

Abstract
Stat3 is an important transcription factor that regulates both proinflammatory and anti-apoptotic pathways in the heart. This study examined the mechanisms of activation of Stat3 in human endothelial cells following hypoxia/reoxygenation (H/R). By expression of constitutively active Rac1 mutant protein, and by RNA silencing of Rac1, we found that Stat3 Y705 and S727 phosphorylation following H/R are dependent on Rac1. Reactive oxygen species produced during H/R, and direct physical association with Rac1 both contribute to Stat3 activation. Stat3 forms a multiprotein complex with Rac1 and PKC in an H/R-dependent manner, which at least in part, appears to regulate Stat3 S727 phosphorylation. Selective inhibition of PKC with calphostin C produces a marked suppression of Stat3 S727 phosphorylation. The association of Stat3 with Rac1 occurs predominantly at the cell membrane, but also inside the nucleus, and occurs through the binding of the coiled-coil domain of Stat3 to the 54 NH2-terminal residues of Rac1. Transfection with a peptide comprising the NH2-terminal 17 amino acid residues of Rac1 inhibits Stat3 S727 phosphorylation after H/R. Thus, Stat3 is activated in endothelial cells by H/R through Rac1-dependent signaling pathways resulting in physical association between Rac1 and Stat3 and the formation of a novel multiprotein complex with PKC.
Keywords: endothelial cells, Stat3, protein kinase C, inflammation, protein-protein interaction, reactive oxygen species
1. Introduction
Ischemia-reperfusion (I/R) causes activation of a number of transcription factors which regulate genes involved in inflammation, apoptosis, and cellular growth and repair [1–3]. Signal transducers and activators of transcription (Stat) proteins have received attention as important gene regulators following I/R [4–6]. Upon activation, Stats form homo- or heterodimers, translocate to the nucleus, and activate transcription by binding to target genes [7]. Within the family of Stats, Stat3 upregulates a number of pro-inflammatory genes in endothelial cells, including cytokines, chemokines, and adhesion molecules [5,6,8,9]. Stat3 has been shown to mediate protection of the heart and other organs against I/R injury [10], and is also essential for the cardioprotection resulting from both pre- and post-ischemic conditioning [11, 12, 13]. Stat3 is therefore an important signaling molecule in the context of I/R, and an understanding of the mechanisms involved in its activation is of considerable interest.
Dimerization and DNA binding of Stat3 require phosphorylation of its Y705 residue, but full transcriptional activity is believed to necessitate phosphorylation of both Y705 and S727 residues [14]. We recently found that phosphorylation of S727 was followed by binding of Stat3 to the transcriptional regulator specificity protein 1 (Sp1), and that this transcriptional complex enhanced the expression of the inflammatory molecule intercellular adhesion molecule-1 (ICAM-1) in endothelial cells following I/R [5]. Interestingly, other downstream actions of activated Stat3 have been described which result in anti-inflammatory effects, mediated through induction of heme oxygenase-1 [15], and Stat3 has also been reported to mediate expression of anti-apoptotic genes in the heart [8,16]. Activation of Stat3 is found in human cancers, and the guanosine triphosphatase Rac1, a subunit of the NADPH-oxidase, is thought to play a role [17]. Stat3 is also activated in several cell types following exposure to growth factors or cytokines, presumably through receptor-related tyrosine phosphorylation, or tyrosine phosphorylation by Janus kinases (JAKs) [18,19]. Rac1 binds to Stat3 in COS-1 and smooth muscle cells treated with growth factors, and appears to regulate the phosphorylation of tyrosine and serine residues [20,21]. However, the domains involved in this important protein-protein interaction have not been determined.
Reactive oxygen species (ROS) have been implicated as a key factor in activation of the JAK-Stat pathway [22,23]. ROS are generated in large quantities during I/R or hypoxia/reoxygenation (H/R) [24], and are also produced in response to cytokines and growth factors [22,25]. The NADPH-oxidase is a major source of ROS in endothelial cells as well as in other cell types [26,27], and its activity is well known to be regulated by Rac proteins [28,29,30]. Thus, Rac1-dependent Stat activation could occur either by physical binding of Rac1 with Stat proteins to facilitate their targeting to specific protein kinases [20], or by Rac1-dependent ROS production, which could indirectly activate different protein kinases, resulting in downstream activation of Stat proteins [31].
Our study is the first to examine the pathways involved in Stat3 activation following hypoxia/reoxygenation. We demonstrate that Rac1 is essential for Stat3 activation in this context, and that both Rac1-induced ROS generation and physical binding between Rac1 and Stat3 are involved. Further, we show that Stat3 activation after H/R is dependent on PKC, which forms a novel multiprotein complex with Rac1 and Stat3.
2. Materials and methods
2.1 cDNA cloning and site-directed mutagenesis
Full-length hStat3α and human wild-type (wt) Rac1 (hRac1-wt) were amplified by RT-PCR from total RNA isolated from human umbilical vein endothelial cells (HUVECs), and cloned using standard methods. Expression constructs of constitutively active (CA) Rac1 (Rac1 G12V) and deletion constructs were made using standard methods. Full-length mStat3α and hStat3α were subcloned and Stat3 deletion constructs were made and amplified using custom designed primers. Accuracy of the reading frame and authenticity of deletions were verified by Western blotting and/or DNA sequencing.
2.2 Cell culture and exposure to hypoxia/reoxygenation
HUVECs were cultured in EGM-2 growth medium (Cambrex). COS-7 and 293 cells were grown in DMEM supplemented with 10% heat-inactivated FBS. Cells were incubated for 2 h at 37°C under normoxic conditions or in ischemia buffer [32] under a hypoxic gas mixture as described [5], and reoxygenated in fresh modified Esumi buffer [20] at 37°.
2.3 Adenoviral infection, and transient and stable transfection
Construction of adenoviruses expressing β-galactosidase or CA Rac1 has been described [33]. The viruses were amplified in HEK 293 cells, followed by purification and measurement of titers. HUVECs or COS-7 cells were infected for 6 h with 50 ICU/cell of viral particles expressing CA Rac1 or β-gal (control virus) and used 36–48 h later. HUVECs were transfected with 20 p moles/ml control siRNA, or siRNA specific for Rac1 [34] or with other constructs and harvested 48–72 h later. Stable transformants were selected with G418 using standard protocols.
2.4 Construction of Rac1 NH2-terminal peptide and expression vectors, and transfection of 293 cells and HUVECs
Peptides were constructed representing the 54 NH2-terminal amino acids of Rac1 to block the interaction between Rac1 and Stat3. cDNA fragments corresponding to two peptides (residues 1-17 (Rac1-17) and 23-54 (Rac1-54)) were transcribed and amplified by R-T PCR and cloned. 293 cells were grown to 70% confluence in 6-well plates, and 0.6μg DNA construct was transfected into each well. After 48 hours, the cells were exposed to hypoxia for 2 h and reoxygenation for 30 min. The cells were then lysed and harvested for Western blotting.
In other experiments, FITC-labeled Rac1-17 was synthesized by ChemPep Inc. (Miami, FL) and 2μg of peptide or control IgG were transfected directly into cultured HUVECs. After 4 h, the cells were exposed to hypoxia for 2 h and reoxygenation for 15 or 30 min. The cells were observed by fluorescent microscopy to document successful transfection and harvested for Western blotting.
2.5 Immunoprecipitation and immunoblotting
Cells were exposed to normoxia or H/R and lysed in IP buffer. Immunoprecipitation and immunoblotting were done as described previously [35].
