

Abstract
Several causative genes for hereditary spastic paraplegia encode proteins with intramembrane hairpin loops that contribute to the curvature of the endoplasmic reticulum (ER), but the relevance of this function to axonal degeneration is not understood. One of these genes is reticulon2. In contrast to mammals, Drosophila has only one widely expressed reticulon orthologue, Rtnl1, and we therefore used Drosophila to test its importance for ER organization and axonal function. Rtnl1 distribution overlapped with that of the ER, but in contrast to the rough ER, was enriched in axons. The loss of Rtnl1 led to the expansion of the rough or sheet ER in larval epidermis and elevated levels of ER stress. It also caused abnormalities specifically within distal portions of longer motor axons and in their presynaptic terminals, including disruption of the smooth ER (SER), the microtubule cytoskeleton and mitochondria. In contrast, proximal axon portions appeared unaffected. Our results provide direct evidence for reticulon function in the organization of the SER in distal longer axons, and support a model in which spastic paraplegia can be caused by impairment of axonal the SER. Our data provide a route to further understanding of both the role of the SER in axons and the pathological consequences of the impairment of this compartment.
INTRODUCTION
Hereditary spastic paraplegias (HSPs) are a group of neurological disorders characterized by retrograde degeneration of long nerve fibres in the corticospinal tracts and posterior columns, sometimes accompanied by additional, mainly neurological symptoms (1,2). The disease mechanisms are largely unknown, but since distal regions of longer axons appear to be worst affected, the disease may reflect problems in trafficking cell components between the cell body and distal axons that can be up to a metre away. Some clues about disease mechanisms come from the identification of over 20 causative HSP genes (SPGs, spastic paraplegia genes) (3). These encode a heterogeneous group of proteins, but the largest single class are intracellular membrane proteins, principally the endosomal or endoplasmic reticulum (ER). Functions ascribed to these proteins (not mutually exclusive) include the inhibition of BMP signalling (4–6), formation of lipid droplets (7–9) or regulation of ER topology (10–13).
At least four autosomal dominant HSPs are caused by mutations in proteins that have a common feature of ER localization, and an intramembrane hairpin loop that can induce or sense the curvature of ER membranes and form oligomeric complexes among themselves and each other (10–15). These hairpin-loop proteins, SPG3A/atlastin1, SPG4/spastin, SPG12/reticulon2 (RTN2) and SPG31/REEP1, contribute to ER topology in a number of ways. Reticulon and REEP proteins share a partly redundant role in the formation of tubular ER, and in the induction of the curvature at the edges of the sheet ER (10,16). ER tubule elongation is proposed to involve both REEP proteins and the microtubule (MT)-severing activity of spastin, which could potentially nucleate new MT elongation and accompanying tubule extension (13,17–19). The GTPase Atlastin1 is thought to mediate the membrane fusion events that maintain the reticular organization of the ER (12,14,20).
Little protein synthesis occurs in axons (21), and consequently they contain very little rough ER (RER) (22); it is at first sight paradoxical that mutations in ER-modelling proteins could be causative for axonal degeneration. However, axons and presynaptic terminals contain the smooth ER (SER), based on ultrastructural evidence and the presence of calcium homeostasis machinery (22–26). Since smooth and tubular ER are broadly equivalent (16), this could explain why axons are sensitive to mutations in proteins that model tubular ER.
One of the major ER-tubulating protein classes is the reticulon family, one of whose members, RTN2, was recently identified as an SPG gene product (15). Here, we used Drosophila to test the effects on the ER and axons of the loss of reticulon function. Drosophila has a single widely expressed reticulon, reticulon1 (Rtnl1), that is an orthologue of reticulons 1–4 in humans. We show that Rtnl1 is required for ER network organization, and that its loss induces an ER stress response. Furthermore, the loss of Rtnl1 leads to abnormalities of an SER marker, the MT cytoskeleton and mitochondria, in the distal axons or presynaptic termini of longer motor axons. Our findings reveal increased susceptibility of posterior axons to the disruption of ER organization and suggest a mechanism by which the loss of hairpin loop proteins gives rise to axonopathy.
RESULTS
Loss of Rtnl1, the Drosophila orthologue of vertebrate reticulons 1–4, causes age-related locomotor deficits
Drosophila has two reticulons, Rtnl1 and Rtnl2, that are orthologous to the four reticulons found in mammals (Fig. 1A) (27,28). We focused on Rtnl1 as most relevant to the function of its mammalian orthologues. Rtnl1 is widely expressed, whereas Rtnl2 expression is restricted to testis and fat body (www.flyatlas.org); moreover, BLASTP searches (not shown) show that Rtnl1 is evolving more slowly than Rtnl2, suggesting that its function is better conserved.
Figure 1.

Loss of Drosophila reticulon causes a progressive locomotor deficit. (A) Dendrogram showing similarity of human and Drosophila reticulon proteins, drawn from a Clustal alignment using the neighbour-joining algorithm in MEGA 5.05. Dashed line indicates fast divergence of Drosophila Rtnl2. (B) The Rtnl1 locus showing proteins encoded by transcripts A-R (Flybase Genome Browser R5.41), location of VDRC RNAi fragments (RNAi) and the Rtnl1::YFP insertion (CPTI00291). (C–E) Confocal sections showing localization of Rtnl1::YFP in epidermal cells overlapping with KDEL (arrowheads in inset; C), in the larval ventral nerve cord (D) where it localizes preferentially in neuropil (arrow) and nerve bundles (arrowheads) in contrast to KDEL, which is concentrated within cell bodies (boxes), and in NMJ presynaptic boutons (E), where it follows the path of the axonal MT marker Futsch (upper panel). (F) VDRC RNAi line (construct GD900) successfully knocks down Rtnl1 expression as observed by PCR amplification of Rp49 and Rtnl1 cDNA from progeny of da-GAL4 crossed to either w1118 (control) or Rtnl1 RNAi flies. (G and H) Loss of Rtnl1 causes an age-dependent climbing deficit. Graphs represent end-to-end distance travelled in 15 s (G) and climbing velocity (H) of Rtnl1 RNAi and control flies (mean ± SEM here and in all subsequent graphs; *P < 0.05, **P < 0.01, ***P < 0.005, otherwise P > 0.05; two-way ANOVA and Bonferroni post-tests; n = 3 independent experiments).
