

Abstract
Aggregated filamentous forms of hyperphosphorylated tau (a microtubule-associated protein) represent pathological hallmarks of Alzheimer's disease (AD) and other tauopathies. While axonal transport dysfunction is thought to represent a primary pathogenic factor in AD and other neurodegenerative diseases, the direct molecular link between pathogenic forms of tau and deficits in axonal transport remain unclear. Recently, we demonstrated that filamentous, but not soluble, forms of wild-type tau inhibit anterograde, kinesin-based fast axonal transport (FAT) by activating axonal protein phosphatase 1 (PP1) and glycogen synthase kinase 3 (GSK3), independent of microtubule binding. Here, we demonstrate that amino acids 2–18 of tau, comprising a phosphatase-activating domain (PAD), are necessary and sufficient for activation of this pathway in axoplasms isolated from squid giant axons. Various pathogenic forms of tau displaying increased exposure of PAD inhibited anterograde FAT in squid axoplasm. Importantly, immunohistochemical studies using a novel PAD-specific monoclonal antibody in human postmortem tissue indicated that increased PAD exposure represents an early pathogenic event in AD that closely associates in time with AT8 immunoreactivity, an early marker of pathological tau. We propose a model of pathogenesis in which disease-associated changes in tau conformation lead to increased exposure of PAD, activation of PP1-GSK3, and inhibition of FAT. Results from these studies reveal a novel role for tau in modulating axonal phosphotransferases and provide a molecular basis for a toxic gain-of-function associated with pathogenic forms of tau.
Introduction
Aggregates of the microtubule-associated protein tau represent major pathological components in Alzheimer's disease (AD) and other tauopathies (Grundke-Iqbal et al., 1986; Wood et al., 1986) and correlate with progressive cognitive decline in AD (Kosik et al., 1986; Arriagada et al., 1992; Giannakopoulos et al., 2003). The identification of tau mutations in hereditary tauopathies directly link tau to neurodegeneration (Goedert and Jakes, 2005). However, the molecular mechanisms behind tau-mediated toxicity remain unclear.
Axonal transport dysfunction is implicated as a critical pathogenic factor in AD and other neurodegenerative diseases (Roy et al., 2005; Morfini et al., 2009). Experiments in isolated squid axoplasm revealed that aggregated WT tau selectively inhibited kinesin-based anterograde fast axonal transport (FAT) at physiological levels, while soluble WT tau monomers did not, even at concentrations >10-fold higher than physiological levels (LaPointe et al., 2009; Morfini et al., 2009). The toxic effect of aggregated tau on anterograde FAT involved activation of protein phosphatase 1 (PP1) and glycogen synthase kinase 3 (GSK3) (LaPointe et al., 2009). Deletion experiments suggested that this effect required amino acids 2–18 of tau (LaPointe et al., 2009). However, these studies did not explain why monomeric, soluble tau did not inhibit FAT.
Findings on the conformation of soluble tau provided a potential explanation for this apparent paradox. Originally, tau was considered a “natively unfolded” protein with little secondary structure in solution (Schweers et al., 1994). However, recent fluorescence resonance energy transfer studies suggest that soluble WT tau monomers adopt a “paperclip” conformation involving close interactions of the C terminus with the microtubule binding repeats (MTBRs) and the N terminus with the C terminus (Jeganathan et al., 2006). Pathological forms of tau appear to alter the paperclip conformation. For example, the AT8 phosphoepitope (Ser199/Ser202/Thr205) found in AD and other tauopathies reduced folding of the N terminus into the paperclip conformation (Jeganathan et al., 2008). Similarly, a tau mutation in a patient with inherited frontotemporal dementia (FTD) results in deletion of the “hinge” region that is crucial for N-terminal folding (Rovelet-Lecrux et al., 2009), rendering this mutant tau protein incapable of assuming the paperclip conformation. These observations suggest that disease-associated tau modifications disrupt the paperclip conformation, but molecular mechanisms linking disruptions in tau conformation to inhibition of FAT remained unknown.
We present evidence indicating that disease-associated modifications in tau that impair the paperclip conformation inhibit anterograde FAT. Results from our experiments suggest that amino acids 2–18 of tau, a domain we refer to as the phosphatase-activating domain (PAD), represent a biologically active protein motif capable of activating the PP1–GSK3 pathway. The relevance of these findings to AD was validated using a novel PAD-specific antibody to document increased PAD exposure early in the course of disease. Based on these results, we propose a model of pathogenesis where modifications in tau, including aggregation, hyperphosphorylation, and FTD-associated mutation all result in increased PAD exposure, activation of the PP1–GSK3 pathway, and inhibition of anterograde FAT. Collectively, our findings provide a common molecular basis for the toxic gain-of-function mechanism associated with biochemically distinct disease-related modifications of tau.
Materials and Methods
Reagents.
The following reagents were purchased from commercial vendors: okadaic acid (catalog #495604, EMD Chemicals) and inhibitor 2 (I-2; catalog #539638, EMD Chemicals); PAD peptide (amino acids 2–18) and scrambled PAD peptide (both with >98% purity) (GenScript); and Complete, EDTA-free Protease Inhibitor tablets (catalog #11873580001, Roche). ING-135 (Kozikowski et al., 2007) was a generous gift from Dr. A. Kozikowski (University of Illinois at Chicago, Chicago, IL).
Recombinant tau proteins.
Tau proteins are numbered according to the largest adult human isoform (ht40; 441 aa) in the CNS. Full-length ht40 (WT tau) was generated from previously described pT7c plasmids (Fig. 1A) (LaPointe et al., 2009). Site-directed mutagenesis (QuikChange II Kit, catalog #200524, Stratagene) was used to create point mutations in full-length tau constructs. Ser199, Ser202, and Thr205 were mutated to glutamic acid (E) to create the AT8 pseudophosphorylated mutant protein (AT8 tau) (Fig. 1B). Amino acids 144–273 (Δ144–273 tau) (Fig. 1C) were deleted by inserting EcoRV restriction sites flanking the appropriate region of the cDNA. Following EcoRV digestion and T4 ligation (New England Biolabs; used according to manufacturer's instructions), the remaining EcoRV site was removed via deletion using the Site-Directed Mutagenesis Kit described above. The 6D tau and 6P tau constructs were generated from previously described pT7c plasmid cDNAs (Fig. 1D,E) (Luo et al., 2004; LaPointe et al., 2009). A 6D protein with amino acids 2–18 deleted (Δ 2–18 6D tau) (Fig. 1F) was created by restriction digestion and ligation of 6D and Δ2–18 tau cDNAs, both of which have been described previously (LaPointe et al., 2009). The tau cDNA plasmid constructs were amplified in XL-1 blue supercompetent Escherichia coli, purified with the Qiagen Miniprep kit, and cDNA sequences were verified by DNA sequencing. T7 express supercompetent E. coli cells were used to express tau constructs, and the proteins were purified through His-tag high-affinity resin and size exclusion chromatography as previously described (Carmel et al., 1996).
Figure 1.

Schematic diagram of the tau constructs used. A, Full-length wild-type ht40 (WT tau) is the largest isoform of tau found in the adult CNS. WT tau contains the PAD motif corresponding to amino acids 2–18 (black box), two alternatively spliced N-terminal exons (E2 and E3), and MTBRs 1–4. B, The AT8 tau protein was pseudophosphorylated at S199, S202, and T205 by mutagenesis of S → E and T → E to mimic the phosphoepitope recognized by the AT8 antibody (Biernat et al., 1992; Goedert et al., 1995) in hyperphosphorylated tau. C, The Δ144–273 tau mutant protein corresponds to a deletion of amino acids 144–273 (the proline-rich region and MTBR1) that was found in a patient with an autosomal-dominant familial form of FTD (Rovelet-Lecrux et al., 2009). D, The 6D tau protein is a noncanonical N-terminal isoform containing amino acids 1–143 with an additional 11 unique C-terminal amino acids (Luo et al., 2004). E, The 6P protein is the same as 6D, except the last 11 C-terminal amino acids are different (Luo et al., 2004). F, The Δ2–18 6D protein has amino acids 2–18 deleted from the N terminus. G, Synthetic peptide composed of the PAD sequence (amino acids 2–18 from WT tau). H, Synthetic peptide composed of the same amino acids as the PAD peptide, but rearranged in a random order.
Squid axoplasm motility assay.
