

Summary
The apicomplexan parasite Toxoplasma gondii is able to suppress nitric oxide production in activated macrophages. A screen of over 6000 T. gondii insertional mutants identified two clones, which were consistently unable to suppress nitric oxide production from activated macrophages. One strain, called 89B7, grew at the same rate as wild-type parasites in naïve macrophages, but unlike wild type, the mutant was degraded in activated macrophages. This degradation was marked by a reduction in the number of parasites within vacuoles over time, the loss of GRA4 and SAG1 protein staining by immunofluorescence assay, and the vesiculation and breakdown of the internal parasite ultrastructure by electron microscopy. The mutagenesis plasmid in the 89B7 clone disrupts the promoter of a 3.4 kb mRNA that encodes a predicted 68 kDa protein with a cleavable signal peptide and a patatin-like phospholipase domain. Genetic complementation with the genomic locus of this patatin-like protein restores the parasites ability to suppress nitric oxide and replicate in activated macrophages. A haemagglutinin-tagged version of this patatin-like protein shows punctate localization into atypical T. gondii structures within the parasite. This is the first study that defines a specific gene product that is needed for parasite survival in activated but not naïve macrophages.
Introduction
Toxoplasma gondii is an obligate intracellular protozoan parasite that invades and survives within a wide variety of host cells including macrophages. T. gondii actively invades host cells independent of phagocytosis and forms a membrane-bound parasitophorous vacuole (PV) that is segregated from endocytic/phagocytic intracellular processes but associates with host cell mitochondria and endoplasmic reticulum (ER). In addition to regulating its association with host cell organelles, T. gondii tachyzoites modulate expression of host genes (Spear et al., 2000), alter intracellular signalling (Denkers et al., 2003), prevent host cell apoptosis (Nash et al., 1998), and protect themselves from the effects of antimicrobial mediators (Kwok et al., 2004). In immunocompetent individuals, T. gondii replication is controlled by an IFN-γ-dependent innate and cell-mediated immune response. However, some of the rapidly replicating form, called tachyzoites, escape killing by the immune response and convert to a slow growing form, called bradyzoites. In cultured murine macrophages, IFN-γ can induce tachyzoites to differentiate into bradyzoites (Bohne et al., 1993).
Production of reactive nitrogen intermediates (RNI) that include nitric oxide (NO) are critical for the control of T. gondii in macrophages activated in vitro (Adams et al., 1990; Murray and Teitelbaum, 1992). NO is also important for the control of T. gondii in vivo in susceptible mice during the establishment of chronic infection (Scharton-Kersten et al., 1997; Schluter et al., 1999) and resolution of ocular infection (Hayashi et al., 1996; Roberts et al., 2000). RNI play a complex role in T. gondii pathogenesis as they can be effectors that limit parasite growth in macrophages (Adams et al., 1990; Murray and Teitelbaum, 1992), induce conversion to bradyzoites (Bohne et al., 1994), cause pathology (Khan et al., 1997) and mediate immunoregulatory functions such as T cell suppression (Bingisser et al., 1998). Intracellular infection with T. gondii inhibits NO production from infected macrophages that are activated by diverse stimuli including IFN-γ, lipopolysaccharide (LPS) and TNF-α (Seabra et al., 2002; Luder et al., 2003). Most importantly, downregulation of inducible NO synthase enables T. gondii to survive and replicate in moderately activated macrophages (Luder et al., 2003). The parasite genes responsible for modulating macrophage functions including NO production are unknown.
Our goal is to identify T. gondii genes that enable the parasite to survive in stress conditions such as activation of infected macrophages. As a first step towards this goal, we created a library of over 6000 T. gondii insertional mutants and performed a screen to identify mutants that failed to inhibit NO production from activated macrophages. Here we describe the isolation of a T. gondii mutant with a defect in its ability to suppress NO production and to survive in activated macrophages. The disrupted gene responsible for this phenotype is T. gondii patatin-like protein (TgPL1), which encodes a 68 kDa protein with a putative signal peptide and patatin-like phospholipase domain.
Results
Identification of mutant strains unable to suppress NO
We created a T. gondii insertional library of over 6000 mutants using restriction enzyme-mediated integration (REMI) to enhance stable integration (Black et al., 1995). These insertional mutants were screened for their ability to suppress NO production in activated RAW macrophages. For this screen, the mutant library was passed from lysed monolayers of fibroblast cells in 96 well plates, to monolayers of RAW macrophages in 96 well plates. Three hours after this transfer, the RAW cells were activated with 100 ng ml−1 LPS and 100 U ml−1 IFN-γ. After 24 h of activation, 50 μl of supernatant was removed from each well and nitrite (a stable downstream product of NO) concentration was determined by Griess reaction (Griess, 1879). We selected mutants that failed to suppress NO production compared with wild-type parasites. To quantify these defects, we infected macrophages with twofold serial dilutions of wild-type or mutant parasites, stimulated with LPS and IFN-γ, and measured nitrite as described above. To quantify the number of viable input parasites used to infect macrophages, a plaque assay in fibroblast cells was performed. Ultimately, two mutant clones were identified (called 89B7 and 77F7) that were not able to suppress NO as well as wild type (Fig. 1).
Fig. 1.
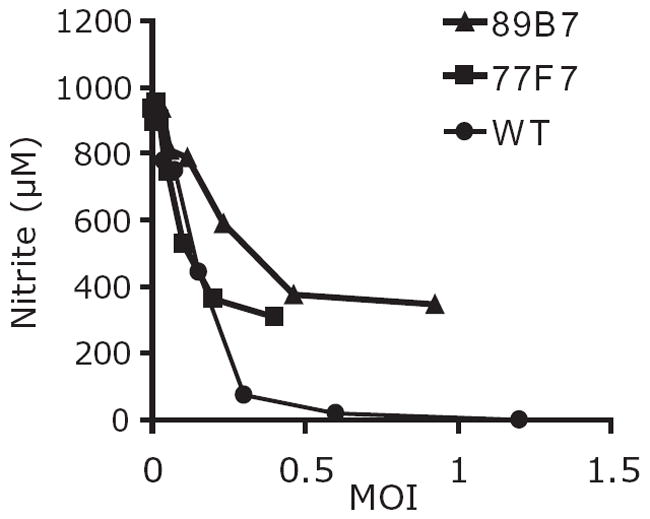
Mutant strains are less efficient than wild-type T. gondii at NO suppression. RAW macrophages were infected with wild-type (WT) or mutant parasites (89B7 or 77F7) prior to stimulation with LPS and IFN-γ. With similar numbers of parasites, macrophages infected with either of the two mutants produced more NO than macrophages infected with wild-type parasites. NO production is expressed as μM nitrite. The graph is from a representative experiment performed with duplicate wells. Similar results were obtained in at least four independent experiments.
Analysis of DNA flanking the insertion site of the mutants
The genomic region adjacent to the insertion site of the mutants was identified by restriction enzyme digestion, ligation and transformation of Escherichia coli. Retrieved T. gondii fragments were sequenced using a primer that extends out from the insertional plasmid and the resulting sequence was compared with the T. gondii genome (http://ToxoDB.org). The insertion plasmid in the 77F7 mutant disrupted a predicted open reading frame TgTwinScan_6824 (same as the draft 3 annotation 44.m5903). The insertion was 110 amino acids downstream from the predicted initiator methionine of this 75 kDa protein. The plasmid inserted into a genomic NotI restriction enzyme site consistent with the creation of the library using NotI REMI. The predicted 77F7 protein had no homologues in the NCBI database and there are no expressed sequence tags (ESTs). The insertion site of the 89B7 mutant is 845 bp upstream of an initiator methionine of protein 44.m02735, draft 3 annotation patatin-like phospholipase domain-containing protein (same as TgTwinScan_5888 and TgTwinScanEt_5062). The insertion site was at a NotI restriction site, 300 bp from the 5′ end of an EST (TgESTzyl52b10.y1). Southern blot analysis of the 89B7 clone shows that the mutagenesis plasmid inserted in a single site within the T. gondii genome. We focused subsequent studies on the 89B7 mutant because patatin-like phospholipases can be secreted bacterial virulence factors [Pseudomonas aeruginosa type III translocated cytotoxin ExoU (Sato and Frank, 2004) and the Legionella pneumophilia type IV translocated protein VipD (Shohdy et al., 2005)].
Characterization of 89B7 parasites in naïve macrophages
To evaluate whether the failure of 89B7 parasites to suppress NO production correlated with decreased survival and replication in macrophages, we first evaluated their integrity in naïve macrophages. Murine bone marrow-derived macrophages were used for immunofluorescence assay (IFA) studies because it was easier to visualize parasite and PV ultrastructure within them compared with more rounded RAW cells. We challenged macrophages with wild-type or 89B7 parasites at a multiplicity of infection (MOI) of 0.5 and determined the number of parasites per PV by IFA at 12, 24 and 36 h after parasite addition. 89B7 parasites displayed similar replication rates to wild-type parasites in naïve macrophages as determined by the mean number of parasites/PV over time (Fig. 2). To determine if the invasion efficiency differed between wild-type and 89B7 parasites in our experimental conditions, we challenged macrophages with parasites and evaluated the number of intracellular and extracellular parasites after 1 h by IFA. Intracellular and extracellular parasites were identified by differential staining of parasites before and after permeabilization. There was no difference in invasion efficiency between wild-type and 89B7 parasites (data not shown). Both wild-type and 89B7 parasites entered macrophages by invasion and not phagocytosis as demonstrated by the absence of lysosomal membrane protein 1 staining at the PV (data not shown).
Fig. 2.