An Rtnl1 exon trap insertion tagged with yellow fluorescent protein (YFP) (Fig. 1B) showed widespread expression in third-instar larvae, with the highest levels observed in neural tissues. Its subcellular localization was seen most clearly in epidermal cells, where it had a reticular distribution, overlapping with labelling of the ER retention signal KDEL (Fig. 1C). In contrast, in the CNS, relatively low levels of Rtnl1::YFP were found in cell bodies where most KDEL labelling is found (Fig. 1D); much higher levels were found in the neuropil (containing axons, dendrites and synapses) and in the nerves that connect the CNS to the periphery, which include motor neuron axons. In motor neuron terminals at the neuromuscular junction (NMJ), Rtnl1::YFP followed the distribution of the axonal MT marker Futsch (homologous to mammalian MAP1B), consistent with the established close association of tubular ER and MTs (Fig. 1E) (13,29). Therefore, Rtnl1::YFP overlaps with the ER, but its distinct distribution in neurons suggests that a smooth tubular ER compartment, marked by Rtnl1::YFP, is found preferentially in axons rather than in cell bodies.
To detect whether the loss of Rtnl1 had any overt phenotypic consequences, we tested for any obvious effects of ubiquitous knockdown of Rtnl1, using an RNAi insertion expressed under the control of da-Gal4, which resulted in almost complete depletion of Rtnl1 (Fig. 1F). Rtnl1 knockdown flies were viable, and an automated three-dimensional tracking system that measured the climbing behaviour of these flies in a negative geotaxis assay (30) showed that they had normal locomotor activity as early adults (Fig. 1G and H). However, as adults aged, they showed a progressive loss of locomotor activity relative to controls (Fig. 1G and H), consistent with a degenerative phenotype caused by Rtnl1 loss.
Rtnl1 is required for normal ER organization and function
In vitro studies have shown that reticulon proteins are required for tubular ER formation and maintenance (10,16). We therefore investigated the effects of Rtnl1 knockdown on ER organization in vivo. The loss of Rtnl1 made the perinuclear pattern of KDEL labelling in epidermal cells more diffuse than the reticular pattern seen in controls, but did not detectably change the overall levels of KDEL labelling (Fig. 2A). In contrast, the distribution of the Golgi marker GM130 was not detectably changed in Rtnl1 knockdown epidermal cells (Fig. 2A). Ultrastructural analysis revealed that the diffuse KDEL labelling in epidermal cells of Rtnl1 knockdown larvae was caused by a striking reorganization of the ER, including a 3-fold increase in the average length of sheet ER profiles, and denser packing of the ribosomal studded sheet ER (Fig. 2B). This could result either from the loss of the curved edges of the sheet ER, to which reticulons contribute (16), or from the conversion of highly curved tubular ER to the sheet ER.
Figure 2.

Loss of Rtnl1 causes expansion of ER sheets and increases the ER stress response. (A) KDEL distribution in third-instar larval epidermal cells is reticular in control larvae but more diffuse in Rtnl1 RNAi larvae; distribution of the Golgi marker GM130 is broadly unaffected. Graph shows KDEL staining intensity per cell as a percentage of control levels (n= 4 independent experiments; ns, not significant, P > 0.7). (B) Electron micrographs show increased length of ER sheets in Rtnl1 RNAi epidermal cells compared with controls. Arrowheads, ER sheets; Nuc, nucleus; M, mitochondrion. The graph shows the quantification of ER sheet cross-sectional length (***P < 0.005; n = 3 independent larvae). (C and D) Single-confocal sections of larval epidermal cells (C) and ventral nerve cord (D) show increased ER stress, measured by increased Xbp1::GFP expression in Rtnl1 RNAi larvae compared with controls and quantified in graphs (**P < 0.01, ***P < 0.005; n = 12–16).
The main signalling mechanism controlling ER homeostasis, including the size and the shape, is the ER unfolded protein response (31). This induces expression of a large number of genes that encode components of the ER-resident folding machinery, stimulate lipid biosynthesis and enlarge the ER (32). We therefore tested whether the expansion of ER sheets observed in Rtnl1 knockdown larvae coincided with an ER stress response, using the ER stress reporter Xbp1::enhanced green fluorescent protein (EGFP) in which EGFP is expressed in frame with Xbp1 only after ER-stress activated splicing occurs (33). Rtnl1 knockdown significantly increased the ER stress response compared with controls, both in epidermal cells and neurons (Fig. 2C and D).
Loss of Rtnl1 leads to defects in longer motor axons
Given that spastic paraplegia mutations lead to axonal degeneration, we wanted to test for tubular ER defects in axons of Rtnl1 knockdown larvae. Since most commonly used ER markers are associated with protein synthesis, traffic or folding, which occurs mainly in the RER in neuronal cell bodies, we set out to identify markers of the SER by identification of Rtnl1::YFP interactors, using mass-spectroscopy, which our earlier findings suggested localized to tubular ER compartments in axons. The highest probability Rtnl1::YFP interactor with predicted ER localization for which an antibody exists was Acyl-CoA synthestase (Acsl), the orthologue of human acyl-CoA synthetases 3 and 4 (34). In primary cultured mammalian neurons, Acsl4 localizes preferentially to tubular ER and is present in axons and dendrites (35,36). Drosophila Acsl staining, which follows long linear profiles within motor axon bundles, shows similar and overlapping localization to Rtnl1 in axons and epidermal cells (Fig. 3A and B). We confirmed by co-immunoprecipitation that Rtnl1 interacts with Acsl (Fig. 3C) and found that Rtnl1 knockdown reduced Acsl protein levels compared with control larvae (Fig. 3D and E). Moreover, Acsl staining is selectively reduced in distal portions of the longer posterior abdominal motor axons, but not in the proximal portions of these axons (Fig. 3F).
Figure 3.

Rtnl1 interacts with the Acyl-CoA synthetase Acsl. Confocal sections showing overlapping localization of Rtnl1::YFP and Acsl in axons (linear staining indicated with arrowheads; A) and epidermal cells (B). (C) Protein lysates from OregonR (control) and Rtnl1::YFP flies were immunoprecipitated with Strep-Tactin Sepharose. Whole lysate (In), unbound (UB) and bound (Out) fractions were analysed by western blot. Loss of Rtnl1 reduces endogenous Acsl protein levels relative to controls in immunoblots of adult lysates (D) and single-confocal sections of larval epidermal cells (E). (F) Anti-Acsl staining of OK6GAL4;UAS-Acsl::myc motor neurons labels structures running along axons (arrowheads). Acsl staining is significantly lowered in posterior but not anterior segments of long motor axons upon knockdown of Rtnl1 (F). Graphs represent Acsl expression levels (ns, not significant, P > 0.2; **P < 0.01; n = 6–8 lysates in D, n = 14–16 larvae in E and F).