Freshly extruded axoplasms from squid giant axons (Loligo pealii; Marine Biological Laboratory, Woods Hole, MA) were used to measure axonal transport, as described previously (Morfini et al., 2007; LaPointe et al., 2009). Tau proteins were diluted in X/2 buffer (175 mm potassium aspartate, 65 mm taurine, 35 mm betaine, 25 mm glycine, 10 mm HEPES, 6.5 mm MgCl2, 5 mm EGTA, 1.5 mm CaCl2, 0.5 mm glucose, and 10 mm ATP, pH 7.4) and perfused into isolated axoplasms at a final concentration of 2 μm. Use of tau proteins at 2 μm was specifically chosen since this is within the physiological levels of tau in brains (Alonso et al., 1996; King et al., 1999) and is within the physiological range of tau monomer/tubulin dimer ratios (Drubin et al., 1985; Morfini et al., 2007; LaPointe et al., 2009). A Zeiss Axiomat microscope equipped with a 100× objective (1.3 numerical aperture) and differential interference contrast optics, equipped for digital video processing and analysis was used to visualize vesicle motility (Brady et al., 1993). Anterograde and retrograde transport were measured by matching vesicle speed to calibrated cursors over 50 min as described previously (Brady et al., 1993; Morfini et al., 2002b).
Preparation of 32P-radiolabeled phosphatase substrate and phosphatase assay in squid axoplasm.
Recombinant GST-c-Jun (75 μg) was incubated with 10 μg of recombinant p38α (a generous gift from Dr. D. Bosco, University of Massachusetts, Amherst, MA) in the presence of 100 μm 32P-radiolabeled ATP in a 20 μl volume. After 1 h at 30°C, the resulting 32P-c-Jun samples were stored at −20°C until used in phosphatase assays.
First, a control experiment was run to confirm that endogenous axoplasmic phosphatases could dephosphorylate 32P-c-Jun. Squid axoplasm was incubated with 32P-c-Jun (1 μg) for 0 min (input) or 30 min (control), and with okadaic acid (1 μm) or I-2 (400 nm) for 30 min. To determine whether PAD affects phosphatase activity, sister axoplasms were extruded on coverglasses and perfused with 35 μl of either PAD or scrambled PAD peptides (10 μm each) in buffer X/2 supplemented with 1 mm ATP plus 1 μg of 32P-cJun. A 4 μl aliquot of perfusate was removed at 10, 20, and 30 min, and was transferred to tubes containing 9 μl of sample buffer. Samples were run by SDS-PAGE, the gels were dried, and 32P incorporation in GST-cJun was analyzed with the Typhoon PhosphorImager.
Kinase assay in isolated squid axoplasm.
Sister axoplasms were extruded on coverglasses and perfused with 30 μl of either PAD or scrambled PAD peptides (10 μm) in buffer X/2 supplemented with 1 mm ATP. After 45 min of incubation, 12 μl aliquots of perfusate containing solubilized axoplasmic proteins were removed from each axon; diluted to 80 μl with 20 mm HEPES, pH 7.4, 3 mm EDTA, 2 μm okadaic acid, and 1/80 protease inhibitor mixture (P8340, Sigma); and kept on ice. Aliquots of diluted perfusates (20 μl) were supplemented with either 200 μm GSK3 substrate peptide (GSM, catalog #12-533, Millipore) or with 50 μm myristoylated alanine-rich C-kinase substrate (MARCKS) (catalog #454880, Calbiochem), and the final volume adjusted to 32 μl with 20 mm HEPES, pH 7.4. Kinase reactions were started by adding 8 μl of 5X kinase buffer (100 mm HEPES, pH 7.4, 250 mm K aspartate, 50 mm MgCl2, and 500 μm 32P-radiolabeled ATP). After 30 min of incubation at room temperature, aliquots were spotted on P81 cellulose paper in duplicate. Papers were washed three times in 100 mm phosphoric acid, and 32P incorporation into peptides was measured by scintillation counting. GSK3 activity was expressed as the ratio of incorporation between GSM and MARCKS peptides for each condition.
Monoclonal antibody production.
Mouse monoclonal antibodies were raised against a synthetic PAD peptide (C2AEPRQEFEVMEDHAGTY18) conjugated to KLH (GenScript) as previously described (Reyes et al., 2008). Briefly, BALB/c female mice (catalog #001026, Jackson Laboratories) were immunized subcutaneously with peptides every 21 d over a 5 month period; splenocytes were then removed and fused to SP2/o myeloma cells. Positive clones that reacted specifically with PAD by ELISA analysis (Reyes et al., 2008) were selected, and then subcloned four times at limiting dilutions and isotyped [tau N-terminal 1 (TNT1) is an IgG1]. Antibody production was accomplished in a CELLine Classic 1000 bioreactor (DC-90005, Sartorius AG) before protein A purification at high pH. The sensitivity and specificity of the TNT1 antibody for PAD in tau was assessed using ELISAs and Western blotting (see Fig. 7A).
Figure 7.
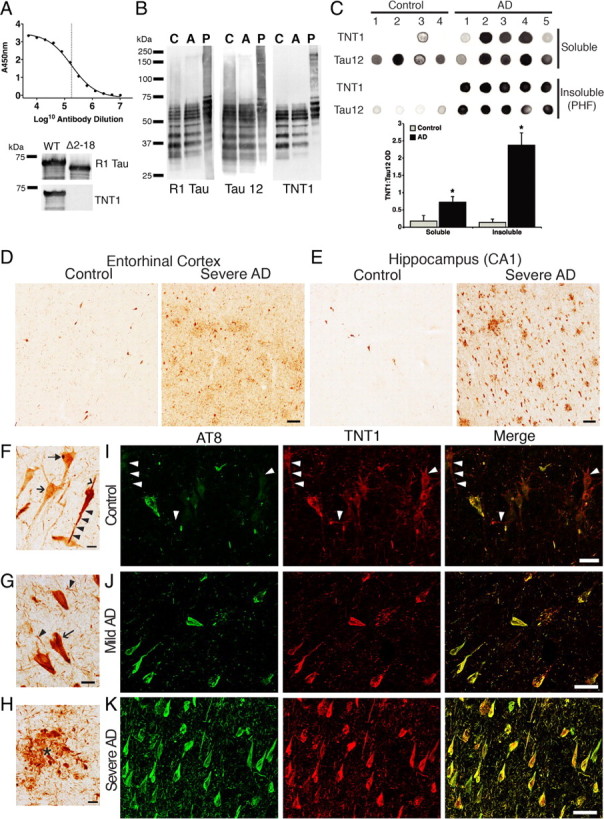
PAD immunoreactivity in AD brains. A, An ELISA titer of the TNT1 antibody using recombinant WT tau indicates a LogEC50 of 5.25 or a 1:180,000 dilution of a 1 mg/ml stock (final concentration, 5.5 ng/ml). TNT1 does not react with tau lacking PAD (amino acids 2–18; Δ2–18) in immunoblots. B, TNT1 labels enriched soluble tau from the frontal cortex of both control (Con Sol) and AD brains (AD Sol) as well as purified paired helical filament tau (AD PHF) from AD frontal cortex in denaturing conditions on Western blots. Comparison of TNT1 blots with Tau12 and R1 tau blots clearly indicates that TNT1 recognizes tau proteins and not other proteins in human brains. C, Dot blots of TNT1 under non-denaturing conditions. TNT1 preferentially labels soluble and PHF tau isolated from AD brains as opposed to tau isolated from control brains, indicating that PAD-exposed tau conformations are more abundant in AD. Quantification of both soluble and insoluble tau fractions is depicted as TNT1:Tau12 optical density (OD) ratios in the bar graph; Tau12 was used to measure total tau. *p ≤ 0.05, unpaired t test. D, E, TNT1 immunohistochemistry in the entorhinal cortex (D) and hippocampus (CA1 region; E) of human Braak stages I–II (control) or V–VI (severe AD) cases. Spatial and temporal patterns of TNT1 staining followed those of Braak staging, starting in the entorhinal cortex (D) and then appearing in the hippocampus (E) before involving temporal cortical gyri in later stages (data not shown). Scale bars, 100 μm. F, TNT1 staining in Braak stage I–II cases. TNT1 diffusely labeled the cytoplasm of pretangle CA2 neurons in the very early stages of inclusion formation (arrow). A subset of diffusely stained neurons contained globular cytoplasmic inclusions (filled arrow). A few neurons contained strongly labeled seemingly less mature tau inclusions that extended into the basal (arrowhead) and apical (black triangle) dendrites. G, H, TNT1 in Braak stages V–VI; note that large amounts of the classical tau inclusions, neurofibrillary tangles (arrow; G), neuropil threads (black triangle; G) and neuritic plaques (*; H), were present in the regions analyzed (CA1 shown). Scale bars, 20 μm. I–K, Representative images of TNT1 (red) and AT8 (green) double-label immunofluorescence in the hippocampus (CA1/CA2 region). I, The TNT1 epitope appears to precede that of AT8 in controls cases (Braak stages I–II). J, Both TNT1 and AT8 continue to accumulate in mild AD (Braak stages III–IV). K, TNT1 and AT8 appear with nearly complete colocalization in severe AD (Braak stages V–VI). Note that when present, AT8 extensively colocalizes with TNT1 at all Braak stages. Scale bars, 50 μm.
