

Abstract
Social behavior dysfunction is a symptomatic element of schizophrenia and autism spectrum disorder (ASD). Although altered activities in numerous brain regions are associated with defective social cognition and perception, the causative relationship between these altered activities and social cognition and perception—and their genetic underpinnings—are not known in humans. To address these issues, we took advantage of the link between hemizygous deletion of human chromosome 22q11.2 and high rates of social behavior dysfunction, schizophrenia and ASD. We genetically manipulated Sept5, a 22q11.2 gene, and evaluated its role in social interaction in mice. Sept5 deficiency, against a high degree of homogeneity in a congenic genetic background, selectively impaired active affiliative social interaction in mice. Conversely, virally guided overexpression of Sept5 in the hippocampus or, to a lesser extent, the amygdala elevated levels of active affiliative social interaction in C57BL/6J mice. Congenic knockout mice and mice overexpressing Sept5 in the hippocampus or amygdala were indistinguishable from control mice in novelty and olfactory responses, anxiety or motor activity. Moreover, post-weaning individual housing, an environmental condition designed to reduce stress in male mice, selectively raised levels of Sept5 protein in the amygdala and increased active affiliative social interaction in C57BL/6J mice. These findings identify this 22q11.2 gene in the hippocampus and amygdala as a determinant of social interaction and suggest that defective social interaction seen in 22q11.2-associated schizophrenia and ASD can be genetically and environmentally modified by altering this 22q11.2 gene.
INTRODUCTION
The ability to socially interact with others has a fundamental biological significance in humans and other species. Its biological role is appreciated in social behavior dysfunctions seen in developmental neuropsychiatric disorders. Social dysfunction is a prodromal and symptomatic element of schizophrenia (1) and autism spectrum disorder (ASD) throughout its developmental course toward adulthood (2).
Individuals with 3 Mb and nested 1.5 Mb hemizygosity of 22q11.2 exhibit extraordinarily high rates of social behavior deficits (3–9), schizophrenia (10–17) and ASD (8,18–22). Moreover, social behavior dysfunction precedes (5)–and its severity is associated with (17,23)–the emergence of schizophrenia among individuals with 22q11.2 hemizygosity. This chromosomal locus is one of many sites of copy number variations associated with ASD (24–33) and schizophrenia (16,33–42).
Because 22q11.2 hemizygous deletions minimally include approximately 30 genes, the precise manner by which individual 22q11.2 genes functionally contribute to the etiology of social interaction deficits, schizophrenia and ASD cannot be ascertained in humans. As this human chromosomal region is conserved in the mouse, a genetic mouse model is one reliable way to circumvent this problem. Previous studies have identified a 200 kb region of 22q11.2 whose gene dose alteration induces social behavior dysfunction and antipsychotic-responsive behavioral abnormalities (43) and prepulse inhibition deficits (44) in mice. Subsequently, we identified Sept5, one of the four encoded genes in the 200 kb region, as a determinant for social interaction in mice (45). Recently, one child has been identified with homozygous deletion of SEPT5 and its adjacent GP1BB, and this patient exhibits deficits in socio-emotional function and language and speech development (46). Although this single case study is consistent with the hypothesis that SEPT5 deficiency causes ASD- and schizophrenia-related phenotypes, more cases are needed to establish the degree of association between this mutation and neuropsychiatric disorders. Moreover, because the two genes are deleted in this individual, whether SEPT5 alone contributes to these various symptoms remains unclear.
In the present study, we took advantage of our identification of the murine Sept5 gene as a risk factor for defective social interaction to further advance our understanding of the role of this 22q11.2 gene in social behavior. Our analysis shows that Sept5 levels in the hippocampus and amygdala act as a determinant of social interaction in mice.
RESULTS
Sept5 deficiency decreases affiliative social interaction
Although our previous study showed that genetic background affects the penetrance of Sept5 deficiency (45), an alternative interpretation is that the phenotypic difference between wild-type (WT) and knockout (KO) mice reflects unequal distributions of background alleles between the genotypes, rather than the genuine impact of Sept5 deficiency. To evaluate the impact of constitutive Sept5 deficiency on social interaction against a much higher level of homogeneous genetic background than our Sept5 KO mice (45), we developed a congenic Sept5-deficient mouse on a C57BL/6J background and tested its behavior. Congenic KO mice showed lower levels of active affiliative social interaction during the second session than WT littermates (Fig. 1A). The phenotype in social interaction did not primarily reflect alterations in non-specific elements of social behavior, as no phenotype was found in their approach behavior to a novel object (Fig. 1B), olfactory functioning in the buried food test (Fig. 1C), anxiety-like behavior in an elevated plus maze (Fig. 1D) and thigmotaxis (Fig. 1E) and general motor activity in the open field (Fig. 1F).
Figure 1.

Effects of genetic Sept5 deficiency on behavior. Data are presented as means ± SEM. (A) Active and passive social interactions. Whether the two groups (KO and WT pairs) differed depended on the type of social behavior (active versus passive) and session (group × session, F1,26= 7.53, P = 0.0109; group × social type × session, F1,26= 7.50, P = 0.011). Although the active social interaction category included both active affiliative and aggressive social behaviors, mice overwhelmingly exhibited active affiliative social interaction (100% in WT and 100% in KO). Asterisks indicate a statistically significant difference of 1%, as determined by the Newman–Keuls comparison. WT, n = 14; KO, n = 14. (B) Time spent contacting a novel object. The two genotype groups were indistinguishable and their contact time equally declined from the first to the second session (genotype, F1,16 = 0.14, P = 0.716; session, F1,16 = 16.67, P = 0.0009; genotype × session, F1,16 = 0.28, P = 0.6036). WT, n = 7; KO, n = 11. (C) Latency to find buried food. The two genotype groups did not differ (t(26) = 1.58, P = 0.1267). WT, n = 13; KO, n = 15. The slightly higher value in the KO group was partly due to one outlier that did not find the food during the test session (see Supplementary Material, Fig. S1). (D) The relative amounts of time spent in the open arms over both open and closed arms in the elevated plus maze (EPM). The two genotype groups were indistinguishable in the relative time spent in (t(23) = 0.39, P = 0.7011) and frequency of visits to (t(23) = 0.16, P = 0.8769, not shown for clarity) the open arms. WT, n = 14; KO, n = 11. (E) Thigmotaxis. The two genotype groups were indistinguishable in time spent in the margin area of the inescapable open field, and this equally increased during the session (genotype, F1,33 = 0.78, P = 0.3832; time, F5,165 = 6.33, P < 0.0001; F5,165 = 0.42, P = 0.8348). WT, n = 19; KO, n = 16. (F) Motor activity in an open field. The two groups were indistinguishable and equally reduced motor activity during the session (genotype, F1,33 = 2.72, P = 0.1089; time, F5,165 = 86.57, P < 0.0001; genotype × time, F10,165= 1.85, P = 0.105). WT, n = 19; KO, n = 16.