2.6 Yeast two-hybrid interaction
Interaction between full-length Rac1 and Stat3 and their segments was examined using the MATCHMAKER two-hybrid system II (Clontech) as described previously [35,36].
2.7 In vitro binding assays
Recombinant GST/Rac1 proteins were expressed and affinity-purified by coupling to glutathione-Sepharose beads as described [35,36]. 35S-labeled Stat3α proteins were translated in vitro. Equal amounts of Stat3 proteins/polypeptides were incubated with 10 μg of GST/Rac1-fusion proteins, washed, fractionated by SDS-PAGE and detected by fluorography.
2.8 Immunofluorescence staining and confocal microscopy
HUVECs were grown on poly-L-lysine coated coverslips and exposed to hypoxia for 2 h and reoxygenation for 15, 30, or 60 min. Cells were fixed with with 4% paraformaldehyde for 10 min, and permeabilized with methanol in −20°C for 10 min. Single or dual immunofluorescence staining was performed using 1:100 dilution of rabbit anti-human p-S727 Stat3 or Stat3 polyclonal antibodies (Cell Signaling Technology, Danvers, MA), goat anti-human PKCζ polyclonal antibody (Santa Cruz Biotechnology, Inc., Santa Cruz, CA) and/or mouse anti-Rac1 mAb (Upstate) as described previously [35,36]. Secondary antibodies included Northern Light donkey anti-rabbit-IgG-NL637 and anti-goat IgG-NL493 ( R&D Systems (Minneapolis, MN). Confocal microscopy was performed using a Carl Zeiss 510 confocal microscope.
2.9 PKCζ knockdown by siRNA
PKCζ siRNA, control siRNA and goat anti-human PKCζ polyclonal antibody were purchased from Santa Cruz Biotechnology, Inc. (Santa Cruz, CA). HUVECs were cultured in 6 well plates to 80% conference. 50 pmoles/mL siRNA or control siRNA were transfected into the cells using Effectene Transfection Reagent (QIAGEN, Inc, Valencia, CA). 48 hours later, the cells were exposed to hypoxia for 2 h and reoxygenation for 30 minutes, and then lysed and analyzed by Western blotting.
2.10 Densitometry and statistical analysis
Chemiluminograms were analyzed by densitometry using the ImageJ software (http://rsbweb.nih.gov/ij/). Band densities were normalized to an internal control for each lane and expressed as a percent of control conditions (defined as 100%). Band densities were then averaged for 3 independent experiments and differences between lanes were analyzed by paired t-test. P values of ≤0.05 were considered statistically significant.
3. Results
3.1 Stat3 phosphorylation following hypoxia-reoxygenation is Rac1 dependent
To examine if Stat3 activation following H/R is regulated through Rac1 activity, we analyzed the effect of exogenously expressed CA Rac1 on Stat3 phosphorylation in HUVECs. Infection of cells with adenoviruses expressing β-gal (control virus) had no effect on phosphorylation status of Stat3 Y705 or S727 compared to uninfected cells in normoxia or following H/R (not shown). Exposure to H/R resulted in an increased level of phosphorylation of both residues in β-gal expressing cells (Fig. 1A,C). Expression of CA Rac1 in these cells during normoxia resulted in increased phosphorylation of Stat3 Y705 and S727, with a further increase in phosphorylation upon exposure to H/R, without a change in expression of Stat3 (Fig. 1A,C). Western blots of cell lysates confirmed that CA Rac1 adenoviruses increased the intracellular level of Rac1 protein (Fig. 1B). Our results are consistent with an earlier report of Rac1-dependent Stat3 activation in EGF-induced COS-1 cells [20]. However, in another study, expression of CA Rac1 was reported not to affect Stat3 Y705 phosphorylation in IL-6-induced HepG2 cells [37].
Figure 1.

Stat3 phosphorylation following hypoxia-reoxygenation is increased by CA Rac1 in HUVECs. (A) CA Rac1 increases Y705 and S727 Stat3 phosphorylation, both in normoxia (N) and after H/R. (B) Expression of CA Rac1 after 48 h infection. (C) Densitometry of Western blots in (A). Values are mean ± SEM, expressed relative to the blot for Ad β-gal under normoxic conditions and averaged for 3 independent experiments. ** p<0.01 vs N for Ad β-gal or vs N for Ad CA Rac1 as indicated.
To further examine whether Rac1 regulates Stat3 phosphorylation, we analyzed the effect of knockdown of Rac1 with siRNA on Stat3 phosphorylation (Fig. 2A,B). siRac1 resulted in a partial inhibition of Stat3 Y705 phosphorylation, but a more marked inhibition of Stat3 S727 phosphorylation, both during normoxia and following H/R.
Figure 2.

(A) Stat3 phosphorylation following hypoxia-reoxygenation is inhibited by siRNA for Rac1. (B) Densitometry of 3 blots, with data normalized for Stat3 (loading control). Values represent mean ± SEM. ** p<0.01 for indicated comparison or comparison to Control siRNA during N.
3.2 Stat3 activation following H/R is mediated through production of ROS
To determine if Stat3 activation is mediated directly by ROS generated upon H/R, we examined the effect of N-acetyl cysteine (NAC), an antioxidant, on the H/R-induced phosphorylation of Stat3 (Fig. 3A,B). Pretreatment of cells with 400 μM NAC for 2 hours resulted in an approximate 50% suppression (p<0.01) of H/R-induced phosphorylation of both Stat3 Y705 and S727 without affecting expression of Stat3. NAC suppressed H/R induced Stat3 phosphorylation at both Y705 and S727 to a similar extent following infection with adenoviruses carrying CA Rac1 (Fig. 3B). Inhibition of Stat3 phosphorylation by NAC also occurred during normoxia but was quantitatively somewhat less. These results suggest that reactive oxygen species generated during H/R, at least in part, are involved in Stat3 activation.
Figure 3.

(A) N-acetyl cysteine (NAC) reduces phosphorylation of both Y705 and S727 Stat3 in HUVECs, both in normoxia (N), and following H/R. (B) Densitometry of 3 blots, with data normalized for Stat3 (loading control). Values represent mean ± SEM. ** p<0.01 for indicated comparison.
3.3 Association of Stat3 with Rac1 and PKC, and Stat3 S727 phosphorylation are augmented following exposure to hypoxia-reoxygenation
To determine if Rac1 activates Stat3 following H/R by directly associating with Stat3, we immunoprecipitated endogenous Stat3 from HUVEC lysates and looked for Rac1 coprecipitation. We found suggestive evidence for increased association between Rac1 and Stat3 following H/R, but we were unable to obtain an adequate amount of Stat3 protein from HUVECs for a definitive result. We therefore performed additional experiments in COS-7 cells stably expressing mStat3α. In these cells, we found an association of Stat3 with Rac1 under normoxic conditions (Fig. 4A, lane 3), which increased substantially (p<0.01) following H/R (Fig 4A, lane 2; Fig. 4D) and even further following H/R with exogenous expression of CA Rac1 (Fig 4A, lane 4; Fig. 4D).
Figure 4.