Tau isolation from human brain tissue.
Fresh frozen frontal cortex from control (n = 4) and severe AD (n = 5) brains was obtained from the Brain Bank of the Cognitive Neurology and Alzheimer's Disease Center at Northwestern University. Heat-stable/acid-soluble tau was extracted based on protocols described before (Reyes et al., 2008). Sarkosyl-insoluble tau [paired helical filaments (PHF)] and Sarkosyl-soluble tau were obtained using methods similar to those in previous reports (Hasegawa et al., 1992).
Western and dot blots.
For Western blot analysis, soluble and insoluble tau samples were separated by 10% SDS-PAGE and transferred to nitrocellulose membranes, as described previously (Reyes et al., 2008). For dot blots, soluble and insoluble tau samples were spotted directly onto nitrocellulose membranes (LaPointe et al., 2009). Both Western and dot blot membranes were blocked with 2% nonfat dry milk in Tris-buffered saline (TBS), pH 7.4, and incubated in primary antibodies overnight at 4°C. The Tau12 (Horowitz et al., 2004) and TNT1 mouse monoclonal antibodies were diluted from 1 mg/ml purified stock solutions to 1:500,000 and 1:300,000, respectively. The rabbit polyclonal antibody, R1 tau (Berry et al., 2004), was diluted 1:300,000 from a 1 mg/ml stock. After rinsing, the membranes were incubated in peroxidase-conjugated horse anti-mouse secondary antibody (PI-2000, Vector Laboratories) for 1 h at room temperature. Reactivity was visualized using ECL substrate (PI32106, Pierce), and Western blots were visualized on Kodak Bio-max XAR film, while dot blots were imaged on the Kodak Imaging Station 4000R for quantification of signal intensity. Data are expressed as the ratio of TNT1/Tau12 intensity.
TNT1 immunohistochemistry.
Tissue sections (40 μm thick) from the entorhinal cortex, hippocampus, inferior temporal gyrus, and superior temporal gyrus of control (Braak stages I–II; n = 4) and severe AD (Braak stages V–VI; n = 4) cases were obtained through the Cognitive Neurology and Alzheimer's Disease Center at Northwestern University. All sections were stained simultaneously to reduce variability in tissue processing. Sections were processed as previously described (Kanaan et al., 2007). The TNT1 primary antibody was diluted 1:400,000 (from 1 mg/ml stock) and incubated with the tissue sections overnight at 4°C. The tissue was incubated in biotinylated goat anti-mouse secondary antibody (diluted 1:500; BA-9200, Vector Laboratories) for 2 h, followed by incubation in ABC solution (PK-6100, Vector Laboratories; used according to manufacturer's instructions) for 1 h. The staining was developed with 3,3′-diaminobenzidine (D5637, Sigma). All steps were performed at room temperature unless otherwise noted, and each step was followed by 6× 10 min rinses in TBS-0.5% Triton X-100. After developing the stain, the tissue sections were mounted and coverslipped. Images were taken using a Nikon Eclipse 90i microscope equipped with a DS-Ri1 camera and Nikon Elements AR 3.10 software. Figures were generated using Adobe Photoshop.
TNT1 and AT8 double-label immunofluorescence.
Tissue sections (as described above) from the hippocampus of control (Braak stages I–II; n = 4), mild AD (Braak stages III–IV; n = 5), and severe AD (Braak stages V-VI; n = 4) cases were processed for double-label immunofluorescence using TNT1 and the AT8 antibody (Biernat et al., 1992; Goedert et al., 1995). All sections were stained simultaneously using similar methods as previously described (Kanaan et al., 2007) to reduce variability in tissue processing. The TNT1 primary antibody was diluted 1:40,000 (from 1 mg/ml stock) and incubated with the tissue sections overnight at 4°C. The tissue was incubated in Alexa Fluor 594 goat anti-mouse IgG1 γ-chain-specific secondary antibody (diluted 1:500; A21125, Invitrogen) for 2 h, followed by blocking uncomplexed secondary antibody sites with 10% mouse serum for 1 h. Then the sections were incubated overnight at 4°C with biotinylated AT8 antibody (MN1020B, Thermo Scientific) diluted at 1:1000. The AT8 antibody was labeled with Alexa Fluor 488-conjugated streptavidin (diluted 1:500; S11223, Invitrogen) for 2 h. Sections were mounted and coverslipped using Vectashield Hardest (H-1400, Vector Laboratories). All steps were performed at room temperature unless otherwise noted, and each step was followed by 6× 10 min rinses in TBS-0.5% Triton X-100. Images were taken using a fluorescence-equipped Nikon Eclipse 90i microscope with a Q-imaging Retiga-SRV Fast1394 camera and Nikon Elements AR 3.10 software (with live deblur function). Figures were generated using Adobe Photoshop.
Statistics.
Each experiment was repeated at least three times (see figures for n values). Comparisons of vesicle motility rates were made using an unpaired t test or a one-way ANOVA, as indicated. The WT tau monomer group was used as the control group since no changes in motility rates are observed in this group compared with unperfused axoplasms (Morfini et al., 2007; LaPointe et al., 2009). When overall significance was reached, the Student–Newman–Keuls post hoc analysis was used to make all possible comparisons. Kinase assays with results were compared using a paired t test (paired by sister axoplasms). Significance was set at p < 0.01 or p < 0.05, as indicated. SigmaStat software (Systat Software) was used for all statistical tests.
Results
6D and 6P tau isoforms inhibit anterograde FAT
The isolated squid axoplasm is an ex vivo model system that allows for direct, quantitative evaluation of different effectors on anterograde (conventional kinesin-dependent) and retrograde (cytoplasmic dynein-dependent) FAT rates by video microscopy. When isolated axoplasm is perfused with aggregated tau, but not soluble tau monomers, there is a selective inhibition of anterograde FAT through an N-terminal-mediated mechanism involving activation of a PP1–GSK3 pathway, phosphorylation of kinesin light chains, and dissociation of kinesin-1 from its transported cargoes (Morfini et al., 2004; LaPointe et al., 2009).
To explore the N-terminal dependence of tau-mediated FAT inhibition, we used the noncanonical N-terminal 6D and 6P tau isoforms (Fig. 1D,E). Both 6D and 6P are identical to WT tau from amino acids 1–143, but lack the proline-rich region, the MTBRs, and C terminus of full-length tau, and each protein contains a unique C-terminal 11 aa sequence (Luo et al., 2004; Andreadis, 2005). Thus, the 6D/6P isoforms can be used as physiological tools for the evaluation of the N terminus of tau in isolation from other functional domains and independent of microtubule binding or self-aggregation because both of these processes require the MTBRs.
Vesicle motility assays in isolated axoplasm confirmed and expanded our initial results with the N-terminal isoforms of tau. As previously reported (Morfini et al., 2007; LaPointe et al., 2009), perfusion of full-length WT tau monomers (2 μm) (Fig. 1A) had no effect on FAT in squid axoplasm (Fig. 2A), while 6D and 6P tau monomers (2 μm) significantly inhibited anterograde FAT when compared with WT tau monomer (Fig. 2B,C) or buffer controls (data not shown). Neither 6D nor 6P tau (Fig. 2D) had an effect on retrograde FAT. Together, these data indicate that the proline-rich region, MTBRs, and the C terminus of full-length tau, as well as the unique 11 aa C termini of 6D and 6P do not play a role in inhibiting anterograde FAT. Since similar levels of FAT inhibition were obtained with 6D and 6P constructs (Fig. 2C,D), which are identical in the first 143 aa, we chose to focus on the 6D tau for further experimentation.
Figure 2.

N-terminal tau isoforms 6D and 6P tau selectively inhibit anterograde FAT. A–D, Vesicle motility assays in isolated squid axoplasm. Individual velocity (micrometers per second) rate measurements (arrowheads) are plotted as a function of time (in minutes). Black arrowheads and lines represent anterograde FAT rates (conventional kinesin-dependent), and gray arrows and lines represent retrograde FAT rates (cytoplasmic dynein-dependent). A, Perfusion with soluble wild-type tau monomers (WT tau) did not affect FAT rates in the anterograde (black triangle) or retrograde (gray triangle) direction. B, In contrast, soluble 6D tau monomers, which cannot bind microtubules, selectively inhibited the rate of anterograde FAT, while retrograde FAT was not significantly affected. C, Quantitative analysis of FAT demonstrates that 6D and 6P (n = 5) tau caused a significant reduction in anterograde FAT when compared with WT tau monomers (*p < 0.01, unpaired t test). D, Retrograde FAT was unaffected by 6D and 6P tau.