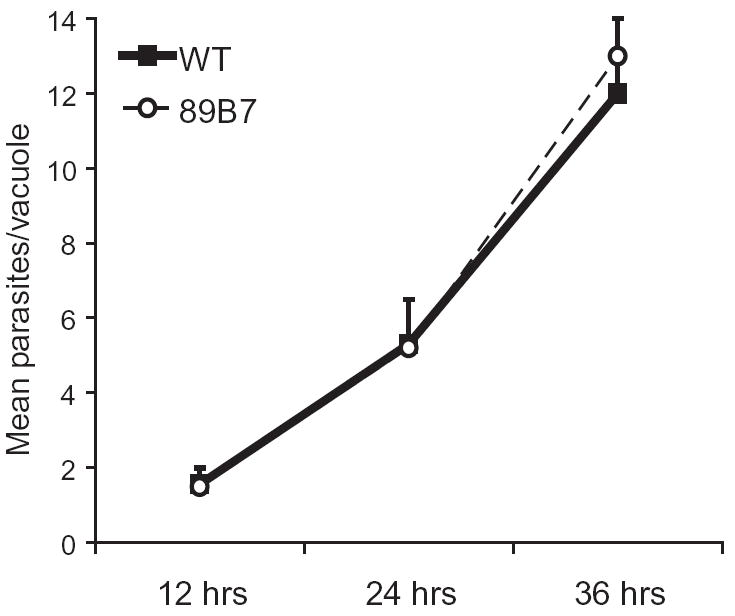
89B7 parasites grow normally in naïve macrophages. The mean number of parasites per vacuole was determined by IFA at 12, 24 and 36 h post infection in naïve bone marrow-derived macrophages. Wild-type (WT) and 89B7 mutant parasites were detected for IFA analysis with serum from a mouse chronically infected with T. gondii. All parasites appeared intact by IFA at each time point. The mean and SD are derived from three counts of 100 PV per time point. The experiment shown is representative of three independent experiments.
Characterization of 89B7 parasites in activated macrophages
As 89B7 invasion and replication in naïve macrophages appeared normal, we evaluated whether they differed from wild-type parasites in their ability to persist in infected macrophages activated with LPS and IFN-γ. Macrophages were challenged with 89B7 or wild-type parasites at a MOI of 0.5. After 3 h, the media were removed and fresh media with or without LPS and IFN-γ were added. At 33 h after activation, cells were fixed, permeabilized and stained to examine the T. gondii by IFA. The number of parasites per vacuole, the ultrastructure of the parasites and the integrity of the PV were evaluated. At 33 h after activation of infected macrophages the majority of wild-type parasites had two to four parasites per vacuole and appeared intact by phase contrast microscopy indicating they were capable of limited replication in activated macrophages (Fig. 3A). In marked contrast, 89B7 parasites either had one parasite per PV at 33 h or had a degraded appearance (Fig. 3A).
Fig. 3.
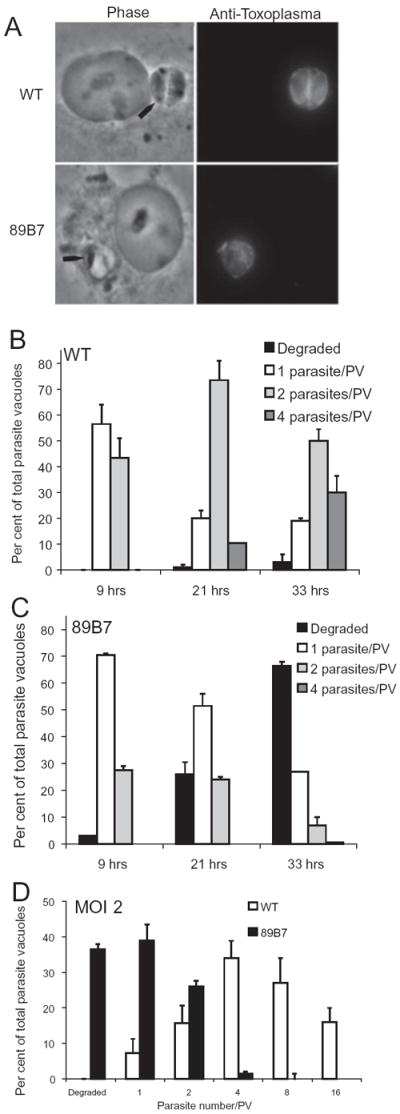
89B7 parasites have a single parasite per vacuole or are degraded following activation of infected macrophages. (A) Phase contrast and IFA shows that wild-type (WT) parasites remain capable of limited replication, while 89B7 parasites have one parasite per PV or are degraded. The black arrows point out the parasites in the phase contrast image. Parasites were detected with serum from a mouse chronically infected with T. gondii at 33 h after activation of infected macrophages. (B and C) Kinetic analysis of wild-type and 89B7 parasites in activated macrophages. Using a MOI of 0.5, the number of parasites per vacuole of wild-type (WT, B) and 89B7 (C) parasites at 9, 21 and 33 h following activation of infected macrophages. Wild-type parasites replicate slowly throughout the course of macrophage activation. In contrast, 89B7 parasites replicate shortly after macrophage activation but then cease replication and appear to be degraded during the time-course. Panel D uses a MOI of 2 and similarly shows the number of WT (white bars) and 89B7 (black bars) parasites per PV 33 h after activation of infected macrophages. The increased MOI increased replication of WT parasites but had only a modest effect on 89B7 parasites. The mean and SD are derived from three counts of 100 PV per time point. Parasites were detected with serum from a mouse chronically infected with T. gondii. The experiment shown is representative of two independent experiments.
To further investigate this limited replication of wild-type parasites and the degraded appearance of the 89B7 parasites in activated macrophages, we monitored parasite replication and integrity during a time-course of infection. Macrophages were infected at a 0.5 MOI, stimulated after 3 h with LPS and IFN-γ, incubated at 37°C for an additional 9, 21 and 33 h. The number of parasites per vacuole was counted for each time point (wild type in Fig. 3B and 89B7 in Fig. 3C). The majority of wild-type parasites replicated throughout the 33 h period following activation as seen by the steady increase in two and four parasite-containing vacuoles. However, approximately 20% had a single parasite per PV at 21 and 33 h. This limited replication in activated macrophages is distinctly different from their robust replication in naïve macrophages (compare Figs 2 and 3B). 89B7 parasites demonstrated only a slight lag in replication compared with wild-type parasites at 9 h post activation but were substantially impaired at 21 and 33 h with the majority of vacuoles containing either one parasite per PV or with a degraded appearance (Fig. 3C). The fact that 25% of 89B7 parasites had two parasites/PV 9 h after activation of infected macrophages indicates that 89B7 remains capable of replication for a short period after macrophage activation but become static/degraded after longer periods of time in activated macrophages. This is also evidenced by the decrease in two parasites per PV at later time points with no corresponding increase in four parasites per PV.
To evaluate the effect of multiplicity of infection on replication of wild-type and 89B7 parasites in activated macrophages, we challenged macrophages with a MOI of 2 (Fig. 3D) and monitored parasite replication and integrity 33 h after macrophage activation. Because these experiments were performed simultaneously with the 0.5 MOI experiment, using the identical parasites and macrophages, they are directly comparable. Replication of wild-type parasites increased substantially when a higher multiplicity of infection was used (compare Fig. 3B and D). Wild-type parasites achieved four, eight and 16 parasites/PV and less than 10% had only one parasite per PV. In contrast, increasing the multiplicity of infection had only a modest effect on replication and integrity of 89B7 parasites.
To evaluate if the impairment of 89B7 parasites in activated macrophages correlated with specific alterations in the parasite, the tubulovesicular network between the parasite and PV membrane (PVM), we stained parasites in naïve and activated macrophages with antibodies to parasite SAG1 (parasite surface), GRA4 (network), GRA7 (PVM), and whole parasites using serum from mice chronically infected with T. gondii. Expression of SAG1 and GRA4 were nearly identical in wild-type and 89B7 parasites in naïve macrophages (Fig. 4A and B). However, 33 h after activation of macrophages, the majority of 89B7 parasites were largely negative for GRA4 expression. Similarly, 28% were negative for both GRA4 and SAG1. In contrast, 89B7 expression of GRA7 was unchanged from wild-type parasites.
Fig. 4.