Stat3 activation following H/R is regulated by association of Stat3 with Rac1 and PKC. (A) In COS-7 cells stably expressing Stat3, the amount of Rac1 coprecipitating with Stat3 increased during H/R (lane 3 vs 2) and following infection with Ad CA Rac1 (lane 4), and the immunoprecipitate also contained PKC and its ζ isoform. (B) In COS-7/Stat3 stable cells, the amounts of Stat3, p-S727 Stat3, and Rac1 co-precipitating with PKC increased during H/R and following infection with Ad CA Rac1. (C) In HUVECs, pretreatment with the specific PKC inhibitor Calphostin C (1 μM) reduced S727 Stat3 phosphorylation during normoxia and H/R, with or without Ad CA Rac1 infection. (D), (E), (F) Densitometry of (A), (B) and (C), respectively, on 3 blots for each panel. Normalization was for the amount of Stat3 in (D), PKC in (E), and for Stat3 (loading control) in (F). Values are mean ± SEM. ** p<0.01 for indicated comparison, or comparison to N.
To identify tyrosine- or serine/threonine-kinases that might associate with Stat3 and catalyze phosphorylation of its Y705 and S727 moieties, we examined co-precipitation of candidate protein kinases with Stat3. Since different isoforms of PKC are activated during H/R [38,39,40], we looked at PKC, and found that it did in fact co-precipitate with Stat3 along with Rac1. The amount of PKC in the complex increased significantly (p<0.01) with exposure of cells to H/R (Fig. 4D; compare lanes 3 and 2 in Fig. 4A) and increased further upon expression of CA Rac1 and exposure to H/R (Fig. 4D; compare lanes 3 and 4 in Fig. 4A). The anti-PKC Ab used in these experiments detects all PKC family members (sc-10800, Santa Cruz Biotech). Further analysis of the Stat3 immunoprecipitates revealed that PKCζ co-precipitates with Stat3, in an H/R-dependent manner (Fig. 4A).
In complementary experiments, we immunoprecipitated PKC (using sc-10800, Santa Cruz Biotech, raised against a C-terminal segment of human PKCα) from COS-7 cells stably expressing mStat3α and examined the immunoprecipitates for co-precipitation of Stat3. Stat3 and Rac1 co-precipitated with PKC in an H/R-dependent manner (Fig. 4B, compare normoxia in lane 3 with H/R in lane 2; quantitatively the increase was about 4-fold for Rac1, but could not be calculated for Stat3 because of the absence of a Stat3 band in normoxia, Fig. 4E). Their association with PKC increased with exogenous expression of CA Rac1 (compare lane 2 with lane 4, Fig. 4B). Quantitatively, association of Rac1 with PKC increased by about 9-fold following H/R and expression of CA Rac1, Fig 4E). In addition, there was a small further increase in Stat3 association with PKC following expression of CA Rac1 and H/R compared to H/R alone (Fig 4B, lane 2 vs. lane 4). Further analysis of the Stat3/PKC immunoprecipitates suggested that S727 phosphorylation of Stat3 increased following H/R and that the increase was even greater with exogenous CA Rac1 expression (Fig. 4B). This finding is consistent with PKC playing a role in Stat3 S727 phosphorylation, and in further support of this idea, pretreatment of HUVECs with a highly specific PKC inhibitor, Calphostin C (1 μM) [41], significantly (p <0.01) suppressed Stat3 S727 phosphorylation in normoxia and following H/R in both control (Ad β-gal infected) and Ad CA Rac1-infected cells (Fig. 4C,F). Pretreatment of cells with another specific PKC inhibitor, chelerythrine [42], also suppressed H/R-induced S727 phosphorylation (not shown). These results suggest that association of Stat3 with Rac1 and PKC may be, at least in part, a novel mechanism of Stat3 activation following H/R.
3.4 PKCζ knockdown by siRNA inhibits Stat3 phosphorylation following hypoxia-reoxygenation
To determine if PKCζ participates in the phosphorylation of Stat3, we transfected HUVECs with PKCζ siRNA or negative control siRNA, and 48 hours later subjected the cells to hypoxia for 2 h and reoxygenation for 30 min or a corresponding period of normoxia (Fig. 5A,B). Knockdown of PKCζ was associated with a reduction in S727-Stat3 phosphorylation, both during normoxia, and following hypoxia-reoxygenation. The results are consistent with the immunoprecipitation experiments (Fig. 4) and support a role for PKCζ in Stat3 S727 phosphorylation.
Figure 5.

(A,B) Knockdown of PKCζ with siRNA reduces S727 Stat3 phosphorylation in HUVECs during normoxia and following hypoxia-reoxygenation. (A) HUVECs were transfected with control siRNA (si-control) or PKCζ siRNA (si-PKCζ), or with no siRNA (Control), lysed, and immunoblotted. (B) 48 h after transfection HUVECs were exposed to hypoxia for 2h and reoxygenation for 30 min, lysed, and immunoblotted. The experiment was done twice with similar results.
(C,D) Following hypoxia-reoxygenation, PKCζ colocalizes with Stat3 in the cytoplasm, but after phosphorylation of S727, Stat3 is found in the nucleus without PKCζ. (C) HUVECs were exposed to hypoxia for 2 h and reoxygenation for 15 min. Cells were fixed and incubated with rabbit anti-Stat3 and goat anti-PKCζ antibodies with secondary anti-rabbit (red) and anti-goat (green) antibodies, and examined by confocal microscopy (63X). a) Stat3, b) PKCζ, c) nuclei (blue), d) merged image, e) enlargement of cell in box to demonstrate cytoplasmic colocalization of Stat3 and PKCζ. (D) HUVECs under normoxia or exposed to hypoxia for 2h and reoxygenation for 60 min were fixed and incubated with rabbit anti-p-S727 Stat3 and goat anti-PKCζ antibodies and detected by secondary antibodies (p-S727 Stat3, red; PKCζ, green), and examined by confocal microscopy (63X). H-R increases cytoplasmic and nuclear p-S727 Stat3, but there is no colocalization with PKCζ. b) merged image showing lack of colocalization, c) separated image of p-S727 Stat3, d) separated image of PKCζ. The confocal microscopy experiments in C and D were each performed twice with similar results.
3.5 Stat3 and PKCζ colocalize in the cytoplasm following hypoxia-reoxygenation
To further examine the association between Stat3 and PKCζ, and determine the subcellular location of their interaction, we examined if Stat3 and PKCζ colocalize by immunofluorescent staining and confocal microscopy (Fig. 5C,D). Following hypoxia and 15 min reoxygenation, (Fig. 5C) Stat3 (red) was localized mainly in the cytoplasm, with some weaker Stat3 staining in the nucleus, while PKCζ (green) was localized exclusively in the cytoplasm. Colocalization (yellow) occurred in the cytoplasm but not the nucleus. After 60 min reoxygenation there was still no nuclear colocalization of Stat3 and PKCζ (not shown). However, we also tracked the location of pS727-Stat3 and interestingly found that after 60 min reoxygenation (Fig. 5D), pS727-Stat3 was located in both the cytoplasm and nucleus, while PKCζ remained nearly completely cytoplasmic (no colocalization seen in either the cytoplasm or nucleus). These results are consistent with cytoplasmic phosphorylation of Stat3 by PKCζ early after reperfusion (Fig. 5C), with subsequent dissociation of phosphorylated Stat3 from PKCζ and passage of activated Stat3 into the nucleus (Fig. 5D).
