

Abstract
α-Syntrophin (α-syn), a scaffold protein, links signaling molecules to the dystrophin–glycoprotein complex. Absence of α-syn from the DGC is known to lead to structurally aberrant neuromuscular junctions (NMJs) with few acetylcholine receptors (AChRs) clustered at synaptic sites. Using α-syn knock-out mice, we show that during the first postnatal week, α-syn is not required for synapse formation. However, at postnatal day 6 (P6)–P7, the structural integrity of the postsynaptic apparatus is altered, the turnover rate of AChRs increases significantly, and the number/density of AChRs is impaired. At the adult α-syn−/− NMJ, the turnover rate of AChRs is ∼4 times faster than wild-type synapses, and most removed receptors are targeted to degradation as few AChRs recycled to synaptic sites. Biochemical analyses show that in muscle cells of adult knock-out α-syn mice, total AChRs and scaffold protein rapsyn are significantly reduced, the 89 kDa and 75 kDa isoforms of tyrosine phosphorylated α-dystrobrevin (α-dbn) 1 (which are required for the maintenance and stability of AChR in α-dbn−/− synapses) are barely detectable. Electroporation of GFP-α-dbn1 in α-syn−/− muscle cells partially restored receptor density, turnover rate, and the structural integrity of the postsynaptic apparatus, whereas expression of rapsyn-GFP failed to rescue the α-syn−/− synaptic phenotype. These results demonstrate that α-syn is required for the maturation and stability of the postsynaptic apparatus and suggest that α-syn may act via α-dbn1.
Introduction
α-Syntrophin (α-syn) is a component of the dystrophin–glycoprotein complex (DGC), which also includes dystrophin/utrophin, dystroglycan, sarcoglycan, sarcospan, and α-dystrobrevin (α-dbn) (Straub and Campbell, 1997; Hack et al., 2000; Dalkilic and Kunkel, 2003). It is thought to function as an adaptor molecule that recruits signaling and structural proteins to the transmembrane DGC. α-syn directly interacts with dystrophin/utrophin, dystrobrevins, neuronal nitric oxide synthase (nNOS), voltage-gated sodium channels, several kinases, aquaporin-4 and other channels and signaling proteins (Kramarcy et al., 1994; Ahn and Kunkel, 1995; Dwyer and Froehner, 1995; Brenman et al., 1996; Froehner et al., 1997; Gee et al., 1998; Adams et al., 2000; Neely et al., 2001).
At mature neuromuscular junctions (NMJs), α-syn is highly concentrated at both the troughs and the crests of the junctional folds and was found to accumulate with acetylcholine receptors (AChRs) at synaptic sites as early as postnatal day 1 (P1) (Kramarcy and Sealock, 2000). Adult animals deficient in α-syn show dramatic changes in the structural organization of the synapses (Adams et al., 2000). AChRs are fewer in number and density, and the pattern in which they are clustered differs dramatically from wild-type mice, with speckles of receptors extending from edges of mutant synapses. Several other proteins of the DGC are either decreased or completely absent from the postsynaptic apparatus (nNOS and utrophin, respectively) (Adams et al., 2000; Kramarcy and Sealock, 2000). However, it remains unknown how the absence of α-syn from synapses reduces receptor density and number in the postsynaptic membrane and at which stage of synaptic development the absence of α-syn begins to impair structural organization of the postsynaptic membrane.
The phenotype of the α-syn−/− NMJ appears very similar to the α-dbn-null mice in many ways. α-dbn−/− NMJs have dramatically reduced levels of AChR, with an abnormal pattern of AChR distribution that resembles the α-syn mutant. In contrast to mice deficient in α-dbn, in which ≈50% of muscle fibers are dystrophic, α-syn-null mice have no sign of muscular dystrophy (Adams et al., 2000; Grady et al., 2000). Previous studies have shown that α-dbn is necessary for tight tethering of AChRs in the postsynaptic membrane, and is required for the maturation of the synapse (Grady et al., 2000; Akaaboune et al., 2002; Grady et al., 2003). However, it is not known whether α-syn is involved in controlling the metabolic stability of AChRs and whether removed or decreased proteins of the DGC from α-syn−/− NMJ mediate the AChR postsynaptic phenotype of α-syn mutants.
In this work, we investigate the effect of the absence of α-syn from the postsynaptic membrane on synapse formation and maturation in vivo. Using a multidisciplinary approach, we show that α-syn is required for synaptic maturation and postsynaptic receptor stability and that α-syn acts in part through α-dbn1, as overexpression of α-dbn1 partially rescues the postsynaptic phenotype of α-syn deficient mice.
Materials and Methods
Animals and confocal microscopy.
In this study, we used non-Swiss albino adult female mice (6–10 weeks old, 25–30 g) that were purchased from Harlan, mice deficient in α-syn that were bred in our animal facility from a pair provided by Dr. Froehner's laboratory, and mice deficient in α-dbn bred from a pair from Dr. Joshua Sanes' laboratory (Harvard University, Cambridge, MA). All animal usage followed methods approved by the University of Michigan Committee on the Use and Care of Animals.
To determine whether α-syn is essential for synapse maturation, wild-type and α-syn mutant mice from P0 to P60 were anesthetized by intraperitoneal injection of 72 mg/kg ketamine and 12 mg/kg xylazine and their sternomastoid muscles were bathed with Alexa Fluor 488-conjugated α-bungarotoxin (BTX-Alexa488; Invitrogen) to label AChRs (30 min, 5 μg/ml). Pups and adult mice were then fixed and NMJs were scanned with a confocal microscope (Leica SP5) using a 100× 1.46 numerical aperture (NA) oil-immersion objective (Leica). The z-stacks were then collapsed and the contrast adjusted with Adobe Photoshop CS2.
Quantitative fluorescence assay.
To determine whether absence of α-syn affects postsynaptic receptor density as synapses develop, the sternomastoid muscle of wild-type and mutant mice from P0 to P60 were exposed and bathed with a saturating dose of BTX-Alexa488 to label all AChRs at superficial synapses (5 μg/ml, 1 h). The nonbound fluorescence was washed out with lactated Ringer's superfusion continuously for 10–15 min and synapses were imaged. The fluorescence intensity of labeled receptors at the NMJ was assayed using a quantitative fluorescence imaging technique, as described by Turney et al. (1996), with minor modifications.
In situ imaging of developing and mature synapses.
The techniques of animal preparation, sternomastoid exposure, and neuromuscular imaging have been previously described in detail (Lichtman et al., 1987; van Mier and Lichtman, 1994; Akaaboune et al., 1999). The anesthetized mouse was placed on its back on the stage of a customized epifluorescence microscope, and superficial NMJs were viewed under a coverslip with a water-immersion objective (Olympus UAPO 20×W3/340 0.7 NA) and a digital CCD camera (Retiga EXi; Qimaging) and imaged (IPLap software; BioVision Technologies). At the end of an imaging session, the incision was closed and the mouse was allowed to recover until reanesthetized and prepared for a second imaging session.
Several different experimental protocols were carried out. In each case, multiple animals were used (typically 5–8). In the first series of experiments, neonatal pups (3 d after birth) of control and mutant mice were injected in the tail with a low dose of BTX-Alexa488 (20 μl, 1 μg/ml). This concentration labels few receptors at the NMJ (<20%). At P4, pups were anesthetized and the sternomastoid muscle was exposed and superficial synapses were imaged immediately (time 0). Twenty-four hours later (P5), the same synapses were reimaged and their fluorescence intensity was measured. When synapses were imaged multiple times, the wound was sutured (7-0 Ethilon monofilament nylon), a local antiseptic was applied to prevent local infection of the neck after each session and the animal allowed to fully recover before the next imaging session.
In the second series of experiments, pups at P4 and P7 were injected with a low dose of BTX-Alexa488 and at P5 and P8, animals were anesthetized, the sternomastoid muscle was exposed and superficial synapses were imaged immediately and reimaged 24 h later (P6 and P9).
In the last series of experiments, the sternomastoid muscle of adult wild-type and mutant mice were bathed with a low dose of BTX-Alexa488 (0.1 μg/ml for 2 min) to label few receptors or injected with a low dose of BTX-Alexa488. The sternomastoid was then washed continuously for 10 min with lactated Ringer's solution. Under this condition of labeling, <30% of receptors were labeled with fluorescent BTX, leaving synapses functional (Akaaboune et al., 1999).
To investigate whether the absence of α-syn affects the insertion of newly synthesized receptors at adult synapses, the sternomastoid muscle was bathed with a single saturating dose of BTX-Alexa488 (5 μg/ml, 1 h). The superficial synapses were imaged and their fluorescence intensities were measured. Three days later, mice were anesthetized, sternomastoid muscles exposed, and the same synapses were imaged and then resaturated with the same fluorescently tagged BTX.
