

Abstract

Aryl and heteroaryl boronic acids and boronate esters are rapidly, often within minutes, transformed into the corresponding phenols by N-oxides in an open flask at ambient temperature. This transformation has broad compatibility with a variety of functional groups.
Phenol is a ubiquitous structural unit found in a vast array of natural products and pharmaceuticals.1 Moreover, it frequently serves as the key synthetic intermediate for construction of more complex structures. Consequently, establishing mild and efficient access to phenols, especially in the presence of polyfunctional groups, is of great significance.
Arylboronic acids and related aromatic/heteroaromatic boronate esters are readily available and have found broad applications in synthetic chemistry.2 Their transformation into phenols is typically accomplished through transition-metal catalyzed hydroxylation or oxidative hydroxylation, but it continues to remain an area of active investigation. For example, Wang et al. and Fu et al. have recently reported copper catalyzed hydroxylations of arylboronic acids at room temperature,3,4 albeit using a stoichiometric strong base (KOH or NaOH) (Scheme 1(a)). Subsequently, alternative base-free or palladium catalyzed conditions, were developed.5 However, these approaches required several hours to complete the conversions. The hydroxylation of arylboronic acids has been achieved without transition metal catalysis, but this requires strong oxidants such as hydrogen peroxide and oxone for success (Scheme 1(b)).6,7 Moreover, the amount of oxidant needed and the reaction conditions (time, temperature) often must be carefully controlled to avoid over-oxidation of sensitive functionality. From a practical point of view, non-metal-mediated processes are much preferred in the pharmaceutical industry, since metal contamination can induce severe concerns and metal removal can be expensive. Herein, we reveal the first N-oxide mediated transformation of arylboronic acids to phenols (Scheme 1(c)). The reaction proceeds rapidly, mostly within a few minutes at ambient temperature and without base. Another notable feature is the compatibility of heteroaryl boronic acids which remains a challenge and has rarely been demonstrated.8
Scheme 1.

Hydroxylation of boronic acids
This report is based on unexpected findings during the exploration of cross-coupling reactions between N,N-dimethyl-4-toluidine N-oxide and boronic acids. Specifically, simple addition together of N-oxide 3 and 4-tert-butylphenylboronic acid 1a resulted in an exothermic reaction that led to the corresponding phenol 2a (Table 1, entry 1). Both are solids at room temperature. Given the nearly complete conversion, we assumed that the temperature rose high enough to melt one or both reagents. The reaction can be conducted at 0.2 M in DCM without compromising efficiency, giving the desired product in high chemical yield (Table 1, entry 2). We also examined other commercially available N-oxides, e.g., NMO and TMAO. Compared with 3, the reactions of 4 and 5 were patently sluggish (Table 1, entries 3 and 4). Notably, the reaction could also be carried out in water with prolonged reaction time, providing a potentially green process (Table 1, entry 5).
Table 1.
Reaction parameters
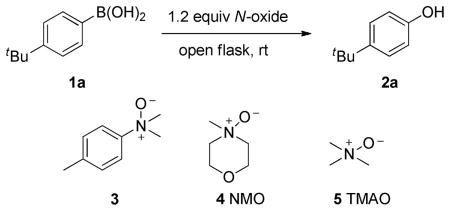
| |||
|---|---|---|---|
| entry | oxidant | condition | result |
| 1 | 3 | no solvent | highly exothermic, conversion > 99% |
| 2 | 3 | DCM (0.2 M) | 1 min, conversion > 99%, isolated yield 97% |
| 3 | 4 | DCM (0.2 M) | 10 min, conversion <10 %; 50 °C for 12 h, isolated yield 90% |
| 4 | 5 | DCM (0.2 M) | 10 min, conversion <10 %; 50 °C for 12 h, isolated yield 86% |
| 5 | 3 | H2O (0.2 M) | 10 h, isolated yield 85% |
The optimized reaction parameters from Table 1 were utilized to explore the substrate scope (Scheme 2). These transformations demonstrated excellent compatibility with a wide range of functional groups, e.g., halide, aldehyde, nitrile, sulfide, ester, nitro, etc. Superior results were observed irrespective of electronic or steric properties of the arylboronic acids. Generally, the conversion of electron-rich arylboronic acids was complete within 1 minute, while electron-deficient arylboronic acids took about 5 minutes or less. Importantly, oxidation-sensitive substituents such as sulfide 2e and aldehydes 2k, 2x tolerated the conditions without suffering over-oxidation. The transformation in the presence of aryl bromides 2g, 2q, 2u and iodide 2h is noteworthy, as the bromide and iodide can be exploited for subsequent functionalization. The chemoselective hydroxylation of boronic acid 2o instead of olefinic epoxidation displayed another undeniable advantage of this methodology. Even the highly crowded 2,6-dimethoxy boronic acid furnished the corresponding phenol 2z in useful yield by extension of the reaction time.
Scheme 2.