3.6 Stat3 and activated Rac1 colocalize at the cell membrane and inside the nucleus following exposure to hypoxia-reoxygenation
To further examine the association between Stat3 and Rac1, we looked for subcellular colocalization of Rac1 and Stat3 (Fig. 6). Since activated Stat3 translocates to the nucleus, cells were fixed within 5 min of reoxygenation to minimize the effects of translocation and determine the intracellular locales of Stat3/Rac1 association. Following exposure of HUVECs infected with Ad β-gal to hypoxia and 5 min reoxygenation, Stat3 was found mainly in the nucleus (red), with weak staining also in the cytoplasm. In these same cells, Rac1 was also localized primarily in the nucleus, with weaker staining at the cell membrane and diffusely in the cytoplasm (green, A1). However, in cells infected with Ad CA Rac1, prominent staining for Rac1 and Stat3 was observed at the cell membrane (arrow), nuclear membrane (arrow head), inside the nucleus, and to a certain extent in the perinuclear region (B, B1). The merged image (B2) showed colocalization of Stat3 and Rac1 (yellow) at the cell membrane (indicated by arrow), nuclear membrane (indicated by arrow head) and inside the nucleus, upon exposure of cells to H/R. There was also weak colocalization in the cytoplasm. The prominent membrane localization of Stat3 and its colocalization with CA Rac1 at the cell membrane observed upon H/R was not apparent in cells kept in normoxia (C, C2). The increased colocalization of CA Rac1 and Stat3 following H/R is consistent with the increased association between Rac1 and Stat3 we observed in immunoprecipitation experiments (Fig. 4).
Figure 6.
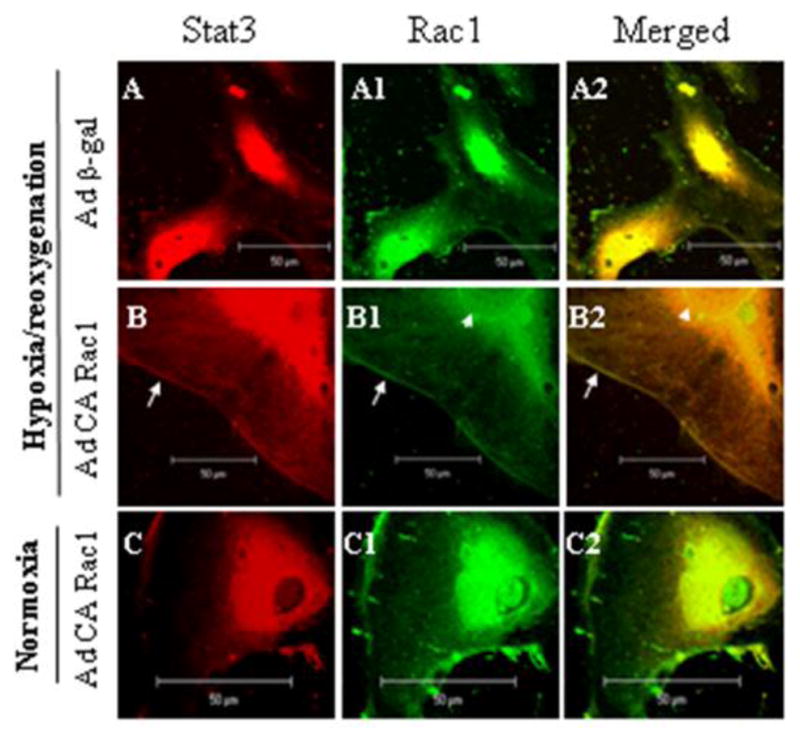
Stat3 and CA Rac1 colocalize following H/R. HUVECs were infected with adenoviruses as in Fig 1, exposed to 2 h hypoxia and 5 min reoxygenation or a corresponding period of normoxia, fixed and processed for dual immunofluorescent labeling with Stat3 (red) and Rac1 (green). Images of 5 nm thick sections were taken by confocal microscopy. Colocalization (yellow color in right panels) is seen predominantly on the cell membrane (arrow) and nuclear membrane (arrow head), and inside the nucleus following H/R and infection with Ad CA Rac1.
3.7 Stat3 and Rac1 interact through the amino acid residues within the coiled-coil domain of Stat3 and the NH2-terminal 54 amino acids of Rac1
To determine if Stat3 and Rac1 interact directly, and to map their interacting domains, we performed yeast two-hybrid assays. As shown in Fig. 7, Stat3 interacted with full-length activated Rac1, and with its AA segments 1-54, 1-122, 1-142, or 1-180, but not with AA segments 40-192, 101-192 or 141-192 (Fig. 7A). Similarly, activated Rac1 interacted with the full-length hStat3α, and its AA segments 107-770 or 131-377, but not with AA segments 1-130, 321-770 or 378-770 (Fig. 7B). Expression of Gal4-BD fusion proteins of full-length CA Rac1 or any of its segments alone, Gal4-AD fusion proteins of full-length Stat3α or its segments alone, or in combination with their complementary Gal4 domain did not activate expression of the reporter genes. These results indicate that the amino acids that sustain Rac1 and Stat3α interaction reside within amino acids 1-54 of Rac1 and the coiled-coiled domain (AA 131-320) of Stat3α.
Figure 7.

The amino acids that sustain Stat3 and Rac1 interaction reside within the coiled-coil domain of Stat3 and the NH2-terminal 54 amino acids of Rac1. (A) CA Rac1 or its segments and Stat3 were coexpressed in S. cerevisiae and assayed for expression of reporter genes. (+) indicates reporter gene expression (interaction between the proteins/peptides) and (−) indicates non-expression (no interaction). S-I, S-II and S–III and the corresponding numbers represent position of molecular switches in the Rac1 protein. (B) Stat3 or its different segments were coexpressed with CA Rac1 in S. cerevisiae and assayed for expression of reporter genes as in (A). C-C = coiled-coil domain, D-B = DNA-binding domain, L-D = linker domain, SH2 = src homology domain 2, T-D = transactivation domain of Stat3.
3.8 Stat3 binds to Rac1 in vitro
To confirm direct interaction between Stat3 and Rac1, we performed in vitro binding assays. As shown in Fig. 8A, CA Rac1 and its different segments were expressed as GST fusion-proteins in bacteria and purified. In vitro binding was performed between GST/Rac1 and 35S-labeled in vitro translated Stat3. Stat3 was found to bind to GST-fusion proteins of CA Rac1, its segments containing AAs 1-54, 1-122, 1-142 and 1-180, but not to AAs 50-192, or GST alone (Fig. 8B). Stat3 segments comprising AAs 1-320 and 131-377 bound to GST/CA-Rac1, but the segment containing AAs 321-770 failed to bind (Fig. 8C), confirming an interaction between the coiled-coil domain of Stat3α and NH2-terminal 54 AAs of Rac1. Simon et al. 2000 [20], implicated the effector domain of Rac1 in Stat3 binding by using effector domain mutants of full-length Rac1, but the interaction was not mapped to the NH2-terminal 54 amino acids of Rac1. In addition, we have, for the first time, identified the coiled-coil domain of Stat3 as the domain that interacts with Rac1.
Figure 8.

Stat3 binds to Rac1 in vitro. (A) Purified GST-Rac1 fusion proteins are shown by Coomassie Blue staining. (B) Ten μg of GST alone or GST-fusion proteins of CA Rac1 or their segments were incubated with in vitro translated [35S]methionine-labeled Stat3α(B), or its amino acid segments (C), (D). All Rac1 fragments bound to Stat3 except for Rac1 aa 50–192. Similarly, Stat3 fragments 1-320 and 131–377 bound to GST/CA Rac1, but fragment 321–770 did not.