Virally guided overexpression of Sept5 in neurons in the hippocampus and amygdala of C57BL/6J mice increases affiliative social interaction
A corollary of our observation with congenic Sept5 KO mice is that heightened expression of Sept5 protein in the mouse brain causes high levels of social interaction. Although Sept5 protein is expressed throughout the mouse and human brain (47,48), we targeted limbic regions for their implicated roles in social behavior. To selectively manipulate Sept5 in distinct brain regions, we constructed a lentiviral vector carrying Sept5 and surgically infused it into the brains of C57BL/6J mice. Compared with enhanced green fluorescent protein (EGFP), Sept5 overexpression in the hippocampus (Fig. 2A) and amygdala (Fig. 3A) increased active affiliative social interaction in C57BL/6J mice. This phenotype was highly selective, as Sept5 overexpression had no effect on novel object exploration (Figs 2B and 3B), latency to find buried food (Figs 2C and 3C), anxiety-related behaviors in the elevated plus maze (Figs 2D and 3D) and thigmotaxis (Figs 2E and 3E) or motor behavior in the open field (Figs 2F and 3F).
Figure 2.

Behavioral effects of virally overexpressed Sept5 in the dorsal hippocampus. (A) Active and passive social interactions. Sept5-infused mice spent more time in active social interaction; there was no difference between the two vector groups in passive social interaction (group, F1,15 = 9.63, P = 0.0073; social type, F1,15 = 162.73, P < 0.0001; session, F1,15 = 47.13, P < 0.0001; group × social type, F1,15 = 6.12, P = 0.0258; group × session, F1,15 = 10.02, P = 0.0064; group × type × session, F1,15 = 3.13, P = 0.097). **Significant at the 1% level, as determined by Newman–Keuls comparisons. Ninety-nine percent and 98% of active social interaction was non-aggressive, active affiliative social interaction in EGFP- and Sept5-infused mice, respectively. EGFP, n = 9; Sept5, n = 8. (B) Time spent contacting a novel object. Sept5 infusion had no effect on this behavior (group, F1,15 = 0.055, P = 0.8177; session, F1,15 = 5.49, P = 0.0333; group × session, F1,15= 2.45, P = 0.1381). EGFP, n = 9; Sept5, n = 8. (C) Latency to find buried food. The two groups did not differ (t(15) = 0.58, P = 0.5707). EGFP, n = 9; Sept5, n = 8. (D) EPM. The two groups did not differ in percent of time spent in (t(11) = 0.90, P = 0.3864) and percent visits to (t(11) = 1.30, P = 0.2186, not shown for clarity) the open arms. EGFP, n = 7; Sept5, n = 6. (E) Thigmotaxis. The two groups were indistinguishable and were stable during the entire session (group, F1,15 = 0.38, P = 0.5461; time, F5,75 = 2.13, P = 0.0709; group × time, F5,75= 0.80, P = 0.5518). EGFP, n = 9; Sept5, n = 8. (F) Motor activity in an open field. The two groups were indistinguishable and stably decreased their locomotor activity during the session (group, F1,15 = 0.76, P = 0.3969; time, F5,75 = 6.76, P < 0.0001; group × time, F5,75 = 0.167, P = 0.974). EGFP, n = 9; Sept5, n = 8.
Figure 3.

Behavioral effects of virally overexpressed Sept5 in the basolateral amygdaloid complex. (A) Active and passive social interactions. Whether EGFP- and Sept5-infused groups differed depended on the type of social behavior (active versus passive) and session (group, F1,16 = 0.67, P = 0.4236; group × type, F1,16 = 0.45, P = 0.5101; group × session, F1,16 = 12.32, P = 0.0029; group × social type × session, F1,16= 9.20, P = 0.0079). Post hoc comparisons showed that, compared with EGFP mice, Sept5-infused mice showed higher levels of active social interaction at the second session (*significant at the 5% level). An analysis of raw data showed that EGFP and Sept5 mice exhibited active affiliative social interaction in 99 and 84%, respectively, in all active social interaction. EGFP, n = 10; Sept5, n = 8. (B) Time spent contacting a novel object. The treatment had no effect on novel object exploration (group, F1,13 = 0.00001, P = 0.998; session, F1,13 = 4.18, P = 0.0616; group × session, F1,13 = 0.38, P = 0.5485). EGFP, n = 9; Sept5, n = 6. (C) Latency to find buried food. The two groups were indistinguishable in this behavior (t(12)= 0.41, P = 0.6884). EGFP, n = 8; Sept5, n = 6. (D) The relative amounts of time spent in the open arms over both open and closed arms of the EPM. The two groups did not differ in percent of time spent in (t(11)= 0.38, P = 0.7079) and percent visits to (t(11)= 0.59, P = 0.5665, not shown for clarity) the open arms. EGFP, n = 7; Sept5, n = 6. (E) Thigmotaxis. The groups were indistinguishable and were stable during the entire session (group, F1,12 = 1.02, P = 0.3334; time, F5,60 = 0.93, P = 0.4708; group × time, F5,60 = 0.19, P = 0.9641). EGFP, n = 7; Sept5, n = 7. (F) Motor activity in an open field. The treatment groups were indistinguishable and stably decreased their locomotor activity during the session (group, F1,12 = 0.38, P = 0.5512; time, F5,60 = 10.08, P < 0.0001; group × time, F5,60 = 0.42, P = 0.8353). EGFP, n = 7; Sept5, n = 7.