Hydroxylation of arylboronic acids
Encouraged by these results, we next investigated the hydroxylation of heteroaryl boronic acids (Scheme 3). Both pyridinyl and quinolinyl boronic acids were readily converted into the corresponding phenols 7a and 7b in good chemical yields. Hydroxylation of pyrrolyl boronic acid 6c followed by tautomerization efficiently generated a usefully synthetic building block 3-pyrrolin-2-one 7c.9 In addition to nitrogen-containing boronic acids, oxygen- and sulfur-containing heteroarenes such as benzofuran 6d and benzothiophene 6e were apt substrates.
Scheme 3.

Hydroxylation of heteroaryl boronic acidsa
a standard conditions: boronic acid 6 (0.2 mmol) and N-oxide 3 (0.24 mmol) in DCM (1 mL), room temperature, open flask.
Remarkably, not only boronic acids but other popular boronic acid surrogates proved suitable for hydroxylation (Scheme 4). Under the standard conditions, various phenyl boronic esters 8a–c as well as potassium phenyltrifluoroborate 8d afforded phenol 2b in satisfactory yield and with slightly slower reaction rates.
Scheme 4.

Hydroxylation of other boronic compoundsa
a standard conditions: boronic compound 8 (0.2 mmol) and N-oxide 3 (0.24 mmol) in DCM (1 mL), room temperature, open flask.
A postulated mechanistic pathway is depicted in Figure 1.10 We speculate that nucleophilic attack of the N-oxide on boronic acid 1 generates key intermediate I. Subsequent migration of the aryl group from boron to oxygen generates boronate ester II. Simultaneous departure of the quaternary ammonium cation provides an important driving force for the N-O bond cleavage. The newly released tertiary amine attacks the boronic ester, facilitating the release of phenoxide anion, which abstracts a proton from the nitrogen-boron complex III furnishing phenol 2 and zwitterion IV that precipitates from solution.11
Figure 1.