3.9 Expression of a Rac1 NH2-terminal peptide comprising Stat3-binding residues suppresses Stat3 S727 phosphorylation following H/R
Since the 54 NH2-terminal residues of Rac1 are required for binding to Stat3, we expressed peptides representing residues 1-17 (Rac1-17) and 23-54 (Rac1-54) in 293 cells to inhibit this interaction and determine the effect on Stat3 phosphorylation following H/R. Cells transfected with Rac1-17 demonstrated decreased Stat3 S727 phosphorylation following H/R (p<0.001, Figure 9A, lower panel). In contrast, Rac1-54 had no significant effect (Fig. 9A). Stat3 S727 phosphorylation was also inhibited when the Rac1-17 peptide was directly transfected into HUVECs exposed to hypoxia for 2 h and reoxygenation for 15 or 30 min (p<0.01, p<0.05, respectively, Fig. 9B, lower panel).
Figure 9.

Transfection of 293 or HUVECs with a Rac1 NH2-terminal peptide blocks Stat3 phosphorylation following H/R. (A) 293 cells were transfected with Rac1-17 (amino acids 1-17) or Rac1-54 (amino acids 23–54). Densitometry of the bands is based on 3 independent observations. H/R increased S727 Stat3 phosphorylation (p = 0.05), and this was inhibited by Rac1-17, but not Rac1-54. (B) HUVECs were transfected directly with Rac1-17 peptide. The upper right panel shows the results of transfection (IgG: red fluorescent dye localized in cytoplasm); FITC-labeled Rac1-17 peptide: green fluorescence localized in nuclei and cytoplasm). The lower panel shows quantitation of 3 experiments. H/R increased S727 Stat3 phosphorylation at 15 min and 30 min (p = 0.014 and 0.035, respectively), and this increase was inhibited by Rac1-17. Mattagajasingh SN et al, “Activation of Stat3 in Endothelial Cells Following Hypoxia-reoxygenation is Mediated by Rac1 and Protein Kinase C”
4. Discussion
Our results strongly support a central role of Rac1 in regulating the activation of the JAK-Stat3 pathway in vascular endothelial cells following H/R. Phosphorylation of both Y705 and S727 residues of Stat3 is clearly regulated by Rac1 in endothelial cells following H/R. In addition, we have made several other new observations: 1) Stat3 associates with Rac1 and isoforms of PKC including PKCζ in a novel multiprotein complex, providing a mechanism for H/R-induced Stat3 S727 phosphorylation; 2) direct binding of Stat3 to Rac1 is mediated by the coiled-coil domain of Stat3 and the NH2-terminal 54 amino acids of Rac1, 3) transfection with a peptide comprising the NH2-terminal 17 amino acid residues of Rac1 inhibits the phosphorylation of Stat3 S727 after H/R, and 4) Stat3 colocalizes with activated Rac1 both at the cell membrane and inside the nucleus following H/R. Thus, following H/R, Rac1 appears to control activation of Stat3 in endothelial cells through multiple Rac-dependent pathways.
We found that Stat3 was associated with Rac1 in quiescent cells, and that the association was increased following H/R, and even more so with expression of CA Rac1 (Fig. 4). Consistent with these results, we observed increased colocalization between Stat3 and Rac1 following H/R (Fig. 6). Our data suggest that the NH2-terminal 54 amino acids of Rac1, which include its GTP-binding and effector domains, are necessary and sufficient for direct binding to the coiled-coil domain of Stat3. These results are consistent with increased association observed between Stat3 and CA Rac1 in IP and immunocolocalization studies. The effector domain of Rac1 undergoes a conformational change upon GTP binding, and CA Rac1 remains in a GTP bound state. Since Rac1 is activated following H/R, it appears that the conformational change in Rac1 effector domain and/or association with other factors upon activation facilitate its association with Stat3. Our finding is consistent with Simon et al [20], who reported that Stat3 binding to Rac1 occurred only when Rac1 was in its activated (GTP-bound) form. The coiled-coil domain of Stat3 serves as an adaptor domain for protein-protein interaction as it organizes in four stranded helical coiled-coils, and has been reported to interact with a number of proteins [43].
We constructed peptides comprising residues 1-17 and 23-54 of the NH2-terminal end of Rac1 to inhibit its interaction with Stat3 and examine the resulting effect on Stat3 phosphorylation. We found that peptide 1-17 did inhibit S727 phosphorylation following H/R in HUVECs and 293 cells, while peptide 23–54 had no effect, suggesting that it is the first 17 amino acids of Rac1 that are involved in Stat3 binding. This finding supports the hypothesis that physical interaction between Rac1 and Stat3 are involved in Stat3 phosphorylation in endothelial cells following H/R. Interestingly, the region from residue 30–40 of Rac1 has been identified as an NH2 –terminal effector site that is required for binding to and activation of the p67phox subunit of the NADPH oxidase [44], resulting in production of ROS. Since peptide 1-17 was effective while peptide 23–54 was not, our results suggest that the peptide inhibited Stat3 S727 phosphorylation by directly preventing Rac1-Stat3 interaction rather than by indirectly inhibiting Rac1-mediated ROS production.
The correlation we observed between S727 phosphorylation and association of Stat3 with PKC and Rac1 suggest that this association may be, at least in part, a mechanism of Stat3 activation following H/R (Fig. 4). We have, for the first time, demonstrated that PKCζ, an aPKC, is one of the isoforms of PKC that associates with Stat3 in an H/R dependent manner, and that knockdown of PKCζ by siRNA inhibits H/R-induced Stat3 S727 phosphorylation (Fig. 5B). However, since Stat3 S727 phosphorylation was significantly inhibited by Calphostin C, which inhibits PKC by binding to the DAG-binding domain [45] present only in cPKCs (isoforms α, βI/II and γ) and nPKCs (isoforms δ, ε, η(Λ), θ and μ), but absent in aPKCs (isoforms ζ, ι, and λ) [46], our data suggest involvement of cPKC and/or nPKC isoforms in addition to PKCζ. Other serine/threonine kinases, such as those in the MAPK signaling pathways may also be involved in Stat3 S727 phosphorylation, as reported previously [45,47]. The contribution of individual kinases will depend on their subcellular localization and proximity to Stat3 and Rac1, and the extent of their activation upon H/R.
Our results indicate a potential role of Rac1 in Stat3 activation in the nucleus. Our observation of nuclear Stat3 in quiescent cells is consistent with earlier reports that unphosphorylated Stat3 was constitutively present in the nucleus and was engaged in regulation of gene expression [48,49]. Although active Rac1 was prominently localized at the cell membrane and inside the nucleus in normoxia, colocalization with Stat3 was observed at the cell and nuclear membranes only after H/R (Fig. 6). The finding of colocalization of Rac1 and Stat3 in the nucleus is consistent with the recent report of Simeone-Penney et al demonstrating a similar finding during PDGF-activation of human airway smooth muscle cells [21]. Transport of activated Stat3 into the nucleus occurs as a complex with GTP-bound Rac1 and MgcRacGAP (male germ cell RacGAP), which contains a nuclear localizing signal (NLS) [50].
Multiple isoforms of the NADPH oxidase are activated in discrete subcellular compartments including membrane ruffles, caveolae, lipid rafts, endosomes and the nucleus [51]. A number of tyrosine and serine/threonine kinases and PKC isoforms that are either constitutively nuclear or that translocate to the nucleus, have also been reported [52]. PKCζ has been shown to be constitutively nuclear, and activate MAPK pathways inside the nucleus during H/R [38]. Interestingly, we found PKCζ to be mainly cytoplasmic. Following H/R, PKCζ physically associates with Stat3 and colocalizes with it in the cytoplasm. Once phosphorylated, however, Stat3 appears to dissociate from PKCζ and travel to the nucleus (Fig 5C, D).