Since Rtnl1::YFP also overlapped strongly with the MT marker Futsch in motor neuron terminals (Fig. 1F), we tested for effects of Rtnl1 knockdown on the axonal cytoskeleton. Rtnl1::YFP could be co-immunoprecipitated with Futsch, suggesting that these proteins might interact directly or in a multiprotein complex (Fig. 4A). In Rtnl1 knockdown larvae, overall levels of Futsch were approximately a third lower than in controls (Fig. 4B), and Futsch was preferentially lost from the distal portions of long motor axons in the posterior abdomen (Fig. 4C). Interestingly, Acsl mutants also lose Futsch staining from posterior NMJs and display a distal bias in axonal transport defects (37). Taken together, this could reflect an increased sensitivity of distal axons for tubular ER defects.
Figure 4.

Rtnl1 interacts with the MT-associated protein Futsch. (A) Protein lysates from OregonR (control) and Rtnl1::YFP flies were immunoprecipitated and blotted as in Figure 3A. Loss of Rtnl1 reduces Futsch protein levels relative to controls in immunoblots of adult lysates (B) and in posterior but not anterior segments of long motor axons (C). Graphs represent Futsch expression levels (ns, not significant, P > 0.44; *P < 0.05, ***P < 0.001; n = 6–8 lysates in B, n = 16–19 larvae in C).
Loss of Rtnl1 causes disrupted mitochondrial organization in posterior NMJs
Since ER forms close contacts with mitochondria (38) and mutations in mitochondrial proteins may also cause HSP (39–41) as do RTN2 mutations (15), we also examined whether Rtnl1 knockdown affected axonal or presynaptic mitochondria. Rtnl1 knockdown did not cause a gross axon transport defect, as judged by axonal levels of the synaptic vesicle marker Csp and by numbers of mitochondria in axons (Fig. 5A). Rtnl1 knockdown also had no effect on mitochondrial number or size in anterior abdominal NMJ boutons at muscles 6/7 (Fig. 5B). However, NMJ boutons at muscles 6/7 in the posterior abdomen contained fewer mitochondria, of greater size, in Rtnl1 knockdown larvae than in controls (Fig. 5C). This suggests that the loss of Rtnl1 disrupts the localization or the fission/fusion balance of mitochondria in boutons of longer motor neurons.
Figure 5.
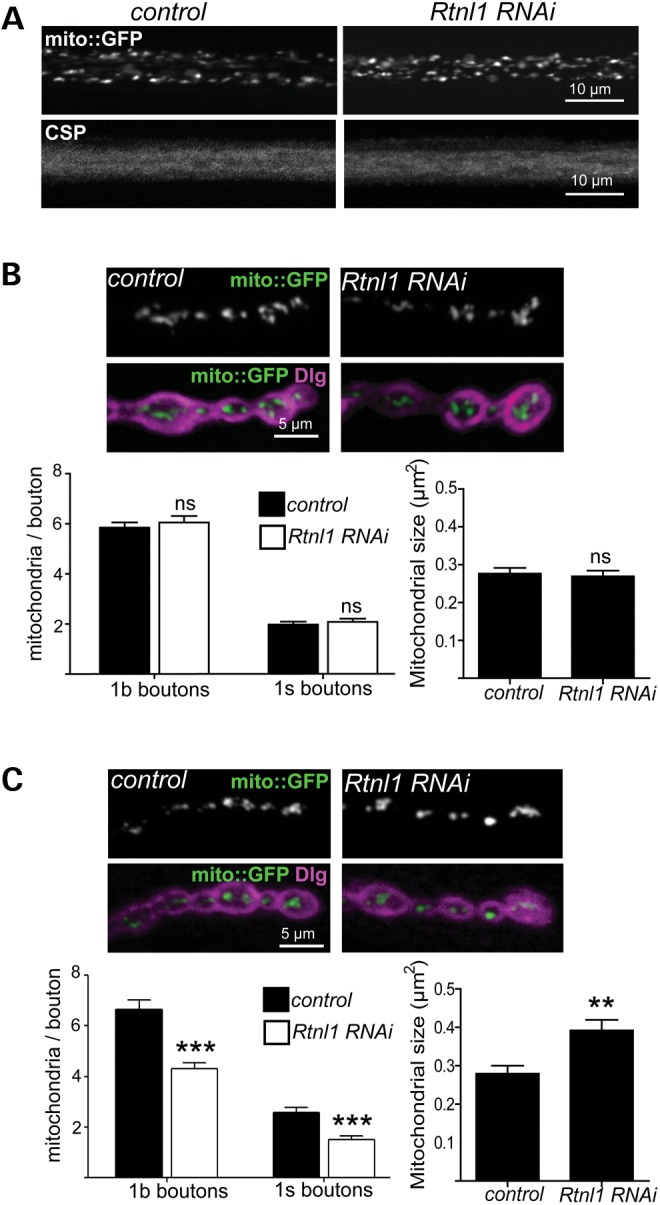
Effects of Rtnl1 loss on motor neuron mitochondria. (A) Single-confocal sections showing no effect of Rtnl1 RNAi (generated using OK6-GAL4 in this figure) on mito::GFP and CSP in posterior larval motor axons. (B and C) Single-confocal sections showing mito::GFP (green) and Dlg (magenta) at 1b boutons of anterior (B) and posterior (C) abdominal NMJs from control and Rtnl1 RNAi larvae. Graphs show the number (in 1b and 1s boutons) and the size (in 1b boutons) of mitochondria in posterior NMJs (ns, not significant; P > 0.5, **P < 0.01, ***P < 0.0005; n = 18–22 NMJs for 1b boutons, n = 12 NMJs for 1s boutons).
DISCUSSION
The hairpin loop protein reticulon, along with REEP, atlastin and spastin family members, is required for ER network organization. The disruption of the ER network, brought about by mutations affecting any one of these families, results in the degeneration of long axons as observed in HSP. However, how mutations in these proteins can specifically target axonal degeneration is not understood. We describe a novel Drosophila model of HSP caused by loss of the orthologue of SPG12, Rtnl1. This is the first animal model providing evidence of an ER phenotype due to the loss of reticulon and shows that major rearrangements of ER morphology do not noticeably affect organismal survival. These findings are consistent with previous studies investigating reticulon function both in the yeast Saccharomyces cervecisiae and in the plant Arabidopsis thaliana (10,16,42,43). Importantly, our model also allows the investigation of the role of tubular ER in axons; the loss of Rtnl1 selectively effects longer axons, suggesting a mechanistic similarity between the cellular phenotypes of Drosophila Rtnl1 knockdown and of spastic paraplegias that are caused by haplo-insufficient loss-of-function or dominant negative alleles of hairpin-loop proteins (15).