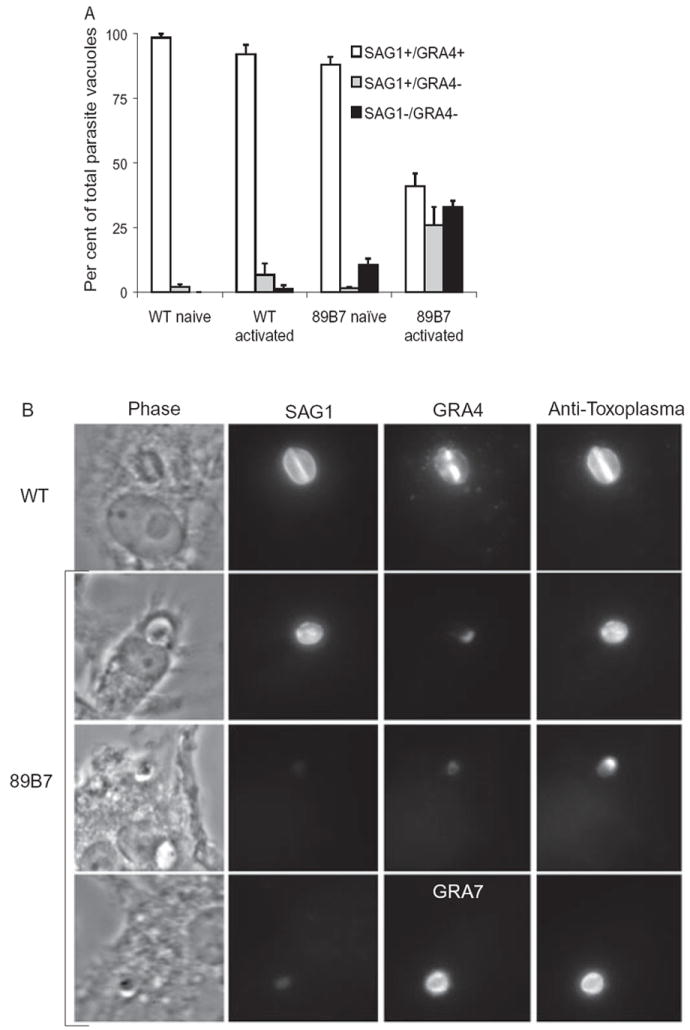
Loss of SAG1 and GRA4, but not GRA7 expression in 89B7 parasites. Degradation/stasis of 89B7 parasites in activated macrophages correlates with decreased expression of SAG1 and GRA4 but no change in GRA7. Panel A shows the per cent of PVs that are SAG1+/GRA4+, SAG1+/GRA4− and SAG1−/GRA4−. Data shown are the mean and SD of three counts of 100 PV each at 33 h post activation or 36 h post challenge of naïve macrophages. Data shown are representative of three independent experiments. Panel B is the representative pictures of SAG1+/GRA4+, SAG1+/GRA4− and SAG1−/GRA4− PVs in activated macrophages. The top panel is wild-type (WT) parasites and the remaining panels are 89B7 parasites. Antibodies to SAG1, GRA4 and GRA7 were used to visualize the parasite surface, the tubulovesicular network and the PVM respectively. Serum from a goat chronically infected with T. gondii (anti-Toxoplasma) and DAPI (not shown) were used to confirm the location of parasites in the absence of SAG1 and GRA4. All pictures are with the 100× objective, in the same experiment, with identical exposure times so a direct comparison of SAG1, GRA4 and GRA7 expression can be made between panels.
We took advantage of the increased resolution achievable by transmission electron microscopy to analyse the structure of 89B7 parasites versus wild-type parasites following macrophage activation. This was important both to confirm the degradation of the 89B7 parasite and to evaluate the process of degradation in greater detail. Because the surface of the 89B7 parasite (SAG1) and the tubulovesicular network (GRA4) was aberrant following activation of infected macrophages, we used GRA7 expression on the PVM to visualize the location of the parasites. Macrophages were infected at a MOI of 1.0 with wild-type or 89B7 parasites and activated as described above. Parasite ultrastructure was evaluated by immuno-electron microscopy 33 h post activation. Wild-type parasites appeared undamaged with intact intracellular organelles and PVM (Fig. 5, WT left panels). In contrast, 89B7 parasites have extensive vesiculation and a breakdown of internal parasite ultrastructure although the parasite membrane is sometimes discernable (Fig. 5 and 89B7 middle and right panels). An array of pictures for the 89B7 mutant is shown to demonstrate the potential sequence of parasite degradation and its heterogeneity. Parasite and PV degradation is extensive in the right panels making it difficult to discern both the parasite and PVM.
Fig. 5.
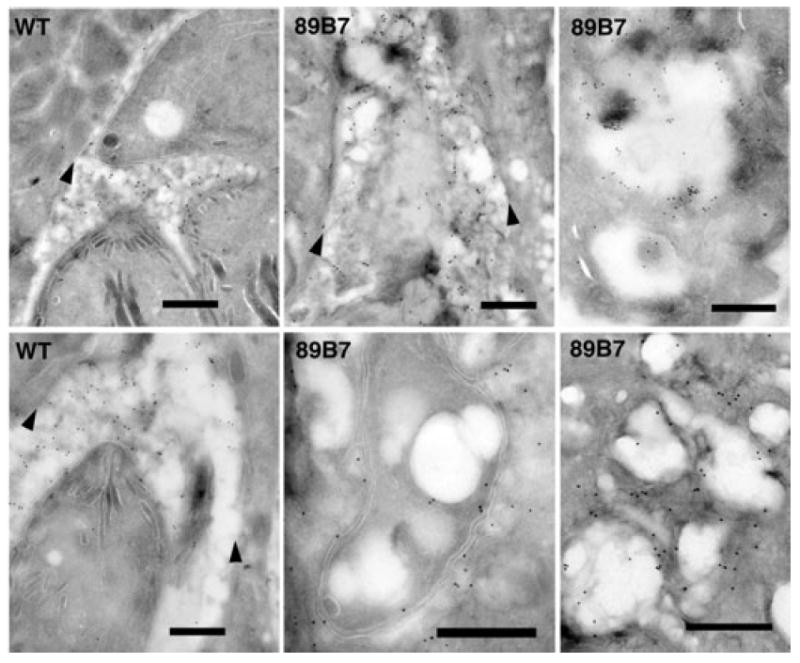
Immuno-electron microscopy shows degradation of 89B7 parasites in activated macrophages. Cryo-immuno-electron microscopy with anti-GRA7 antibody (18 nm colloidal gold) of wild-type (WT) or 89B7 parasites (89B7) 33 h after activation of infected macrophages. Wild-type parasites are intact with defined micronemes and rhoptries (left panels). 89B7 parasites are vesiculated and their internal ultrastructure undetectable (centre panels) or are completely degraded (right panels). Arrowheads demark the location of the PVM. The scale bar is equal to 500 nm on all images.
Characterization of 89B7 parasites in vivo
As 89B7 is the first T. gondii strain identified as defective in its ability to survive and replicate in activated macrophages in vitro, we wanted to investigate its potential to cause disease in mice in both acute and chronic infections. To simulate acute infection, intraperitoneal (I.P.) injection of mice was performed with a dose that was above the lethal dose 50% (LD50) for wild-type Prugniaud parasites in CBA/J mice (~1 × 105 parasites). When 4 × 105 of wild-type or 89B7 parasites were inoculated I.P., no differences in the time to death were seen. We then examined chronic infection with a non-lethal dose of 2 × 104 wild-type or 89B7 parasites and found no differences in the total number of cysts per brain after 22 days of infection. We concluded that even though the 89B7 strain is severely defective within in vitro stimulated macrophages, its ability to infect mice is not affected.
The protein predicted to be disrupted in 89B7 contains a patatin-like domain
The draft 3 annotation in ToxoDB has annotated 44.m02735 as Patatin-like phospholipase domain-containing protein. A global analysis search of Pfam and conserved domain databases (NCBI) with the 44.m02735 protein detected significant homology to patatin-like phospholipase domains (PF01734) from residue 287 to 532 with an E-value of 1.4 × 10−6 (E < 0.05 is significant). Position-specific iterated and pattern-hit initiated BLAST (NCBI) indicated the protein has homology to a predicted secreted alpha beta hydrolase in two related parasites, Cryptosporidium parvum and Cryptosporidium hominis (E-value = 10−14). It also had lesser homology to numerous patatin proteins (E-value = 10−5). To determine if this predicted protein contained the conserved patatin-like phospholipase domains, we aligned its patatin domain with 57 other members of the patatin family using NCBI conserved domain database searches (cdart and rpsblast: Fig. 6). Patatin-like proteins, such as cPLA2, share three conserved regions important for phospholipase activity (Dessen et al., 1999; Hirschberg et al., 2001). Region 1 consists of a glycine-rich region with a lysine or arginine residue (G-G-X-R/K), and in 12 of the 57 seed proteins (including ExoU), it can be expanded to (L-V-L-X-G-G-G-A-R/K). This region is conserved in our 89B7 protein and is predicted to function as an oxyanion hole to stabilize the negative charge that develops during substrate cleavage after nucleophilic attack by the catalytic serine. Region 2 includes a hydrolase motif containing the catalytic serine (G-X-S-X-G). In lipases, the hydrolase motif lies in a tight turn between a β strand and α helix, exposing the catalytic serine at the tip of the ‘nucleophilic elbow’. The small side-chains of the glycines in the motif allow for this arrangement. Unexpectedly, in the 89B7 disrupted protein has a glycine residue in place of the conserved serine residue. Region 3 containing the catalytic aspartate residue (D-X-G/A) is conserved in the 89B7 disrupted protein. Site-directed mutagenesis has shown that the first two glycines in the oxyanion hole, the serine in the hydrolase motif, and the aspartate in the third motif are all important for catalytic activity (Sato et al., 2003; Rabin and Hauser, 2005). Given that in our 89B7 protein is missing the key catalytic serine, it is highly unlikely to have phospholipase activity. Thus, we have designated this gene as TgPL1 (T. gondii patatin-like protein 1).
Fig. 6.

Alignment of TgPL1 with the patatin family shows the absence of a catalytic serine. The catalytic triad domains from TgPL1 and members of the patatin-like PLA2 family (labelled Patatins) were aligned using NCBI rpsblast. Identical amino acids are in bold. Underlined sequences designate the putative catalytic domains for the cdart seed sequences. Numbers on the right side correlate to the amino acid locations in TgPL1.
TgPL1 mRNA analysis
To determine if the insertion disrupted the TgPL1 mRNA in the mutant, we performed Northern Blot analysis on total RNA from wild-type and 89B7 parasites. Northern blot analysis confirmed that a 3.4 kb mRNA was present in wild-type parasites but reduced in abundance in 89B7 mutant (Fig. 7A) compared with an α-tubulin loading control (Fig. 7B). We confirmed the donor and acceptor sites for the single 663 bp intron that was predicted by all of the gene models on ToxoDB. 5′ and 3′ RACE was performed to determine the ends of the mRNA. The 5′ untranslated region (UTR) was 720 bp, while the 3′UTR was 640 bp, which correlates with the mRNA size seen by Northern blot. The insertion site was 130 bp upstream of the start of transcription, suggesting that we have disrupted the TgPL1 promoter in the 89B7 mutant strain, which reduces but does not eliminate the mRNA.