Stat3 has received recent attention for its cytoprotective effects unrelated to gene transcription. Tyrosine 705-phosphorylated Stat3 has been shown to promote phosphorylation of survival proteins in the Reperfusion Injury Salvage Kinase (RISK) pathway, including Akt, ERK2, and GSK3β, in anoxic-reoxygenated chick hearts [53]. A pool of Stat3 located in the mitochondria has been described with a direct, non-transcriptional role in regulation of the electron transport chain [54]. Overexpression of transcriptionally inactive Stat3 in mitochondria attenuates damage to the mitochondria during cell stress, with decreased production of ROS and retention of cytochrome c [54]. Mitochondrial Stat3 appears to contribute to cytoprotection by stimulating respiration and inhibiting mitochondrial permeability transition pore opening [55]. Stat3 has also been shown to protect against postpartum cardiomyopathy, although this may occur through transcriptional regulation of ROS scavenging enzymes like manganese superoxide dismutase [56].
Because of the pleiotropic effects of Rac1 in Stat3 activation, it has been difficult to elucidate the functional significance of Rac1-Stat3 binding. Stat3 may be recruited to kinase signaling complexes through its association with Rac1, and the kinase(s) may then be activated in physical proximity to Stat3 by factors such as Rac-effectors or Rac1-mediated ROS. Alternatively, GTP-bound Rac1, by binding to Stat3, might bring a conformational change in the Stat3 molecule, or provide coupling energy that favors binding of other factors such as protein kinases to the Stat3 molecule. This combined activation by Rac1 GTPase and protein kinases may be required for complete and highly specific activation of Stat3 and may be analogous to simultaneous activation of WASP by GTP-bound Cdc42 and the tyrosine kinase, Lck [57]. Thus, Rac1 and Stat3, in association with other factors, could establish redox-active signaling platforms in different cellular compartments, including the nucleus, that can serve as a sensor of cellular redox status, and bring rapid alterations in cellular redox-responsive gene expression.
A potential study limitation is the use of human umbilical vein endothelial cells for most of our experiments. It is possible that our results would have been different if we had used human arterial or microvascular endothelial cells, or endothelial cells from another species. However, some of our experiments were also performed in COS-7 and 293 cells (of monkey and human embryonic kidney origin, respectively), and yielded results similar to HUVECs (see Fig. 9), In addition, results consistent with our findings have been reported in a number of other cell types, including human airway [21] and rat smooth muscle cells [31].
Highlights.
Stat3 activation by hypoxia/reoxygenation is mediated by both Rac1 and PKC.
Activation of Stat3 occurs through a multiprotein complex with Rac1 and PKC.
Stat3 binds through its coiled-coil domain to the 54 NH2-terminal residues of Rac1.
A peptide of the 17 NH2-terminal amino acids of Rac1 can inhibit Stat3 activation.
Acknowledgments
We are thankful to Dr. James Darnell, Jr. for kindly providing us with mStat3α wt and mutant expression vectors.
Funding
This study was supported by Program Project Grant HL-65608 to LCB from the National Heart, Lung and Blood Institute, and from start-up funds from the Cardiovascular Institute, University of Pittsburgh, to SNM.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Das DK. Redox regulation of cardiomyocyte survival and death. Antioxid Redox Signal. 2001;3:23–37. doi: 10.1089/152308601750100461. [DOI] [PubMed] [Google Scholar]
- 2.Stein AB, Tang XL, Guo Y, Xuan YT, Dawn B, Bolli R. Delayed adaptation of the heart to stress: late preconditioning. Stroke. 2004;35(11 Suppl 1):2676–9. doi: 10.1161/01.STR.0000143220.21382.fd. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Valen G. Signal transduction through nuclear factor kappa B in ischemia-reperfusion and heart failure. Basic Res Cardiol. 2004;99:1–7. doi: 10.1007/s00395-003-0442-7. [DOI] [PubMed] [Google Scholar]
- 4.Soond SM, Latchman DS, Stephanou A. STAT signalling in the heart and cardioprotection. Expert Rev Mol Med. 2006;8:1–16. doi: 10.1017/S1462399406000032. [DOI] [PubMed] [Google Scholar]
- 5.Yang XP, Irani K, Mattagajasingh S, Dipaula A, Khanday F, Ozaki M, Fox-Talbot K, Baldwin WM, 3rd, Becker LC. Signal transducer and activator of transcription 3alpha and specificity protein 1 interact to upregulate intercellular adhesion molecule-1 in ischemic-reperfused myocardium and vascular endothelium. Arterioscler Thromb Vasc Biol. 2005;25:1395–400. doi: 10.1161/01.ATV.0000168428.96177.24. [DOI] [PubMed] [Google Scholar]
- 6.Yang XP, Mattagajasingh S, Su S, Chen G, Cai Z, Fox-Talbot K, Irani K, Becker LC. Fractalkine upregulates intercellular adhesion molecule-1 in endothelial cells through CX3CR1 and the Jak Stat5 pathway. Circ Res. 2007;101:1001–8. doi: 10.1161/CIRCRESAHA.107.160812. [DOI] [PubMed] [Google Scholar]
- 7.Shuai K, Horvath CM, Huang LH, Qureshi SA, Cowburn D, Darnell JE., Jr Interferon activation of the transcription factor Stat91 involves dimerization through SH2-phosphotyrosyl peptide interactions. Cell. 1994;76:821–8. doi: 10.1016/0092-8674(94)90357-3. [DOI] [PubMed] [Google Scholar]
- 8.Wang M, Zhang W, Crisostomo P, Markel T, Meldrum KK, Fu XY, Meldrum DR. Endothelial STAT3 plays a critical role in generalized myocardial proinflammatory and proapoptotic signaling. Am J Physiol Heart Circ Physiol. 2007;293:H2101–8. doi: 10.1152/ajpheart.00125.2007. [DOI] [PubMed] [Google Scholar]
- 9.Wung BS, Hsu MC, Wu CC, Hsieh CW. Resveratrol suppresses IL-6-induced ICAM-1 gene expression in endothelial cells: Effects on the inhibition of STAT3 phosphorylation. Life Sciences. 2005;78:389–97. doi: 10.1016/j.lfs.2005.04.052. [DOI] [PubMed] [Google Scholar]
- 10.Boengler K, Hilfiker-Kleiner D, Drexler H, Heusch G, Schulz R. The myocardial JAK/STAT pathway: from protection to failure. Pharmacol Ther. 2008;120:172–85. doi: 10.1016/j.pharmthera.2008.08.002. [DOI] [PubMed] [Google Scholar]