Although the disruption of SER functions, particularly in axons, is a prime candidate step to link the dysfunction of hairpin-loop HSP gene products to axon degeneration, studying the SER within axons has been challenging. In mature neurons, commonly used ER markers, including antibodies against KDEL (Fig. 1D) and PDI (21), localize preferentially to cell bodies rather than axons, likely reflecting the roles of these proteins in the protein biosynthetic region of the RER. However, Rtnl1::YFP and the tubular ER protein Acsl were highly expressed in mature motor axons. Here, they follow the distribution of axonal MT markers, reflecting the association of the ER with MTs (13,29). This association is required for the regulation of ER tubule dynamics via several mechanisms, the most common of which is ER sliding, whereby ER tubules bind to the shaft of an existing MT and slide along it as they grow (Fig. 6A) (38). Since the loss of Rtnl1 leads to partial loss of the tubular ER marker Acsl specifically from the distal portions of longer motor axons, the loss or impairment of SER function in longer axons could underlie the axonal pathology of spastic paraplegia (Fig. 6B). We suggest that there may be only incomplete loss of tubular ER in Rtnl1 knockdown axons due to partial functional redundancy between reticulon and other hairpin-loop family members, as occur in yeast (10).
Figure 6.
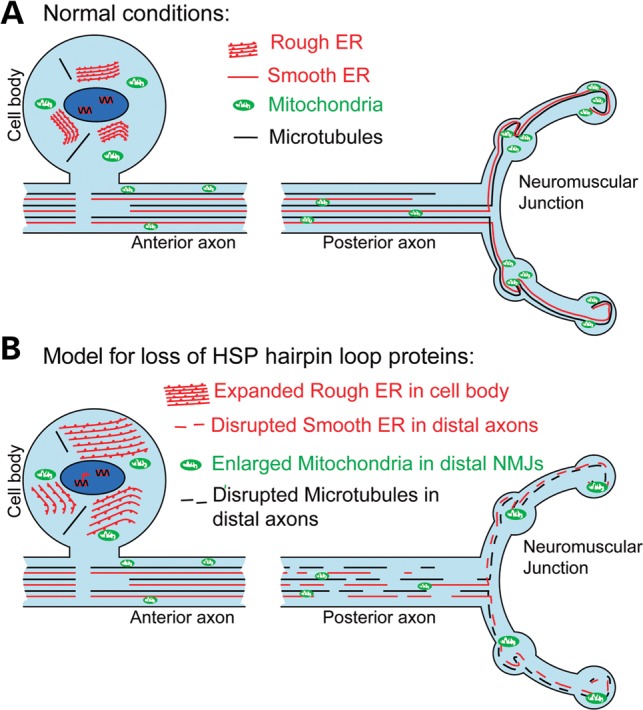
Model of hairpin loop protein dysfunction in motor neuron axonopathy. (A) The schematic diagram represents motor neurons under ‘normal’ or non-diseased conditions. Ribosomal studded RER sheets predominate within the neuronal cell body, whereas tubular SER runs the length of the axon closely associated with the MT cytoskeleton (13,21,29). In addition, the ER has extensive contact points with mitochondria which are required for lipid biosynthesis, calcium homeostasis and mitochondrial division (38,46). (B) Mutation or depletion of any one of the hairpin loop proteins causes disruption of the ER network (either less extensive and/or discontinuous) and degeneration of long motor axons as observed in HSP. Our findings have revealed an increased susceptibility of posterior axons to the disruption of ER organization, resulting in the disruption of tubular SER, MT cytoskeleton and mitochondria. This suggests that the loss of tubular ER from long motor axons may be the mechanism by which the loss of hairpin loop protein function gives rise to motor neuron axonopathy.
Mitochondria and the ER have extensive contact points that are crucial for calcium homoeostasis and lipid biosynthesis (38). These contact points are mediated in part by the mitochondrial fusion protein Mfn2 and are stably maintained during ER sliding along MTs (44,45). The loss of Rtnl1 from motor neurons resulted in fewer, larger mitochondria within boutons of posterior, but not anterior, NMJs (Fig. 5). ER tubules appear to play a role in defining the position of mitochondrial division sites (46); therefore, mitochondrial fission at terminals of the longer motor axons could be more sensitive to ER tubule disruption, and the reduction in mitochondria in posterior axons could thus be a consequence of ER defects caused by loss of Rtnl1 (Fig. 6B). Furthermore, mitochondrial dysfunction is also implicated in some forms of HSP: mutations in a number of mitochondrial proteins can cause HSP (39–41); a number of other HSP proteins, spatacsin, spastizin and spartin (SPG11, SPG15 and SPG20, respectively) are reported to localize at least in part to mitochondria (47–49); and a possible mitochondrial fission defect has been reported in at least one SPG31 patient (50). Our observations indicate the possibility of mechanisms that may link the pathogenesis of HSP proteins involved in ER morphogenesis and mitochondrial dysfunction. It is worth noting however that in addition to mitochondria, the ER contacts other membranous organelles such as endosomes and mutlivesicular bodies as well as the plasma membrane (44,51,52). These interactions may also be affected by disruption of tubular ER.
Our results provide the first direct evidence of SER defects in mature axons in a loss-of-function SPG genotype. They also provide a route to further study the roles of the SER in normal neurons, impairment of which may lead to degeneration. These roles potentially include local effects on processes such as mitochondrial function and calcium homeostasis, but also long-distance communication along SER tubules, which extend throughout neurons, as in the ‘neuron within a neuron’ model (53). The study of HSP model flies in which SER organization is impaired promises to illuminate both the function of this fascinating compartment and the cell physiological processes that may be impaired by its disruption, and that may contribute to progressive axonal degeneration that spreads proximally from distal axonal ends.
MATERIALS AND METHODS
Fly stocks
The Rtnl1 exon trap line CPTI-001291 contains FLAG and StrepII tags in addition to Venus YFP (http://flybase.org/reports/FBal0262134.html). For knockdown experiments, either the UAS-Rtnl1-RNAi line 7866 (construct GD900, which has no predicted off-targets) or the w1118 control stock, 60000 (obtained from the Vienna Drosophila RNAi Center, www.vdrc.at) (54), was crossed with either da-GAL4 (55) or OK6-GAL4 (56); da-GAL4 was used except where indicated otherwise. Other fly stocks used were xbp1::EGFP (33), PBac{RB3.WH3}AcslKO (37), P{PZ}Acsl05847 (34), P{UAS-Acsl.715.MycC}3 (34) and mito::GFP (P{UAS-mito-HA-GFP.AP}3) (57).