Although there were occasional dorsal spreads of EGFP, along needle tracks, within the somatosensory cortex of the hippocampus- and amygdala-infusion groups (Supplementary Material, Fig. S2A and B), selective infusion of the vector into the somatosensory cortex (Supplementary Material, Fig. S2C) had no effect on any of these behaviors (Fig. 4A and C–F), except that Sept5 infusion into this cortical area facilitated novel object approach (Fig. 4B).
Figure 4.

Behavioral effects of virally overexpressed Sept5 in the somatosensory cortex. (A) Active and passive social interactions. Sept5 infused into the somatosensory cortex had no effect on social interaction (group, F1,17 = 0.37, P = 0.5535; social type, F1,17 = 115.75, P < 0.0001; session, F1,17 = 1.42, P = 0.2495; group × social type, F1,17 = 0.65, P = 0.4306; group × session, F1,17 = 0.03, P = 0.8603; group × type × session, F1,17= 0.46, P = 0.5077). Ninety-eight percent and 100% of active social interaction was non-aggressive, affiliative social interaction in EGFP- and Sept5-infused mice, respectively. EGFP, n = 10; Sept5, n = 9. (B) Time spent contacting a novel object. Sept5 infusion into the somatosensory cortex raised levels of approach toward a novel object (group, F1,17 = 5.87, P = 0.0269; session, F1,17 = 2.48, P = 0.1338; group × session, F1,17 = 0.09, P = 0.7683). EGFP, n = 10; Sept5, n = 9. (C) Latency to find buried food. Sept5 expression in the somatosensory cortex had no effect on this test of olfactory functioning (t(17) = 1.19, P = 0.2496). EGFP, n = 10; Sept5, n = 9. (D) EPM. The two groups did not differ in percent of time spent in (t(12) = 0.85, P = 0.4112) and percent visits to (t(12) = 0.55, P = 0.5921) the open arms. EGFP, n = 7; Sept5, n = 7. (E) Thigmotaxis. Sept5 overexpression in the somatosensory cortex had no effect on thigmotaxis (group, F1,14 = 0.56, P = 0.4673; time, F5,70 = 1.53, P = 0.1919; group × time, F5,70 = 1.01, P = 0.4205). EGFP, n = 8; Sept5, n = 8. (F) Motor activity in an open field. Sep5 overexpression in the somatosensory cortex had no effect on locomotor activity (group, F1,14 = 0.12, P = 0.7366; time, F5,70 = 31.11, P < 0.0001; group × time, F5,70 = 0.548, P = 0.7392). EGFP, n = 8; Sept5, n = 8.
Gene expression, as identified by EGFP, was centered in the CA2/CA3 of the dorsal hippocampus (Fig. 5A; Supplementary Material, Fig. S2A), the basolateral amygdaloid complex (Fig. 5C; Supplementary Material, Fig. S2B) or the somatosensory cortex (Supplementary Material, Fig. S2C). Consistent with the relatively higher levels of CamkIIα promoter activity in the hippocampus than in the amygdala (49), more wide-spread gene expression was seen in the former than the latter region (Fig. 5A and C). Gene expression was observed in neurons, as labeled by NeuN (Fig. 5B and D).
Figure 5.
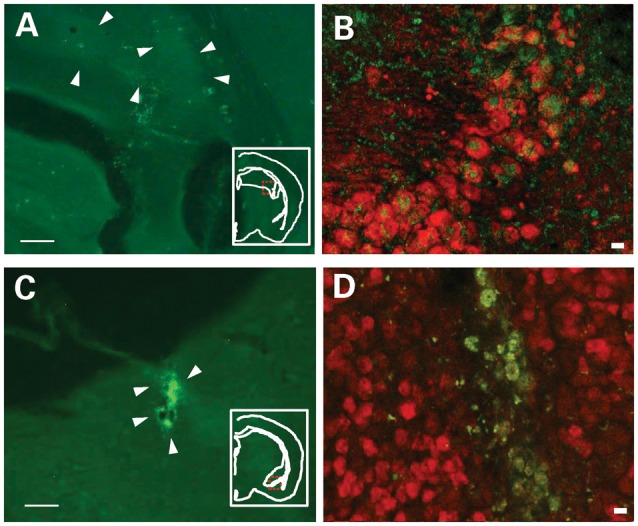
(A and C) EGFP expression, indicated by arrows, in mice infused with PCamkIIα-Sept5-IRES-EGFP into the hippocampus (A) and amygdala (C). Because Sept5 staining does not distinguish endogenous Sept5 and exogenously expressed Sept5, EGFP was used to identify areas where Sept5 was overexpressed. Insets indicate the area from which the image was taken; the viral particles were expressed in the dorsal hippocampus and the basolateral amygdaloid complex. Scale bar: 500 μm. (B and D) Confocal image of EGFP (green) and NeuN (red) in the CA2/3 region of the hippocampus (B) and basolateral amygdaloid complex (D). Scale bar: 10 μm.
Post-weaning individual housing raises Sept5 levels in the amygdala and increases active affiliative social interaction in C57BL/6J mice
Individuals with identical 22q11.2 hemizygosity vary in the presence, onset and severity of social dysfunctions and developmental neuropsychiatric disorders (5–9). Environmental factors, such as stress, have been suggested to contribute to the onset of neuropsychiatric disorders and behavioral/neurocognitive deficits (50), but it is difficult to isolate the precise environmental factor in humans. Unlike rats, male mice are highly territorial and intolerant against same-sex conspecifics, and frequently exhibit inter-individual aggression when group-housed (51). We took advantage of the well-documented effect of post-weaning individual housing to reduce anxiety and increase affiliative social interaction in male mice (52–55).