Fig. 7.
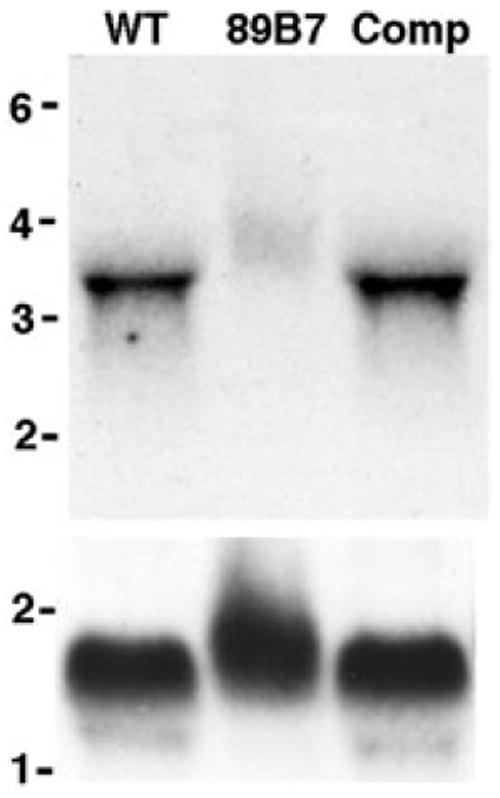
Northern blot analysis of wild-type, 89B7 and an 89B7-complemented strain. The mRNA is 3.4 kb in wild-type parasites (WT), reduced in 89B7 parasites (89B7), and restored in the complemented strain (Comp). Parasites were counted prior to RNA isolation and RNA from 3 × 107 parasites was loaded per lane. The blot was probed for TgPL1 (A) and then striped and reprobed for α-tubulin (B). Molecular weight markers are expressed in kb.
Genetic complementation restores parasite survival in activated macrophages
To complement the 89B7 mutant, we introduced a genomic construct containing the native TgPL1 promoter, because regulation of the TgPL1 protein may be important in T. gondii. We PCR amplified the genomic region of the TgPL1 gene including 2.4 kb of sequence upstream of the transcription start site and 400 bp downstream of the polyadenylation site. We expressed this genomic construct in the 89B7 mutant and confirmed that it restored expression of TgPL1 mRNA by Northern blot analysis (Fig. 7A). We then compared the phenotype of wild-type, 89B7, and complemented parasites in macrophages at 0.5 MOI, and examined them by IFA 33 h post activation. The number of vacuoles containing one, two or four parasites as well as the number that appeared degraded, were counted in three independent blinded studies. Complementation with the transgene protected the 89B7 mutant from degradation in activated macrophages and restored its ability to slowly replicate (Fig. 8A). Southern blot analysis of the complemented strain (Comp) showed a single insertion of the plasmid (data not shown). To ensure that the phenotype the complemented strain was not a position effect due to where the plasmid inserted into the genome, we examined the selected populations from two different electroporations for their ability to protect 89B7 from degradation in activated macrophages (Fig. 8A, Pop 1 and Pop 2). These selected populations showed reduced degradation and increased replication compared with the mutant 89B7 strain. Southern blot analysis showed that each of these selected populations contained several clones with independent insertion sites (data not shown).
Fig. 8.
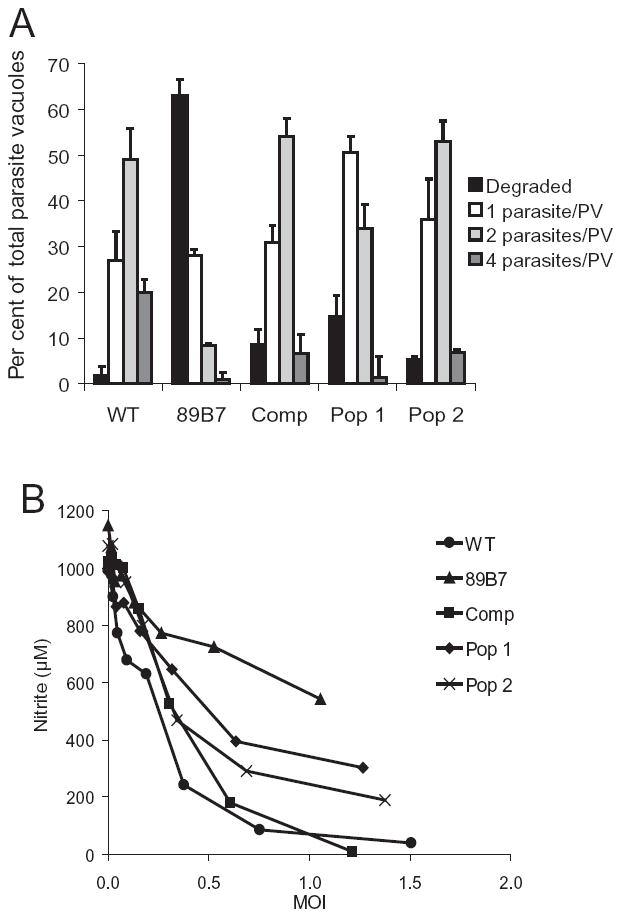
Complementation with TgPL1 restores 89B7 survival in activated macrophages and suppression of NO. Panel A shows that the addition of TgPL1 restores the ability of the 89B7 strain to survive in activated macrophages. Parasite replication (grey bars) versus degradation (black bars) was quantified for wild-type (WT), 89B7, 89B7-complemented clone (Comp), and 89B7-complemented populations (Pop 1 and Pop 2). The graph shows the number of parasites per PV 33 h after activation of infected macrophages. Numbers are the mean and SD from three counts of 100 PV each. The experiment is representative of three independent experiments. Comp, Pop 1 and Pop 2 are from independent electroporations. Panel B shows that the addition of TgPL1 enhances the ability of 89B7 parasites to suppress NO production from activated macrophages. The data shown are a representative experiment from a total of three experiments.
We also examined the ability of the complemented clone, and the selected populations to restore the ability of the 89B7 strain to suppress NO in activated macrophages. As shown in Fig. 8B, the NO suppression of the complemented clone was similar to wild type. Likewise, the two independent selected populations enhanced the ability of 89B7 to suppress NO production. While this enhancement was not to wild-type levels of suppression, it is expected that some false positive clones exist in the selected populations (population 1 likely has more false positives than population 2). Similarly, we found that the expression of GRA4 and SAG1 was restored in 89B7-complemented clone in activated macrophages (data not shown). Taken together, these results confirm that the TgPL1 gene, disrupted in the 89B7 mutant strain, is responsible for the lack of NO suppression and degraded appearance of the 89B7 parasites in activated macrophages.
Size and initial localization of TgPL1 protein
To examine the protein size and location of the TgPL1 protein, we expressed it with a carboxyl-terminal haemag-glutinin epitope (HA) from the endogenous TgPL1 promoter used for the complementation experiments. Western immunoblotting revealed a 68 kDa protein that reacted specifically to the anti-HA antibodies (data not shown). This size agrees with all of the ToxoDB gene models except TgTigrScan_5707, which predicts a truncated 58 kDa form of the protein. Although TgGLEAN_3693 and TgGlmHMM_3250 both predict proteins that begin at the third in frame methionine, this disparity will not correlate with a change in protein size. Starting at the third methionine creates a patatin-like protein without an N-terminal signal sequence.
We examined the localization of TgPL1-HA expressed from its endogenous promoter in extracellular and intracellular parasites within human foreskin fibroblast cells (HFFs). TgPL1-HA was dispersed throughout the parasite cytoplasm in a punctate lattice in extracellular and intracellular parasites that was distinct from parasite dense granules as indicated by GRA4 staining (Fig. 9). The fact that TgPL1-HA location does not change between extracellular and intracellular parasites suggests that its localization was independent of secretory events associated with parasite invasion. Within intracellular parasites, TgPL1-HA remained localized within the parasite and was not apparent in the PV or in the host cell cytosol. We were concerned that trafficking of TgPL1-HA might be impaired due to the HA-tag, so we compared the location of TgPL1-HA with the ER marker BiP. In T. gondii, BiP localizes most prominently to a cup-like region anterior to the apical end of the nucleus (seen in Fig. 9), while the rest of the ER is concentrated posterior to the nucleus (Joiner and Roos, 2002). Figure 9 shows that TgPL1-HA does not colocalize with BiP and is not concentrated to the posterior side of the nucleus, thus the TgPL1-HA location is distinct from the ER. Because patatin-like proteins have been implicated as storage vacuoles in plants, we examined if TgPL1-HA localizes to the T. gondii characterized storage vesicles, the acidocalcisomes. When acidocalcisomes were visualized by staining for T. gondii vacuolar proton pyrophosphatase α-VP1 (Luo et al., 2001) it was clear that TgPL1-HA does not localize to acidocalcisomes (Fig. 9). We also determined that TgPL1-HA localization was distinct from micronemes (MIC2) and rhoptries (Rhop 2.3.4). Finally, we analysed TgPL1-HA localization in naïve and activated macrophages. TgPL1 expression was similar in each cell type and did not appear to be altered by macrophage activation (Fig. 9). Independent of host cell type, TgPL1-HA expression was also distinct from mitochondria visualized by Mitotracker Red (Molecular Probes). Based on these analyses of TgPL1-HA localization, TgPL1 may localize to a unique punctate network within the parasite.