Semi-quantitative PCR
Total RNA was purified from five male flies (<3 days post-eclosion) collected in TRIzol reagent (Invitrogen, Paisley, UK). cDNA was produced using the SuperScript III First Strand kit (Invitrogen, UK), 50 µm oligo-dT primer and 2 µg of DNase-treated RNA. Amplification of Rp49 mRNA was used to control for cDNA concentration in the PCR reaction. Primers used were: Rp49-L: CCGACCACGTTACAAGAACTCTC; Rp49-R: CGCTTCAAGGGACAGTATCTGA; Rtnl1-L: ACGTAGGAGCACCGCGAACG; and Rtnl1-R: CTGAAGGCGACGGTGCCGAA. PCR conditions were 95°C for 30 s, 60°C for 30 s and 72°C for 1 min, repeated for 20 cycles for Rp49 and 25 cycles for Rtnl1. At least six different samples for each genotype were analysed. PCR products were visualized with ethidium bromide on a gel, and band intensities quantified using ImageJ.
Immunoprecipitation and immunoblot
Soluble protein lysates were purified from male flies (<3 days post-eclosion) by homogenization in 1% Triton X-100, 25 mm HEPES (pH 7.4), 150 mm NaCl, 2 mm EDTA and Protease Inhibitor Cocktail (Sigma, Poole, UK), followed by centrifugation at 15 000g for 30 min at 4°C. Immunoprecipitation was carried out with Strep-Tactin Sepharose (IBA, Göttingen, Germany). Briefly, protein samples (Input) were incubated overnight at 4°C with Strep-Tactin Sepharose, and unbound and bound proteins were collected by centrifugation. For immunoblot, protein samples were separated on polyacrylamide minigels and electrophoretically transferred to nitrocellulose membranes (Bio-Rad, Hercules, CA, USA). The nitrocellulose was blocked in 5% non-fat milk in 10 mm Tris–HCl, 100 mm NaCl and 0.05% Tween-20 and incubated with primary antibodies overnight at 4°C. Primary antibodies used were against: FLAG (F1804; Sigma), Acsl (34), α-tubulin (T9026; Sigma) and Futsch (22C10; Hybridoma) (58). After incubation with HRP (horseradish peroxidase)-coupled secondary antibodies and SuperSignal Chemiluminescent Substrate (Pierce), bands were visualized by autoradiography. Band intensities were determined using ImageJ, and levels from knockdown samples normalized to control samples on the same film.
Histology and immunomicroscopy
Third-instar larvae were dissected in chilled Ca2+-free HL3 solution (59), fixed in 4% formaldehyde in PBS for 30 min and permeabilized in 0.1% Triton X-100 in PBS. Primary antibodies used were as for immunoblotting and against: GFP (ab6556; Abcam, Cambridge, UK), KDEL (MAC256; Abcam), GM130 (Ab32337; Abcam), HRP (P7899; Sigma), Discs-large (4F3) (60) and cysteine string protein (DCSP-2 (6D6) (61). The nuclear stain used was TO-PRO-3 iodide (T3605; Invitrogen). Fixed preparations were mounted in Vectashield (Vector Laboratories) and viewed using a Nikon CSi confocal head mounted on an Eclipse 90i microscope. Motor axons were imaged passing through segment A2 (‘anterior’) and segment A6 (‘posterior’). NMJs were imaged at muscle 6/7 of segment A3 (‘anterior’) and segment A6 (‘posterior’). Images were acquired using a 60×/1.4NA objective and the Nikon EZC1 3.90 software, and imported to ImageJ. Brightness, contrast and colour channels were adjusted using Photoshop (Adobe).
Image analysis
For fluorescence intensity analysis, control and mutant larvae were stained and washed in the same tube, with one genotype tail-clipped for identification. Mean grey intensity for regions of interest was quantified in ImageJ, using at least three independent experiments, each with at least four larvae. Mitochondria were counted manually, blind to genotype. Type 1b and 1s boutons were differentiated by intensity of Dlg staining.
Electron microscopy
Third-instar larvae were dissected in chilled Ca2+-free HL3 solution and fixed in 3% glutaraldehyde, 0.3% hydrogen peroxide and 2 mm sodium EDTA for 4 h at 4°C. After four washes in 0.1 m HEPES buffer, larva were post-fixed in 1% osmium ferricyanide (Oxchem, Edinburgh, UK) in buffer at room temperature for 1 h. After rinsing in de-ionized water, samples were stained in 2% uranyl acetate in maleate buffer, dehydrated in an ethanol series, followed by acetonitrile and then infiltrated with Quetol epoxy resin (Agar Scientific, Stansted, UK) and cured at 60°C for 48 h. Sections were cut on a Leica Ultracut UCT ultra-microtome at 70 nm, using a diamond knife, and contrasted with uranyl acetate and lead citrate. They were viewed using a Tecnai G2 electron microscope operated at 120 kV, and an AMT XR60B camera running the Deben software. EM images were collected from three larvae per genotype, with at least five epidermal cells imaged per larva. Images were blinded for genotype before ER branch length was determined in ImageJ.
Locomotor assay
Locomotor assays were carried out as described (30), using 10 flies per vial, 3 vials per experiment and 3 independent experiments. Flies were kept at 25°C and assayed every 3 days. Climbing ability was determined over 15 s in a vertical glass vial (10 cm length, 2.5 cm diameter). All trajectories were quantified for their end-to-end distance and their velocity.
Statistical analysis
Data were exported to GraphPad Prism 5 (GraphPad Software, Inc.) for statistical analysis. Statistical significance was determined using Student's t-test, except for analysis of climbing, where two-way ANOVA and Bonferroni post hoc analyses were used.
ACKNOWLEDGEMENTS
We thank Z. Wang, H.D. Ryoo, T.E. Rusten, J. Roote, the Developmental Studies Hybridoma Bank and the Bloomington and Vienna Drosophila Stock Centres for antibodies and stocks. We thank J. Skepper and J. Powell for help with EM, K. Lilley for support with mass spectrometry analysis, and D. Crowther and A. Mok for assistance with the fly climbing assay.
Conflict of Interest statement. None declared.
FUNDING
N.C.O'S. was supported by Marie Curie Individual Fellowship 236777. E.R. is a Wellcome Trust Senior Research Fellow in Clinical Science (grant 082381) and is supported by a strategic award from the Wellcome Trust to CIMR (grant 079895). Funding to pay the Open Access publication charges for this article was provided by the Wellcome Trust.