To investigate whether absence of α-syn has any effect on the recycling of AChRs, sternomastoid muscles of adult wild-type and α-syn-null mice were labeled with biotinylated α-bungarotoxin (BTX-biotin) (5 μg/ml, 1 h; Invitrogen) followed by a saturating dose of Alexa Fluor 488-tagged streptavidin (strept-Alexa488) (10 μg/ml, 3 h; Invitrogen), as described previously in detail by Bruneau et al. (2005). Briefly, after 3 d of initial labeling (to allow more internalization of AChRs), the mouse was anesthetized and the sternomastoid muscle was imaged, then incubated with strept-Alexa488 (10 μg/ml, 3 h) to label the recycled receptors; later, superficial synapses were reimaged and their fluorescence intensities were measured to determine the contribution of recycled AChRs to total receptors at wild-type and mutant synaptic sites. Control experiments for ruling out dissociation of streptavidin from biotin on the surface of the muscle cells were described in our previous work (Bruneau et al., 2005; Bruneau and Akaaboune, 2006; Martinez-Pena y Valenzuela et al., 2010). Based on the fact that after internalization of AChR-BTX-biotin/streptavidin complexes inside the muscle, Alexa-labeled streptavidin dissociates rapidly from AChR-BTX-biotin within internal vesicles (likely endosomes), and on the extracellular surface the rate of dissociation of Alexa-labeled streptavidin from biotin was too slow to be detected even after several days, we can assess recycling by adding fresh Alexa-streptavidin hours or days after an initial saturating dose.
To determine the effects of the absence of α-syn on half-life of preexisting and recycled pools, the sternomastoid muscle was exposed and bathed with BTX-biotin (5 μg/ml, 30 min) to label AChR following by a saturating dose of strept-Alexa488 (10 μg/ml, 3 h) (Bruneau et al., 2005; Bruneau and Akaaboune, 2006). Three days later, the animal was anesthetized and the sternomastoid was exposed and bathed with Alexa Fluor 594-tagged streptavidin (10 μg/ml, 3 h; Invitrogen) to specifically label the recycled AChRs, as has been previously described in detail (Bruneau et al., 2005). Superficial doubly labeled synapses (red, recycled; and green, “preexisting”) were imaged and reimaged several days later and their fluorescence intensities were assayed.
Western blot.
Sternomastoid muscles were removed from wild-type and α-syn-null mice, minced, and homogenized on ice in a buffer containing 50 mm HEPES, pH 7.4, 2 mm EDTA, 250 mm sucrose, 150 mm NaCl, 1% NP-40, 0.25% sodium deoxycholate. Homogenates were shaken for 30 min and centrifuged at 10,000 × g for 10 min, giving a supernatant (S) and a pellet (P). The supernatant was collected and proteins were quantified using Pierce BCA protein assay. Identical amounts of proteins were separated by 12% SDS-PAGE and transferred onto a polyvinylidene fluoride membrane. The membranes were bathed in PBS pH 7.4, 2% skimmed milk, 0.05% Tween 20, blocking solution for 1 h, incubated in either monoclonal anti-rapsyn (R2029, clone 1234; Sigma) diluted at 0.5 μg/ml, anti-dystrobrevin (610766 Clone 23; BD Biosciences) diluted at 1:4000, or anti-α1-AChR subunit (MAB210; MMS-609R; Covance) diluted at 1:5000 in PBS plus 0.05% Tween 20 at 4°C overnight. The membranes were then incubated for 1 h in HRP-conjugated goat anti-rat or donkey anti-mouse secondary antibody at 1:20,000 (Jackson ImmunoResearch). After extensive washing, the membranes were incubated with SuperSignal West Femto Maximum Sensitivity Substrate (Pierce).
To determine the amount of receptors on the surface of muscle cells of mutant and wild-type mice, sternomastoid muscles were exposed and bathed with 5 μg/ml BTX-biotin for 1 h and receptors bound to BTX-biotin were pulled down using NeutrAvidin-coated agarose beads.
To determine the amount of intracellular AChR, sternomastoid muscles were bathed in 5 μg/ml unlabeled BTX for 1 h to saturate all surface AChRs and nonbound unlabeled BTX was extensively washed out. Muscles were then removed, homogenized and muscle lysates were incubated with 300 nm BTX-biotin for 1 h at 4°C. Receptors were then isolated using NeutrAvidin-coated agarose beads from 300 μg of total proteins.
In both intracellular and surface isolated receptors, the NeutrAvidin beads were collected by brief centrifugation and washed three times with homogenization buffer without detergents. The beads were resuspended in 75 μl of 2× LDS buffer (Invitrogen) and boiled for 5 min to release proteins, which were separated on 12% SDS-PAGE and transferred onto a polyvinylidene fluoride membrane. After blocking in 2% nonfat milk, the membranes were incubated in rat anti-α1-AChR subunit at 4°C overnight. The membranes were then incubated for 1 h in HRP-conjugated goat anti-rat secondary antibody at 1:10,000 (Jackson ImmunoResearch). After extensive washing, the membranes were incubated with SuperSignal West Pico Substrate (Pierce). Immunoblot quantitation was performed by scanning films and using the NIH ImageJ software.
Quantitative reverse transcriptase-PCR analysis of mRNA expression.
RNA was isolated using TRIzol (Invitrogen) from pairs of mouse sternomastoid muscles. Reverse transcription into cDNA was performed using 1 μg of total cellular RNA, 50 pmol of oligo(dT)20 primer (Invitrogen) and SuperScript III Reverse Transcriptase (Invitrogen). Quantitative real-time PCR was performed with 1 μl of cDNA product and the Power SYBR Green PCR Master Mix (Applied Biosystems), using a 7500 Fast Real Time PCR System (Applied Biosystems). We used the following primers, designed to pair with different exons of mouse α1-AChR subunit (Chrna1; NM_007389.4): forward 5′-gaggaccaccgtgagattgt-3′ and reverse 5′-aatcgacccattgctgtttc-3′ (product size, 121 bp); rapsyn (Rapsn; NM_009023.3): forward 5′-ctcagatgcctgcaaaacaa-3′ and reverse 5′-aggttgtgtggaaacccaag-3′ (product size, 111 bp); α-dbn1 (Dtna1; NM_207650.3): forward 5′-ctccggctcctcagacag-3′ and reverse 5′-ggcagatgctgaacggatg-3′ (product size, 195 bp). Expression was normalized to WT using the 2−ΔΔCt method (Livak and Schmittgen, 2001) and glyceraldehyde-3-phosphate dehydrogenase (Gapdh; NM_008084.2) as an internal reference, with primers 5′-aactttggcattgtggaagg-3′ and 5′-ggatgcagggatgatgttct-3′ (product size, 132 bp).
Immunofluorescence.
To determine whether absence of α-syn affects chromatin modification and transcriptional activation of postsynaptic nuclei, the sternomastoid muscle of wild-type and mutant adult mice were exposed and bathed with a saturating dose of BTX-Alexa488 (Invitrogen) to label all superficial synapses (5 μg/ml, 30 min). The nonbound fluorescence was washed out with lactated Ringer's superfusion and the animals were perfused with 4% formaldehyde. After extraction and dissociation into thin fiber bundles, the sternomastoid muscle was permeabilized with 1% Triton X-100 Tris-buffered saline (TBS) for 30 min and then washed twice with TBS. Fiber bundles were immersed and incubated overnight in rabbit anti-phospho (Ser10)-acetyl (Lys14)-histone H3 (Millipore), diluted 1:500 in 1% bovine serum albumin (BSA) TBS. After washing with TBS, fiber bundles were incubated with DyLight 594-conjugated goat anti-rabbit IgG (Jackson Immunoresearch), diluted 1:1000 in 1% BSA TBS for 1 h. After washing with TBS, fibers were further dissociated, mounted in ProLong with DAPI (Invitrogen) and imaged with a confocal microscope (Leica SP5). The z-stacks were then collapsed and the contrast adjusted with Photoshop.
Electroporation of GFP-α-dbn1, rapsyn-GFP, and α-syn-GFP into the sternomastoid muscle and confocal microscopy.
Adult mice deficient in α-syn (20–28 g) were anesthetized, the sternomastoid muscle was surgically exposed and 10 μg of plasmid driving exogenous expression of GFP-α-dbn1, GFP-α-dbn1-P3− (three tyrosine phosphorylation sites at Y685, Y693 and Y710 mutated to phenylalanine), rapsyn-GFP, or α-syn-GFP was added (all constructs were driven by a CMV promoter). The plasmid solution was layered over the muscle surface as described by Bruneau and Akaaboune (2010). Gold electrodes were placed parallel to the muscle fibers on either side of the muscle, and eight monopolar square-wave pulses were applied perpendicularly to the long axis of the muscle. Following electroporation, the mouse was sutured and allowed to recover in a heated recovery chamber. One to 2 weeks later, the animal was reanesthetized and Alexa Fluor 594-labeled α-bungarotoxin (BTX-Alexa594) was added to the sternomastoid muscle to label AChRs (1 h). The animal was then perfused transcardially with 2% formaldehyde and the sternomastoid muscle was removed, mounted, scanned with a confocal microscope (Leica SP5), and imaged. The z-stacks were then collapsed and the contrast adjusted with Photoshop.