- 11.Goodman MD, Koch SE, Afzal MR, Butler KL. STAT subtype specificity and ischemic preconditioning in mice: is STAT-3 enough? Am J Physiol Heart Circ Physiol. 2011;300:H522–6. doi: 10.1152/ajpheart.00231.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Boengler K, Buechert A, Heinen Y, Roeskes C, Hilfiker-Kleiner D, Heusch G, Schulz R. Cardioprotection by ischemic postconditioning is lost in aged and STAT3-deficient mice. Circ Res. 2008;102:131–5. doi: 10.1161/CIRCRESAHA.107.164699. [DOI] [PubMed] [Google Scholar]
- 13.Heusch G, Musiolik J, Gedik N, Skyschally A. Mitochondrial STAT3 activation and cardioprotection by ischemic postconditioning in pigs with regional myocardial ischemia/reperfusion. Circ Res. 2011;109:xxx–xxx. doi: 10.1161/CIRCRESAHA.111.255604. (Epub ahead of print) [DOI] [PubMed] [Google Scholar]
- 14.Levy DE, Darnell JE., Jr Stats: transcriptional control and biological impact. Nat Rev Mol Cell Biol. 2002;3:651–62. doi: 10.1038/nrm909. [DOI] [PubMed] [Google Scholar]
- 15.Freitas A, Alves-Filho JC, Secco DD, Neto AF, Ferreira SH, Barja-Fidalgo C, Cunha FQ. Heme oxygenase/carbon monoxide-biliverdin pathway down regulates neutrophil rolling, adhesion and migration in acute inflammation. Br J Pharmacol. 2006;149:345–54. doi: 10.1038/sj.bjp.0706882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Hilfiker-Kleiner D, Hilfiker A, Fuchs M, Kaminski K, Schaefer A, Schieffer B, Hillmer A, Schmiedl A, Ding Z, Podewski E, Podewski E, Poli V, Schneider MD, Schulz R, Park JK, Wollert KC, Drexler H. Signal transducer and activator of transcription 3 is required for myocardial capillary growth, control of interstitial matrix deposition, and heart protection from ischemic injury. Circ Res. 2004;95:187–195. doi: 10.1161/01.RES.0000134921.50377.61. [DOI] [PubMed] [Google Scholar]
- 17.Raptis L, Arulanandam R, Geletu M, Turkson J. The R(h)oads to Stat3: Stat3 activation by the Rho GTPases. Exp Cell Res. 2011;317:1787–95. doi: 10.1016/j.yexcr.2011.05.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.David M, Wong L, Flavell R, Thompson SA, Wells A, Larner AC, Johnson GR. STAT activation by epidermal growth factor (EGF) and amphiregulin. Requirement for the EGF receptor kinase but not for tyrosine phosphorylation sites or JAK1. J Biol Chem. 1996;271:9185–8. doi: 10.1074/jbc.271.16.9185. [DOI] [PubMed] [Google Scholar]
- 19.Vignais ML, Sadowski HB, Watling D, Rogers NC, Gilman M. Platelet-derived growth factor induces phosphorylation of multiple JAK family kinases and STAT proteins. Mol Cell Biol. 1996;16:1759–69. doi: 10.1128/mcb.16.4.1759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Simon AR, Vikis HG, Stewart S, Fanburg BL, Cochran BH, Guan KL. Regulation of STAT3 by direct binding to the Rac1 GTPase. Science. 2000;290:144–7. doi: 10.1126/science.290.5489.144. [DOI] [PubMed] [Google Scholar]
- 21.Simeone-Penney MC, Severgnini M, Rozo L, Takahashi S, Cochran BH, Simon AR. PDGF-induced human airway smooth muscle cell proliferation requires STAT3 and the small GTPase Rac1. Am J Physiol Lung Cell Mol Physiol. 2008;294:L698–704. doi: 10.1152/ajplung.00529.2007. [DOI] [PubMed] [Google Scholar]
- 22.Simon AR, Rai U, Fanburg BL, Cochran BH. Activation of the JAK-STAT pathway by reactive oxygen species. Am J Physiol. 1998;275:C1640–52. doi: 10.1152/ajpcell.1998.275.6.C1640. [DOI] [PubMed] [Google Scholar]
- 23.Lei C, Deng J, Wang B, Cheng D, Yang Q, Dong H, Xiong L. Reactive oxygen species scavenger inhibits STAT3 activation after transient focal cerebral ischemia-reperfusion injury in rats. Anesth Analg. 2011;113:153–9. doi: 10.1213/ANE.0b013e31821a9fbe. [DOI] [PubMed] [Google Scholar]
- 24.McCord JM. Oxygen-derived free radicals in postischemic tissue injury. N Engl J Med. 1985;312:159–63. doi: 10.1056/NEJM198501173120305. [DOI] [PubMed] [Google Scholar]
- 25.Finkel T. Oxygen radicals and signaling. Curr Opin Cell Biol. 1998;10:248–53. doi: 10.1016/s0955-0674(98)80147-6. [DOI] [PubMed] [Google Scholar]
- 26.Babior BM. NADPH oxidase: an update. Blood. 1999;93:1464–76. [PubMed] [Google Scholar]
- 27.Sirker A, Zhang M, Shah AM. NADPH oxidases in cardiovascular disease: insights from in vivo models and clinical studies. Basic Res Cardiol. 2011;106:735–47. doi: 10.1007/s00395-011-0190-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Abo A, Pick E, Hall A, Totty N, Teahan CG, Segal AW. Activation of the NADPH oxidase involves the small GTP-binding protein p21rac1. Nature. 1991;353:668–70. doi: 10.1038/353668a0. [DOI] [PubMed] [Google Scholar]
- 29.Bokoch GM. Regulation of the human neutrophil NADPH oxidase by the Rac GTP- binding proteins. Curr Opin Cell Biol. 1994;6:212–8. doi: 10.1016/0955-0674(94)90138-4. [DOI] [PubMed] [Google Scholar]
- 30.Van Buul JD, Fernandez-Borja M, Anthony EC, Hordijk PL. Expression and localization of NOX2 and NOX4 in primary human endothelial cells. Antioxid Redox Signal. 2005;7:308–17. doi: 10.1089/ars.2005.7.308. [DOI] [PubMed] [Google Scholar]
- 31.Pelletier S, Duhamel F, Coulombe P, Popoff MR, Meloche S. Rho family GTPases are required for activation of Jak/STAT signaling by G protein-coupled receptors. Mol Cell Biol. 2003;23:1316–33. doi: 10.1128/MCB.23.4.1316-1333.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Stephanou A, Brar BK, Scarabelli TM, Jonassen AK, Yellon DM, Marber MS, Knight RA, Latchman DS. Ischemia-induced STAT-1 expression and activation play a critical role in cardiomyocyte apoptosis. J Biol Chem. 2000;275:10002–8. doi: 10.1074/jbc.275.14.10002. [DOI] [PubMed] [Google Scholar]
- 33.Deshpande SS, Angkeow P, Huang J, Ozaki M, Irani K. Rac1 inhibits TNF-alpha-induced endothelial cell apoptosis: dual regulation by reactive oxygen species. FASEB J. 2000;14:1705–14. doi: 10.1096/fj.99-0910com. [DOI] [PubMed] [Google Scholar]
- 34.Deroanne CF, Hamelryckx D, Ho TT, Lambert CA, Catroux P, Lapière CM, Nusgens BV. Cdc42 downregulates MMP-1 expression by inhibiting the ERK1/2 pathway. J Cell Sci. 2005;118:1173–83. doi: 10.1242/jcs.01707. [DOI] [PubMed] [Google Scholar]