Post-weaning individual housing selectively raised Sept5 protein levels in the amygdala (Fig. 6A and B). No alteration was seen in the prefrontal cortex, nucleus accumbens, caudate-putamen, hippocampus, ventral tegmental area (VTA) and substantia nigra.
Figure 6.

Effects of post-weaning housing conditions on Sept5 protein levels in the brain of C57BL/6J mice. Western blot analysis of Sept5 protein is shown. (A) Representative blots are shown for each region. (B) Quantitative analysis of Sept5 levels. Data are presented as means ± SEM. Although the overall housing effect was not significant (F1,26 = 0.93, P = 0.3438), there was a significant interaction between housing and the brain region (F6,156= 5.26, P < 0.0001). This interaction was due to a significantly higher level of Sept5 in the amygdala of individually housed mice, as determined by post hoc comparisons (**significant at the 1% level). G, n = 7–14 mice for each region; I, n = 8–10 mice for each region. A sample from each mouse was used up to two to three times for all regions except for the SN and VTA; samples from the SN and VTA were used only once for each mouse. G, group housing; I, individual housing. PFC, prefrontal cortex; NA, nucleus accumbens; CP, caudate-putamen; AMY, amygdala; HIP, hippocampus; VTA, ventral tegmental area; SN, substantia nigra.
Post-weaning individual housing increased active affiliative social interaction (Fig. 7A), but had no effect on a novel object approach (Fig. 7B) or the buried food search (Fig. 7C). In contrast, individual housing decreased anxiety-related behaviors in the elevated plus maze (Fig. 7D) and thigmotaxis in an inescapable open field (Fig. 7E), and elevated basal levels of motor activity in the open field, but not the rate of habituation of motor activity (Fig. 7F).
Figure 7.

Behavioral effects of post-weaning housing conditions. Data are presented as means ± SEM. GH, group housing; IH, individual housing. (A) Active and passive social interactions. Main effects were significant for housing conditions (F1,33 = 37.43, P < 0.0001), social interaction type (F1,33= 248.27, P < 0.0001) and session (F1,33 = 8.24, P = 0.0071). The only significant interaction was that between housing conditions and social interaction type (F1,33 = 25.80, P < 0.0001). Active social interaction was predominantly active affiliative social interaction in group-housed (98%) and individually housed (91%) mice. Post hoc comparisons indicated that individually housed mice spent more time in active social interaction than group-housed mice. Asterisks indicate statistically significant differences (1%) from GH mice as determined by the Newman–Keuls comparison. GH, n = 18; IH, n = 17. (B) Time spent contacting a novel object. Mice under the two housing conditions spent an indistinguishable amount of time interacting with a novel object, and equally habituated (housing, F1,27 = 0.98, P = 0.3323; session, F1,27 = 30.18, P < 0.0001; genotype × session, F1,27 = 2.36, P = 0.136). GH, n = 15; IH, n = 14. (C) Latency to find buried food. Mice under the two housing conditions were indistinguishable in the latency to find buried food (t(13) = 0.72, P = 0.4873). GH, n = 7; IH, n = 8. (D) The relative amounts of time spent in the open arms over both open and closed arms in the elevated plus maze. Individually housed mice spent more time in (t(34) = 2.25, P = 0.0307) and visited more frequently (t(34) = 2.20, P = 0.0349, not shown for clarity) the open arms than group-housed mice. GH, n = 20; IH, n = 16. (E) Thigmotaxis. Although there was no overall housing effect on the time spent in the margin (F1,46 = 2.84, P = 0.099) and neither group altered their time in the margin over time (time, F5,230 = 1.96, P = 0.0862), there was a significant interaction between the two factors (F5,230 = 4.14, P = 0.0013). This interaction was due to a significantly higher level of margin time in individually housed mice at the last time point. **Significant at the 1% level, as determined by Newman–Keuls post hoc comparisons. GH, n = 25; IH, n = 23. (F) Motor activity in an open field. Individually housed mice traveled more distance than group-housed mice, and both groups equally reduced their motor activity during the 30 min session (housing, F1,46 = 8.86, P = 0.0046; time, F5,230 = 106.84, P < 0.0001; housing × time, F5,230 = 1.28, P = 0.2721). GH, n = 25; IH, n = 23.
DISCUSSION
Our results provide three lines of evidence to support the hypothesis that Sept5, a 22q11.2 gene, is a determinant for affiliative social interaction in mice. First, constitutive homozygosity of Sept5 reduced levels of active affiliative social interaction in congenic KO mice compared with WT mice. Second, virally guided overexpression of Sept5 in the hippocampus or amygdala enhanced social interaction in C57BL/6J mice. The impact of Sept5 alterations on active affiliative social interaction was highly selective and was not attributable to alterations in novel object exploration, olfactory investigation, anxiety-related behaviors or motor activity. Third, post-weaning individual housing, a condition known to reduce stress in male mice, selectively elevated Sept5 protein levels in the amygdala and increased active affiliative social interaction.
Defective social interaction is the primary symptom of ASD and a prodromal and symptomatic element of schizophrenia (1,5,17,23). Although a repetitive behavioral tendency is another symptomatic element of these neuropsychiatric disorders, neither non-congenic Sept5-deficient mice (45) nor congenic Sept5 KO mice (data not shown) exhibited such a behavioral trait in spontaneous alternation or rewarded alternation. Given that heterozygosity of Tbx1, another 22q11.2 gene, causes both social interaction deficits and a repetitive behavioral trait (56), we suggest that more than one 22q11.2 gene impact multiple or single symptomatic elements of 22q11.2-associated schizophrenia and ASD.
