

Abstract
Cholate-CoA ligase (CCL) and bile acid-CoA: amino acid N-acyltransferase (BAAT) sequentially mediate bile-acid amidation. Defects can cause intrahepatic cholestasis. Distinction has required gene sequencing. We assessed potential clinical utility of immunostaining of liver for CCL and BAAT. Using commercially available antibodies against BAAT and CCL, we immunostained liver from an infant with jaundice, deficiency of amidated bile acids, and transcription-terminating mutation in BAAT. CCL was normally expressed. BAAT expression was not detected. Immunostaining may facilitate diagnosis in bile-acid amidation defects.
Keywords: Amidation, Bile acid-CoA, Amino acid N-acyltransferase, Cholate-CoA ligase, Cholestasis, Conjugation, Electrospray ionisation-mass spectroscopy, Immunohistochemistry, Liver, Neonatal hepatitis, SLC27A5, Transmission electron microscopy
INTRODUCTION
Conjugated bile acids are detergents essential in facilitating the enteric absorption of fats and of fat-soluble substances, among them several vitamins. They are synthesised in hepatocytes, which expel them into the lumen of the bile canaliculus via the transport protein bile salt export pump (BSEP). Conjugated bile acids are partially deconjugated during passage through the intestine. Bile acids are recovered from chyme and returned to the liver in portal venous blood; unconjugated bile acids are reconjugated with glycine or taurine within hepatocytes before re-export into canalicular bile[1]. That errors in synthesis or transport of bile acids may lead to intrahepatic cholestasis is well known[1,2]. Errors in conjugation of bile acids with amino acids (“amidation”), a two-step process, also may cause usual cholestasis, with jaundice, but can also produce anicteric cholestasis manifest as malabsorption of fat-soluble vitamins, growth retardation, and pruritus. Few descriptions are published of children with bile-acid amidation defects and of mutations in genes that encode the enzymes responsible for amidation. Diagnosis can be elusive[3]. We present our correlation of immunohistochemical, clinical-biochemistry, and genetic findings in a child with a bile-acid amidation defect and suggest approaches to diagnosis.
CASE REPORT
The first child of consanguine British parents of Pakistani heritage was born at term after an uncomplicated pregnancy. She was well until age 7 wk, when jaundice and pale stools were noted. Her liver and biliary tract were sonographically unremarkable. Clinical-laboratory findings included conjugated hyperbilirubinemia (total:direct bilirubin 106:67 μmol/L) with elevated serum alanine aminotransferase activity (378 IU/L), normal-range gamma-glutamyl transpeptidase (GGT) activity (30 IU/L), and hypercholanaemia (total serum bile acids 282 μmol/L, expected < 14). Liver biopsy at age 8 wk found hepatocellular and canalicular cholestasis, with slight portal-tract fibrosis and a “neonatal hepatitis” pattern of focal giant-cell change of hepatocytes, with haemopoiesis. Canalicular-margin expression of BSEP and of ectoenzymes (alanyl aminopeptidase, carcinoembryonic antigen, GGT) was normal. Transmission electron microscopy found changes of hepatocellular and canalicular cholestasis, without “Byler bile” or other abnormalities. Urine and plasma analysed by electrospray ionisation-mass spectroscopy[1,3] contained di- and tri-hydroxy C24 bile acids, without amidated forms (Figure 1). A defect in cholate-CoA ligase (CCL) or bile acid-CoA: amino acid N-acyltransferase (glycine N-choloyltransferase; BAAT) was hypothesised.
Figure 1.