6D Tau inhibits FAT by activating a PP1–GSK3 signaling cascade
Results from our previous work (LaPointe et al., 2009) prompted us to evaluate whether aggregated tau and 6D tau inhibited anterograde FAT through a common mechanism. To this end, we coperfused axoplasms with 6D tau and pharmacological inhibitors of either PP1 or GSK3. Coperfusion of 6D tau with I-2 (50 nm), a specific PP1 inhibitor, completely prevented the effects of 6D tau on anterograde FAT (Fig. 3A,C). Similarly, coperfusion of 6D tau with okadaic acid (200 nm), a less specific inhibitor of the major serine/threonine phosphatases (including both PP1 and PP2), prevented the inhibition of anterograde FAT elicited by 6D and 6P tau (data not shown). Furthermore, coperfusion of 6D tau and the GSK3-specific pharmacological inhibitor ING-135 (100 nm) (Kozikowski et al., 2007; LaPointe et al., 2009) completely blocked the inhibitory effects of 6D tau on anterograde FAT (Fig. 3B,C). Retrograde transport was not significantly affected in any experiment (Fig. 3D). Together, these data demonstrate that, as posited for aggregated tau (LaPointe et al., 2009), short N-terminal isoforms of tau inhibit anterograde FAT by a mechanism involving activation of PP1 and GSK3 that is independent of microtubule binding.
Figure 3.

6D tau inhibits anterograde FAT via activation of a PP1–GSK3 cascade. A, Coperfusion of 6D tau and I-2, a specific PP1 inhibitor, prevented inhibition of anterograde (black triangle) FAT elicited by 6D (compare with Fig. 2B). B, Coperfusion of 6D tau and ING-135, a specific GSK3 inhibitor, similarly prevented inhibition of anterograde FAT. C, Quantitative analysis of FAT shows that inhibition of PP1 (via I-2) or inhibition of GSK3 (via ING-135) completely prevents the anterograde FAT inhibition elicited by 6D tau (one-way ANOVA, *p < 0.01 versus WT Tau, 6D+I-2, 6D+ING-135). D, Retrograde FAT (gray triangle, retrograde) was unaffected by 6D, regardless of whether inhibitors were present.
PAD is necessary and sufficient for anterograde FAT inhibition
Aggregated tau lacking amino acids 2–18 or PAD (Δ2–18) did not inhibit anterograde FAT (LaPointe et al., 2009), suggesting that this domain plays a critical role in the activation of the PP1–GSK3 pathway. To determine whether this domain mediates the inhibitory effects of 6D tau monomer on FAT, axoplasms were perfused with a recombinant 6D tau protein lacking PAD (Δ2–18 6D tau; 2 μm) (Fig. 1F). As observed with Δ2–18 tau aggregates (LaPointe et al., 2009), monomeric Δ2–18 6D tau showed no effect on FAT (Fig. 4A,D), demonstrating that PAD is necessary for 6D tau-mediated inhibition of anterograde FAT.
Figure 4.

PAD is necessary and sufficient to trigger anterograde FAT inhibition. A, Perfusion of Δ2–18 6D into isolated axoplasm did not affect FAT in either direction (black triangle, anterograde; gray triangle, retrograde). B, In contrast, perfusion of the PAD peptide, which corresponds to amino acids 2–18 in WT tau, caused a reduction of anterograde, but not retrograde FAT rates. C, A scrambled version of the PAD peptide was used as a control, and when applied to squid axoplasms did not affect FAT. D, Quantitative analysis of FAT rates indicates that PAD peptide significantly inhibits anterograde FAT (one-way ANOVA, *p < 0.01), while deletion of the PAD sequence in 6D (Δ2–18 6D) and scrambling the PAD peptide (Scram PAD) completely prevented inhibition of anterograde FAT. E, Retrograde FAT rates were unaffected in all groups tested.
We next sought to determine whether PAD is sufficient to induce anterograde FAT inhibition or whether other tau domains are also required. Isolated axoplasms were perfused with a synthetic PAD peptide (AEPRQEFEVMEDHAGTY) (Fig. 1G) or with a scrambled control PAD peptide (AQEMDRVYEGEAPHTFE) (Fig. 1H) at the same concentration (2 μm). The PAD peptide selectively inhibited anterograde FAT (Fig. 4B,D), but not retrograde FAT (Fig. 4B,E). In contrast, the scrambled PAD peptide had no effect on FAT in either direction (Fig. 4C–E). Together, these data demonstrate that the PAD in the N-terminal region of tau is both necessary and sufficient to induce inhibition of anterograde FAT.
PAD inhibits FAT through a PP1–GSK3 signaling cascade
We next evaluated whether FAT inhibition induced by the PAD peptide involved activation of PP1 and GSK3 as observed with 6D tau (above) and aggregated tau (LaPointe et al., 2009). Again, coperfusion of the PAD peptide with either I-2 (50 nm) (Fig. 5A) or ING-135 (100 nm) (Fig. 5B) completely prevented anterograde FAT inhibition. Anterograde (Fig. 5C) and retrograde (Fig. 5D) FAT rates observed after coperfusion of the tau PAD peptide with either I-2 or ING-135 were indistinguishable from those observed after perfusion of soluble WT tau.
Figure 5.

PAD peptide inhibits anterograde FAT by activation of the PP1–GSK3 cascade. A, Coperfusion of PAD peptide with I-2, a specific PP1 inhibitor, had no effect on anterograde (black triangle) or retrograde (gray triangle) FAT in squid axoplasm (compare with Fig. 4B). B, Coperfusion of PAD peptide with ING-135, a specific GSK3 inhibitor, similarly prevented inhibition of anterograde FAT by PAD. C, Quantitative analysis of FAT reveals that PAD peptide alone specifically and significantly inhibits anterograde FAT (one-way ANOVA, *p > 0.01), while addition of either PP1 or GSK3 inhibitors completely prevented the reduction in anterograde FAT rates induced by the PAD peptide. D, Retrograde FAT rates were unaffected in all groups tested. E, 32P-c-Jun (a phosphatase substrate) undergoes dephosphorylation by endogenous axoplasmic phosphatases after 30 min. Axoplasms were perfused with 32P-c-Jun alone (Ctrl) or with 32P-c-Jun and either okadaic acid (OK, 1 μm) or I-2 (400 nm). Aliquots were taken immediately after perfusion (0′) and 30 min later (30′), and analyzed by autoradiography. F, 32P-c-Jun was dephosphorylated to a greater extent in PAD-perfused axoplasms than its scrambled PAD-perfused “sister” counterpart, suggesting increased activation of endogenous axoplasmic phosphatases by PAD. Sister axoplasms from the same squid were perfused with 32P-c-Jun and either scrambled PAD (Scr) or PAD peptides, and analyzed by autoradiography (n = 2 sets of sister axoplasms).
The effect of the PAD peptide on the activity of endogenous axoplasmic phosphatases was evaluated using a phosphatase activity assay (Fig. 5E,F). In this assay, a recombinant GST-tagged c-Jun construct was phosphorylated with p38 kinase in the presence of γ32P-ATP for use as a phosphatase substrate. Isolated axoplasms were perfused with radiolabeled c-Jun (32P-c-Jun), and aliquots of the perfusates were collected at various time points and separated by SDS-PAGE. Autoradiograph analysis showed a marked reduction in 32P-c-Jun signal after 30 min of incubation, indicating the action of axonal phosphatases. The reduction was completely abolished by okadaic acid and, to a lesser extent, by I-2, suggesting dephosphorylation of 32P-c-Jun by axoplasmic serine-threonine phosphatases (Fig. 5E). Next, two sister axoplasms were prepared from the same squid, and each was perfused with 32P-c-Jun and either the 2–18 or scrambled 2–18 peptides. Consistent with results from vesicle motility assays, 32P-c-Jun was dephosphorylated to a greater extent in PAD peptide-perfused axoplasms than their scrambled peptide-perfused counterpart (Fig. 5F), suggesting that the PAD peptide induced activation of endogenous axoplasmic phosphatases.
Kinase activity assays were used to determine the effect of the PAD peptide on axoplasmic GSK3. Sister axoplasms were treated with PAD peptide or scrambled peptide, and then aliquots of perfusate containing axoplasmic proteins were incubated with 32P-ATP and either a GSK3 (GSM peptide) or protein kinase C (MARCKS peptide) substrate. A trend of elevated GSK3 activity (∼32%) relative to PKC activity (p = 0.28, paired t test; n = 3) was observed for axoplasms incubated with the PAD peptide (mean = 4.9 ± 2.5), compared with those incubated with scrambled peptide (mean = 3.7 ± 1.7), which supports the results from vesicle motility assays. Reduced sensitivity to changes in GSK3 activity may likely be a result of PAD affecting only a fraction of all available axoplasmic GSK3 enzymes. Collectively, these data indicate that PAD is both necessary and sufficient to inhibit anterograde FAT by activating the PP1–GSK3 cascade.