Results
Effect of α-syntrophin deficiency on the maturation of developing synapses
To examine at which stage of synaptic development the absence of α-syn begins to alter normal postsynaptic apparatus development, sternomastoid muscles of α-syn-null and wild-type mice at different stages of development (P0, P5, P7, P15, and P60) were bathed with BTX-Alexa488 to label AChRs, then fixed, and imaged. High resolution confocal images of synapses show no significant differences in AChR distribution in mutant NMJs at P0 or P5, but abnormal patterning of AChRs was evident at P7 and became more pronounced at P15 where obvious patchy aggregates and extension of receptors from edges were observed. This abnormal patterning of AChR distribution persists and even worsens at mature synapses (P60) (Fig. 1A). Careful analyses of mutant and wild-type synapses revealed that in mutants, the transition of synapses from plaque-like to perforated and to branched receptor distribution was delayed and the size of synapses remains relatively small compared with wild type (Fig. 1B,C). These observations suggest that α-syn is required for the maturation of the postsynaptic apparatus.
Figure 1.

Absence of α-syn delays the maturation of the synapse. A, Sternomastoid muscles of mutant and wild-type mice at different stages of development were bathed with BTX-Alexa488 to label AChRs and synapses were imaged with confocal microscope. Note that synapses at P5 of wild type and mutant look similar (there is no obvious abnormality in the pattern of receptor distribution). However, at P7, AChRs appear to have a patchy distribution in mutant synapses compared with wild-type synapses. At P15 and P60, the structural organization (many small, high-density aggregates of AChRs distributed both at the crests and troughs of junction folds, with frequent speckled extensions of AChRs from the edges) of mutant synapses is different from wild type at the same age. Arrows indicate synaptic perforations and asterisks, synaptic invaginations, as quantified in C. Scale bars, 10 μm. B, Graph shows the area occupied by AChRs as labeled with fluorescent BTX-Alexa488. Note that the overall synaptic area of mutant synapses is smaller than wild type as synapses mature. Each data point represents the mean and SEM of the area of several junctions. C, Graph shows fewer perforations/invaginations in developing mutant synapses than wild type, indicating that the maturation process is somewhat delayed in mutant synapses. Each bar represents the mean and SD of several junctions. ***p < 0.0001.
Next, we examined whether the abnormal patterning of receptor distribution in α-syn−/− NMJ is accompanied by alterations in the density and number of AChRs at synaptic sites. To do this, sternomastoid muscles from different developmental stages (P0–P60) were labeled with BTX-Alexa488 to saturate all receptors. Superficial synapses were then imaged and their fluorescence intensity was assayed. The density of AChRs at different stages of development was expressed as a percentage of the fluorescence present at mature wild-type NMJs. As shown in Figure 2, no significant difference in receptor density between wild-type and α-syn−/− NMJs was seen at P0 (WT: 13 ± 3% SD, n = 23; α-syn−/−: 12.5 ± 2% SD, n = 16) and P5 (WT: 24.6 ± 5% SD, n = 21; α-syn−/−: 24.2, ± 6% SD, n = 37). At P7, however, the density of AChRs at α-syn−/− NMJs is (25 ± 6% SD, n = 20), 33% lower than the density of AChRs at wild-type synapses (37 ± 9% SD, n = 20) of the same age (Fig. 2). In contrast to wild-type synapses, in which the density of AChRs continues to increase as synapses mature, the density of AChRs at α-syn−/− NMJs reaches a low maximum at P15 and remains unchanged thereafter (Fig. 2). We also quantified the number of AChRs by calculating the product of average density and synaptic area, since during normal development of the synapse, both the size and average density within synaptic area increase. After postnatal day 7 and as synapses mature, the total number of receptors at synaptic sites begins to diverge from wild-type synapses and remains low in α-syn mice compared with wild type (data not shown). Together these results indicate that α-syn is required for the increase of postsynaptic receptor density as synapses mature.
Figure 2.
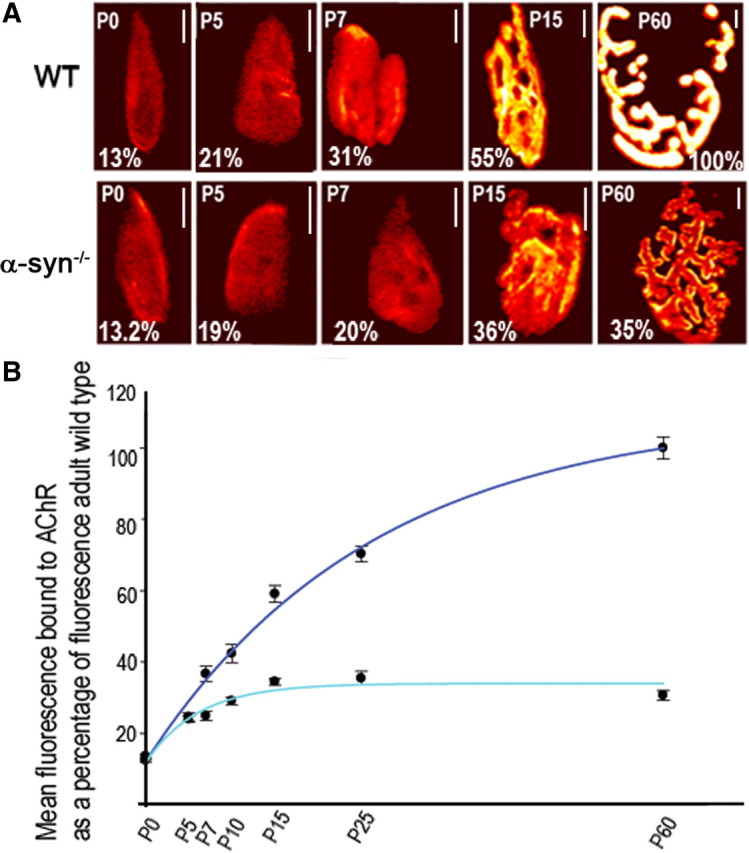
The density of postsynaptic receptors at the neuromuscular junction of α-syn-null mice is dramatically affected as synapses mature. AChRs in sternomastoid muscles of control and mutant mice were labeled with BTX-Alexa488 until their saturation. Superficial synapses were imaged and their fluorescence intensity was measured and normalized to fluorescence intensity of adult wild-type synapses. A, Examples of neuromuscular junctions imaged at different stages of development using a quantitative fluorescence assay. These pseudo-color images provide a linear representation of the density of AChRs (white-yellow, high density; red-black, low density). B, Quantification of the density of AChRs using the same approach shown in A. Each data point represents the mean and SEM of several NMJs. Note that at P0 and P5, the density of postsynaptic receptors is the same between wild-type and mutant synapses. At wild-type synapses, the density of postsynaptic receptors increases steadily but the density of postsynaptic receptors at mutant synapses rises slowly and remains low as synapses mature. Scale bars, 10 μm.
AChR dynamics in developing synapses of mice lacking α-syntrophin
Having found that absence of α-syn from NMJs impairs postsynaptic receptor density and distribution as early as P7, we asked whether failure to increase the density and number of postsynaptic receptors is accompanied by an accelerated rate of receptor loss from the postsynaptic membrane. To test this, labeled AChRs on the sternomastoid of α-syn−/− and wild-type pups at P4 were imaged and then reimaged 24 h later. Twenty-four hours after initial imaging, fluorescence intensity of the labeled receptors had decreased 39 ± 8% SD (n = 26) in mutant synapses [t1/2 ≈ 34 h] and 35 ± 8% SD (n = 20) in wild-type synapses [t1/2 ≈ 38 h] (p > 0.05) (Fig. 3A). This result indicates that absence of α-syn has no significant effect on receptor turnover rate during the first 5 d after birth. However, at 6 d after birth, fluorescence intensity of the labeled receptors had decreased 54 ± 6% SD (n = 18) over 24 h in mutant synapses [t1/2 ≈ 21 h] and only 36 ± 6% SD (n = 25) in wild-type synapses [t1/2 ≈ 37 h] over the same period (p < 0.0001) (Fig. 3B). At P9, fluorescence intensity of the labeled AChR decreased 48 ± 6% SD (n = 24) [t1/2 ≈ 26 h] in the α-syn−/− synapses, but only 26 ± 6% SD (n = 20) [t1/2 ≈ 55 h] in wild-type synapses over 24 h (p < 0.0001) (Fig. 3C). This indicates that α-syn is involved in receptor removal from postsynaptic sites as synapses mature.
Figure 3.