Fig. 9.
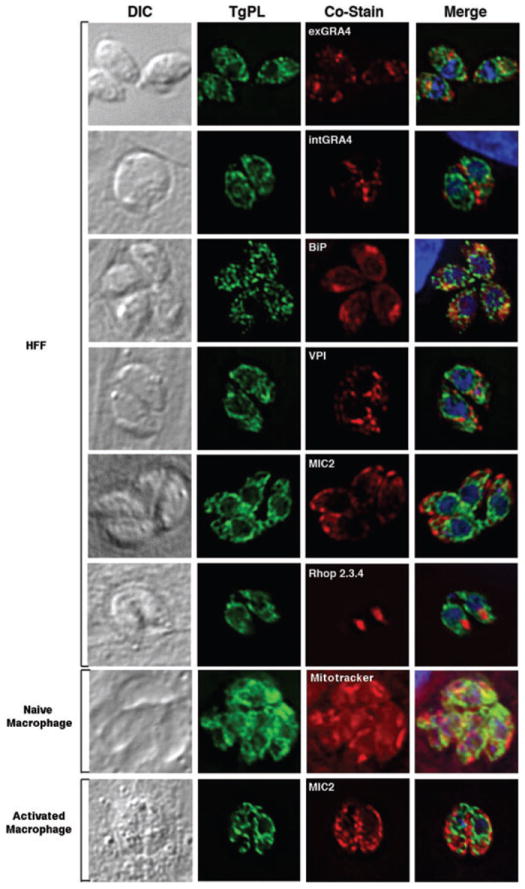
Colocalization studies with a HA-tagged TgPL1. Extracellular parasites are shown in the first panel and all other panels display intracellular parasites. HA-tagged TgPL1 was restricted to the parasite and did not colocalize with dense granules (GRA4), ER (BiP), acidocalcisomes (VP1), or secretory organelles including micronemes (MIC2) and rhoptries (Rhop 2.3.4). TgPL1 expression was also distinct from mitochondria (Mitotracker), and was not altered by macrophage activation.
Discussion
A common feature of intracellular parasites is their ability to create a specialized replicative niche within host cells, providing them with a relatively safe haven even in the midst of an inhospitable microenvironment. In the case of T. gondii, this ability allows it to use motile phagocytic cells to disseminate into the brain to establish chronic infection and avoid degradation (Courret et al., 2006). Although it is clear that T. gondii tachyzoites modulate signalling cascades in infected macrophages (Denkers and Butcher, 2005) and dendritic cells (McKee et al., 2004), the parasite genes that modulate innate immune cell effectiveness have not been identified. In this study, we have performed the first forward genetic screen in a parasite to uncover genes required for stress conditions such as activation of infected macrophages. We identified TgPL1, a previously uncharacterized T. gondii gene as important for parasite survival in activated but not naïve macrophages. Parasites deficient in TgPL1 failed to replicate and were largely degraded in response to activation of infected macrophages. This correlated with a failure to suppress NO production. Control of NO production is important for parasite viability in activated macrophages in cell culture as moderate levels of NO restrict parasite growth and allow conversion to bradyzoites (Bohne et al., 1994) while high concentrations of NO are more likely to induce parasite stasis or death (Luder et al., 2003). It is important to note that our results indicate that TgPL1 is not required for parasite invasion, survival or replication in naïve macrophages or in fibroblast cells. This is the first T. gondii gene identified as important for parasite survival in activated innate immune cells. This is also the first description of a patatin-like protein in any parasite.
Although T. gondii replicates extremely well in naïve macrophages, its fate in activated macrophages can depend on the activation stimuli, its timing, the parasite to macrophage ratio, and the type/differentiation state of the macrophages. Cell culture macrophages require stimulation with both IFN-γ and LPS or TNF-α to suppress parasite replication through production of RNI (Sibley et al., 1991; Adams et al., 1990). T. gondii infection also suppresses macrophage NO production enhancing parasite replication in moderately activated macrophages (Luder et al., 2003). However, high doses of IFN-γ and LPS strongly suppress growth of intracellular parasites (Luder et al., 2003). Stimulation of CD40 also suppresses parasite replication in CD40 positive macrophages through mechanisms independent of IFN-γ (Andrade et al., 2005; 2006). Of key importance, suppression of parasite replication does not appear to prevent parasite dissemination in monocytes and dendritic cells during infection as phagocytes with single parasites transport parasites to the brain where they resume replication prior to cyst formation (Courret et al., 2006).
In contrast to suppression of intracellular parasites, macrophage T. gondii cidal activity can be induced by priming mice for 7 days with a uracil auxotrophic strain of T. gondii (Ling et al., 2006). Such primed macrophages degrade wild-type parasites through a mechanism dependent on the IFN-γ-inducible GTPase IGTP, PI3 kinase and independent of RNI. Priming macrophages with IFN-γ and TNF-α or CD40 prior to parasite invasion can also induce T. gondii cidal activity in vitro (Sibley et al., 1985 1991; Andrade et al., 2006).
Our goal was to identify parasite genes important for surviving environmental stress such as that induced by macrophage activation. In our macrophage activation model, we first allowed parasites to invade naïve bone marrow-derived macrophages and establish their PV. We then identified macrophage activation conditions delivered 3 h after parasite invasion that suppressed but did not prevent the growth of wild-type parasites. Consequently, we used a significantly reduced concentration of LPS and IFN-γ then a previous study that showed complete suppression of parasite replication in activated macrophages (Luder et al., 2003). Our studies led to the identification of the 89B7 mutant that is impaired in its ability to suppress RNI production from infected macrophages. Additional analysis revealed that unlike wild parasites that replicate slowly in activated macrophages, the 89B7 mutant is largely degraded.
Transmission electron microscopy and IFA analysis showed degradation of the 89B7 mutant induced by activation of infected macrophages was accompanied by vesiculation and disruption of the internal structure of the parasite. In contrast, the PVM generally appeared structurally intact as shown by GRA7 expression on the PVM. IFA analysis also showed aberrant expression of GRA4 in the T. gondii tubulovesicular network and a loss of expression of the surface marker SAG1 following macrophage activation. This sequence of events is reminiscent of wild-type parasite degradation within in vivo primed effector macrophages in which parasite vesiculation, disruption and stripping of the parasite plasma membrane occurs (Ling et al., 2006). Future investigation will determine if TgPL1 is important for modulating macrophage activation or if it aids parasite resistance to environmental alterations induced by macrophage activation and parasite degradation leads to an inability to suppress RNI production. The initial failure to detect a role for TgPL1 in pathogenesis may reflect the broad host cell range of T. gondii, heterogeneity in phagocyte activation in vivo or the limited role of RNI for control of acute infection compared with other macrophage effectors such as IGTP. It also indicates that survival in inhospitable host cells during infection is a multifactorial process.
Although TgPL1 protein has limited homology to known proteins it had the greatest degree of homology to a predicted alpha beta hydrolase in both C. parvum and C. hominis. TgPL1 shares homology with a subgroup of alpha beta hydrolases characterized as patatin-like phospholipases. Patatins are a multigene family of vacuolar glycoproteins of approximately 40 kDa in plants that are believed to function as a storage protein (Mignery et al., 1988; Senda et al., 1996; Hirschberg et al., 2001). Storage proteins in plants are proteins whose major role is to accumulate nitrogen, sulphur or carbon that enable plants to survive periods of adverse conditions between growing seasons (Shewry, 2003). They are located in specialized protein storage vacuoles (Robinson et al., 2005), stored as an inactive enzyme and are activated by environmental stress or pathogenic infection (Hirschberg et al., 2001).
Other members of the patatin-like phospholipase family includes multiple bacterial virulence factors including the Pseudomonas aeruginosa type III translocated cytotoxin ExoU (Sato and Frank, 2004) and the Legionella pneumophilia type IV translocated protein VipD (Shohdy et al., 2005). Deletions outside of the patatin domain of ExoU and VipD alter the virulence properties of these two proteins suggesting that bacterial patatin-like phospholipases have functions that aid pathogenesis in addition to their predicted phospholipase activity. It has also been noted that pathogenic bacteria are more likely to express multiple patatin-like proteins compared with non-pathogenic strains (Banerji and Flieger, 2004). As we did not detect TgPL1 outside the parasite and its sequence suggests it lacks phospholipase activity, we believe it is unlikely to function similar to bacterial virulence proteins, which are secreted into host cells and whose activity requires phospholipase activity. Because of the limited homology of TgPL1, it is possible that its function is unrelated to its incomplete patatin domain.
Our current hypothesis is that TgPL1 functions as a storage protein, like patatin in potato tubers, to aid parasite survival during physiologic stress conditions such as macrophage activation. First, it clearly is important for T. gondii survival following macrophage activation but is not required for invasion or replication/survival in naïve macrophages. Our localization studies also are more consistent with a storage protein rather than secreted virulence factor because TgPL1 stays within the parasite and has a unique punctate distribution though out the parasite’s cytosol. It is also not a virulence factor for acute or early chronic infection. Although TgPL1 is predicted to have a cleavable signal peptide, it does not localize to previously characterized T. gondii secretory organelles, the parasite tubulovesicular network, the PVM, host cell cytosol, and perhaps most surprising, the acidocalcisomes. The absence of a catalytic serine is unlikely to interfere with its storage function as the majority of storage proteins have no apparent lipase activity. In fact, replacement of a catalytic serine residue with glycine in TgPL1 may be evolutionarily advantageous to T. gondii as it would avoid intrinsic phospholipase A2 activity that could generate proinflammatory lipid mediators.