Disease-associated changes in tau cause inhibition of FAT
Together, the above data suggest that increased exposure of PAD in the context of aggregated WT tau and 6D/6P tau isoforms, results in PP1-GSK3 activation and anterograde FAT inhibition, whereas PAD exposure is limited in soluble WT tau. Consistent with this notion, a paperclip conformation has been shown for soluble, monomeric WT tau (Jeganathan et al., 2006). This conformation involves intramolecular interactions between the C terminus and the MTBR, and between the extreme N terminus (containing PAD) and the C terminus. Folding of tau into the paperclip conformation may therefore limit exposure of PAD, preventing it from activating the PP1–GSK3 cascade. This model predicts that disease-related tau modifications in monomeric tau that alter the paperclip conformation may result in abnormal exposure of PAD and should be toxic to FAT. To test this possibility, we evaluated the effects of two disease-associated forms of tau, termed “AT8 tau” and “Δ144–273 tau” (Fig. 1B,C), on FAT.
Pseudophosphorylation of tau at the AT8 epitope (comprised of Ser199, Ser202, and Thr205) reduces the folding of the N terminus into the paperclip conformation (Jeganathan et al., 2008), suggesting that phosphorylation of tau at these sites could increase PAD exposure. To determine whether AT8 phosphorylated tau monomers could inhibit anterograde FAT, we perfused AT8 pseudophosphorylated tau [Ser199Glu/Ser202Glu/Thr205Glu (AT8 tau)] (Fig. 1B) into isolated squid axoplasms. Soluble AT8 tau monomers inhibited anterograde FAT (Fig. 6A,C), while retrograde transport was unaffected (Fig. 6A,D). These data indicate that phosphorylation of tau at the AT8 epitope, which is associated with hyperphosphorylation of tau in AD and other tauopathies, renders soluble monomeric tau capable of inhibiting anterograde FAT.
Figure 6.

Tau monomers with disease-associated modifications inhibit anterograde FAT. A, Perfusion of squid axoplasm with soluble tau monomers containing mutations S199E, S202E, and T205E (phosphomimicking the AT8 epitope seen in hyperphosphorylated tau; AT8 tau) causes a reduction in anterograde FAT (black triangle), but not retrograde (gray triangle) FAT. B, Similarly, perfusion of squid axoplasm with soluble Δ144–273 tau monomer (mimicking a deletion mutation associated with a case of FTD) results in a reduction in anterograde FAT, but not retrograde FAT. C, Quantitative analysis of FAT rates demonstrates that AT8 tau monomers and Δ144–273 tau monomers significantly inhibit anterograde FAT, when compared with WT tau monomers (one-way ANOVA, *p < 0.01). D, Retrograde FAT rates remained unaffected for all pathogenic tau monomers tested.
A novel mutation in the tau gene associated with a case of FTD was recently reported (Rovelet-Lecrux et al., 2009). This tau mutation results in deletion of amino acids 144–273 encompassing the proline-rich domain and the first MTBR (Δ144–273 tau) (Fig. 1C). Deletion of these tau domains should result in constitutive exposure of PAD because they play a pivotal role in the intramolecular folding of the N terminus. Consistent with our model, Δ144–273 tau monomers significantly inhibited anterograde FAT (Fig. 6B,C), while retrograde FAT remained unaffected (Fig. 6B,D).Together, these data indicate that disease-associated modifications and mutations in tau that increase exposure of PAD promote activation of the PP1–GSK3 pathway and inhibition of anterograde FAT.
Increased exposure of PAD is an early event in AD
Results from isolated axoplasm experiments suggested a molecular basis for tau toxicity. To evaluate the relevance of these findings in human disease, we generated a novel monoclonal antibody, termed TNT1, which specifically recognizes PAD. The TNT1 antibody exhibits a very high affinity (EC50 = 5.5 ng/ml) for WT tau containing PAD (Fig. 7A) and does not react with tau missing PAD (Δ2–18 tau) in ELISAs [WT tau: mean absorbance at 450 nm (A450) = 2.4 ± 0.03; Δ2–18 tau: mean A450 = 0.003 ± 0.004] and immunoblots (Fig. 7A). Using the TNT1 antibody, we examined the availability of PAD in AD and control brains. First, we probed samples of soluble and insoluble tau isolated from the frontal cortex of control and severe AD cases using immunoblots. The pattern of TNT1 reactivity on Western blots was consistent with that of tau isolated from human brain as indicated by the Tau12 (Horowitz et al., 2004) and R1 tau antibodies (Berry et al., 2004) (two well characterized tau-specific antibodies), confirming the specificity of TNT1 for tau protein and not non-tau proteins in human brains (Fig. 7B). Under these denaturing conditions, TNT1 reacted similarly with soluble tau from controls and AD samples, as well as with insoluble tau from AD samples (Fig. 7B).
To further explore the characteristics of TNT1 reactivity, we analyzed soluble and insoluble tau fractions using dot blots, which like Western blots are solid-state assays, but unlike Western blots the proteins are not denatured. In dot blots, TNT1 showed a significantly increased selectivity for both soluble and insoluble tau isolated from AD, as opposed to control brains (Fig. 7C). The lack of distinction between control and AD tau in denaturing conditions by TNT1, but significant selectivity for AD tau over control tau in non-denaturing conditions, highlights that TNT1 reactivity is indicative of PAD exposure, which is dependent upon maintaining protein conformation. Thus, these results suggest that PAD is more readily available in the native state of tau derived from AD brains, compared with tau derived from control brains, and TNT1 immunoreactivity can be used as an indicator of PAD exposure.
Immunohistochemistry with TNT1 was performed on tissue sections from controls (Braak stages I–II; n = 4) and severe AD (Braak stages V–VI; n = 4) cases to determine when and where PAD exposure occurs in the disease process. Qualitative evaluation of sections from the entorhinal cortex (Fig. 7D) and hippocampus (Fig. 7E), as well as the inferior and superior temporal gyri (data not shown) indicated that TNT1 reactivity followed the typical pattern of Braak staging (Braak and Braak, 1991). In age-matched control cases, TNT1 diffusely labeled neurons in the earliest stages of tau deposition (e.g., early pretangle neurons), and a subset of these neurons contained small, darkly stained, globular intracellular inclusions (Fig. 7F). A small number of neurons in control cases exhibited strong TNT1 immunoreactivity of seemingly immature tangle-like inclusions that extended into both apical and basal dendrites (Fig. 7F). With progression into severe AD, there was a remarkable increase in the amount of TNT1 immunoreactive inclusions (Fig. 7D,E), which included neurofibrillary tangles (Fig. 7G), neuropil threads (Fig. 7G), and neuritic plaques (Fig. 7H). These data indicated that increased PAD exposure, as revealed by TNT1 immunoreactivity, occurs early in AD and remains present throughout the disease process.
We next determined the relationship between PAD exposure and AT8 phosphorylation in tau since this modification rendered soluble tau monomers toxic to FAT (see above). Tissue sections from the hippocampus of controls (Braak stages I–II; n = 4), mild AD (Braak stages III–IV; n = 5), and severe AD (Braak stages V-VI; n = 4) were double stained with TNT1 and AT8 antibodies using immunofluorescence (Fig. 7I–K). Qualitative observations indicated that TNT1 immunoreactivity might appear slightly before the development of AT8 phosphorylation in the course of AD. Numerous TNT1-positive structures (e.g., cell bodies and threads) in control cases (Fig. 7I) did not appear to be AT8 positive. As pathology progressed into mild AD (Fig. 7J), the amount of both TNT1 and AT8 increased. Finally, in severe AD stages (Fig. 7K) both TNT1 and AT8 were abundant and exhibited nearly complete colocalization. It should be noted that when present, AT8 immunoreactivity was almost always colocalized with TNT1 in all cases analyzed. Together, these data suggest that increased PAD exposure represents an early event in AD pathogenesis and that AT8 may not be required for PAD exposure in situ.
Discussion
Recently, we demonstrated that WT tau aggregates inhibit anterograde FAT by activating a PP1–GSK3 signaling cascade through a mechanism requiring the N terminus of tau (LaPointe et al., 2009). Extending those observations, we have identified a tau domain comprised of amino acids 2–18 that is both necessary and sufficient to activate the PP1–GSK3 cascade and inhibit anterograde FAT. These effects suggest a novel function for tau and define a new functional motif in tau, the PAD. Inhibition of anterograde FAT by the 6D and 6P isoforms of tau and a synthetic PAD peptide demonstrates that this effect does not require tau binding to microtubules or tau aggregation. Our results demonstrate that exposure of PAD is a critical factor mediating the ability of pathogenic forms of tau to inhibit FAT. Consistent with this hypothesis, two disease-associated tau modifications that increase PAD exposure inhibited anterograde FAT as soluble monomers. Further supporting the role of PAD exposure as an important disease-related event, the novel PAD-specific antibody TNT1 selectively labeled both soluble and insoluble tau isolated from AD brains, but not that from controls. Moreover, qualitative observations of TNT1 immunoreactivity in human AD tissue sections suggested that PAD exposure represents an early pathological event that closely associates in time with AT8 phosphorylation. These data have important implications regarding the role of tau in disease pathogenesis and provide a mechanism through which disease-associated modifications and/or mutations in tau lead to a toxic gain-of-function in AD and other tauopathies.