Absence of α-syn accelerates the loss rate of AChRs at developing synapses. A, Neonatal pups of wild-type and mutant mice aged 3 d after birth were injected with a low dose of BTX-Alexa488 in their tails (<20% of AChR were labeled), and 24 h later they were anesthetized and synapses on the sternomastoid muscle were imaged immediately (time 0) and reimaged 24 h later. Example of two views of the same neuromuscular synapses of mutant and wild type imaged and reimaged at 0 and 24 h later. The total fluorescence intensity (a measure of the total number of AChRs) was expressed as 100% at the initial labeling (time 0) and normalized to this on the successive view (24 h). Graph summarizes the data from many synapses using the approach in A. Note that the remaining fluorescently labeled AChR in mutant synapses was not significantly different from wild-type synapses. B, Example of two images of wild-type and mutant NMJs of pups 6 d old at time 0 and 24 h later. Graph shows data from many synapses. Note that the loss of fluorescence from labeled AChR is increased in mutant synapses compared with wild type. C, At P9, the loss of fluorescence of labeled AChR from wild-type synapses averaged at 26% after 24 h, whereas the loss of fluorescence from α-syn−/− synapses was 48%. Note that α-syn deficiency dramatically decreases the half-life of AChRs. Each data point represents the mean percentage of fluorescence intensity (±SD) at each view. Scale bars, 10 μm. ***p < 0.0001.
AChR dynamics at mature synapses of mice lacking α-syntrophin
The observation that α-syn−/− synapses bear fewer AChRs than controls prompted us to examine the possible mechanisms that lead to decreased AChR density at postsynaptic sites. We investigated this issue in adult mutant mice. First, we examined whether absence of α-syn from adult synapses decreases the half-life of AChRs. To examine this, sternomastoid muscles were labeled with a nonsaturating dose of fluorescent BTX (<30% of receptors were labeled, leaving synapses functional) and the loss of fluorescently tagged AChRs was monitored over the next 24 h. We found that the fluorescence intensity of the labeled receptors had decreased 8 ± 3% SD (n = 31) of original fluorescence in wild-type synapses [t1/2 ≈ 9 d] and 30 ± 7% SD (n = 25) of original fluorescence in mutant synapses [t1/2 ≈ 2 d] (Fig. 4A). Therefore, the half-life of AChRs at α-syn−/− synapses is ∼4 times faster than AChR at wild-type synapses.
Figure 4.

Dynamics of AChRs at mature neuromuscular junctions deficient in α-syn. A, Adult wild-type and mutant mice were injected with a low dose of BTX-Alexa488 in their tails (<30% of AChRs were labeled, so synapses are still functional), and 24 h later they were anesthetized and synapses on the sternomastoid muscle were imaged immediately and reimaged 24 h later. Example of two views of the same neuromuscular synapses of mutant and wild type, imaged and reimaged at 0 and 24 h later. Graph summarizes the data from many synapses using the approach in A. Note that the loss rate of labeled AChRs from mutant synapses was significantly higher than that of wild-type synapses. B, The sternomastoid muscles of mice deficient in α-syn were labeled with a saturating dose of BTX-Alexa488 and superficial synapses were immediately imaged. Three days later, the same synapses were located, reimaged and the loss of their fluorescence intensity was measured (72 h remaining). A second saturating dose of BTX-Alexa488 was added to label new AChRs that had been inserted during the 72 h and synapses were imaged again (72 h newly inserted). The total fluorescence intensity was expressed as 100% at the initial labeling and normalized to this on each successive view. Graph summarizes the data obtained from many junctions. C, The sternomastoid muscles of mice deficient in α-syn and wild-type mice were labeled with a saturating dose of BTX-biotin followed by a saturating dose of strept-Alexa488 and superficial synapses were imaged. The total fluorescence intensity was expressed as 100% at the initial labeling and normalized to this on each successive view. To assess the amount of recycled receptors that had been reinserted between time 0 and 3 d, strept-Alexa488 was then added to the muscle and the same synapses were reimaged. Graph summarizes the amount of recycled receptors present at synaptic sites, obtained from many junctions. Each bar represents the mean percentage of original fluorescence intensity ± SD. D, Absence of α-syn increases the removal rates of preexisting and recycled AChRs from the same synapse as assayed by loss of fluorescence intensities compared with wild-type synapses. The total fluorescence intensity of each AChR pool was normalized to 100% at initial imaging. Pseudo-color images provide a linear representation of the density of AChRs. Scale bars, 10 μm. ***p < 0.0001. E, Graph shows loss of fluorescently labeled preexisting and recycled AChRs from several synapses. Note that removal of AChRs from both pools is faster in the absence of α-syn. Each data point represents the mean percentage of original fluorescence intensity ± SEM.
Next, we used the loss rate to calculate the insertion rate of new AChRs into the postsynaptic membrane. Knowing that the size of mature synapses remains relatively constant for at least several weeks, the sternomastoid muscle was bathed with a single saturating dose of BTX-Alexa488 to label all AChRs on the surface of the muscle cell, and superficial synapses were imaged immediately (time 0). Three days later, the same synapses were located, reimaged and the loss of their fluorescence intensity was measured. A second saturating dose of BTX-Alexa488 was added to label new AChRs and synapses were imaged again. We found that most lost receptors were replaced with new AChRs, indicating that the rate of insertion of new receptors into the postsynaptic membrane was not impaired by the absence of α-syn (Fig. 4B).
Then, we explored whether a defect in the number of recycled receptors could account for the low number and density of receptors at α-syn−/− NMJs, since this pool of receptors contributes to the density and number of AChRs in the postsynaptic membrane (Bruneau et al., 2005; Bruneau and Akaaboune, 2006). The sternomastoid muscle of mutant and wild-type animals was labeled with BTX-biotin, followed by a saturating dose of strept-Alexa488, as described previously (Bruneau et al., 2005) and superficial synapses were imaged immediately (time 0). Three days after the initial labeling (to allow for a sizable amount of receptor internalization), the same synapses were imaged to measure the loss of fluorescence and then the sternomastoid muscle was bathed with strept-Alexa488 to label recycled AChRs (receptors that had lost their initial strept-Alexa488 tag while retaining BTX-biotin during the process of internalization and reinsertion in the postsynaptic membrane). The same synapses were reimaged and the percentage of recycled AChRs at mutant synapses was measured and compared with wild-type synapses. We found that only 9 ± 4% SD (n = 21) of the original fluorescence had recovered at mutant synapses, while the recovery was 22 ± 3% SD (n = 23) in wild-type synapses (Fig. 4C). This result indicates that in the absence of α-syn, fewer recycled receptors are clustered in synaptic sites.
Having shown that AChRs are removed faster from mutant than wild-type synapses, next we asked which of the receptor pools [recycled receptors and/or not yet internalized AChRs (preexisting)] is affected by the absence of α-syn. To address this, AChRs in the sternomastoid muscle of wild-type and mutant mice were sequentially labeled with BTX-biotin followed by a single saturating dose of (green) strept-Alexa488 as described by Bruneau et al. (2005). Three days after the initial labeling, the sternomastoid muscle was bathed with Alexa Fluor 594-tagged streptavidin (red) to specifically label recycled AChRs and doubly labeled synapses were immediately imaged (time 0) and reimaged 3 d later. The fluorescence intensity of labeled recycled AChRs at wild-type and α-syn−/− synapses decreased to 22 ± 6% SD (n = 16) and 15 ± 3% SD (n = 22) of the original fluorescence, respectively (p < 0.0001), while the fluorescence intensity of labeled preexisting AChRs decreased to 59 ± 6% SD (n = 21) in wild-type synapses and to 42 ± 8% SD (n = 18) in mutant synapses (p < 0.0001) (Fig. 4D,E). This result indicates that the stability of both recycled and preexisting AChRs is affected by the absence of α-syn from synapses.
We also asked whether a mistargeting of the insertion of receptors into nonsynaptic zones could also account for the low number/density of receptors at synaptic sites of mice deficient in α-syn. To test this, the sternomastoid muscles of mutant and wild-type mice were labeled with BTX-biotin. Receptors on the muscle surface were isolated with NeutrAvidin-coated beads, and analyzed by Western blot using anti-α1-AChR subunit antibody (MAB210). If absence of α-syn specifically impairs the insertion of AChRs into the postsynaptic membrane, the amount of receptors on the surface should be smaller in mutant than wild type and greater in the internal pool of mutant than that of wild type. Conversely, if loss of α-syn mistargets receptors into nonsynaptic zones, the amount of receptors on the surface of muscle cells should be comparable to wild type. Western blot analysis of receptors reveals the presence of few receptors in both surface and internal compartments of mutant muscle cells compared with wild type (Fig. 5A,B). This result was further confirmed by Western blot analysis of total homogenates (membrane and internal fractions) from mutant and wild-type muscle cells using rat anti-α1-AChR subunit, showing that the amount of total AChRs is significantly reduced in mutant mice compared with wild type (Fig. 5A,B).
Figure 5.
