JM, Mietzner TA, Cornelissen CN, Fitzgerald MC, Crumbliss AL. Metallomics. 2009;1:249–255. doi: 10.1039/b902860a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Kenney CD, Cornellisen CN. J Bacteriol. 2002;188:6138–6145. doi: 10.1128/JB.184.22.6138-6145.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Usher KC, Ozkan E, Gardner KH, Deisenhofer J. Proc Natl Acad Sci, USA. 2001;98:10676–10681. doi: 10.1073/pnas.181353398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Ma L, Kaserer W, Annamalai R, Scott DC, Jin B, Jiang X, Xiao Q, Maymani H, Massis LM, Ferreira LCS, Newton SMC, Klebba PE. J Biol Chem. 2007;28:397–406. doi: 10.1074/jbc.M605333200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Roulhac PL, Powell KD, Dhungana S, Weaver KD, Mietzner TA, Crumbliss AL, Fitzgerald MC. Biochemistry. 2004;43:15767–15774. doi: 10.1021/bi0481848. [DOI] [PubMed] [Google Scholar]
- 50.Roulhac PL, Weaver KD, Adhikari P, Anderson DS, DeArmond PD, Mietzner TA, Crumbliss AL, Fitzgerald MC. Biochemistry. 2008;47:4298–4305. doi: 10.1021/bi701188x. [DOI] [PubMed] [Google Scholar]
- 51.Motekaitis RJ, Martell AE. J Coord Chem. 1994;31:67–78. [Google Scholar]
- 52.Laemmli UK. Nature. 1970;227:680–685. doi: 10.1038/227680a0. [DOI] [PubMed] [Google Scholar]
- 53.Myers JK, Pace CN, Scholtz JM. Biochemistry. 1997;36:10923–10929. doi: 10.1021/bi9707180. [DOI] [PubMed] [Google Scholar]
- 54.Powell KD, Fitzgerald MC. Anal Chem. 2001;73:3300–3304. doi: 10.1021/ac0100805. [DOI] [PubMed] [Google Scholar]
- 55.Tetin SY, Hazlett TL. Methods. 2000;20:341–361. doi: 10.1006/meth.1999.0927. [DOI] [PubMed] [Google Scholar]
- 56.Tetin SY, Rumbley CA, Hazlett TL, Voss EW., Jr Biochemistry. 1993;32:9011–9017. doi: 10.1021/bi00086a005. [DOI] [PubMed] [Google Scholar]
- 57.Bordoli L, Kiefer F, Arnold K, Benkert P, Battey J, Schwede T. Nat Protoc. 2009;4:1–13. doi: 10.1038/nprot.2008.197. [DOI] [PubMed] [Google Scholar]
- 58.Kelley LA, Sternberg MJ. Nat Protoc. 2009;4:363–71. doi: 10.1038/nprot.2009.2. [DOI] [PubMed] [Google Scholar]
- 59.Roy A, Kucukural A, Zhang Y. Nat Protoc. 2010;5:725–38. doi: 10.1038/nprot.2010.5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Colovos C, Yeates TO. Protein Sci. 1993;2:1511–9. doi: 10.1002/pro.5560020916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Laskoswki RA, MacArthur MW, Moss DS, Thorton JM. J App Crystallog. 1993;26:283–291. [Google Scholar]
- 62.Cobessi D, Meksem A, Brillet K. Proteins. 2010;78:286–94. doi: 10.1002/prot.22539. [DOI] [PubMed] [Google Scholar]
- 63.Laskowski RA. Nucleic Acids Res. 2009;37:D355–9. doi: 10.1093/nar/gkn860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Ghaemaghami S, Fitzgerald MC, Oas TG. Proc Natl Acad Sci, USA. 2000;97:8296–8301. doi: 10.1073/pnas.140111397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Powell KD, Ghaemaghami S, wang MZ, Ma L, Oas TG, Fitzgerald MC. J Am Chem Soc. 2002;124:10256–10257. doi: 10.1021/ja026574g. [DOI] [PubMed] [Google Scholar]
- 66.Fitzgerald MC, Tang L, Hooper ED. Comprehensive Analytical Chemistry, Elsevier Science. 2008;52:127–149. [Google Scholar]
- 67.Libertini LJ, Small EW. Biophys J. 1985;47:765–772. doi: 10.1016/S0006-3495(85)83979-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Brillet K, Journet L, Celia H, Paulus L, Stahl A, Pattus F, Cobessi D. Structure. 2007;15:1383–91. doi: 10.1016/j.str.2007.08.013. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.