REFERENCES
- 1.Reid E. Science in motion: common molecular pathological themes emerge in the hereditary spastic paraplegias. J. Med. Genet. 2003;40:81–86. doi: 10.1136/jmg.40.2.81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Fink J.K. Hereditary spastic paraplegia. Curr. Neurol. Neurosci. Rep. 2006;6:65–76. doi: 10.1007/s11910-996-0011-1. [DOI] [PubMed] [Google Scholar]
- 3.Blackstone C., O'Kane C.J., Reid E. Hereditary spastic paraplegias: membrane traffic and the motor pathway. Nat. Rev. Neurosci. 2011;12:31–42. doi: 10.1038/nrn2946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Wang X., Shaw W.R., Tsang H.T., Reid E., O'Kane C.J. Drosophila spichthyin inhibits BMP signaling and regulates synaptic growth and axonal microtubules. Nat. Neurosci. 2007;10:177–185. doi: 10.1038/nn1841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Tsang H.T., Edwards T.L., Wang X., Connell J.W., Davies R.J., Durrington H.J., O'Kane C.J., Luzio J.P., Reid E. The hereditary spastic paraplegia proteins NIPA1, spastin and spartin are inhibitors of mammalian BMP signalling. Hum. Mol. Genet. 2009;18:3805–3821. doi: 10.1093/hmg/ddp324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Fassier C., Hutt J.A., Scholpp S., Lumsden A., Giros B., Nothias F., Schneider-Maunoury S., Houart C., Hazan J. Zebrafish atlastin controls motility and spinal motor axon architecture via inhibition of the BMP pathway. Nat. Neurosci. 2010;13:1380–1387. doi: 10.1038/nn.2662. [DOI] [PubMed] [Google Scholar]
- 7.Szymanski K.M., Binns D., Bartz R., Grishin N.V., Li W.P., Agarwal A.K., Garg A., Anderson R.G., Goodman J.M. The lipodystrophy protein seipin is found at endoplasmic reticulum lipid droplet junctions and is important for droplet morphology. Proc. Natl Acad. Sci. USA. 2007;104:20890–20895. doi: 10.1073/pnas.0704154104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Eastman S.W., Yassaee M., Bieniasz P.D. A role for ubiquitin ligases and Spartin/SPG20 in lipid droplet turnover. J. Cell Biol. 2009;184:881–894. doi: 10.1083/jcb.200808041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Edwards T.L., Clowes V.E., Tsang H.T., Connell J.W., Sanderson C.M., Luzio J.P., Reid E. Endogenous spartin (SPG20) is recruited to endosomes and lipid droplets and interacts with the ubiquitin E3 ligases AIP4 and AIP5. Biochem. J. 2009;423:31–39. doi: 10.1042/BJ20082398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Voeltz G.K., Prinz W.A., Shibata Y., Rist J.M., Rapoport T.A. A class of membrane proteins shaping the tubular endoplasmic reticulum. Cell. 2006;124:573–586. doi: 10.1016/j.cell.2005.11.047. [DOI] [PubMed] [Google Scholar]
- 11.Hu J., Shibata Y., Voss C., Shemesh T., Li Z., Coughlin M., Kozlov M.M., Rapoport T.A., Prinz W.A. Membrane proteins of the endoplasmic reticulum induce high-curvature tubules. Science. 2008;319:1247–1250. doi: 10.1126/science.1153634. [DOI] [PubMed] [Google Scholar]
- 12.Orso G., Pendin D., Liu S., Tosetto J., Moss T.J., Faust J.E., Micaroni M., Egorova A., Martinuzzi A., McNew J.A., Daga A. Homotypic fusion of ER membranes requires the dynamin-like GTPase atlastin. Nature. 2009;460:978–983. doi: 10.1038/nature08280. [DOI] [PubMed] [Google Scholar]
- 13.Park S.H., Zhu P.P., Parker R.L., Blackstone C. Hereditary spastic paraplegia proteins REEP1, spastin, and atlastin-1 coordinate microtubule interactions with the tubular ER network. J. Clin. Invest. 2010;120:1097–1110. doi: 10.1172/JCI40979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Hu J., Shibata Y., Zhu P.P., Voss C., Rismanchi N., Prinz W.A., Rapoport T.A., Blackstone C. A class of dynamin-like GTPases involved in the generation of the tubular ER network. Cell. 2009;138:549–561. doi: 10.1016/j.cell.2009.05.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Montenegro G., Rebelo A.P., Connell J., Allison R., Babalini C., D'Aloia M., Montieri P., Schüle R., Ishiura H., Price J., et al. Mutations in the ER-shaping protein human Reticulon2 cause axonal degeneration in hereditary spastic paraplegia type 12. J. Clin. Invest. 2012;122:538–544. doi: 10.1172/JCI60560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Shibata Y., Shemesh T., Prinz W.A., Palazzo A.F., Kozlov M.M., Rapoport T.A. Mechanisms determining the morphology of the peripheral ER. Cell. 2010;143:774–788. doi: 10.1016/j.cell.2010.11.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Evans K.J., Gomes E.R., Reisenweber S.M., Gundersen G.G., Lauring B.P. Linking axonal degeneration to microtubule remodeling by Spastin-mediated microtubule severing. J. Cell Biol. 2005;168:599–606. doi: 10.1083/jcb.200409058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Roll-Mecak A., Vale R.D. The Drosophila homologue of the hereditary spastic paraplegia protein, spastin, severs and disassembles microtubules. Curr. Biol. 2005;15:650–655. doi: 10.1016/j.cub.2005.02.029. [DOI] [PubMed] [Google Scholar]
- 19.Sanderson C.M., Connell J.W., Edwards T.L., Bright N.A., Duley S., Thompson A., Luzio J.P., Reid E. Spastin and atlastin, two proteins mutated in autosomal-dominant hereditary spastic paraplegia, are binding partners. Hum. Mol. Genet. 2006;15:307–318. doi: 10.1093/hmg/ddi447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Bian X., Klemm R.W., Liu T.Y., Zhang M., Sun S., Sui X., Liu X., Rapoport T.A., Hu J. Structures of the atlastin GTPase provide insight into homotypic fusion of endoplasmic reticulum membranes. Proc. Natl Acad. Sci. USA. 2011;108:3976–3981. doi: 10.1073/pnas.1101643108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Rolls M.M., Hall D.H., Victor M., Stelzer E.H., Rapoport T.A. Targeting of rough endoplasmic reticulum membrane proteins and ribosomes in invertebrate neurons. Mol. Cell. Biol. 2002;13:1787–1791. doi: 10.1091/mbc.01-10-0514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Rolls M.M., Satoh D., Clyne P.J., Henner A.L., Uemura T., Doe C.Q. Polarity and intracellular compartmentalization of Drosophila neurons. Neural Dev. 2007;2:7. doi: 10.1186/1749-8104-2-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Broadwell R.D., Cataldo A.M. The neuronal endoplasmic reticulum: its cytochemistry and contribution to the endomembrane system. J. Comp. Neurol. 1984;230:231–248. doi: 10.1002/cne.902300208. [DOI] [PubMed] [Google Scholar]