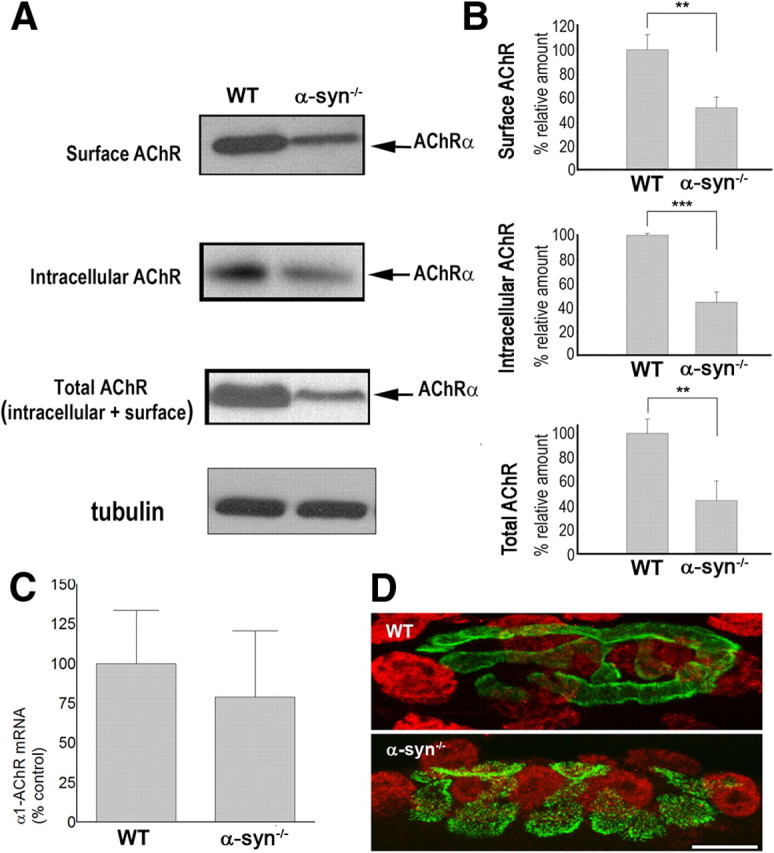
Absence of α-syn reduces both surface and internal receptors. A, Receptors on the surface of sternomastoid muscles of mutant and wild-type mice were saturated with BTX-biotin, pulled down with NeutrAvidin-agarose beads and immunoblotted with anti-α1-AChR subunit. To assess the amount of AChRs in the internal compartment, sternomastoid muscles of mutant and wild-type mice were incubated with unlabeled BTX to saturate all surface receptors; then muscle homogenates were incubated with BTX-biotin to bind AChRs in the internal pool, pulled down with NeutrAvidin-agarose beads and probed for α1-AChR subunit. Total amount of AChRs (on the surface and in the internal compartment) was probed by Western blotting of homogenates from sternomastoid muscles of mutant and wild-type mice. Tubulin was used as a loading control. B, Quantitative analysis of the amounts of AChRs on the surface, in the internal pool, and the total of mutant and wild-type muscle cells. At least three independent repetitions were performed in each experiment. Data are shown as mean ± SD; n = 5. C, Real-time PCR of α1-AChR subunit mRNA expression levels are similar in sternomastoid muscles of WT and α-syn−/−; error bars are SD with n = 3 mice. D, Transcriptional activation is similar in subsynaptic nuclei of WT and α-syntrophin knock-out (α-syn−/−) mice. Muscle fibers were stained with Alexa Fluor 488-BTX (green) for the neuromuscular junction and anti-phospho (Ser10)-acetyl (Lys14)-histone H3 (red). Representative images are shown. Scale bar, 10 μm. **p < 0.01; ***p < 0.001.
This finding prompted us to test whether the low number of receptor is a consequence of a low level of AChR transcripts. To test this, the mRNA levels of α1-AChR subunit from mutant and wild type were measured by real-time reverse transcriptase (RT)-PCR using glyceraldehyde 3-phosphate dehydrogenase as an internal control. We found that AChR mRNA (α1 subunit) expression level was similar between mutants and controls, indicating that loss of α-syn has no effect on receptor gene expression (Fig. 5C). These results were further confirmed by immunofluorescence with antibodies against phospho (Ser10)-acetyl (Lys14)-histone H3 (Fig. 5D), which indicated normal activity of subsynaptic nuclei gene expression. Together, these results strongly imply that α-syn is involved in controlling the stability and trafficking of AChRs at multiple points during the secretory process.
Absence of α-syntrophin sharply reduced AChR-associated scaffold protein levels in muscle cells
Next, we investigated whether loss of α-syn also affects the expression levels of other postsynaptic scaffolding proteins, such as α-dbn and rapsyn. Previous studies have shown that the concentration of α-dbn was reduced at synaptic sites of muscle fibers deficient in α-syn (Adams et al., 2000), but it was not clear whether absence of α-syn leads to a reduction in total amount of α-dbn in the muscle cells and/or a decrease in recruitment of α-dbn to synapses. To examine this, the sternomastoid muscle was removed and muscle homogenates were examined by Western blot analysis using antibodies against α-dbn. We found that isoforms of α-dbn1 and 2 expressed by the muscle were reduced. As shown in Figure 6, the 89 kDa isoform of α-dbn1 was reduced >85% of the control, and the 75 kDa isoform was undetectable. It is worth mentioning that both α-dbn1 isoforms have been shown to be phosphorylated, but the 75 kDa isoform is more heavily phosphorylated, and contains more phosphotyrosine than the 89 kDa isoform in the presence of the tyrosine phosphatase inhibitor, pervanadate (Nawrotzki et al., 1998). This severe reduction is not due to low levels of α-dbn transcript, as quantitative RT-PCR shows that mRNA levels were not affected (Fig. 6C). The concentration of α-dbn 2 (nonphosphorylated isoform) was reduced by half (Fig. 6A,B). Rapsyn, a scaffolding protein required for AChR aggregation, is also reduced in mutant syntrophin mice. As displayed in Figure 6, both Western blot and quantitative RT-PCR show that rapsyn was reduced by half compared with wild type (Fig. 6), indicating that α-syn is involved in controlling rapsyn gene expression (see Discussion).
Figure 6.

Absence of α-syn induces a severe reduction of α-dbn isoforms. Equal amounts (50 μg) of protein from total sternomastoid muscle extracts of α-syn mutant and wild-type mice were separated on SDS-polyacrylamide gels, and subjected to immunoblotting with a pan-α-dbn antibody. A, Blots show a severe reduction of 89 kDa α-dbn and undetectable 75 kDa α-dbn isoforms in α-syn mutant compared with wild type, whereas 59 kDa α-dbn2 isoform is reduced to ∼50% of wild-type levels. Rapsyn was reduced to ∼50% of wild type. B, Graphs show quantification of Western blots. C, Graph shows that real-time PCR of α-dbn1 mRNA expression levels are similar in sternomastoid muscles of WT and α-syntrophin knock-out (α-syn−/−), whereas rapsyn mRNA expression level was reduced by half in α-syn knock-out compared with wild type. Error bars are SD with n = 3 mice. **p < 0.01; ***p < 0.001.
To investigate whether the phenotype of α-syn−/− NMJ might be caused by loss of recruited scaffold proteins, we electroporated GFP-α-dbn1 into the sternomastoid muscle of mice deficient in α-syn. One to 2 weeks later, mice were perfused, muscles were removed, and AChRs were labeled with fluorescent BTX-Alexa594. As shown in Figure 7A, GFP-α-dbn1 was concentrated at synaptic sites of electroporated deficient muscle cells. High resolution images show that GFP-α-dbn1 constructs partially restored the AChR distribution shape (categories 2 and 3; Fig. 7B). The restoration was more pronounced in some synapses than other, most likely depending on the expression level of constructs (Fig. 7B). However, we found no significant difference in AChR pattern at synapses expressing GFP-α-dbn1 or GFP-tyrosine-mutated dystrobrevin isoform (Fig. 7A). Conversely, overexpression of rapsyn-GFP alone in α-syn−/− muscle fibers does not restore the normal synaptic phenotype (Fig. 7A,B), nor does it change the distribution of AChR in WT synapses (data not shown) (Bruneau et al., 2010).
Figure 7.