The demonstration that PAD is a region within tau that can modulate the PP1–GSK3 cascade has significant implications for tau pathology and suggests a novel biological role for tau regulating the activity of phosphotransferases and anterograde FAT (Fig. 8). Tau has been shown to target PP1 to microtubules and can directly interact with PP1 (Liao et al., 1998). Amino acids 5–8, located within PAD, comprise one of the proposed PP1 binding sites in tau (Liao et al., 1998). Additionally, tau and GSK3β have been copurified as part of a high-molecular-weight complex in association with purified microtubules (Sun et al., 2002). Thus, in addition to microtubule stabilization, tau may bind and target phosphotransferases to the vicinity of microtubules (Shahani and Brandt, 2002) and regulate FAT through modulation of these enzymes (Fig. 8A), a functional scheme that may be disrupted in disease (Fig. 8B).
Figure 8.

Proposed role of PAD in anterograde fast axonal transport regulation and in axonal transport dysfunction in disease. A, Tau is normally localized to microtubules where, in addition to stabilizing microtubules, it may regulate local cargo delivery through PAD-mediated activation of the PP1–GSK3 cascade. When tau is folded into the paperclip conformation, membrane-bound cargoes are transported by conventional kinesin along microtubules because PAD is hidden. In contrast, cargo is delivered at appropriate locations where tau is in an extended conformation and PAD is exposed. The exposure of PAD activates PP1-GSK3, leading to phosphorylation of kinesin light chains and release of cargoes. B, In disease states, tau aggregations and/or modifications expose PADs in tau that no longer binds to microtubules, promoting aberrant activation of the PP1–GSK3 cascade, increased inhibition of anterograde FAT, and ultimately neuron dysfunction/degeneration. Abnormal presentation of the biologically active PAD motif represents a critical factor in determining whether disease-associated forms of tau inhibit anterograde FAT.
Deficits in FAT have previously been implicated in the neuronal dysfunction associated with dysferopathies such as AD, non-AD tauopathies, and other neurodegenerative diseases (Morfini et al., 2002a, 2009; Roy et al., 2005). In AD brains, dystrophic neurites, synaptic loss, and protein mislocalization (Scheff et al., 1990; Dessi et al., 1997) are all consistent with FAT deficits. Accordingly, studies in various animal models of AD and tauopathies have reported alterations in axonal transport (e.g., axonal swellings, synaptic loss, and impaired vesicle transport) (for review, see Higuchi et al., 2006). Interestingly, tau appears to play a critical role in amyloid-β-mediated axonal transport disruption, as genetically removing tau mitigates the effects of treating neurons with amyloid-β oligomers (Vossel et al., 2010). These studies suggested a link among tau, axonal dysfunction, and neurodegenerative disease; however, until this report the molecular mechanisms linking deficits in FAT to disease-related modifications of tau had remained unclear (Morfini et al., 2002a).
Several studies on the role for tau in axonal transport dysfunction proposed a mechanism involving direct interference of tau with the binding of conventional kinesin to microtubules (Ebneth et al., 1998; Seitz et al., 2002; Vershinin et al., 2007). However, overexpression of human tau in transgenic mice did not affect FAT rates in the optic nerve (Yuan et al., 2008), and levels of soluble WT tau 20-fold greater than endogenous levels of tau had no effect on FAT in squid axoplasm (Morfini et al., 2007). Studies on aggregated tau (LaPointe et al., 2009) and the current data provide evidence for an alternative mechanism independent of microtubule binding that does not require aggregation. Specifically, many of the tau species examined in these studies do not effectively bind microtubules (e.g., tau aggregates, 6D/6P tau, PAD peptide, and FTD Δ144–273 tau), and many were used as monomers (e.g., 6D/6P tau, PAD peptide, FTD Δ144–273 tau, and AT8 tau). Moreover, we have established that tau inhibits anterograde FAT by activating the PP1–GSK3 cascade with nearly all of the tau species tested thus far. Together, these data clearly demonstrate that tau-mediated disruption of anterograde FAT via the PP1–GSK3 cascade is independent of microtubule binding and does not require aggregation.
Based on our prior studies (LaPointe et al., 2009), it was unclear why monomeric WT tau failed to affect FAT even though the PAD motif was present. Tau was originally thought to exist in an extended random coil state based on spectroscopic studies (Mandelkow et al., 2007). However, fluorescence resonance energy transfer studies indicated that soluble monomeric WT tau adopts a folded paperclip conformation (Jeganathan et al., 2006, 2008), that may prevent PAD from activating the PP1–GSK3 cascade. Conversely, conformations of tau that increase exposure of PAD should promote inhibition of anterograde FAT.
We tested this hypothesis using numerous forms of tau in which the accessibility of PAD is increased. In tau aggregates, both termini of tau are believed to maintain a random coil structure extending from the filament core formed by the MTBRs (Barghorn et al., 2004). Evidence also suggests aggregated tau favors an Alz50-type conformation (Carmel et al., 1996), in which the N terminus is in close contact with the MTBRs. However, the results presented here suggest that the extreme N terminus of tau remains accessible in aggregated tau. The 6D and 6P isoforms of tau lack the MTBRs and C terminus (Luo et al., 2004) necessary for the paperclip conformation, leaving PAD constitutively available, and the PAD peptide is composed of only amino acids 2–18. Similarly, a recombinant pseudophosphorylated AT8 tau exhibits reduced folding of the N terminus into the paperclip conformation (Jeganathan et al., 2008). Finally, deletion of amino acids 144–273 in the FTD tau construct should dramatically reduce or eliminate the ability of the N terminus to fold into the paperclip, as this deletion removes the proline-rich hinge region involved in N-terminal folding. Consistent with our hypothesis, all of these “PAD-exposed” tau species inhibited anterograde FAT; thus, exposure of PAD provides a common mechanism of toxicity for biochemically heterogeneous forms of pathogenic tau.
Highlighting the relevance of results obtained from studies in isolated squid axoplasm, a PAD-specific antibody (TNT1) that preferentially recognizes disease-related forms of tau confirmed the importance of PAD exposure in human disease pathogenesis. Data obtained from immunostaining studies in human brain tissue indicated that increased PAD exposure occurs very early in the process of tau inclusion formation and that AT8 is closely associated with PAD exposure during AD progression. Importantly, quantitative analyses are required to confirm our observations and conclusively determine the time course of PAD exposure in relation to other tau modifications during the progression of disease in humans. However, in combination with our squid axoplasm data, it is reasonable to assume that AT8 modification abundantly found in AD (Braak et al., 1994) and other tauopathies (Takahashi et al., 2002; Sakai et al., 2006; Shiarli et al., 2006) may facilitate PAD exposure and tau-mediated FAT dysfunction in situ. Additionally, our data suggest a cyclical relationship between PAD exposure and AT8 phosphorylation in which the AT8 modification might result, at least in part, from the increased GSK3 activation triggered by abnormally exposed PAD, since S199, S202, and T205 are GSK3β phosphorylation sites.
Since neurons are thought to maintain function for years while bearing tau inclusions (Morsch et al., 1999), it is reasonable to assume the existence of compensatory mechanisms that render pathogenic forms of tau innocuous. Phosphorylation of tyrosine 18 (within PAD) by kinases such as fyn (Lee et al., 2004) or N-terminal cleavage of tau (Horowitz et al., 2004; Sengupta et al., 2006) may represent such protective mechanisms. Jeganathan et al. (2008) demonstrated that combinations of phosphorylation events have different effects on tau folding. While AT8 alone reduces N-terminal folding, AT8 in combination with the AT100 (T212/S214) and PHF-1 (S396/S404) phosphorylation epitopes causes tau to fold into a more compact paperclip conformation. Regulation of tau conformation through protein–protein or intraprotein interactions as well as through modifications like phosphorylation or proteolysis might be part of a complex set of events that regulate tau function, including microtubule binding, modulation of phosphotransferase-based signaling pathways, and anterograde FAT.
Although tau has been recognized as a potential therapeutic target in AD and tauopathies, choosing therapeutic targets and testable outcome measures has proven difficult without an understanding of the mechanisms through which tau can influence disease progression. The identification of PAD and its effects on phosphotransferase activity and FAT regulation allow us to propose specific targets for intervention. For instance, reducing the activities of PP1 and/or GSK3 may reduce the toxic potential of pathogenic forms of tau. Alternatively, therapeutic strategies aimed at directly blocking PAD exposure and preventing it from activating the PP1–GSK3 cascade may provide yet another viable point of intervention.