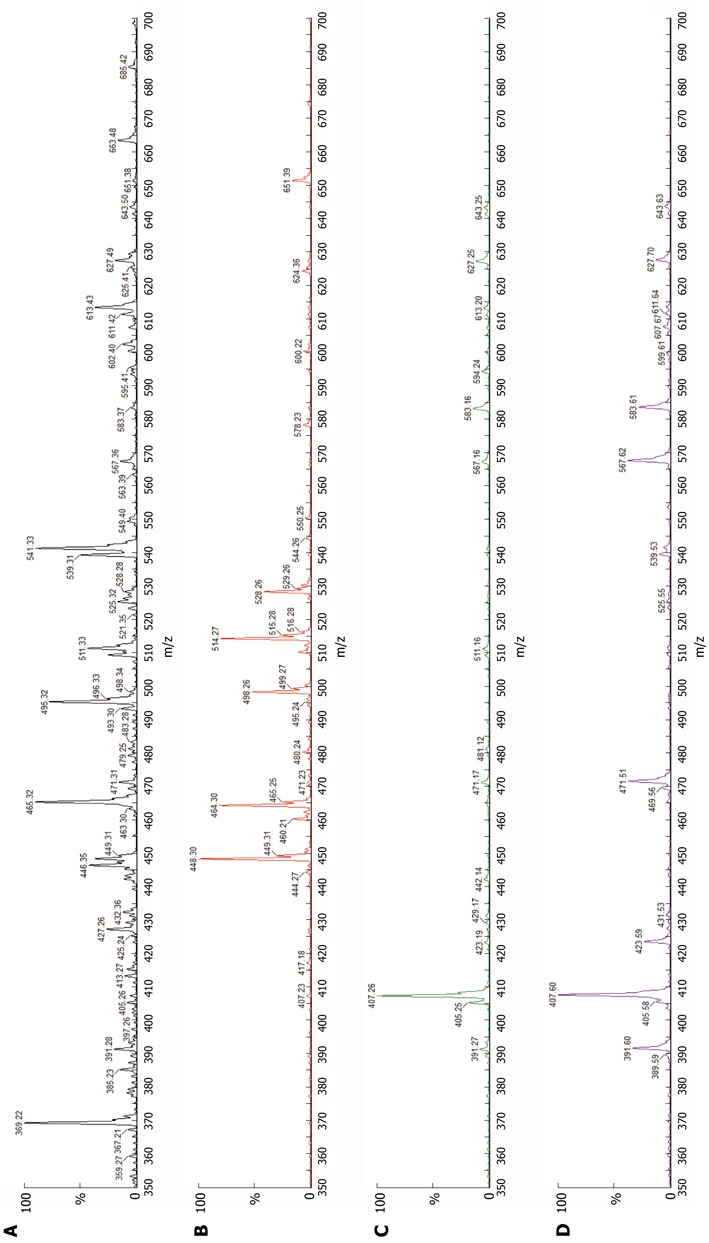
Bile acid mass spectra, urine. A: Normal (neither icterus nor known amidation defect); B: Deficiency of bile salt export pump (BSEP) (intrahepatic cholestasis with icterus); C: Deficiency of cholate-CoA ligase (described elsewhere[3]); D: Deficiency of bile acid-CoA: amino acid N-acyltransferase (patient). Abscissa, mass-charge ratio (m/z); ordinate, intensity of ion current as percentage of intensity of most abundant peak in spectrum (%). From left to right, principal peak sites and the species represented are: 391: Unamidated chenodeoxycholic acid (CDCA); 407: Unamidated cholic acid (CA); 448: Glycine-conjugated CDCA; 464: Glycine-conjugated CA; 471: Unamidated CDCA sulphate; 487: Unamidated CA sulphate; 498: Taurine-conjugated CDCA; 514: Taurine-conjugated CA; 528: Glycine-conjugated CDCA sulphate; 567: Unamidated CDCA glucuronide; 583: Unamidated CA glucuronide; 613: 27-Nor-cholestane pentol glucuronide; 627: Cholestane pentol glucuronide; 643: Cholestane hexol glucuronide. In normal urine (A), bile-acid and bile-alcohol peaks are just detectable. In urine from a patient with BSEP deficiency (B), increased excretion of glycine- and taurine-conjugated bile acids is apparent. Large quantities of non-conjugated species are present in both cholate-CoA ligase deficiency (C) and BAAT deficiency (D); differences between the two in size and distribution of this and other peaks are not apparent.