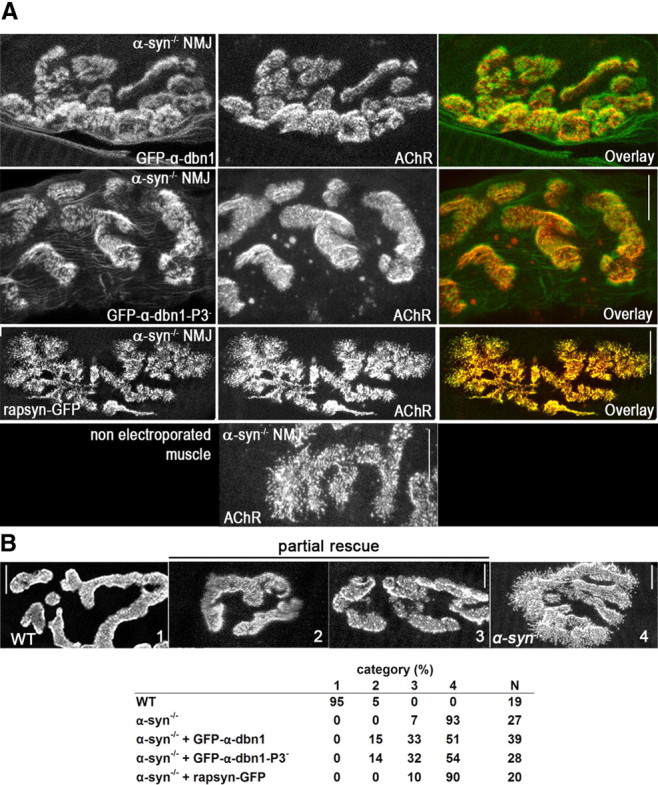
Electroporation of α-dbn1 partially rescues the pattern of AChR distribution at α-syn−/− NMJ. A, The sternomastoid muscle of adult α-syn-null mice was electroporated with GFP-α-dbn1 or GFP-α-dbn1 with three tyrosine phosphorylation sites mutated to phenylalanine (GFP-α-dbn1–P3−). One to 2 weeks later, the mouse was perfused with 2% formaldehyde, and receptors were labeled with BTX-Alexa594. Neuromuscular synapses on electroporated and nonelectroporated muscles were imaged with the confocal microscope. Representative high resolution images show that GFP-constructs are expressed and localized at postsynaptic sites. Expression of either α-dbn1 and α-dbn1–P3− in α-syn−/− muscle partially restored the AChR pattern of distribution. Note that synapses in muscle cells deficient in α-syn electroporated with GFP-α-dbn1 appear to have relatively smooth borders with few patchy clusters of AChR and less extensions of receptor beyond the edge. However, nonelectroporated α-syn−/− muscle synapses exhibit hot spots of receptors and fragmented branch borders with many speckles. Rapsyn-GFP was similarly electroporated into adult α-syn knock-out muscles. While rapsyn-GFP is found at postsynaptic sites, rapsyn-GFP does not rescue the synaptic phenotype in muscle cells deficient in α-syn. B, Synapses were categorized according to their AChR distribution, ranging from category 1, showing well defined troughs and synaptic borders, where most WT synapses are found, to 4, with markedly disorganized and patchy AChR distribution and fuzzy synaptic borders, typical of α-syn−/− synapses. Categories 2 and 3 show intermediate phenotypes, as exemplified by the synapses shown. The table shows the frequency distribution (in percentages) for each category per group, as well as the number of synapses analyzed (N). Scale bars, 10 μm.
To analyze in more detail the rescue of α-syn−/− synaptic phenotype by GFP-α-dbn1, we examined first whether expression of GFP-α-dbn1 rescues the density of postsynaptic receptors of α-syn−/− NMJs. To do this, the sternomastoid muscle of mice deficient in α-syn was electroporated with GFP-α-dbn1 and 7 d later, the sternomastoid was bathed with a saturating dose of BTX-Alexa594 to label all receptors. Superficial synapses of electroporated with GFP-α-dbn1 and nonelectroporated muscle fibers from the same sternomastoid were imaged and the fluorescence intensity of labeled AChRs was measured. The density of AChRs (expressed as a percentage of the fluorescence present at mature wild-type NMJ) at α-syn−/− NMJ overexpressing GFP-α-dbn1 (66 ± 4% of WT synapses, n = 26 NMJs) is 78% higher than the density of AChRs at nonelectroporated synapses (31 ± 6% SD, n = 20 NMJs), but lower than wild type (Fig. 8A). Next, we examined whether overexpression of GFP-α-dbn1 increases the half-life of AChRs at α-syn−/− NMJ. Twenty-four hours after initial imaging, fluorescence intensity of the labeled receptors had decreased 16 ± 3% SD (n = 16 NMJs) in mutant synapses expressing GFP-α-dbn1 [t1/2 ≈ 4 d], compared with 31 ± 5% SD (n = 7 NMJs) in nonelectroporated synapses [t1/2 ≈ 2 d] (p < 0.0001) and 8 ± 3% SD [t1/2 ≈ 9 d] (n = 9 NMJs) in wild-type synapses (p < 0.0001) (Fig. 8B). Finally, we tested whether the number of recycled receptor has increased in α-syn−/− NMJs expressing GFP-α-dbn1. We found that, 3 d after initial labeling, 74% more AChRs recycled back into mutant synapses expressing GFP-α-dbn1 (n = 10 NMJs), when compared with nonelectroporated fibers (n = 12 NMJs) (Fig. 8C). These results indicate that α-dbn1 partially rescued synaptic phenotype including AChR half-life, recycling and postsynaptic density. Conversely, overexpression of α-syn-GFP into the sternomastoid muscle of mice deficient in α-dbn, which have a NMJ phenotype similar to that of α-syn−/− NMJ, does not restore the normal synaptic phenotype (Fig. 8D). Also, overexpression of α-dbn in wild-type synapses does not increase receptor density (data not shown). Together, these results suggest that the synaptic effects of α-dbn and α-syn are not merely additive. Not all proteins lost from α-syn-null synapses are involved in the increase of the turnover rate of receptor. In mice lacking utrophin, which is highly concentrated at wild-type synaptic sites, but lost in the absence of α-syn, the AChR turnover rate is unaffected (data not shown) (Akaaboune et al., 2002).
Figure 8.

Electroporation of α-dbn1 partially rescues the density, stability and recycling of AChRs at expressing α-syn−/− NMJs. A, The sternomastoid muscle of adult α-syn-null mice was electroporated with GFP-α-dbn1. One week later, AChRs were labeled with a saturating dose of BTX-Alexa594 and superficial synapses, with or without GFP-α-dbn1, from the same sternomastoid muscle were imaged. Fluorescence intensity was measured and normalized to WT synapses. Total fluorescence was normalized to WT synapses. Graph summarizes the data from many synapses. B, One week after electroporation of GFP-α-dbn1, AChRs were labeled with BTX-Alexa594, and superficial synapses, with or without GFP-α-dbn1, from the same sternomastoid muscle were imaged immediately (time 0) and reimaged 24 h later. Note that there are more remaining fluorescently tagged AChRs in synapses electroporated with GFP-α-dbn than in nonelectroporated synapses, though comparably less than WT NMJs. Graph summarizes the data from many synapses. C, Sternomastoid muscles from α-syn−/− mice were electroporated with GFP-α-dbn1. One week later, surface AChRs were labeled with BTX-biotin, followed by a saturating dose of unlabeled streptavidin, then bathed with strept-Alexa594 to label the AChRs recycled for 3 d. Graph summarizes the data from many synapses. Each bar represents the mean percentage of fluorescence intensity ± SD. ***p < 0.0001. D, The sternomastoid muscle of adult α-dbn-null mice was electroporated with α-syn-GFP, AChRs were labeled with BTX-Alexa594, and imaged with a confocal microscope. Note that α-syn-GFP does not rescue the α-dbn−/− synaptic phenotype. Scale bars, 10 μm.
Discussion
The importance of the DGC in muscle integrity is well established. However, little is known about the role of this complex in the molecular dynamics of receptors at postsynaptic sites in vivo. In this study, we examined the mechanisms by which absence of α-syn leads to fewer receptors at mature synaptic sites and whether α-syn is required for synaptic development. The main results are as follows: (1) absence of α-syn dramatically destabilizes AChRs at mature synapses; (2) the AChR recycling pathway is impaired in the absence of α-syn; (3) postsynaptic defects coincide with an abnormal shift in turnover rate of AChR in developing synapses; (4) absence of α-syn leads to a dramatic reduction in the total amount of AChRs, rapsyn, and α-dbn in muscle cells; (5) AChR transcript levels are not affected by lack of α-syn; and (6) electroporation of GFP-α-adn1 into α-syn−/− muscle fibers restores to some extent the NMJ phenotype. Together, these results establish a role for α-syn in the maintenance and stability of AChRs in vivo.
Previous studies have shown that NMJs of α-syn−/− muscle contain few receptors, raising the possibility that α-syn may be critical for the maintenance of receptor density and stability. The present work shows that synapses lacking α-syn have a significantly increased turnover rate of AChRs and drastically reduced density of AChRs both in developing and mature synapses. This high turnover is not a consequence of muscular dystrophy, since no sign of dystrophy is seen in muscle fibers (Adams et al., 2000).
α-Syntrophin is a component of the DGC, which is present in both the troughs and the crests of the junctional folds (Kramarcy and Sealock, 2000), and links signaling proteins to the DGC (Brenman et al., 1996; Gee et al., 1998). However, no direct link between α-syn and AChR has been reported so far, raising the question of how α-syn might control the density/number and stability of receptors at the crest of the postjunctional folds. It is possible that absence of α-syn from the complex may change the conformation of the DGC and its association with other postsynaptic components, leading to instability of the complex as a whole, which in turn may affect the stability and/or tethering of AChRs into the postsynaptic membrane. At NMJs deficient in α-syn, several components of the DGC are either reduced or completely lost (Adams et al., 2000). For example, the total amount of α-dbn in muscle (Fig. 6) is reduced, and utrophin and nNOS are completely lost from synaptic sites (Adams et al., 2000). From our studies, it appears that absence of α-syn has no effect on the gene expression of α-dbn, since we see no difference in α-dbn transcript levels between α-syn-null and wild-type animals. This raises the question of how loss of α-syn might control the stability and trafficking of α-dbn from the ER into the cell surface. This issue warrants further investigation, particularly with regard to signaling processes associated with the DGC, such as those involving nNOS.