Plausible mechanism
In conclusion, a N-oxide mediated, mild and practical hydroxylation of boronic acids and boronate esters has been developed. The reactions are conducted in an open flask at room temperature, and most of them are complete within a few minutes. This transformation has broad functional group compatibility for both electron-rich and - deficient substituents. In addition to arylboronic acids, heteroaryl boronic acids are also tolerated in this methodology.
Significantly, this transformation provides a mechanistic inspiration which prompted us to pursue other transition-metal free functionalizations of boronic acids based upon the same concept. These results will be revealed in due course.
Supplementary Material
Acknowledgments
Financial support was provided by the Robert A. Welch Foundation (GL625910) and the NIH (GM31278).
Footnotes
Supporting Information Available. Experimental procedures, characterization data, and 1H, 13C spectra of compounds. This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.(a) Tyman JHP. Synthetic and Natural Phenols. Elsevier; New York: 1996. [Google Scholar]; (b) Rappoport Z. The Chemistry of Phenols. Wiley-VCH; Weinheim: 2003. [Google Scholar]
- 2.(a) Diederich F, Stang PJ. Metal-catalyzed Cross-coupling Reactions. Wiley-VCH; Weinheim: 1998. [Google Scholar]; (b) Hall DG. Boronic Acids: Preparation and Applications in Organic Synthesis and Medicine. Wiley-VCH; Weinheim: 2007. [Google Scholar]; (c) Suzuki A. Angew Chem, Int Ed. 2011;50:6722. doi: 10.1002/anie.201101379. [DOI] [PubMed] [Google Scholar]
- 3.Xu J, Wang X, Shao C, Su D, Cheng G, Hu Y. Org Lett. 2010;12:1964. doi: 10.1021/ol1003884. [DOI] [PubMed] [Google Scholar]
- 4.Yang H, Li Y, Jiang M, Wang J, Fu H. Chem Eur J. 2011;17:5652. doi: 10.1002/chem.201003711. [DOI] [PubMed] [Google Scholar]
- 5.(a) Inamoto K, Nozawa K, Yonemoto M, Kondo Y. Chem Commun. 2011;47:11775. doi: 10.1039/c1cc14974a. [DOI] [PubMed] [Google Scholar]; (b) Kaboudin B, Abedi Y, Yokomatsu T. Eur J Org Chem. 2011:6656. [Google Scholar]; (c) Chowdhury AD, Mobin SM, Mukherjee S, Bhaduri S, Lahiri GK. Eur J Inorg Chem. 2011:3232. [Google Scholar]
- 6.(a) Webb KS, Levy D. Tetrahedron Lett. 1995;36:5117. [Google Scholar]; (b) Simon J, Salzbrunn S, Prakash GKS, Petasis NA, Olah GA. J Org Chem. 2001;66:633. doi: 10.1021/jo0015873. [DOI] [PubMed] [Google Scholar]; (c) Travis BR, Ciaramitaro BP, Borhan B. Eur J Org Chem. 2002:3429. [Google Scholar]; (d) Prakash GKS, Chacko S, Panja C, Thomas TE, Gurung L, Rasul G, Mathew T, Olah GA. Adv Synth Catal. 2009;351:1567. [Google Scholar]
- 7.Other hydroxylations of boronic acids, see: Kianmehr E, Yahyaee M, Tabatabai K. Tetrahedron Lett. 2007;48:2713.Hosoi K, Kuriyama Y, Inagi S, Fuchigami T. Chem Commun. 2010;46:1284. doi: 10.1039/b914093j.Zou YQ, Chen JR, Liu XP, Lu LQ, Davis RL, Jørgensen KA, Xiao WJ. Angew Chem Int Ed. 2012;51:784. doi: 10.1002/anie.201107028.
- 8.An example of hydroxylation of organotrifluoroborates, see: Molander GA, Cavalcanti LN. J Org Chem. 2011;76:623. doi: 10.1021/jo102208d.
- 9.For selected examples, see: Shepherd NE, Tanabe H, Xu Y, Matsungaga S, Shibasaki M. J Am Chem Soc. 2010;132:3666. doi: 10.1021/ja1002636.Shao C, Yu HJ, Wu NY, Tian P, Wang R, Feng CG, Lin GQ. Org Lett. 2011;13:788. doi: 10.1021/ol103054a.Lin L, Zhang J, Ma X, Fu X, Wang R. Org Lett. 2011;13:6410. doi: 10.1021/ol202713f.Huang H, Jin Z, Zhu K, Liang X, Ye J. Angew Chem, Int Ed. 2011;50:3232. doi: 10.1002/anie.201008255.
- 10.For similar mechanism, see: Kabalka GW, Wadgaonkar PP, Shoup TM. Organometallics. 1990;9:1316.
- 11.The hydrolyzed product ArNMe2 of zwitterion IV can be observed in the reaction.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.