BAAT, encoding BAAT, was sequenced by routine techniques using DNA from peripheral-blood leucocytes[4]. The results indicated that the patient was homozygous for the previously undescribed mutation c.415C > T/p.R139X in BAAT. Solute carrier family 27 (fatty acid transporter), member 5 (SLC27A5), encoding CCL, was not sequenced. Immunostaining with antibodies against CCL (HPA007292; Sigma, St Louis, MO, United States) and BAAT (ab97455; Abcam, Cambridge, United Kingdom), conducted using manufacturer-recommended protocols, found good expression of CCL and no expression of BAAT (Figure 2); controls, including liver of age-matched infants with cholestasis due to extrahepatic biliary atresia and to failure of BSEP expression, marked appropriately (reaction conditions described on request). The patient, now aged 7 years, receives ursodeoxycholic acid and fat-soluble vitamin preparations and despite slight hypercholanaemia (total serum bile acids 77 μmol/L) is clinically well (height and weight 25th percentile for age, with good progress in school). Her only sibling, a sister, is without signs or symptoms of liver disease. The parents have declined genetic study of the sibling.
Figure 2.
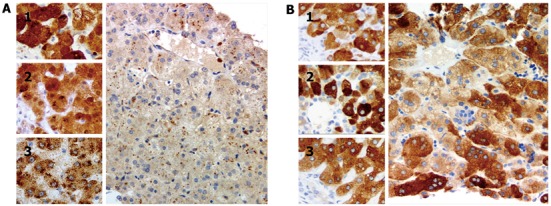
Photomicrographs of liver immunostained for enzymes that subserve bile-acid amidation. A: Main image: Core needle biopsy specimen, patient liver (age 57 d); inset, control livers (1, infant, matched for age with patient, with cholestasis and extrahepatic biliary atresia; 2, infant, matched for age with patient, with intrahepatic cholestasis in setting of absence of bile salt export pump (BSEP) expression; and 3, adult without cholestasis). The patient liver lacks immunohistochemically demonstrable bile acid-CoA: amino acid N-acyltransferase (BAAT). Granular cytoplasmic marking, suggestive of peroxisomal location, is found in (3); marking is more diffuse in the livers of age-matched infants; B: Tissue specimens as in (A). Diffuse cytoplasmic marking for cholate-CoA ligase (CCL) is seen in patient liver (main image) and in control livers (1, infant, matched for age with patient, with cholestasis and extrahepatic biliary atresia; 2, infant, matched for age with patient, with intrahepatic cholestasis in setting of absence of BSEP expression; and 3, adult without cholestasis). A, anti-BAAT antibody, and B, anti-CCL antibody (see principal text for sources); in all images, reaction development with EnVision proprietary technique (Dako UK, Ely, United Kingdom), haematoxylin counterstain, original magnification 200 ×.
DISCUSSION
Bile salts within the biliary-tract and small-bowel lumina exist principally as glycyl or tauryl conjugates of primary bile acids. Bacteria within chyme and faeces deconjugate and dehydroxylate bile salts. The resulting secondary bile acids are taken up by the bowel and passed into venous effluent. They are then absorbed by hepatocytes and again conjugated with glycine or taurine in the process of amidation; newly synthesised bile acids also undergo amidation in the liver[1].