- 24.Burton P.R., Laveri L.A. The distribution, relationships to other organelles, and calcium-sequestering ability of smooth endoplasmic reticulum in frog olfactory axons. J. Neurosci. 1985;5:3047–3060. doi: 10.1523/JNEUROSCI.05-11-03047.1985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Hartter D.E., Burton P.R., Laver L.A. Distribution and calcium-sequestering ability of smooth endoplasmic reticulum in olfactory axon terminals of frog brain. Neurosci. 1987;23:371–386. doi: 10.1016/0306-4522(87)90297-1. [DOI] [PubMed] [Google Scholar]
- 26.Shimizu H., Fukaya M., Yamasaki M., Watanabe M., Manabe T., Kamiya H. Use-dependent amplification of presynaptic Ca2+ signaling by axonal ryanodine receptors at the hippocampal mossy fiber synapse. Proc. Natl Acad. Sci. USA. 2008;105:11998–12003. doi: 10.1073/pnas.0802175105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Wakefield S., Tear G. The Drosophila reticulon, Rtnl-1, has multiple differentially expressed isoforms that are associated with a sub-compartment of the endoplasmic reticulum. Cell. Mol. Life Sci. 2006;63:2027–2038. doi: 10.1007/s00018-006-6142-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Röper K. Rtnl1 is enriched in a specialized germline ER that associates with ribonucleoprotein granule components. J. Cell Sci. 2007;120:1081–1092. doi: 10.1242/jcs.03407. [DOI] [PubMed] [Google Scholar]
- 29.Waterman-Storer C.M., Salmon E.D. Endoplasmic reticulum membrane tubules are distributed by microtubules in living cells using three distinct mechanisms. Curr. Biol. 1998;8:798–806. doi: 10.1016/s0960-9822(98)70321-5. [DOI] [PubMed] [Google Scholar]
- 30.Jahn T.R., Kohlhoff K.J., Scott M., Tartaglia G.G., Lomas D.A., Dobson C.M., Vendruscolo M., Crowther D.C. Detection of early locomotor abnormalities in a Drosophila model of Alzheimer's disease. J. Neurosci. Methods. 2011;197:186–189. doi: 10.1016/j.jneumeth.2011.01.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Ron D., Walter P. Signal integration in the endoplasmic reticulum unfolded protein response. Nat. Rev. Mol. Cell Biol. 2007;8:519–529. doi: 10.1038/nrm2199. [DOI] [PubMed] [Google Scholar]
- 32.Sriburi R., Jackowski S., Mori K., Brewer J.W. XBP1: a link between the unfolded protein response, lipid biosynthesis, and biogenesis of the endoplasmic reticulum. J. Cell Biol. 2004;167:35–41. doi: 10.1083/jcb.200406136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Ryoo H.D., Domingos P.M., Kang M.J., Steller H. Unfolded protein response in a Drosophila model for retinal degeneration. EMBO J. 2007;26:242–252. doi: 10.1038/sj.emboj.7601477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Zhang Y., Chen D., Wang Z. Analyses of mental dysfunction-related ACSl4 in Drosophila reveal its requirement for Dpp/BMP production and visual wiring in the brain. Hum. Mol. Genet. 2009;18:3894–3905. doi: 10.1093/hmg/ddp332. [DOI] [PubMed] [Google Scholar]
- 35.Piccini M., Vitelli F., Bruttini M., Pober B.R., Jonsson J.J., Villanova M., Zollo M., Borsani G., Ballabio A., Renieri A. FACL4, a new gene encoding long-chain acyl-CoA synthetase 4, is deleted in a family with Alport syndrome, elliptocytosis, and mental retardation. Genomics. 1998;47:350–358. doi: 10.1006/geno.1997.5104. [DOI] [PubMed] [Google Scholar]
- 36.Meloni I., Parri V., De Filippis R., Ariani F., Artuso R., Bruttini M., Katzaki E., Longo I., Mari R., Bellan C., et al. The XLMR gene ACSL4 plays a role in dendritic spine architecture. Neuroscience. 2009;159:657–669. doi: 10.1016/j.neuroscience.2008.11.056. [DOI] [PubMed] [Google Scholar]
- 37.Liu Z., Huang Y., Zhang Y., Chen D., Zhang Y.Q. Drosophila Acyl-CoA synthetase long-chain family member 4 regulates axonal transport of synaptic vesicles and is required for synaptic development and transmission. J. Neurosci. 2011;31:2052–2063. doi: 10.1523/JNEUROSCI.3278-10.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.de Brito O.M., Scorrano L. An intimate liaison: spatial organization of the endoplasmic reticulum–mitochondria relationship. EMBO J. 2010;29:2715–2723. doi: 10.1038/emboj.2010.177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Casari G., De Fusco M., Ciarmatori S., Zeviani M., Mora M., Fernandez P., De Michele G., Filla A., Cocozza S., Marconi R., Dürr A., et al. Spastic paraplegia and OXPHOS impairment caused by mutations in paraplegin, a nuclear-encoded mitochondrial metalloprotease. Cell. 1998;93:973–983. doi: 10.1016/s0092-8674(00)81203-9. [DOI] [PubMed] [Google Scholar]
- 40.Hansen J.J., Dürr A., Cournu-Rebeix I., Georgopoulos C., Ang D., Nielsen M.N., Davoine C.S., Brice A., Fontaine B., Gregersen N., Bross P. Hereditary spastic paraplegia SPG13 is associated with a mutation in the gene encoding the mitochondrial chaperonin Hsp60. Am. J. Hum. Genet. 2002;70:1328–1332. doi: 10.1086/339935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Verny C., Guegen N., Desquiret V., Chevrollier A., Prundean A., Dubas F., Cassereau J., Ferre M., Amati-Bonneau P., Bonneau D., et al. Hereditary spastic paraplegia-like disorder due to a mitochondrial ATP6 gene point mutation. Mitochondrion. 2011;11:70–75. doi: 10.1016/j.mito.2010.07.006. [DOI] [PubMed] [Google Scholar]