The absence of utrophin from α-syn−/− NMJ is likely not responsible for the rapid turnover rates of AChR at α-syn−/− NMJ. The turnover rate of AChR was no faster in utrophin-null than in control mice (Akaaboune et al., 2002). Interestingly, however, in α-adbn−/− synapses, which bear few receptors and have a higher turnover rate of AChRs (Grady et al., 2000; Akaaboune et al., 2002), α-syn appears to be unaltered and remains highly concentrated at the NMJ (Grady et al., 1999). Thus, it is conceivable that accelerated turnover rate of receptors in the α-syn mutant may be secondary to the dramatic reduction of α-dbn at the NMJ and muscle cells. Due to the fact that full-length phosphorylated α-dbn isoforms are severely reduced in α-syn mutants, that α-syn−/− and α-dbn−/− NMJ have similar phenotypes, and that α-dbn isoforms are able to rescue α-dbn mutant synapses (Grady et al., 2003), it is plausible that the NMJ phenotype and receptor stability in α-syn−/− are controlled by α-dbn alone or synergistically with other factors. If the α-syn−/− NMJ phenotype is caused by α-dbn reduction, then we expect at least some sign of muscular dystrophy, which is not seen in α-syn−/− mice. Perhaps the presence of α-dbn2, which is less reduced (Fig. 5), may be sufficient to prevent muscular dystrophy in these mutant mice. Consistent with this, previous results have shown that transgenic expression of either α-dbn2 or 1 isoforms in α-dbn−/− mice, in which muscular dystrophy was observed (Grady et al., 1999), is sufficient to rescue the dystrophic phenotype (Grady et al., 2003).
Our present data argue that the α-syn−/− NMJ phenotype results, at least in part, from α-dbn reduction, since electroporation of exogenous of α-dbn restores synaptic distribution of AChR to some extent. In contrast to α-dbn−/− NMJs, in which overexpression of phosphorylatable α-dbn1 is able to fully rescue the synaptic phenotype, phosphorylated and non-tyrosine phosphorylated α-dbn1 isoforms partially rescue the syn−/− NMJ phenotype in the same way, possibly because α-syn is required for the tethering and/or the signaling pathway of α-dbn phosphorylation.
Deficiencies in certain DGC components do not cause defects in the NMJ (Duclos et al., 1998; Hack et al., 1998). Interestingly, only components that control localization of nNOS at the NMJ are involved in the maintenance of the NMJ structure and mice deficient in components that induce removal of nNOS from synapses exhibit structural defects of NMJ similar to those observed in nNOS-null mice (Shiao et al., 2004). This suggests that nNOS may be the common factor that contributes to defects in NMJ missing DGC molecules. nNOS is lost from synapses deficient in α-syn and α-dbn, which raises the possibility that nNOS along with α-dbn may be involved in the stability of receptors at the postsynaptic sites. Indeed, expression of a nNOS transgene can partially rescue NMJ phenotype in α-syn−/− extensor digitorum longus and quadriceps muscles (Shiao et al., 2004).
Results presented here show that the absence of α-syn induces abnormal AChR distribution and a decrease in AChR density and stability at ∼P7. α-Syntrophin is expressed at the postsynaptic membrane at birth (Kramarcy and Sealock, 2000), but its absence from synapses has no effect on receptor distribution or stability during the first postnatal week, suggesting that α-syn is not required for the early steps of synapse maturation and it becomes functionally relevant as synapse matures. While the mechanism by which absence of α-syn may affect AChR stability and distribution only after the first week is not known, it is possible that such events might be triggered by loss of α-dbn, nNOS and/or other proteins. In support of this, α-dbn−/− mice show a decrease in AChR density and abnormal patterning of AChR distribution as early as 7 d postnatal (Grady et al., 2000). Thus, it is conceivable that a partial loss of α-dbn in the α-syn mutant may initiate AChR instability during synaptic development, leading to a decline in the density of AChRs as development proceeds.
One interesting finding of the current study is that the contribution of receptor recycling to total receptors present at α-syn−/− NMJs was very low compared with mature wild-type NMJs, where a significant number of recycled AChRs contribute to the steady-state of the postsynaptic receptor density (Bruneau et al., 2005). This raises the question of how α-syn may regulate the trafficking and degradation of internalized AChR. Previous studies have shown that α-dbn interacts with several cytoskeletal proteins, including the cargo-binding domain of the kinesin heavy chain (Ceccarini et al., 2005) and intermediate filaments (Mizuno et al., 2001; Newey et al., 2001). Kinesin motor proteins and other cytoskeleton elements have been found to be involved in the trafficking and delivery of receptors to synaptic sites (Setou et al., 2002). Thus it is possible that alteration in AChR recycling may involve α-dbn, since this later is dramatically reduced in α-syn mice.
Here we show that loss of α-syn is also associated with a reduction in rapsyn concentration, a 43 kDa protein that is critical for receptor clustering (Gautam et al., 1995). Conversely, AChR are necessary for the clustering of rapsyn in both mammals and fish (Ono et al., 2001, 2004; Bruneau et al., 2008). A threshold density of AChR likely provides a critical signal that enables proteins of the DGC and intracellular scaffold to remain aggregated. However, it is possible that α-syn reduces the levels of rapsyn, which then controls receptor number and density. Consistent with this, previous reports showed that rapsyn and receptors are trafficked in the same intracellular vesicle from the ER to cell surface (Marchand et al., 2002). However, in our experiments, overexpression of rapsyn-GFP was unable to restore the α-syn−/− synaptic phenotype. While the mechanism by which absence of α-syn leads to instability of AChRs remains largely unknown, the present study implies that the effect of α-syn on AChR is in part mediated by α-dbn1. Understanding how DGC components are involved in regulating the postsynaptic receptor number/density at the NMJ will be the focus of future study.
Footnotes
This work was supported by the National Institutes of Health (M.A.) and partially by the National Science Foundation (M.A). We thank the members of the M. Akaaboune, J. Kuwada, R. Hume, C. Collins, and H. Xu laboratories (University of Michigan, Ann Arbor, MI) for thoughtful comments and discussions. We are also grateful to Dr. Richard Hume (University of Michigan, Ann Arbor, MI) for the input and comments on this manuscript.