Sept5 was the first 22q11.2 gene whose deficiency was found to reduce levels of affiliative social interaction in mice (45). Our previous study showed that social interaction deficits were present in mice with a mixed genetic background of CD1, 129X1/SvJ and 129S1, but were absent when the genetic background was shifted toward 129X1/SvJ (45). Our present data show that after achieving a highly homogeneous genetic background with C57BL/6J mice, congenic Sept5 KO mice still exhibit affiliative social interaction deficits. Moreover, overexpression of Sept5, against the co-isogenic genetic background, in the brains of C57BL/6J mice elevated levels of affiliative social interaction. These observations lend further support to the hypothesis that Sept5 is indeed a determinant of social interaction and that genetic background influences its phenotypic penetrance. This provides a plausible explanation for the clinical observation that not all individuals with identical 22q11.2 hemizygous deletions exhibit social behavior deficits, and those with this phenotype exhibit varying degrees of phenotypic severity (5–9).
A decrease in time spent in social interaction is usually seen from the first to second sessions and is thought to reflect recognition memory. C57BL/6J mice infused with EGFP alone showed this habituation, but congenic Sept5 WT mice instead showed higher levels of active social interaction in the second session than in the first session, despite the fact that congenic WT mice carry a C57BL/6J genetic background. As even after 10 generations of back-crossing congenic WT mice and the donor inbred strain are not genetically identical (57), allelic differences between congenic WT mice and C57BL/6J mice might have contributed to the baseline difference.
Virally guided overexpression of Sept5 in the hippocampus increased the overall level of active affiliative social interaction, and EGFP- and Sept5-infused mice showed similar rates of habituation. In contrast, virally guided overexpression of Sept5 in the amygdala elevated levels of active affiliative social interaction in the second, but not first session. The reason for this selective elevation of social interaction in the second session in the amygdala group is not clear. One possibility is that, as the stimulus mice exhibit less active social interaction, mice in the Sept5 amygdala group reactively exhibit more active social interaction. However, our data do not support this interpretation; although such a trend would show as a negative correlation in time spent in social interaction between Sept5 amygdala mice and stimulus mice, there was a statistically non-significant trend of ‘positive’ correlation in time spent in active social interaction at the second session between the two groups (r2 = 0.330). Another possibility is that the Sept5 amygdala group had a heightened level of anxiety during the first session and exhibited a higher level of social interaction when anxiety presumably subsided by the second session. Although we did not see any phenotypic difference between the EGFP and Sept5 amygdala groups in anxiety-related traits in the elevated plus maze, anxiety seen in this task—where the source of anxiety is thought to originate from an open area and height—is not identical to what is expected in social interaction. Given that Sept5 elevations in these two limbic structures did not affect social interaction in the same way, more work is needed to identify the precise manner through which these structures mediate social behavior.
Post-weaning individual housing selectively raised Sept5 levels in the amygdala. This is consistent with our observation that Sept5 overexpression in the amygdala increases basal levels of affiliative social interaction. However, virally guided overexpression of Sept5 in the hippocampus also increased social interaction, but the housing condition did not induce a detectable increase in Sept5 levels in this structure. Our virally guided overexpression of Sept5 was confined to the dorsal hippocampus but was sufficient to raise levels of social interaction. As our hippocampal tissue for western blotting included the ventral and dorsal hippocampus and subiculum, it might not be sufficiently sensitive to detect highly local up-regulation of Sept5 within the hippocampus.
Although post-weaning individual housing and virally guided overexpression selectively increased Sept5 in the amygdala, these two treatments did not produce the identical behavioral effects. Moreover, constitutive sept5 deficiency in KO mice and environmentally elevated Sept5 in the amygdala did not induce clear-cut opposing behavioral phenotypes. These results are hardly surprising, as constitutive Sept5 deficiency and post-weaning individual housing are likely to induce many non-identical molecular alterations in the brain. In fact, this environmental manipulation, unlike selective overexpression of Sept5 in the amygdala, additionally induced many behavioral signs of reduced anxiety and stress. Notwithstanding these differences, Sept5 might be one of the converging points through which genetic deficiency and stress-related environmental factors impact social behaviors. One way to ameliorate social interaction deficits in 22q11.2 hemizygous patients could be to elevate the expression of Sept5 from the remaining copy of 22q11.2 by reducing stress levels.
The hippocampus is structurally altered in both ASD (58,59) and 22q11.2 hemizygous patients (60). The amygdala has been implicated as a site whose activity alteration is associated with defective social cognition in individuals with ASD and schizophrenia (61) and with 22q11.2 hemizygous deletions (62). While performing tasks that require social perception, altered activation patterns have been noted in the fusiform cortex and amygdala of individuals with 22q11.2 hemizygosity (62). A future challenge is to identify the precise network of structures through which Sept5 acts as a determinant for social cognition.
‘Connectivity’ is an emerging theoretical construct to explain diverse phenotypes seen in patients with schizophrenia and ASD (63–65). Synaptic contact can be considered a structural basis of connectivity between neurons. Sept5-containing filaments, together with syntaxin-1A, are localized at presynaptic terminals, and Sept5 is thought to regulate neurotransmitter release at synapses together with the SNARE complex (66–68). Moreover, in vitro knockdown of Sept5 has been shown to alter axon length and dendritic complexity (69,70). Proteins located at the trans-synaptic (i.e. neurexins) and post-synaptic (neuroligin and Shank3) sites are implicated in ASD- and schizophrenia-related phenotypes in other genetic mouse models (71). Our data add Sept5 as a pre-synaptic contributor to the synaptopathology of schizophrenia and ASD and support the notion that the symptomatic elements of schizophrenia and ASD stem from improperly formed or functioning synapses (71).