TgPL1 is the first patatin-like protein isolated in protozoa parasites. Although TgPL1 is unlikely to have phospholipase activity due to the absence of a catalytic serine domain, a text search of the T. gondii genome indicates that there are four other predicted proteins with patatin-like domains (TwinScan_1934, 2456, 3015 and 5787) in addition to TgPL1. These proteins all have a catalytic serine residue at the predicted site for phospholipase activity. We examined these other T. gondii patatin-like proteins using PSORT and SignalP and found that only TgPL1 has a predicted signal peptide (data not shown). Additional studies will be required to evaluate if these patatin-like proteins share conserved functions in T. gondii invasion, intracellular survival and pathogenesis.
In this study we identify a new patatin-like protein in T. gondii that is important for parasite survival following activation of infected macrophages. We show that it is localized within the parasite in a potentially novel cytosolic/vesicular network. Our hypothesis is that integrity of the parasite and PV during physiologic stress (e.g. macrophage activation) may require the parasite be able to accumulate and withdraw stored resources such as proteins and lipids through a process dependent on TgPL1. If this is the case it is likely that parasite degradation is responsible for the failure of the 89B7 mutant to suppress NO production. However, it is also possible that the absence of TgPL1 affects other proteins the traffic to the host cell to modulate RNI production. Further studies on TgPL1 will help to understand how T. gondii survives harsh intracellular environments.
Experimental procedures
T. gondii strains and cell culture
The Prugniaud strain of T. gondii deleted in hypoxanthine-xanthine-guanosine phosphoribosyl transferase (ΔHPT) and containing a signature-tag (Knoll et al., 2001) was used for insertional mutagenesis. As stable transfection of this ΔHPT strain was performed with a construct containing the HPT gene, ‘wild-type’ Prugniaud was used for all phenotypic analysis. All parasite strains were maintained in monolayers of human HFFs in standard T. gondii culture conditions (Mordue and Sibley, 1997). The monocytic/macrophage RAW264.7 cell line (ATCC) was passaged every 2 days and cultured in RPMI supplemented with 10% fetal bovine serum (FBS). Murine bone marrow-derived macrophages were isolated from CBA/J mice (JAX) as previously described (Mordue and Sibley, 1997) and grown in RPMI with 10% FBS.
Insertional mutagenesis and creation of library
Sixty parasite clones, each containing a unique DNA signature-tag, were insertionally mutagenized by electroporation with 10 μg of pLJK47 linearized with NotI (Knoll et al., 2001). Stable integration into the genome was enhanced by REMI with either NotI, DpnII or EcoRI (Black et al., 1995). Parasites were selected in 50 μg ml−1 mycophenolic acid and 50 μg ml−1 xanthine, and cloned by limiting dilution. Two or more separate electroporations were performed for each of the 60 signature-tagged clones and a minimum of 100 clones were isolated for each of the tagged strains. The parasites were maintained in HFFs in 96 well plates.
Screen for inability of T. gondii to inhibit macrophage NO
For the initial screen, an equal volume of parasites was passed from lysed monolayers of HFFs in 96 well plates into 96 well plates containing low-passage RAW cell monolayers. Parasites were allowed to infect RAW cells for 3 h, then fresh media with 100 ng ml−1 LPS (E. coli, Sigma Immunochemicals) and 100 U ml−1 IFN-γ (eBioscience, San Diego, CA) was added. IFN-γ and LPS were stored frozen in single use aliquots and LPS was sonicated immediately before use to ensure that the micelles were disrupted. After 24 h, 50 μl of supernatant was removed from each well and nitrite concentration was determined by Griess reaction (Mordue and Sibley, 2003). All reagents for the Griess reaction were obtained from Sigma Immunochemicals, St Louis, MO. Wild-type or mutant parasite suppression of NO was evaluated by comparing NO production from uninfected, naïve and stimulated macrophages. Mutants were selected that had nitrite levels similar to uninfected activated macrophages. Twenty-four mutants were selected and re-evaluated. In this screen, plaque assays in HFFs were performed simultaneously so that NO production could be equalized based on the number of viable input parasites. Four insertional mutants were evaluated in a final screen with twofold serial dilutions of wild-type or mutant parasites to generate a dose–response curve. Each experiment had duplicate wells and was repeated four times. To calculate the MOI for Fig. 8B, we determined the number of macrophages per well by simultaneously plating two extra wells of macrophages at the beginning of the experiment, then at the time of infection, we scraped the wells and counted the number of macrophages.
Acute and chronic mouse infections
For acute mouse infection experiments, two 9-week-old CBA/J mice received 4 × 105 I.P. of either 89B7 or wild-type Prugniaud strains. Plaque assays were performed immediately after the inoculation to confirm the number of viable parasites that were injected. When mice were moribund (severely hunched, not moving, and >20% weight loss) they were euthanized. For chronic infections, two 9-week-old CBA/J mice received 2 × 104 I.P. of either 89B7 or wild-type parasites, plaque assays were performed, and 22 days after inoculation (early chronic infection) the mice were sacrificed. Their brains were removed, ground with a mortar and pestle, fixed with 3.0% formaldehyde for 20 min, and permeabilized and blocked in 0.2% Triton X-100 with 3.0% bovine serum albumin for 30 min. Tissue cysts were stained with Fluorescein labelled Dolichos biflorus agglutinin (Vector Laboratories), and three 5 μl samples mounted and cyst numbers counted by fluorescence microscopy. All experiments were repeated twice.
Isolation of DNA flanking the insertion site
Genomic T. gondii DNA was prepared as previously described (Medina-Acosta and Cross, 1993) and digested with HindIII or NcoI restriction enzymes to cut once within the insertional plasmid and unknown sites in adjacent genomic DNA. After digestion, the DNA was ligated, transformed into GC10 Thunderbolt Electrocompetent bacteria (Gene Choice, Frederick, MD), and selected for kanamycin resistance. The DNA flanking the insertion site was sequenced and compared with the T. gondii genome database (http://ToxoDB.org).
TgPL1 mRNA analysis
Total RNA was isolated using Ultraspec according to the manufacturer’s instructions (BIOTECX Laboratories Inc.). cDNA was synthesized from wild-type Prugniaud parasites using random hexamers and SuperScript™ III (Invitrogen). The predicted intron splice junction was confirmed by PCR of cDNA with primers 5′-GCTCTGCCACATTCTCGTTC-3′ and 5′-TTGCCTCTTCGTCCATAGGC-3′. The 1.2 kb amplified product was cloned using pCR-TOPO (Invitrogen), and used as a probe for Northern and Southern analyses. The 5′ end of the mRNA was identified using 5′ RLM RACE (Ambion, Austin, TX) with nested gene specific primers 5′-TACAAACCCCGCAAACTGTCCCTC-3′ and 5′-AAACAAACCACTTCCCCGCTGC-3′. The 3′ end was identified using cDNA synthesized with a poly-T-anchor oligonucleotide and nested PCR using 5′-CCGGAATTCGGTACCTCTCAGA-3′ as the anchor sequence, and gene specific primers 5′-TAAGCAGTCGCCACTCCAAGAGAGG-3′ and 5′-TCTGCGGACCCTTTTGTTGT-3′.
Complementation of 89B7
A complementation construct was created containing the entire 6.75 kb TgPL1 genomic locus (including 2.4 kb upstream of the 5′ end of the mRNA and 400 bp downstream of the polyadenylation site). This was PCR amplified in two pieces using genomic DNA as the template, Accuprime Pfx polymerase (Invitrogen), and primers 5′-CAGCAGAAACGCAGATTATG-3′ and 5′-GGTAAGAGCCACAGTCCACAT-3′ for the first half and primers 5′-TGCTGTTCTCCTTCTGTATGG-3′ and 5′-TGGGGGCAAATGCTTATC-3′ for the second half. PCR products were cloned using pCR-TOPO (Invitrogen), digested with EcoRV (from pCR-TOPO) and SacI for the first half, and SacI and SpeI for the second half, then subcloned into EcoRV and SpeI digested pBC SK+ (Stratagene) with dihydrofolate reductase from DHFR-TS (Donald and Roos, 1993) blunted into the NotI site, creating pEndoTgPL1. The 89B7 mutant was transfected with 10 μg linearized pEndoTgPL1 and selected with 1 μM pyrimethamine. Parasites were cloned by limiting dilution and examined by Southern Blot. All clones had an identical band pattern by Southern, indicting that only one independent insertion into the genome occurred (Comp). For the selected complementation populations (Pop 1 and Pop 2, Fig. 8), two independent electroporations were performed with 50 μg linearized pEndoTgPL1 and selected with 1 μM pyrimethamine. Populations were analysed by Southern blot for independent insertion events into the genome.
HA epitope tag construct
The TgPL1 open reading frame was PCR amplified with a NsiI site at the presumed initiator methionine and an in frame HA-tag followed by a PacI site at stop codon using primers 5′-ATGCATGGTAACAGCGTGTCATTGCGA-3′ and 5′-TTAATTAAGCGTAGTCCGGGACGTCGTACGGGTAAGACTCTTCAGACTTTGCCTCCTCGTCC-3′. This 2 kb product was cloned using pCR-TOPO (Invitrogen), and subcloned into PacI digested, NsiI partially digested pT/230 (Soldati and Boothroyd, 1995) creating pTubTgPL1-HA. To create pEndoTgPL1-HA, the endogenous promoter was subcloned from the complementation construct pEndoTgPL1 by digestion with EcoRV and SacI, and ligated into pTubTgPL1-HA digested with HindIII (then blunted with Klenow), and partially digested SacI. Stable co-transfections were performed into wild-type Prugniaud parasites with 2 μg linearized DHFR-TS and either 20 μg linearized pTubTgPL1-HA or pEndoTgPL1-HA, and selected with 1 μM pyrimethamine. TgPL1-HA overexpressed from the pTubTgPL1-HA construct had a distinct ER location (colocalization with BiP and concentration around the nucleus), thus only the TgPL1-HA expressed from its endogenous promoter was used for the localization studies.