References
- Adams ME, Kramarcy N, Krall SP, Rossi SG, Rotundo RL, Sealock R, Froehner SC. Absence of alpha-syntrophin leads to structurally aberrant neuromuscular synapses deficient in utrophin. J Cell Biol. 2000;150:1385–1398. doi: 10.1083/jcb.150.6.1385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ahn AH, Kunkel LM. Syntrophin binds to an alternatively spliced exon of dystrophin. J Cell Biol. 1995;128:363–371. doi: 10.1083/jcb.128.3.363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Akaaboune M, Culican SM, Turney SG, Lichtman JW. Rapid and reversible effects of activity on acetylcholine receptor density at the neuromuscular junction in vivo. Science. 1999;286:503–507. doi: 10.1126/science.286.5439.503. [DOI] [PubMed] [Google Scholar]
- Akaaboune M, Grady RM, Turney S, Sanes JR, Lichtman JW. Neurotransmitter receptor dynamics studied in vivo by reversible photo-unbinding of fluorescent ligands. Neuron. 2002;34:865–876. doi: 10.1016/s0896-6273(02)00739-0. [DOI] [PubMed] [Google Scholar]
- Brenman JE, Chao DS, Gee SH, McGee AW, Craven SE, Santillano DR, Wu Z, Huang F, Xia H, Peters MF, Froehner SC, Bredt DS. Interaction of nitric oxide synthase with the postsynaptic density protein PSD-95 and alpha1-syntrophin mediated by PDZ domains. Cell. 1996;84:757–767. doi: 10.1016/s0092-8674(00)81053-3. [DOI] [PubMed] [Google Scholar]
- Bruneau EG, Akaaboune M. The dynamics of recycled acetylcholine receptors at the neuromuscular junction in vivo. Development. 2006;133:4485–4493. doi: 10.1242/dev.02619. [DOI] [PubMed] [Google Scholar]
- Bruneau EG, Akaaboune M. Dynamics of the rapsyn scaffolding protein at the neuromuscular junction of live mice. J Neurosci. 2010;30:614–619. doi: 10.1523/JNEUROSCI.4595-09.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bruneau EG, Brenner DS, Kuwada JY, Akaaboune M. Acetylcholine receptor clustering is required for the accumulation and maintenance of scaffolding proteins. Curr Biol. 2008;18:109–115. doi: 10.1016/j.cub.2007.12.029. [DOI] [PubMed] [Google Scholar]
- Bruneau E, Sutter D, Hume RI, Akaaboune M. Identification of nicotinic acetylcholine receptor recycling and its role in maintaining receptor density at the neuromuscular junction in vivo. J Neurosci. 2005;25:9949–9959. doi: 10.1523/JNEUROSCI.3169-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ceccarini M, Torreri P, Lombardi DG, Macchia G, Macioce P, Petrucci TC. Molecular basis of dystrobrevin interaction with kinesin heavy chain: structural determinants of their binding. J Mol Biol. 2005;354:872–882. doi: 10.1016/j.jmb.2005.09.069. [DOI] [PubMed] [Google Scholar]
- Dalkilic I, Kunkel LM. Muscular dystrophies: genes to pathogenesis. Curr Opin Genet Dev. 2003;13:231–238. doi: 10.1016/s0959-437x(03)00048-0. [DOI] [PubMed] [Google Scholar]
- Duclos F, Straub V, Moore SA, Venzke DP, Hrstka RF, Crosbie RH, Durbeej M, Lebakken CS, Ettinger AJ, van der Meulen J, Holt KH, Lim LE, Sanes JR, Davidson BL, Faulkner JA, Williamson R, Campbell KP. Progressive muscular dystrophy in alpha-sarcoglycan-deficient mice. J Cell Biol. 1998;142:1461–1471. doi: 10.1083/jcb.142.6.1461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dwyer TM, Froehner SC. Direct binding of Torpedo syntrophin to dystrophin and the 87 kDa dystrophin homologue. FEBS Lett. 1995;375:91–94. doi: 10.1016/0014-5793(95)01176-f. [DOI] [PubMed] [Google Scholar]
- Froehner SC, Adams ME, Peters MF, Gee SH. Syntrophins: modular adapter proteins at the neuromuscular junction and the sarcolemma. Soc Gen Physiol Ser. 1997;52:197–207. [PubMed] [Google Scholar]
- Gautam M, Noakes PG, Mudd J, Nichol M, Chu GC, Sanes JR, Merlie JP. Failure of postsynaptic specialization to develop at neuromuscular junctions of rapsyn-deficient mice. Nature. 1995;377:232–236. doi: 10.1038/377232a0. [DOI] [PubMed] [Google Scholar]
- Gee SH, Madhavan R, Levinson SR, Caldwell JH, Sealock R, Froehner SC. Interaction of muscle and brain sodium channels with multiple members of the syntrophin family of dystrophin-associated proteins. J Neurosci. 1998;18:128–137. doi: 10.1523/JNEUROSCI.18-01-00128.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grady RM, Grange RW, Lau KS, Maimone MM, Nichol MC, Stull JT, Sanes JR. Role for alpha-dystrobrevin in the pathogenesis of dystrophin-dependent muscular dystrophies. Nat Cell Biol. 1999;1:215–220. doi: 10.1038/12034. [DOI] [PubMed] [Google Scholar]
- Grady RM, Zhou H, Cunningham JM, Henry MD, Campbell KP, Sanes JR. Maturation and maintenance of the neuromuscular synapse: genetic evidence for roles of the dystrophin–glycoprotein complex. Neuron. 2000;25:279–293. doi: 10.1016/s0896-6273(00)80894-6. [DOI] [PubMed] [Google Scholar]
- Grady RM, Akaaboune M, Cohen AL, Maimone MM, Lichtman JW, Sanes JR. Tyrosine-phosphorylated and nonphosphorylated isoforms of alpha-dystrobrevin: roles in skeletal muscle and its neuromuscular and myotendinous junctions. J Cell Biol. 2003;160:741–752. doi: 10.1083/jcb.200209045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hack AA, Ly CT, Jiang F, Clendenin CJ, Sigrist KS, Wollmann RL, McNally EM. Gamma-sarcoglycan deficiency leads to muscle membrane defects and apoptosis independent of dystrophin. J Cell Biol. 1998;142:1279–1287. doi: 10.1083/jcb.142.5.1279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hack AA, Groh ME, McNally EM. Sarcoglycans in muscular dystrophy. Microsc Res Tech. 2000;48:167–180. doi: 10.1002/(SICI)1097-0029(20000201/15)48:3/4<167::AID-JEMT5>3.0.CO;2-T. [DOI] [PubMed] [Google Scholar]
- Kramarcy NR, Sealock R. Syntrophin isoforms at the neuromuscular junction: developmental time course and differential localization. Mol Cell Neurosci. 2000;15:262–274. doi: 10.1006/mcne.1999.0823. [DOI] [PubMed] [Google Scholar]
- Kramarcy NR, Vidal A, Froehner SC, Sealock R. Association of utrophin and multiple dystrophin short forms with the mammalian M(r) 58,000 dystrophin-associated protein (syntrophin) J Biol Chem. 1994;269:2870–2876. [PubMed] [Google Scholar]
- Lichtman JW, Magrassi L, Purves D. Visualization of neuromuscular junctions over periods of several months in living mice. J Neurosci. 1987;7:1215–1222. doi: 10.1523/JNEUROSCI.07-04-01215.1987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Livak KJ, Schmittgen TD. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) Method. Methods. 2001;25:402–408. doi: 10.1006/meth.2001.1262. [DOI] [PubMed] [Google Scholar]
- Marchand S, Devillers-Thiéry A, Pons S, Changeux JP, Cartaud J. Rapsyn escorts the nicotinic acetylcholine receptor along the exocytic pathway via association with lipid rafts. J Neurosci. 2002;22:8891–8901. doi: 10.1523/JNEUROSCI.22-20-08891.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martinez-Pena y Valenzuela I, Mouslim C, Akaaboune M. Calcium/calmodulin kinase II-dependent acetylcholine receptor cycling at the mammalian neuromuscular junction in vivo. J Neurosci. 2010;30:12455–12465. doi: 10.1523/JNEUROSCI.3309-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mizuno Y, Thompson TG, Guyon JR, Lidov HG, Brosius M, Imamura M, Ozawa E, Watkins SC, Kunkel LM. Desmuslin, an intermediate filament protein that interacts with alpha-dystrobrevin and desmin. Proc Natl Acad Sci U S A. 2001;98:6156–6161. doi: 10.1073/pnas.111153298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nawrotzki R, Loh NY, Ruegg MA, Davies KE, Blake DJ. Characterisation of alpha-dystrobrevin in muscle. J Cell Sci. 1998;111:2595–2605. doi: 10.1242/jcs.111.17.2595. [DOI] [PubMed] [Google Scholar]
- Neely JD, Amiry-Moghaddam M, Ottersen OP, Froehner SC, Agre P, Adams ME. Syntrophin-dependent expression and localization of Aquaporin-4 water channel protein. Proc Natl Acad Sci U S A. 2001;98:14108–14113. doi: 10.1073/pnas.241508198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Newey SE, Howman EV, Ponting CP, Benson MA, Nawrotzki R, Loh NY, Davies KE, Blake DJ. Syncoilin, a novel member of the intermediate filament superfamily that interacts with alpha-dystrobrevin in skeletal muscle. J Biol Chem. 2001;276:6645–6655. doi: 10.1074/jbc.M008305200. [DOI] [PubMed] [Google Scholar]
- Ono F, Higashijima S, Shcherbatko A, Fetcho JR, Brehm P. Paralytic zebrafish lacking acetylcholine receptors fail to localize rapsyn clusters to the synapse. J Neurosci. 2001;21:5439–5448. doi: 10.1523/JNEUROSCI.21-15-05439.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ono F, Mandel G, Brehm P. Acetylcholine receptors direct rapsyn clusters to the neuromuscular synapse in zebrafish. J Neurosci. 2004;24:5475–5481. doi: 10.1523/JNEUROSCI.0851-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Setou M, Seog DH, Tanaka Y, Kanai Y, Takei Y, Kawagishi M, Hirokawa N. Glutamate-receptor-interacting protein GRIP1 directly steers kinesin to dendrites. Nature. 2002;417:83–87. doi: 10.1038/nature743. [DOI] [PubMed] [Google Scholar]
- Shiao T, Fond A, Deng B, Wehling-Henricks M, Adams ME, Froehner SC, Tidball JG. Defects in neuromuscular junction structure in dystrophic muscle are corrected by expression of a NOS transgene in dystrophin-deficient muscles, but not in muscles lacking alpha- and beta1-syntrophins. Hum Mol Genet. 2004;13:1873–1884. doi: 10.1093/hmg/ddh204. [DOI] [PubMed] [Google Scholar]
- Straub V, Campbell KP. Muscular dystrophies and the dystrophin-glycoprotein complex. Curr Opin Neurol. 1997;10:168–175. doi: 10.1097/00019052-199704000-00016. [DOI] [PubMed] [Google Scholar]
- Turney SG, Culican SM, Lichtman JW. A quantitative fluorescence-imaging technique for studying acetylcholine receptor turnover at neuromuscular junctions in living animals. J Neurosci Methods. 1996;64:199–208. doi: 10.1016/0165-0270(95)00135-2. [DOI] [PubMed] [Google Scholar]
- van Mier P, Lichtman JW. Regenerating muscle fibers induce directional sprouting from nearby nerve terminals: studies in living mice. J Neurosci. 1994;14:5672–5686. doi: 10.1523/JNEUROSCI.14-09-05672.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]