MATERIALS AND METHODS
Mice
Male C57BL/6J mice (Jackson Laboratories, Bar Harbor, ME, USA) and congenic Sept5 KO and WT littermates were used. Congenic Sept5 KO and WT mice were developed by backcrossing the original Sept5 heterozygous mice (72) to C57BL/6J mice for 10 generations. Male Sept5 WT, HT and KO littermates were housed together in cages (27 cm × 16 cm × 12.5 cm). Male C57BL/6J mice, housed in a group of three to four mice, were infused with an EGFP lentiviral vector or a Sept5-EGFP lentiviral vector (see Lentiviral vector and Surgery in what follows). Mice were given a week to recover before being tested. For the groups that were used to evaluate the impact of post-weaning housing conditions, 3-week-old male, C57BL/6J mice were randomly assigned to either group (n = 3–4) housing or individual housing and they were used for behavioral testing or killed for protein analysis after 6 weeks in these housing conditions. Mice remained in their housing condition until the end of testing. All mice were given access to food and water ad libitum and kept under a 14 h:10 h light/dark cycle, unless otherwise specified. Animal handling and use followed a protocol approved by the Animal Care and Use Committee of Albert Einstein College of Medicine, in accordance with National Institutes of Health guidelines.
Behavioral testing
Behavioral testing began at 2 months of age. This age point in mice represents the developmental phase after achieving sexual maturation around 1 month of age and before reaching mature adulthood at 3 months of age; many biological processes in the brain continue to change during this period (56,73). Mice were tested in a battery of assays based on our standard procedure (45,56,73–75).
Congenic Sept5 mice and C57BL/6J mice were tested in the following order: social interaction, elevated plus maze and open field; two separate groups were tested either on novel object recognition or the buried food test. C57BL/6J mice were tested in the following order: social interaction, novel object recognition, open field, elevated plus maze and buried food test. A randomized order of assays is problematic, as a more stressful task affects performance on subsequent tasks (76). Therefore, tasks were given in order from less to more stressful, with a 1-day interval between tests so that repeated testing did not affect phenotypic expression. All testing was done during the light phase of the cycle. An hour before testing, mice were brought into a room outside the testing room. An observer was blinded to genotype and experimental groups.
Social interaction
We followed our protocol (45,56,73). Two mice were taken from their home cages and individually placed in new, separate cages for 30 min. Mice were next placed together in a new, third cage for two 5 min sessions separated by a 30 min interval: the second session evaluated recognition memory. Pairings were always made between a WT mouse and a KO mouse taken from different litters housed in different cages. For the viral overexpression experiment, group-housed, non-operated C57BL/6J mice were paired with EGFP- or Sept5-infused mice housed in different cages. For the housing experiment, group-housed C57BL/6J mice were paired with C57BL/6J mice that were individually or group-housed in different cages. Social behaviors were scored for active and passive behaviors by experimenters blinded to housing condition. Active behaviors included aggressive (i.e. tail rattle, bites, kicks, sideways offense, boxing, wrestling, mounting and pursuit) and affiliative (i.e. olfactory investigation and allogrooming) behaviors. Passive behaviors were escape, leap, side-by-side position and submissive posture. Time spent in aggressive, affiliative and passive interactions were analyzed separately.
Elevated plus maze
A mouse was placed on the central platform of the elevated plus maze facing one of the open arms. Behavior was recorded for 5 min. An observer blinded to the experimental condition rated the recordings. We analyzed the percentage of time spent in and of entrances to the open arms over the total time in and entrances to both the open and closed arms.
Locomotor activity
Four identical, automated open-field apparatuses (Truscan, Coulbourn Instruments, PA, USA) were used to measure spontaneous locomotor activity and thigmotaxis. Each 30 min session was analyzed in six 5 min bins. To assess horizontal locomotor activity and thigmotaxis, distance traveled (cm) in the entire open field (26 cm × 26 cm) and time spent (s) in the marginal area along the walls (a 4 cm band extending from the wall), respectively, were analyzed.
Novel object contact
At the beginning of testing, an individual mouse was placed in a new home cage for 30 min. The mouse was then moved into another home cage that contained a modified falcon tube (3 cm diameter × 8.5 cm length) during two 5 min sessions with a 30 min intersession interval. All activity was recorded by a digital camera placed above the home cage. An observer blinded to experimental condition scored the amount of time each mouse spent contacting the object. Sniffing and physical contact with the object were used for analysis.
Buried food test
For 2 consecutive days, a piece of the stimulus food (Honey Teddy Grahams, Nabisco, East Hanover, NJ, USA) was placed in their home cages in the presence of their normal mouse chow. On Day 3, normal chow was removed from the cage top and testing began 20–22 h later. A mouse was placed in a novel home cage containing 3 cm deep bedding (testing cage). After 5 min, the mouse was placed in another clean home cage. The stimulus food was buried about 1 cm beneath the 3 cm deep bedding in the test cage, and the mouse was then returned to the test cage. The latency to start consumption of the stimulus food was recorded. If the mouse did not find the stimulus food in 5 min, testing was terminated and that mouse received a latency of 300 s.
Lentiviral vector
The adult form of the mouse Sept5_v1 gene (77) (RIKEN, Yokohama, Japan) was inserted into a lentiviral vector containing an internal ribosome entry site (IRES) and EGFP. Sept5 was polymerase chain reaction (PCR)-amplified with primers with attB1/attB2 and introduced into a pDONR-221 vector by BP clonase reaction (Invitrogen, Grand Island, NY, USA). The IRES-EGFP sequence (Clontech, Mountain View, CA, USA) was amplified with primers with attB2/attB3 and introduced into a pDONR-P2R-P3 vector (Invitrogen) by BP reaction. The promoter region of calcium/calmodulin-dependent protein kinase II alpha (CamkIIα), a neuron-specific protein (78), was PCR-amplified with primers with attB4/attB1r and introduced into a pDONR-P4P1r vector (Invitrogen) by BP reaction. The CamkIIα promoter element, Sept5, and IRES-EGFP were introduced into a pLenti6.4/R4R2/V5-DEST vector (Invitrogen). A plasmid was generated by inserting PCamkIIα-Sept5-IRES-EGFP or PCamkIIα-IRES-EGFP. Using 293T cells transfected with the plasmid, we confirmed that Sept5 protein is expressed (Supplementary Material, Fig. S3A). Viral particles were generated by co-transfection of 293FT cells (Invitrogen) by the pLenti6.4/R4R2/V5-DEST-based plasmid-containing PCamkIIα-Sept5-IRES-EGFP or PCamkIIα-IRES-EGFP and three packaging helper plasmids, using the calcium phosphate method (79). Seventy-two hours after transfection, viral particles were collected and resuspended in phosphate-buffered saline. Viral titers were estimated by counting surviving cells following blasticidin.