Immunofluorescence analysis
One-week-old bone marrow-derived macrophages were dislodged with cold calcium and magnesium-free PBS, and plated on glass coverslips at a concentration of 2 × 105 cells ml−1 to 50% confluency. After setting overnight, macrophages were infected with T. gondii parasites, which were allowed to invade for 3 h. They were then stimulated with LPS and IFN-γ as described above, or left unstimulated. At 12, 24 or 36 h after infection, cells were fixed and permeabilized as described above. Parasites were visualized using serum from mice chronically infected with T. gondii, followed by goat anti-mouse Alexa Fluor 488 (Molecular Probes, Seattle, WA). The number of parasites per vacuole and the number of degraded vacuoles were counted in approximately 25 random fields (at least 100 vacuoles total) using a Zeiss inverted Axiovert 200 microscope. The samples were coded to prevent counting bias. For analysis of GRA4, GRA7 and SAG1 expression, infected macrophages were fixed with 3% formaldehyde, and stained with rabbit antibodies to GRA7 or GRA4, mouse monoclonal antibodies to SAG1 (Clone TP3, Abcam, Cambridge, MA or clone P30/3 MorphoSys US Inc. Biogenesis, Kingston, NH), and a goat polyclonal antisera to T. gondii (Fitzgerald Industries International, Concord, MA). Secondary antibodies used for detection were Alexa Flour 594 anti-rabbit, Alexa Flour 488 anti-mouse and Alexa Flour 675 anti-goat (Molecular Probes). Samples were mounted using Vecta-Shield mounting media with DAPI (Vector Laboratories, Burlingame, CA). These IFA studies were visualized using a Zeiss inverted Axiovert 200 motorized microscope with a 100× objective (PlanApo 1.4 na oil PH3 objective), Zeiss filter sets 31, 34, 38 and 50 and Axiovision 4.3 software. Pictures were taken with the Zeiss Axiocam MRM cool CCD camera.
For the immunolocalization studies with intracellular parasites, confluent HFFs on glass coverslips were infected with TgPL1-HA expressed from its endogenous promoter containing parasites for approximately 24 h. For studies with extracellular parasites, lysed TgPL1-HA containing parasites were dried on the slide. For localization within naïve macrophages, murine bone marrow-derived macrophages were infected at a MOI of 0.5, allowed to grow for 24 h, then with 0.5 μg ml−1 of Mitotracker Red CMXRos (Molecular Probes) as described previously (Sinai et al., 1997). For localization within activated macrophages, bone marrow-derived macrophages were infected at a MOI of 0.5, and then IFN-γ and LPS were added as described above and grown for an additional 33 h. All samples were fixed in 3% formaldehyde. HA-tagged proteins were detected using an anti-HA antibody (71-5500 from Zymed or 16B12 from Covance) and colocalized with against MIC2, ROP 2.3.4., GRA4, VP1 and BiP. Specific antibody staining was visualized with the appropriate Alexa Fluor 488- and Alexa Fluor 633-conjugated goat anti-IgG secondary reagents (Molecular Probes). Samples were mounted using VectaShield mounting media with DAPI (Vector Laboratories). Serial image stacks (0.2 micron Z-increment) were collected at 100× (PlanApo oil immersion 1.4 na) on a motorized Zeiss Axioplan IIi equipped with a rear-mounted excitation filter wheel, a triple pass (DAPI/FITC/Texas Red) emission cube, differential interference contrast optics, and a Hamamatsu ORCA-AG CCD camera. Fluorescence images were deconvolved by a constrained iterative algorithm, pseudocoloured, and merged using OpenLabs 4.0 software (Improvision, Lexington, MA).
Electron microscopy
For ultrastructural analysis, infected murine bone marrow-derived macrophages were activated after 3 h as described above and allowed to grow for an additional 33 h. At that time, they were fixed in 4% paraformaldehyde/0.01% glutaraldehyde in 200 mM phosphate buffer pH 7.2 for 40 min at 24°C. Samples were then embedded in 10% gelatin and infiltrated overnight with 2.3 M sucrose/20% polyvinylpyrrolidone in PIPES/MgCl2 at 4°C. Samples were trimmed, frozen in liquid nitrogen, and sectioned with a Leica Ultracut UCT cryoultramicrotome (Leica Microsystems, Bannockburn, IL). 70 nm sections were blocked with 5% FBS/5% NGS for 30 min and subsequently incubated with rabbit anti-GRA7 antibody (1:2000) for 1 h at room temperature. Sections were then washed in block buffer and probed with 18 nm colloidal gold-conjugated anti-rabbit IgG (1:30) (Jackson ImmunoResearch Laboratories, West Grove, PA) for 1 h. Sections were washed in PIPES buffer followed by an extensive water rinse, and stained with 0.3% uranyl acetate/1.7% methyl cellulose. Samples were viewed with a JEOL 1200EX transmission electron microscope (JEOL USA, Peabody, MA). All labelling experiments were conducted in parallel with controls omitting the primary antibody. The scale bar is equal to 500 nm on all images.
Acknowledgments
We sincerely thank David Sibley, Peter Bradley, Jean Francois Dubremetz, Jay Bangs, Silva Moreno and Isabelle Coppens for generously supplying antibodies; David Roos for the DHFR-TS plasmid; Ned Ruby for helpful discussions; and Jay Bangs for the use of the Zeiss Axioplan IIi. We especially thank Wandy Beatty for the expert electron microscopy studies. This research was supported by the Burroughs Wellcome Fund Career Award 992908 (L.J.K.), National Institutes of Health (NIH), Award A1054603 (L.J.K.), and NIH National Research Service Award GM072125 (C.M.T.).
References
- Adams LB, Hibbs JB, Jr, Taintor RR, Krahenbuhl JL. Microbiostatic effect of murine-activated macrophages for Toxoplasma gondii. Role for synthesis of inorganic nitrogen oxides from 1-arginine. J Immunol. 1990;144:2725–2729. [PubMed] [Google Scholar]
- Andrade RM, Portillo JA, Wessendarp M, Subauste CS. CD40 signaling in macrophages induces activity against an intracellular pathogen independently of gamma interferon and reactive nitrogen intermediates. Infect Immun. 2005;73:3115–3123. doi: 10.1128/IAI.73.5.3115-3123.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andrade RM, Wessendarp M, Gubbels MJ, Striepen B, Subauste CS. CD40 induces macrophage anti-Toxoplasma gondii activity by triggering autophagy-dependent fusion of pathogen-containing vacuoles and lysosomes. J Clin Invest. 2006;116:2366–2377. doi: 10.1172/JCI28796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Banerji S, Flieger A. Patatin-like proteins: a new family of lipolytic enzymes present in bacteria? Microbiology. 2004;150:522–525. doi: 10.1099/mic.0.26957-0. [DOI] [PubMed] [Google Scholar]
- Bingisser RM, Tilbrook PA, Holt PG, Kees UR. Macrophage-derived nitric oxide regulates T cell activation via reversible disruption of the Jak3/STAT5 signaling pathway. J Immunol. 1998;160:5729–5734. [PubMed] [Google Scholar]
- Black M, Seeber F, Soldati D, Kim K, Boothroyd JC. Restriction enzyme-mediated integration elevates transformation frequency and enables co-transfection of Toxoplasma gondii. Mol Biochem Parasitol. 1995;74:55–63. doi: 10.1016/0166-6851(95)02483-2. [DOI] [PubMed] [Google Scholar]
- Bohne W, Heesemann J, Gross U. Induction of bradyzoite-specific Toxoplasma gondii antigens in gamma interferon-treated mouse macrophages. Infect Immun. 1993;61:1141–1145. doi: 10.1128/iai.61.3.1141-1145.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bohne W, Heesemann J, Gross U. Reduced replication of Toxoplasma gondii is necessary for induction of bradyzoite-specific antigens: a possible role for nitric oxide in triggering stage conversion. Infect Immun. 1994;62:1761–1767. doi: 10.1128/iai.62.5.1761-1767.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Courret N, Darche S, Sonigo P, Milon G, Buzoni-Gatel D, Tardieux I. CD11c and CD11b expressing mouse leukocytes transport single Toxoplasma gondii tachyzoites to the brain. Blood. 2006;107:309–316. doi: 10.1182/blood-2005-02-0666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Denkers EY, Butcher BA. Sabotage and exploitation in macrophages parasitized by intracellular protozoans. Trends Parasitol. 2005;21:35–41. doi: 10.1016/j.pt.2004.10.004. [DOI] [PubMed] [Google Scholar]
- Denkers EY, Kim L, Butcher BA. In the belly of the beast: subversion of macrophage proinflammatory signalling cascades during Toxoplasma gondii infection. Cell Microbiol. 2003;5:75–83. doi: 10.1046/j.1462-5822.2003.00258.x. [DOI] [PubMed] [Google Scholar]