Two enzymes, CCL [International Union of Biochemistry and Molecular Biology European Commission (EC) 6.2.1.7] in cytoplasm and BAAT (EC 2.3.1.65) in peroxisomes, act sequentially to amidate bile acids[1,3]. Experimental data conflict on whether non-amidated bile acids are poorer substrates for BSEP, the protein that transports bile salts against a concentration gradient from hepatocyte cytoplasm into bile-canaliculus lumen, than are amidated bile acids[5,6]. Non-conjugated bile acids may back-diffuse from canalicular lumen into hepatocytes and into the space of Disse, impeding secretory efficiency, and excess of bile acids within hepatocytes may cause injury[4]. Data on back-diffusion of non-amidated bile acids from bile-duct lumen into cholangiolar venous effluent, with drainage into portal-vein radicles and presentation to the liver lobule of such bile acids in excess (“cholehepatic shunting”), are not available. However, this process may conduce to hepatocellular overload with bile acids, and hence to hepatocellular injury.
Intrahepatic cholestasis has been described in association with amidation defects. Anicteric cholestasis, manifest as failure to thrive with malabsorption and fat-soluble vitamin deficiency, was found in Amish children homozygous for the mutation c.226A > G/p.M76V in BAAT[4]. A preliminary description of children with three additional different mutations in BAAT, all in homozygous form, has appeared[7]. Homozygous mutation in SLC27A5 (c.1012C > T/p.H338Y) also has been associated with intrahepatic cholestasis in a prematurely born infant[3]. To what extent exogenous factors - ontogenic immaturity, parenteral alimentation, various drugs, or a genetic background that includes mutation in other cholestasis-associated genes[3] - conduce to jaundice with amidation defects, expected per se to result in anicteric cholestasis[8], remains to be determined. Our patient, who developed icterus, was born at term, was not subjected to parenteral alimentation, and received no drug treatment recognised as capable of precipitating cholestasis. Were intrahepatocytic concentrations of bile acids elevated (4), leading to damage to organelles and to cellular processes, with jaundice a non-specific sequela? The factors that predisposed to icterus in our patient must remain matter for speculation.
As reported in patients with defects of bile-acid amidation[3,4], cholestasis in our patient was not associated with elevations in serum GGT activity even when conjugated hyperbilirubinemia was present. Normal-range serum GGT activity is a feature of disorders of bile-acid synthesis as well as of bile-acid amidation. It also characterises certain forms of intrahepatic cholestasis ascribed to deficiency of BSEP expression or function, abnormalities in canalicular-membrane composition, and abnormalities of intracellular trafficking of canalicular-membrane components[2]. Evidence indicating such disorders, either clinical or histopathologic, was not identified in our patient.
Little histopathologic information is available concerning hepatobiliary disease in bile-acid amidation deficiency. Whether the appearances of BAAT deficiency differ from those of CCL deficiency in routinely stained sections, for example, is still an open question. The same is true for differences in ultrastructural features; too few instances have been described to allow discrimination. As urine and plasma bile-acid spectra and chromatograms in amidation-deficiency disease do not vary between CCL deficiency and BAAT deficiency (personal observations, PTC; cf. Figure 1), screening of both SLC27A5 and BAAT for variations from canonical sequence is at present necessary to distinguish between forms of amidation defect.
Our findings demonstrate that immunohistochemical study can detect absence of BAAT expression. They correlated well with genetic findings that suggested that BAAT synthesis was not to be expected in our patient. Lack of BAAT expression thus may constitute a strong indication for evaluation of BAAT for mutation.
We believe that immunohistochemical screening of liver tissue for CCL and BAAT expression may identify instances of bile-acid amidation deficiency disease in patients with normal-range serum GGT activity and either icteric or anicteric cholestasis. To do so admittedly may not identify all such instances; defects in protein function without defects in protein expression are to be expected, particularly with mutations that substitute one amino-acid residue for another[3]. However, immunostaining is routinely practiced, whereas only a few laboratories speciate bile acids in plasma and urine or offer analysis of genes that encode enzymes involved in bile-acid handling. In addition, liver biopsy continues to be an approach widely used in the evaluation of neonatal cholestasis, permitting repeated interrogation of liver-biopsy material as application of diagnostic algorithms suggests candidate disorders. Results on immunostaining of liver-biopsy or hepatectomy specimens from patients with hepatobiliary disease and normal-range serum GGT activity thus may guide both clinical-biochemistry and genetic investigations, speeding definitive diagnosis.