Footnotes
This work was supported by NIH Grants T32 AG020506-07 (N.M.K.); AG09466 (L.I.B.); and NS23868, NS23320, and NS41170 (S.T.B.); as well as 2007/2008 MBL Summer Research Fellowships and an ALS/CVS Therapy Alliance grant (G.M.). We thank Alena Leitman, Sarah Berth, Agnieska Kaminska, Lisa Baker, Meryl Horn, Alex Sturbaum, and Yuka Atagi of the Brady and Morfini laboratories, who participated in our research at the Marine Biological Laboratory (Woods Hole, MA). We would also like to thank Juan Reyes for preparing some of the human tau samples and John Hardy (University College, London, UK) for drawing our attention to the Δ144-273 FTD mutation.
The authors declare no competing financial interests.
References
- Alonso AC, Grundke-Iqbal I, Iqbal K. Alzheimer's disease hyperphosphorylated tau sequesters normal tau into tangles of filaments and disassembles microtubules. Nat Med. 1996;2:783–787. doi: 10.1038/nm0796-783. [DOI] [PubMed] [Google Scholar]
- Andreadis A. Tau gene alternative splicing: expression patterns, regulation and modulation of function in normal brain and neurodegenerative diseases. Biochim Biophys Acta. 2005;1739:91–103. doi: 10.1016/j.bbadis.2004.08.010. [DOI] [PubMed] [Google Scholar]
- Arriagada PV, Growdon JH, Hedley-Whyte ET, Hyman BT. Neurofibrillary tangles but not senile plaques parallel duration and severity of Alzheimer's disease. Neurology. 1992;42:631–639. doi: 10.1212/wnl.42.3.631. [DOI] [PubMed] [Google Scholar]
- Barghorn S, Davies P, Mandelkow E. Tau paired helical filaments from Alzheimer's disease brain and assembled in vitro are based on beta-structure in the core domain. Biochemistry. 2004;43:1694–1703. doi: 10.1021/bi0357006. [DOI] [PubMed] [Google Scholar]
- Berry RW, Sweet AP, Clark FA, Lagalwar S, Lapin BR, Wang T, Topgi S, Guillozet-Bongaarts AL, Cochran EJ, Bigio EH, Binder LI. Tau epitope display in progressive supranuclear palsy and corticobasal degeneration. J Neurocytol. 2004;33:287–295. doi: 10.1023/B:NEUR.0000044190.96426.b9. [DOI] [PubMed] [Google Scholar]
- Biernat J, Mandelkow EM, Schröter C, Lichtenberg-Kraag B, Steiner B, Berling B, Meyer H, Mercken M, Vandermeeren A, Goedert M, Mandelkow E. The switch of tau protein to an Alzheimer-like state includes the phosphorylation of two serine-proline motifs upstream of the microtubule binding region. EMBO J. 1992;11:1593–1597. doi: 10.1002/j.1460-2075.1992.tb05204.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Braak E, Braak H, Mandelkow EM. A sequence of cytoskeleton changes related to the formation of neurofibrillary tangles and neuropil threads. Acta Neuropathol. 1994;87:554–567. doi: 10.1007/BF00293315. [DOI] [PubMed] [Google Scholar]
- Braak H, Braak E. Neuropathological stageing of Alzheimer-related changes. Acta Neuropathol. 1991;82:239–259. doi: 10.1007/BF00308809. [DOI] [PubMed] [Google Scholar]
- Brady ST, Richards BW, Leopold PL. Assay of vesicle motility in squid axoplasm. Methods Cell Biol. 1993;39:191–202. doi: 10.1016/s0091-679x(08)60171-5. [DOI] [PubMed] [Google Scholar]
- Carmel G, Mager EM, Binder LI, Kuret J. The structural basis of monoclonal antibody Alz50's selectivity for Alzheimer's disease pathology. J Biol Chem. 1996;271:32789–32795. doi: 10.1074/jbc.271.51.32789. [DOI] [PubMed] [Google Scholar]
- Dessi F, Colle MA, Hauw JJ, Duyckaerts C. Accumulation of SNAP-25 immunoreactive material in axons of Alzheimer's disease. Neuroreport. 1997;8:3685–3689. doi: 10.1097/00001756-199712010-00006. [DOI] [PubMed] [Google Scholar]
- Drubin DG, Feinstein SC, Shooter EM, Kirschner MW. Nerve growth factor-induced neurite outgrowth in PC12 cells involves the coordinate induction of microtubule assembly and assembly-promoting factors. J Cell Biol. 1985;101:1799–1807. doi: 10.1083/jcb.101.5.1799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ebneth A, Godemann R, Stamer K, Illenberger S, Trinczek B, Mandelkow E. Overexpression of tau protein inhibits kinesin-dependent trafficking of vesicles, mitochondria, and endoplasmic reticulum: implications for Alzheimer's disease. J Cell Biol. 1998;143:777–794. doi: 10.1083/jcb.143.3.777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giannakopoulos P, Herrmann FR, Bussière T, Bouras C, Kövari E, Perl DP, Morrison JH, Gold G, Hof PR. Tangle and neuron numbers, but not amyloid load, predict cognitive status in Alzheimer's disease. Neurology. 2003;60:1495–1500. doi: 10.1212/01.wnl.0000063311.58879.01. [DOI] [PubMed] [Google Scholar]
- Goedert M, Jakes R. Mutations causing neurodegenerative tauopathies. Biochim Biophys Acta. 2005;1739:240–250. doi: 10.1016/j.bbadis.2004.08.007. [DOI] [PubMed] [Google Scholar]
- Goedert M, Jakes R, Vanmechelen E. Monoclonal antibody AT8 recognises tau protein phosphorylated at both serine 202 and threonine 205. Neurosci Lett. 1995;189:167–169. doi: 10.1016/0304-3940(95)11484-e. [DOI] [PubMed] [Google Scholar]
- Grundke-Iqbal I, Iqbal K, Quinlan M, Tung YC, Zaidi MS, Wisniewski HM. Microtubule-associated protein tau. A component of Alzheimer paired helical filaments. J Biol Chem. 1986;261:6084–6089. [PubMed] [Google Scholar]
- Hasegawa M, Morishima-Kawashima M, Takio K, Suzuki M, Titani K, Ihara Y. Protein sequence and mass spectrometric analyses of tau in the Alzheimer's disease brain. J Biol Chem. 1992;267:17047–17054. [PubMed] [Google Scholar]
- Higuchi M, Saido TC, Suhara T. Animal models of tauopathies. Neuropathology. 2006;26:491–497. doi: 10.1111/j.1440-1789.2006.00685.x. [DOI] [PubMed] [Google Scholar]
- Horowitz PM, Patterson KR, Guillozet-Bongaarts AL, Reynolds MR, Carroll CA, Weintraub ST, Bennett DA, Cryns VL, Berry RW, Binder LI. Early N-terminal changes and caspase-6 cleavage of tau in Alzheimer's disease. J Neurosci. 2004;24:7895–7902. doi: 10.1523/JNEUROSCI.1988-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jeganathan S, von Bergen M, Brutlach H, Steinhoff HJ, Mandelkow E. Global hairpin folding of tau in solution. Biochemistry. 2006;45:2283–2293. doi: 10.1021/bi0521543. [DOI] [PubMed] [Google Scholar]
- Jeganathan S, Hascher A, Chinnathambi S, Biernat J, Mandelkow EM, Mandelkow E. Proline-directed pseudo-phosphorylation at AT8 and PHF1 epitopes induces a compaction of the paperclip folding of Tau and generates a pathological (MC-1) conformation. J Biol Chem. 2008;283:32066–32076. doi: 10.1074/jbc.M805300200. [DOI] [PubMed] [Google Scholar]
- Kanaan NM, Kordower JH, Collier TJ. Age-related accumulation of Marinesco bodies and lipofuscin in rhesus monkey midbrain dopamine neurons: relevance to selective neuronal vulnerability. J Comp Neurol. 2007;502:683–700. doi: 10.1002/cne.21333. [DOI] [PubMed] [Google Scholar]
- King ME, Ahuja V, Binder LI, Kuret J. Ligand-dependent tau filament formation: implications for Alzheimer's disease progression. Biochemistry. 1999;38:14851–14859. doi: 10.1021/bi9911839. [DOI] [PubMed] [Google Scholar]
- Kosik KS, Joachim CL, Selkoe DJ. Microtubule-associated protein tau (τ) is a major antigenic component of paired helical filaments in Alzheimer disease. Proc Natl Acad Sci U S A. 1986;83:4044–4048. doi: 10.1073/pnas.83.11.4044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kozikowski AP, Gaisina IN, Yuan H, Petukhov PA, Blond SY, Fedolak A, Caldarone B, McGonigle P. Structure-based design leads to the identification of lithium mimetics that block mania-like effects in rodents. Possible new GSK-3beta therapies for bipolar disorders. J Am Chem Soc. 2007;129:8328–8332. doi: 10.1021/ja068969w. [DOI] [PubMed] [Google Scholar]