Surgery
Male C57BL/6J mice were anesthetized with sodium pentobarbital [40 mg/kg, intraperitoneal (i.p.)] and were then placed in a stereotaxic apparatus (MyNeuroLab, Richmond, IL, USA). A 2.5 μl Hamilton syringe (Hamilton, Reno, NV, USA) with a 30-gauge needle was used to inject 0.3 μl of the EGFP lentiviral vector (2.30 × 106 TU/ml) or the Sept5-EGFP lentiviral vector (6.40 × 106 TU/ml) for a period of 5 min and the needle was left in place for another 5 min. Bilateral injections were made into the hippocampus (anterior-posterior (AP), −1.82 mm; medial-lateral (ML), 2.25 mm; dorsal-ventral (DV), 2.8 mm), amygdala (AP, −1.82 mm; ML, 3.0 mm; DV, 5.5 mm) or somatosensory cortex (AP, −1.82 mm; ML, 3.0 mm; DV, 2.0 mm). Mice were given a week to recover before being tested.
Immunohistochemistry
After behavioral analysis was completed, animals were anesthetized with sodium pentobarbital (62.5 mg/kg, i.p.) and perfused with saline followed by 4% paraformaldehyde. Brains were coronally sliced at 50 µm, covering the entire antero-posterior extent of the hippocampus and amygdala, and were examined for EGFP. A separate set of sections were immunohistochemically stained for NeuN (mouse anti-NeuN antibody, 1:200, Millipore) as a marker of neuron together with Texas-red-conjugated anti-mouse IgG (1:500, Jackson ImmunoResearch, West Grove, PA, USA) or Alexa594-conjugated anti-mouse IgG (1:500, Invitrogen) secondary antibody. Signals were analyzed under an epi-fluorescent microscope (Zeiss) and a confocal microscope (Leica).
To evaluate the extent of virally expressed Sept5, we used a small set of Sept5 KO mice. KO mice received infusions of the Sept5-EGFP lentiviral vector into the hippocampus or basolateral amygdaloid complex and killed 1 week later. One set of sections were mounted and examined for EGFP. A separate set of adjacent sections were immunohistochemically stained with either a goat polyclonal Sept5 antibody (1:1000, Santa Cruz Biotechnology, Santa Cruz, CA, USA) or a mouse monoclonal Sept5 antibody (1:10 000, Assay Designs, Ann Arbor, MI, USA), following our standard Nickel-intensified DAB-staining procedure (80–82). EGFP and Sept5 signals were analyzed under an epi-fluorescent microscope (Zeiss) and a light microscope (Zeiss), respectively. The extents of EGFP and Sept5 protein signals overlapped (Supplementary Material, Fig. S2B–E), indicating that EGFP reliably represents the extent of virally expressed Sept5.
Western blot
Cervical dislocation was used to sacrifice mice for western blotting. The following brain areas were dissected out in saline on ice: prefrontal cortex, nucleus accumbens, caudate-putamen, amygdala, hippocampus, substantia nigra and VTA. Tissues were homogenized by sonication in electrophoretic mobility shift assay buffer. Sixty micrograms of sample, as determined by Bradford assay, was loaded into a 10% acrylamide gel. Transferred proteins on membranes were labeled with a goat polyclonal Sept5 antibody (1:200, Santa Cruz Biotechnology), followed by a donkey anti-goat horseradish peroxidase-conjugated IgG secondary antibody (1:5000, Santa Cruz Biotechnology). A rat monoclonal anti-tubulin antibody (1:5000, Abcam, Cambridge, MA, USA) was used in conjunction with goat anti-rat horseradish peroxidase-conjugated IgG secondary antibody (1:5000, Santa Cruz Biotechnology) to determine the amount of protein loaded. For the detection of Sept5 in 293T cells transfected with the plasmid, a mouse anti-Septin 5 (SP18) monoclonal antibody (1:200, Santa Cruz Biotechnology) and anti-mouse horseradish peroxidase-conjugated IgG secondary antibody (1:10 000, Vector) were used. Specific bands were visualized using SuperSignal West (Thermo Scientific, Waltham, MA, USA). Absorption was then measured using a GS-700 densitometer (Bio-Rad, Hercules, CA, USA) with the Quantity One software. The levels of Sept5 were normalized to tubulin and percentages of levels over standard housing were calculated for analysis.
Statistical analysis
All data are presented as the mean ± standard error of the mean (SEM). Statistical significance was determined by analysis of variance followed by Newman–Keuls post hoc comparisons. When two groups were compared, Student's t-test was used. The minimum level of significance was set at 5%.
SUPPLEMENTARY MATERIAL
Supplementary Material is available at HMG online.
ACKNOWLEDGEMENTS
We thank Dr Dan Scott for blinding experimental groups.
Conflict of Interest statement. None declared.
FUNDING
This work was supported by the National Institutes of Health (HD05311), National Alliance for Research on Schizophrenia and Depression Independent Investigator Award and the Maltz Foundation to N.H.