- Dessen A, Tang J, Schmidt H, Stahl M, Clark JD, Seehra J, Somers WS. Crystal structure of human cytosolic phospholipase A2 reveals a novel topology and catalytic mechanism. Cell. 1999;97:349–360. doi: 10.1016/s0092-8674(00)80744-8. [DOI] [PubMed] [Google Scholar]
- Donald RG, Roos DS. Stable molecular transformation of Toxoplasma gondii: a selectable dihydrofolate reductase-thymidylate synthase marker based on drug-resistance mutations in malaria. Proc Natl Acad Sci USA. 1993;90:11703–11707. doi: 10.1073/pnas.90.24.11703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Griess P. Bemerkungen zu der abhandlung der H.H. Weselsky und Benedikt ‘Ueber einige azoverbindungen’. Chem Ber. 1879;12:426–428. [Google Scholar]
- Hayashi S, Chan CC, Gazzinelli RT, Pham NT, Cheung MK, Roberge FG. Protective role of nitric oxide in ocular toxoplasmosis. Br J Ophthalmol. 1996;80:644–648. doi: 10.1136/bjo.80.7.644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hirschberg HJ, Simons JW, Dekker N, Egmond MR. Cloning, expression, purification and characterization of patatin, a novel phospholipase A. Eur J Biochem. 2001;268:5037–5044. doi: 10.1046/j.0014-2956.2001.02411.x. [DOI] [PubMed] [Google Scholar]
- Joiner KA, Roos DS. Secretory traffic in the eukaryotic parasite Toxoplasma gondii: less is more. J Cell Biol. 2002;157:557–563. doi: 10.1083/jcb.200112144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khan IA, Schwartzman JD, Matsuura T, Kasper LH. A dichotomous role for nitric oxide during acute Toxoplasma gondii infection in mice. Proc Natl Acad Sci USA. 1997;94:13955–13960. doi: 10.1073/pnas.94.25.13955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knoll LJ, Furie GL, Boothroyd JC. Adaptation of signature-tagged mutagenesis for Toxoplasma gondii: a negative screening strategy to isolate genes that are essential in restrictive growth conditions. Mol Biochem Parasitol. 2001;116:11–16. doi: 10.1016/s0166-6851(01)00295-x. [DOI] [PubMed] [Google Scholar]
- Kwok LY, Schluter D, Clayton C, Soldati D. The antioxidant systems in Toxoplasma gondii and the role of cytosolic catalase in defence against oxidative injury. Mol Microbiol. 2004;51:47–61. doi: 10.1046/j.1365-2958.2003.03823.x. [DOI] [PubMed] [Google Scholar]
- Ling YM, Shaw MH, Ayala C, Coppens I, Taylor GA, Ferguson DJ, Yap GS. Vacuolar and plasma membrane stripping and autophagic elimination of Toxoplasma gondii in primed effector macrophages. J Exp Med. 2006;203:2063–2071. doi: 10.1084/jem.20061318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luder CG, Algner M, Lang C, Bleicher N, Gross U. Reduced expression of the inducible nitric oxide synthase after infection with Toxoplasma gondii facilitates parasite replication in activated murine macrophages. Int J Parasitol. 2003;33:833–844. doi: 10.1016/s0020-7519(03)00092-4. [DOI] [PubMed] [Google Scholar]
- Luo S, Vieira M, Graves J, Zhong L, Moreno SN. A plasma membrane-type Ca(2+)-ATPase co-localizes with a vacuolar H(+)-pyrophosphatase to acidocalcisomes of Toxoplasma gondii. EMBO J. 2001;20:55–64. doi: 10.1093/emboj/20.1.55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McKee AS, Dzierszinski F, Boes M, Roos DS, Pearce EJ. Functional inactivation of immature dendritic cells by the intracellular parasite Toxoplasma gondii. J Immunol. 2004;173:2632–2640. doi: 10.4049/jimmunol.173.4.2632. [DOI] [PubMed] [Google Scholar]
- Medina-Acosta E, Cross GA. Rapid isolation of DNA from trypanosomatid protozoa using a simple ‘miniprep’ procedure. Mol Biochem Parasitol. 1993;59:327–329. doi: 10.1016/0166-6851(93)90231-l. [DOI] [PubMed] [Google Scholar]
- Mignery GA, Pikaard CS, Park WD. Molecular characterization of the patatin multigene family of potato. Gene. 1988;62:27–44. doi: 10.1016/0378-1119(88)90577-x. [DOI] [PubMed] [Google Scholar]
- Mordue DG, Sibley LD. Intracellular fate of vacuoles containing Toxoplasma gondii is determined at the time of formation and depends on the mechanism of entry. J Immunol. 1997;159:4452–4459. [PubMed] [Google Scholar]
- Mordue DG, Sibley LD. A novel population of Gr-1+-activated macrophages induced during acute toxoplasmosis. J Leukoc Biol. 2003;74:1015–1025. doi: 10.1189/jlb.0403164. [DOI] [PubMed] [Google Scholar]
- Murray HW, Teitelbaum RF. 1-arginine-dependent reactive nitrogen intermediates and the anti-microbial effect of activated human mononuclear phagocytes. J Infect Dis. 1992;165:513–517. doi: 10.1093/infdis/165.3.513. [DOI] [PubMed] [Google Scholar]
- Nash PB, Purner MB, Leon RP, Clarke P, Duke RC, Curiel TJ. Toxoplasma gondii-infected cells are resistant to multiple inducers of apoptosis. J Immunol. 1998;160:1824–1830. [PubMed] [Google Scholar]
- Rabin SD, Hauser AR. Functional regions of the Pseudomonas aeruginosa cytotoxin ExoU. Infect Immun. 2005;73:573–582. doi: 10.1128/IAI.73.1.573-582.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roberts F, Roberts CW, Ferguson DJ, McLeod R. Inhibition of nitric oxide production exacerbates chronic ocular toxoplasmosis. Parasite Immunol. 2000;22:1–5. doi: 10.1046/j.1365-3024.2000.00259.x. [DOI] [PubMed] [Google Scholar]
- Robinson DG, Oliviusson P, Hinz G. Protein sorting to the storage vacuoles of plants: a critical appraisal. Traffic. 2005;6:615–625. doi: 10.1111/j.1600-0854.2005.00303.x. [DOI] [PubMed] [Google Scholar]
- Sato H, Frank DW. ExoU is a potent intracellular phospholipase. Mol Microbiol. 2004;53:1279–1290. doi: 10.1111/j.1365-2958.2004.04194.x. [DOI] [PubMed] [Google Scholar]
- Sato H, Frank DW, Hillard CJ, Feix JB, Pankhaniya RR, Moriyama K, et al. The mechanism of action of the Pseudomonas aeruginosa-encoded type III cytotoxin, ExoU. EMBO J. 2003;22:2959–2969. doi: 10.1093/emboj/cdg290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scharton-Kersten TM, Yap G, Magram J, Sher A. Inducible nitric oxide is essential for host control of persistent but not acute infection with the intracellular pathogen Toxoplasma gondii. J Exp Med. 1997;185:1261–1273. doi: 10.1084/jem.185.7.1261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schluter D, ckert-Schluter M, Lorenz E, Meyer T, Rollinghoff M, Bogdan C. Inhibition of inducible nitric oxide synthase exacerbates chronic cerebral toxoplasmosis in Toxoplasma gondii-susceptible C57BL/6 mice but does not reactivate the latent disease in T. gondii-resistant BALB/c mice. J Immunol. 1999;162:3512–3518. [PubMed] [Google Scholar]
- Seabra SH, de Souza W, Damatta RA. Toxoplasma gondii partially inhibits nitric oxide production of activated murine macrophages. Exp Parasitol. 2002;100:62–70. doi: 10.1006/expr.2001.4675. [DOI] [PubMed] [Google Scholar]
- Senda K, Yoshioka H, Doke N, Kawakita K. A cytosolic phospholipase A2 from potato tissues appears to be patatin. Plant Cell Physiol. 1996;37:347–353. doi: 10.1093/oxfordjournals.pcp.a028952. [DOI] [PubMed] [Google Scholar]
- Shewry PR. Tuber storage proteins. Ann Bot (Lond) 2003;91:755–769. doi: 10.1093/aob/mcg084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shohdy N, Efe JA, Emr SD, Shuman HA. Pathogen effector protein screening in yeast identifies Legionella factors that interfere with membrane trafficking. Proc Natl Acad Sci USA. 2005;102:4866–4871. doi: 10.1073/pnas.0501315102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sibley LD, Weidner E, Krahenbuhl JL. Phagosome acidification blocked by intracellular Toxoplasma gondii. Nature. 1985;315:416–419. doi: 10.1038/315416a0. [DOI] [PubMed] [Google Scholar]
- Sibley LD, Adams LB, Fukutomi Y, Krahenbuhl JL. Tumor necrosis factor-alpha triggers antitoxoplasmal activity of IFN-gamma primed macrophages. J Immunol. 1991;147:2340–2345. [PubMed] [Google Scholar]
- Sinai AP, Webster P, Joiner KA. Association of host cell endoplasmic reticulum and mitochondria with the Toxoplasma gondii parasitophorous vacuole membrane: a high affinity interaction. J Cell Sci. 1997;110(Part 17):2117–2128. doi: 10.1242/jcs.110.17.2117. [DOI] [PubMed] [Google Scholar]
- Soldati D, Boothroyd JC. A selector of transcription initiation in the protozoan parasite Toxoplasma gondii. Mol Cell Biol. 1995;15:87–93. doi: 10.1128/mcb.15.1.87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spear W, Chan D, Coppens I, Johnson RS, Giaccia A, Blader IJ. The host cell transcription factor hypoxia-inducible factor 1 is required for Toxoplasma gondii growth and survival at physiological oxygen levels. Cell Microbiol. 2000;8:339–352. doi: 10.1111/j.1462-5822.2005.00628.x. [DOI] [PubMed] [Google Scholar]