- LaPointe NE, Morfini G, Pigino G, Gaisina IN, Kozikowski AP, Binder LI, Brady ST. The amino terminus of tau inhibits kinesin-dependent axonal transport: implications for filament toxicity. J Neurosci Res. 2009;87:440–451. doi: 10.1002/jnr.21850. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee G, Thangavel R, Sharma VM, Litersky JM, Bhaskar K, Fang SM, Do LH, Andreadis A, Van Hoesen G, Ksiezak-Reding H. Phosphorylation of tau by fyn: implications for Alzheimer's disease. J Neurosci. 2004;24:2304–2312. doi: 10.1523/JNEUROSCI.4162-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liao H, Li Y, Brautigan DL, Gundersen GG. Protein phosphatase 1 is targeted to microtubules by the microtubule-associated protein Tau. J Biol Chem. 1998;273:21901–21908. doi: 10.1074/jbc.273.34.21901. [DOI] [PubMed] [Google Scholar]
- Luo MH, Tse SW, Memmott J, Andreadis A. Novel isoforms of tau that lack the microtubule-binding domain. J Neurochem. 2004;90:340–351. doi: 10.1111/j.1471-4159.2004.02508.x. [DOI] [PubMed] [Google Scholar]
- Mandelkow E, von Bergen M, Biernat J, Mandelkow EM. Structural principles of tau and the paired helical filaments of Alzheimer's disease. Brain Pathol. 2007;17:83–90. doi: 10.1111/j.1750-3639.2007.00053.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morfini G, Pigino G, Beffert U, Busciglio J, Brady ST. Fast axonal transport misregulation and Alzheimer's disease. Neuromolecular Med. 2002a;2:89–99. doi: 10.1385/NMM:2:2:089. [DOI] [PubMed] [Google Scholar]
- Morfini G, Szebenyi G, Elluru R, Ratner N, Brady ST. Glycogen synthase kinase 3 phosphorylates kinesin light chains and negatively regulates kinesin-based motility. EMBO J. 2002b;21:281–293. doi: 10.1093/emboj/21.3.281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morfini G, Szebenyi G, Brown H, Pant HC, Pigino G, DeBoer S, Beffert U, Brady ST. A novel CDK5-dependent pathway for regulating GSK3 activity and kinesin-driven motility in neurons. EMBO J. 2004;23:2235–2245. doi: 10.1038/sj.emboj.7600237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morfini G, Pigino G, Mizuno N, Kikkawa M, Brady ST. Tau binding to microtubules does not directly affect microtubule-based vesicle motility. J Neurosci Res. 2007;85:2620–2630. doi: 10.1002/jnr.21154. [DOI] [PubMed] [Google Scholar]
- Morfini GA, Burns M, Binder LI, Kanaan NM, LaPointe N, Bosco DA, Brown RH, Jr, Brown H, Tiwari A, Hayward L, Edgar J, Nave KA, Garberrn J, Atagi Y, Song Y, Pigino G, Brady ST. Axonal transport defects in neurodegenerative diseases. J Neurosci. 2009;29:12776–12786. doi: 10.1523/JNEUROSCI.3463-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morsch R, Simon W, Coleman PD. Neurons may live for decades with neurofibrillary tangles. J Neuropathol Exp Neurol. 1999;58:188–197. doi: 10.1097/00005072-199902000-00008. [DOI] [PubMed] [Google Scholar]
- Reyes JF, Reynolds MR, Horowitz PM, Fu Y, Guillozet-Bongaarts AL, Berry R, Binder LI. A possible link between astrocyte activation and tau nitration in Alzheimer's disease. Neurobiol Dis. 2008;31:198–208. doi: 10.1016/j.nbd.2008.04.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rovelet-Lecrux A, Lecourtois M, Thomas-Anterion C, Le Ber I, Brice A, Frebourg T, Hannequin D, Campion D. Partial deletion of the MAPT gene: a novel mechanism of FTDP-17. Hum Mutat. 2009;30:E591–E602. doi: 10.1002/humu.20979. [DOI] [PubMed] [Google Scholar]
- Roy S, Zhang B, Lee VM, Trojanowski JQ. Axonal transport defects: a common theme in neurodegenerative diseases. Acta Neuropathol. 2005;109:5–13. doi: 10.1007/s00401-004-0952-x. [DOI] [PubMed] [Google Scholar]
- Sakai K, Piao YS, Kikugawa K, Ohara S, Hasegawa M, Takano H, Fukase M, Nishizawa M, Kakita A, Takahashi H. Corticobasal degeneration with focal, massive tau accumulation in the subcortical white matter astrocytes. Acta Neuropathol. 2006;112:341–348. doi: 10.1007/s00401-006-0093-5. [DOI] [PubMed] [Google Scholar]
- Scheff SW, DeKosky ST, Price DA. Quantitative assessment of cortical synaptic density in Alzheimer's disease. Neurobiol Aging. 1990;11:29–37. doi: 10.1016/0197-4580(90)90059-9. [DOI] [PubMed] [Google Scholar]
- Schweers O, Schönbrunn-Hanebeck E, Marx A, Mandelkow E. Structural studies of tau protein and Alzheimer paired helical filaments show no evidence for beta-structure. J Biol Chem. 1994;269:24290–24297. [PubMed] [Google Scholar]
- Seitz A, Kojima H, Oiwa K, Mandelkow EM, Song YH, Mandelkow E. Single-molecule investigation of the interference between kinesin, tau and MAP2c. EMBO J. 2002;21:4896–4905. doi: 10.1093/emboj/cdf503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sengupta S, Horowitz PM, Karsten SL, Jackson GR, Geschwind DH, Fu Y, Berry RW, Binder LI. Degradation of tau protein by puromycin-sensitive aminopeptidase in vitro. Biochemistry. 2006;45:15111–15119. doi: 10.1021/bi061830d. [DOI] [PubMed] [Google Scholar]
- Shahani N, Brandt R. Functions and malfunctions of the tau proteins. Cell Mol Life Sci. 2002;59:1668–1680. doi: 10.1007/PL00012495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shiarli AM, Jennings R, Shi J, Bailey K, Davidson Y, Tian J, Bigio EH, Ghetti B, Murrell JR, Delisle MB, Mirra S, Crain B, Zolo P, Arima K, Iseki E, Murayama S, Kretzschmar H, Neumann M, Lippa C, Halliday G, et al. Comparison of extent of tau pathology in patients with frontotemporal dementia with Parkinsonism linked to chromosome 17 (FTDP-17), frontotemporal lobar degeneration with Pick bodies and early onset Alzheimer's disease. Neuropathol Appl Neurobiol. 2006;32:374–387. doi: 10.1111/j.1365-2990.2006.00736.x. [DOI] [PubMed] [Google Scholar]
- Sun W, Qureshi HY, Cafferty PW, Sobue K, Agarwal-Mawal A, Neufield KD, Paudel HK. Glycogen synthase kinase-3beta is complexed with tau protein in brain microtubules. J Biol Chem. 2002;277:11933–11940. doi: 10.1074/jbc.M107182200. [DOI] [PubMed] [Google Scholar]
- Takahashi M, Weidenheim KM, Dickson DW, Ksiezak-Reding H. Morphological and biochemical correlations of abnormal tau filaments in progressive supranuclear palsy. J Neuropathol Exp Neurol. 2002;61:33–45. doi: 10.1093/jnen/61.1.33. [DOI] [PubMed] [Google Scholar]
- Vershinin M, Carter BC, Razafsky DS, King SJ, Gross SP. Multiple-motor based transport and its regulation by Tau. Proc Natl Acad Sci U S A. 2007;104:87–92. doi: 10.1073/pnas.0607919104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vossel KA, Zhang K, Brodbeck J, Daub AC, Sharma P, Finkbeiner S, Cui B, Mucke L. Tau reduction prevents Abeta-induced defects in axonal transport. Science. 2010;330:198. doi: 10.1126/science.1194653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wood JG, Mirra SS, Pollock NJ, Binder LI. Neurofibrillary tangles of Alzheimer disease share antigenic determinants with the axonal microtubule-associated protein tau (tau) Proc Natl Acad Sci U S A. 1986;83:4040–4043. doi: 10.1073/pnas.83.11.4040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yuan A, Kumar A, Peterhoff C, Duff K, Nixon RA. Axonal transport rates in vivo are unaffected by tau deletion or overexpression in mice. J Neurosci. 2008;28:1682–1687. doi: 10.1523/JNEUROSCI.5242-07.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]