- 35.Mattagajasingh SN, Huang SC, Hartenstein JS, Snyder M, Marchesi VT, Benz EJ. A nonerythroid isoform of protein 4.1R interacts with the nuclear mitotic apparatus (NuMA) protein. J Cell Biol. 1999;145:29–43. doi: 10.1083/jcb.145.1.29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Mattagajasingh SN, Huang SC, Hartenstein JS, Benz EJ., Jr Characterization of the interaction between protein 4.1R and ZO-2. A possible link between the tight junction and the actin cytoskeleton. J Biol Chem. 2000;275:30573–85. doi: 10.1074/jbc.M004578200. [DOI] [PubMed] [Google Scholar]
- 37.Schuringa JJ, Dekker LV, Vellenga E, Kruijer W. Sequential activation of Rac-1, SEK-1/MKK-4, and protein kinase Cdelta is required for interleukin-6-induced STAT3 Ser-727 phosphorylation and transactivation. J Biol Chem. 2001;276:27709–15. doi: 10.1074/jbc.M009821200. [DOI] [PubMed] [Google Scholar]
- 38.Mizukami Y, Kobayashi S, Uberall F, Hellbert K, Kobayashi N, Yoshida K. Nuclear mitogen-activated protein kinase activation by protein kinase c zeta during reoxygenation after ischemic hypoxia. J Biol Chem. 2000;275:19921–7. doi: 10.1074/jbc.M907901199. [DOI] [PubMed] [Google Scholar]
- 39.Neckár J, Marková I, Novák F, Nováková O, Szárszoi O, Ost’ádal B, Kolár F. Increased expression and altered subcellular distribution of PKC-delta in chronically hypoxic rat myocardium: involvement in cardioprotection. Am J Physiol Heart Circ Physiol. 2005;288:H1566–72. doi: 10.1152/ajpheart.00586.2004. [DOI] [PubMed] [Google Scholar]
- 40.Ping P, Zhang J, Qiu Y, Tang XL, Manchikalapudi S, Cao X, Bolli R. Ischemic preconditioning induces selective translocation of protein kinase C isoforms epsilon and eta in the heart of conscious rabbits without subcellular redistribution of total protein kinase C activity. Circ Res. 1997;81:404–14. doi: 10.1161/01.res.81.3.404. [DOI] [PubMed] [Google Scholar]
- 41.Kobayashi E, Nakano H, Morimoto M, Tamaoki T. Calphostin C (UCN-1028C), a novel microbial compound, is a highly potent and specific inhibitor of protein kinase C. Biochem Biophys Res Commun. 1989;159:548–53. doi: 10.1016/0006-291x(89)90028-4. [DOI] [PubMed] [Google Scholar]
- 42.Herbert JM, Augereau JM, Gleye J, Maffrand JP. Chelerythrine is a potent and specific inhibitor of protein kinase C. Biochem Biophys Res Comm. 1990;172:993–999. doi: 10.1016/0006-291x(90)91544-3. [DOI] [PubMed] [Google Scholar]
- 43.Calò V, Migliavacca M, Bazan V, Macaluso M, Buscemi M, Gebbia N, Russo A. STAT proteins: from normal control of cellular events to tumorigenesis. J Cell Physiol. 2003;197:157–68. doi: 10.1002/jcp.10364. [DOI] [PubMed] [Google Scholar]
- 44.Diekmann D, Nobes CD, Burbelo PD, Abo A, Hall A. Rac GTPase interacts with GAPs and target proteins through multiple effector sites. EMBO J. 1995;14:5297–5305. doi: 10.1002/j.1460-2075.1995.tb00214.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Galdiero M, Vitiello M, D’Isanto M, Raieta K, Galdiero E. STAT1 and STAT3 phosphorylation by porins are independent of JAKs but are dependent on MAPK pathway and plays a role in U937 cells production of interleukin-6. Cytokine. 2006;36:218–28. doi: 10.1016/j.cyto.2006.12.003. [DOI] [PubMed] [Google Scholar]
- 46.Alvi F, Idkowiak-Baldys J, Baldys A, Raymond JR, Hannun YA. Regulation of membrane trafficking and endocytosis by protein kinase C: emerging role of the pericentrion, a novel protein kinase C-dependent subset of recycling endosomes. Cell Mol Life Sci. 2007;64:263–70. doi: 10.1007/s00018-006-6363-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Lim CP, Cao X. Regulation of Stat3 activation by MEK kinase 1. J Biol Chem. 2001;276:21004–11. doi: 10.1074/jbc.M007592200. [DOI] [PubMed] [Google Scholar]
- 48.Meyer T, Gavenis K, Vinkemeier U. Cell type-specific and tyrosine phosphorylation- independent nuclear presence of STAT1 and STAT3. Exp Cell Res. 2002;272:45–55. doi: 10.1006/excr.2001.5405. [DOI] [PubMed] [Google Scholar]
- 49.Reich NC, Liu L. Tracking STAT nuclear traffic. Nat Rev Immunol. 2006;6:602–12. doi: 10.1038/nri1885. [DOI] [PubMed] [Google Scholar]
- 50.Kawashima T, Bao YC, Minoshima Y, Nomura Y, Hatori T, Hori T, Fukagawa T, Fukada T, Takahashi N, Nosaka T, Inoue M, Sato T, Kukimoto-Niino M, Shirouzu M, Yokoyama S, Kitamura T. A Rac GTPase-activating protein, MgcRacGAP, is a nuclear localizing signal-containing nuclear chaperone in the activation of STAT transcription factors. Mol Cell Biol. 2009;29:1796–813. doi: 10.1128/MCB.01423-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Li JM, Shah AM. Intracellular localization and preassembly of the NADPH oxidase complex in cultured endothelial cells. J Biol Chem. 2002;277:19952–60. doi: 10.1074/jbc.M110073200. [DOI] [PubMed] [Google Scholar]
- 52.Martelli AM, Evangelisti C, Nyakern M, Manzoli FA. Nuclear protein kinase C. Biochim Biophys Acta. 2006;1761:542–51. doi: 10.1016/j.bbalip.2006.02.009. [DOI] [PubMed] [Google Scholar]
- 53.Pedretti S, Raddatz E. STAT3α interacts with nuclear GSK3beta and cytoplasmic RISK pathway and stabilizes rhythm in the anoxic-reoxygenated embryonic heart. Basic Res Cardiol. 2011;106:355–69. doi: 10.1007/s00395-011-0152-5. [DOI] [PubMed] [Google Scholar]
- 54.Szczepanek K, Chen Q, Larner AC, Lesnefsky EJ. Cytoprotection by the modulation of the mitochondrial electron transport chain: the emerging role of mitochondrial STAT3. Mitochondrion. 2011 Sep 10; doi: 10.1016/j.mito.2011.08.011. (Epub ahead of print) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Boengler K, Hilfiker-Kleiner D, Heusch G, Schulz R. Inhibition of permeability transition pore opening by mitochondrial STAT3 and its role in myocardial ischemia/reperfusion. Basic Res Cardiol. 2010;105:771–85. doi: 10.1007/s00395-010-0124-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Hilfiker-Kleiner D, Kaminski K, Podewski E, Bonda T, Schaefer A, Silwa K, Forster O, Quint A, Landmesser U, Doerries C, Luchtefeld M, Poli V, Schneider MD, Balligand J-L, Desjardins F, Ansari A, Struman I, Nguyen NQN, Zschemisch NH, Klein G, Heusch G, Schulz R, Hilfiker A, Drexler H. A cathepsin D-cleaved 16 kDa form of prolactin mediates postpartum cardiomyopathy. Cell. 2007;128:589–600. doi: 10.1016/j.cell.2006.12.036. [DOI] [PubMed] [Google Scholar]
- 57.Torres E, Rosen MK. Protein-tyrosine kinase and GTPase signals cooperate to phosphorylate and activate Wiskott-Aldrich syndrome protein (WASP)/neuronal WASP. J Biol Chem. 2006;281:3513–20. doi: 10.1074/jbc.M509416200. [DOI] [PubMed] [Google Scholar]