ACKNOWLEDGMENTS
We thank Rayner A, chief biomedical scientist, and Starling C, biomedical scientist, of the histopathology laboratories at the Institute of Liver Studies, King’s College Hospital, for their work in establishing and validating the immunohistochemical techniques used in this study, and Wagner B, chief biomedical scientist, Electron Microscopy Unit, Histopathology Department, Northern General Hospital, Sheffield, United Kingdom, for technical and consultative ultrastructural studies.
Footnotes
Supported by Great Ormond Street Hospital Children’s Charity, to Clayton PT; National Institutes of Health; and Grant R01 DK58214, to Bull LN
Peer reviewers: Bruno Stieger, Professor, Division of Clinical Pharmacology and Toxicology, Department of Medicine, University Hospital, 8091 Zurich, Switzerland; Dr. Karel van Erpecum, Gastroenterology and Hepatology, University Hospital Utrecht, 3508GA Utrecht, The Netherlands
S- Editor Gou SX L- Editor A E- Editor Zhang DN
References
- 1.Clayton PT. Disorders of bile acid synthesis. J Inherit Metab Dis. 2011;34:593–604. doi: 10.1007/s10545-010-9259-3. [DOI] [PubMed] [Google Scholar]
- 2.Knisely AS, Gissen P. Trafficking and transporter disorders in pediatric cholestasis. Clin Liver Dis. 2010;14:619–633. doi: 10.1016/j.cld.2010.08.001. [DOI] [PubMed] [Google Scholar]
- 3.Chong CP, Mills PB, McClean P, Gissen P, Bruce C, Stahlschmidt J, Knisely AS, Clayton PT. Bile acid-CoA ligase deficiency--a new inborn error of bile acid metabolism. J Inherit Metab Dis. 2012;35:521–530. doi: 10.1007/s10545-011-9416-3. [DOI] [PubMed] [Google Scholar]
- 4.Carlton VE, Harris BZ, Puffenberger EG, Batta AK, Knisely AS, Robinson DL, Strauss KA, Shneider BL, Lim WA, Salen G, et al. Complex inheritance of familial hypercholanemia with associated mutations in TJP2 and BAAT. Nat Genet. 2003;34:91–96. doi: 10.1038/ng1147. [DOI] [PubMed] [Google Scholar]
- 5.Noé J, Stieger B, Meier PJ. Functional expression of the canalicular bile salt export pump of human liver. Gastroenterology. 2002;123:1659–1666. doi: 10.1053/gast.2002.36587. [DOI] [PubMed] [Google Scholar]
- 6.Mita S, Suzuki H, Akita H, Hayashi H, Onuki R, Hofmann AF, Sugiyama Y. Vectorial transport of unconjugated and conjugated bile salts by monolayers of LLC-PK1 cells doubly transfected with human NTCP and BSEP or with rat Ntcp and Bsep. Am J Physiol Gastrointest Liver Physiol. 2006;290:G550–G556. doi: 10.1152/ajpgi.00364.2005. [DOI] [PubMed] [Google Scholar]
- 7.Heubi JE, Setchell KD, Rosenthal P, Shah S, Buckley D, Jha P, Zhang W, Potter CJ, Suskind D, Bull LN. Oral glycocholic acid treatment of patients with bile acid amidation defects improves growth and fat-soluble vitamin absorption [abstr] Hepatology. 2009;50 Suppl 4:895A. [Google Scholar]
- 8.Hofmann AF, Strandvik B. Defective bile acid amidation: predicted features of a new inborn error of metabolism. Lancet. 1988;2:311–313. doi: 10.1016/s0140-6736(88)92359-8. [DOI] [PubMed] [Google Scholar]