- 42.Tolley N., Sparkes I.A., Hunter P.R., Craddock C.P., Nuttall J., Roberts L.M., Hawes C., Pedrazzini E., Frigerio L. Overexpression of a plant reticulon remodels the lumen of the cortical endoplasmic reticulum but does not perturb protein transport. Traffic. 2008;9:94–102. doi: 10.1111/j.1600-0854.2007.00670.x. [DOI] [PubMed] [Google Scholar]
- 43.Sparkes I., Tolley N., Aller I., Svozil J., Osterrieder A., Botchway S., Mueller C., Frigerio L., Hawes C. Five Arabidopsis reticulon isoforms share endoplasmic reticulum location, topology, and membrane-shaping properties. Plant Cell. 2010;22:1333–1343. doi: 10.1105/tpc.110.074385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Friedman J.R., Webster B.M., Mastronarde D.N., Verhey K.J., Voeltz G.K. ER sliding dynamics and ER-mitochondrial contacts occur on acetylated microtubules. J. Cell Biol. 2010;190:363–375. doi: 10.1083/jcb.200911024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.de Brito O.M., Scorrano L. Mitofusin 2 tethers endoplasmic reticulum to mitochondria. Nature. 2008;456:605–610. doi: 10.1038/nature07534. [DOI] [PubMed] [Google Scholar]
- 46.Friedman J.R., Lackner L.L., West M., DiBenedetto J.R., Nunnari J., Voeltz G.K. ER tubules mark sites of mitochondrial division. Science. 2011;334:358–362. doi: 10.1126/science.1207385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Lu J., Rashid F., Byrne P.C. The hereditary spastic paraplegia protein spartin localises to mitochondria. J. Neurochem. 2006;98:1908–1919. doi: 10.1111/j.1471-4159.2006.04008.x. [DOI] [PubMed] [Google Scholar]
- 48.Murmu R.P., Martin E., Rastetter A., Esteves T., Muriel M.P., El Hachimi K.H., Denora P.S., Dauphin A., Fernandez J.C., Duyckaerts C., et al. Cellular distribution and subcellular localization of spatacsin and spastizin, two proteins involved in hereditary spastic paraplegia. Mol. Cell. Neurosci. 2011;47:191–202. doi: 10.1016/j.mcn.2011.04.004. [DOI] [PubMed] [Google Scholar]
- 49.Yao A., Jin S., Li X., Liu Z., Ma X., Tang J., Zhang Y.Q. Drosophila FMRP regulates microtubule network formation and axonal transport of mitochondria. Hum. Mol. Genet. 2011;20:51–63. doi: 10.1093/hmg/ddq431. [DOI] [PubMed] [Google Scholar]
- 50.Goizet C., Depienne C., Benard G., Boukhris A., Mundwiller E., Sole G., Coupry I., Pilliod J., Martin-Negrier M., Fedirko E., et al. REEP1 mutations in SPG31: frequency, mutational spectrum, and potential association with mitochondrial morpho-functional dysfunction. Hum. Mutat. 2011;32:1118–1127. doi: 10.1002/humu.21542. [DOI] [PubMed] [Google Scholar]
- 51.Wu M.M., Buchanan J., Luik R.M., Lewis R.S. Ca2+ store depletion causes STIM1 to accumulate in ER regions closely associated with the plasma membrane. J. Cell Biol. 2006;174:803–813. doi: 10.1083/jcb.200604014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Eden E.R., White I.J., Tsapara A., Futter C.E. Membrane contacts between endosomes and ER provide sites for PTP1B-epidermal growth factor receptor interaction. Nat. Cell Biol. 2010;12:267–272. doi: 10.1038/ncb2026. [DOI] [PubMed] [Google Scholar]
- 53.Berridge M.J. Neuronal calcium signalling. Neuron. 1998;21:13–26. doi: 10.1016/s0896-6273(00)80510-3. [DOI] [PubMed] [Google Scholar]
- 54.Dietzl G., Chen D., Schnorrer F., Su K.C., Barinova Y., Fellner M., Gasser B., Kinsey K., Oppel S., Scheiblauer S., et al. A genome-wide transgenic RNAi library for conditional gene inactivation in Drosophila. Nature. 2007;448:151–156. doi: 10.1038/nature05954. [DOI] [PubMed] [Google Scholar]
- 55.Perrin L., Bloyer S., Ferraz C., Agrawal N., Sinha P., Dura J.M. The leucine zipper motif of the Drosophila AF10 homologue can inhibit PRE-mediated repression: implications for leukemogenic activity of human MLL-AF10 fusions. Mol. Cell. Biol. 2003;23:119–130. doi: 10.1128/MCB.23.1.119-130.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Aberle H., Haghighi A.P., Fetter R.D., McCabe B.D., Magalhaes T.R., Goodman C.S. wishful thinking encodes a BMP type II receptor that regulates synaptic growth in Drosophila. Neuron. 2002;33:545–558. doi: 10.1016/s0896-6273(02)00589-5. [DOI] [PubMed] [Google Scholar]
- 57.Pilling A.D., Horiuchi D., Lively C.M., Saxton W.M. Kinesin-1 and Dynein are the primary motors for fast transport of mitochondria in Drosophila motor axons. Mol. Biol. Cell. 2006;17:2057–2068. doi: 10.1091/mbc.E05-06-0526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Fujita S.C., Zipursky S.L., Benzer S., Ferrus A., Shotwell S.L. Monoclonal antibodies against the Drosophila nervous system. Proc. Natl Acad. Sci. USA. 1982;79:7929–7933. doi: 10.1073/pnas.79.24.7929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Stewart B.A., Atwood H.L., Renger J.J., Wang J., Wu C.F. Improved stability of Drosophila larval neuromuscular preparations in haemolymph-like physiological solutions. J. Comp. Physiol. A. 1994;175:179–191. doi: 10.1007/BF00215114. [DOI] [PubMed] [Google Scholar]
- 60.Parnas D., Haghighi A.P., Fetter R.D., Kim S.W., Goodman C.S. Regulation of postsynaptic structure and protein localization by the Rho-type guanine nucleotide exchange factor dPix. Neuron. 2001;32:415–424. doi: 10.1016/s0896-6273(01)00485-8. [DOI] [PubMed] [Google Scholar]
- 61.Zinsmaier K.E., Eberle K.K., Buchner E., Walter N., Benzer S. Paralysis and early death in cysteine string protein mutants of Drosophila. Science. 1994;263:977–980. doi: 10.1126/science.8310297. [DOI] [PubMed] [Google Scholar]