References
- 1.Penn D.L., Corrigan P.W., Bentall R.P., Racenstein J.M., Newman L. Social cognition in schizophrenia. Psychol. Bull. 1997;121:114–132. doi: 10.1037/0033-2909.121.1.114. [DOI] [PubMed] [Google Scholar]
- 2.Sigman M. The Emanuel Miller Memorial Lecture 1997. Change and continuity in the development of children with autism. J. Child Psychol. Psychiatry. 1998;39:817–827. [PubMed] [Google Scholar]
- 3.Swillen A., Devriendt K., Legius E., Eyskens B., Dumoulin M., Gewillig M., Fryns J.P. Intelligence and psychosocial adjustment in velocardiofacial syndrome: a study of 37 children and adolescents with VCFS. J. Med. Genet. 1997;34:453–458. doi: 10.1136/jmg.34.6.453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Golding-Kushner K.J., Weller G., Shprintzen R.J. Velo-cardio-facial syndrome: language and psychological profiles. J. Craniofac. Genet. Dev. Biol. 1985;5:259–266. [PubMed] [Google Scholar]
- 5.Baker K.D., Skuse D.H. Adolescents and young adults with 22q11 deletion syndrome: psychopathology in an at-risk group. Br. J. Psychiatry. 2005;186:115–120. doi: 10.1192/bjp.186.2.115. [DOI] [PubMed] [Google Scholar]
- 6.Heineman-de Boer J.A., Van Haelst M.J., Cordia-de H.M., Beemer F.A. Behavior problems and personality aspects of 40 children with velo-cardio-facial syndrome. Genet. Couns. 1999;10:89–93. [PubMed] [Google Scholar]
- 7.Kiley-Brabeck K., Sobin C. Social skills and executive function deficits in children with the 22q11 deletion syndrome. Appl. Neuropsychol. 2006;13:258–268. doi: 10.1207/s15324826an1304_7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Niklasson L., Rasmussen P., Oskarsdottir S., Gillberg C. Chromosome 22q11 deletion syndrome (CATCH 22): neuropsychiatric and neuropsychological aspects. Dev. Med. Child Neurol. 2002;44:44–50. doi: 10.1017/s0012162201001645. [DOI] [PubMed] [Google Scholar]
- 9.Woodin M., Wang P.P., Aleman D., Donald-McGinn D., Zackai E., Moss E. Neuropsychological profile of children and adolescents with the 22q11.2 microdeletion. Genet. Med. 2001;3:34–39. doi: 10.1097/00125817-200101000-00008. [DOI] [PubMed] [Google Scholar]
- 10.Shprintzen R.J., Goldberg R., Golding-Kushner K.J., Marion R.W. Late-onset psychosis in the velo-cardio-facial syndrome. Am. J. Med. Genet. 1992;42:141–142. doi: 10.1002/ajmg.1320420131. [DOI] [PubMed] [Google Scholar]
- 11.Pulver A.E., Nestadt G., Goldberg R., Shprintzen R.J., Lamacz M., Wolyniec P.S., Morrow B., Karayiorgou M., Antonarakis S.E., Housman D. Psychotic illness in patients diagnosed with velo-cardio-facial syndrome and their relatives. J. Nerv. Ment. Dis. 1994;182:476–478. doi: 10.1097/00005053-199408000-00010. [DOI] [PubMed] [Google Scholar]
- 12.Murphy K.C., Jones L.A., Owen M.J. High rates of schizophrenia in adults with velo-cardio-facial syndrome. Arch. Gen. Psychiatry. 1999;56:940–945. doi: 10.1001/archpsyc.56.10.940. [DOI] [PubMed] [Google Scholar]
- 13.Bassett A.S., Hodgkinson K., Chow E.W., Correia S., Scutt L.E., Weksberg R. 22q11 deletion syndrome in adults with schizophrenia. Am. J. Med. Genet. 1998;81:328–337. [PMC free article] [PubMed] [Google Scholar]
- 14.Gothelf D., Presburger G., Zohar A.H., Burg M., Nahmani A., Frydman M., Shohat M., Inbar D., Aviram-Goldring A., Yeshaya J., et al. Obsessive-compulsive disorder in patients with velocardiofacial (22q11 deletion) syndrome. Am. J. Med. Genet. B Neuropsychiatr. Genet. 2004;126:99–105. doi: 10.1002/ajmg.b.20124. [DOI] [PubMed] [Google Scholar]
- 15.Karayiorgou M., Morris M.A., Morrow B., Shprintzen R.J., Goldberg R., Borrow J., Gos A., Nestadt G., Wolyniec P.S., Lasseter V.K. Schizophrenia susceptibility associated with interstitial deletions of chromosome 22q11. Proc. Natl Acad. Sci. USA. 1995;92:7612–7616. doi: 10.1073/pnas.92.17.7612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Bassett A.S., Marshall C.R., Lionel A.C., Chow E.W., Scherer S.W. Copy number variations and risk for schizophrenia in 22q11.2 deletion syndrome. Hum. Mol. Genet. 2008;17:4045–4053. doi: 10.1093/hmg/ddn307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Debbane M., Glaser B., David M.K., Feinstein C., Eliez S. Psychotic symptoms in children and adolescents with 22q11.2 deletion syndrome: neuropsychological and behavioral implications. Schizophr. Res. 2006;84:187–193. doi: 10.1016/j.schres.2006.01.019. [DOI] [PubMed] [Google Scholar]
- 18.Fine S.E., Weissman A., Gerdes M., Pinto-Martin J., Zackai E.H., Donald-McGinn D.M., Emanuel B.S. Autism spectrum disorders and symptoms in children with molecularly confirmed 22q11.2 deletion syndrome. J. Autism Dev. Disord. 2005;35:461–470. doi: 10.1007/s10803-005-5036-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Vorstman J.A., Morcus M.E., Duijff S.N., Klaassen P.W., Heineman-de Boer J.A., Beemer F.A., Swaab H., Kahn R.S., van Engeland H. The 22q11.2 deletion in children: high rate of autistic disorders and early onset of psychotic symptoms. J. Am. Acad. Child Adolesc. Psychiatry. 2006;45:1104–1113. doi: 10.1097/01.chi.0000228131.56956.c1. [DOI] [PubMed] [Google Scholar]
- 20.Antshel K.M., Aneja A., Strunge L., Peebles J., Fremont W.P., Stallone K., Abdulsabur N., Higgins A.M., Shprintzen R.J., Kates W.R. Autistic spectrum disorders in velo-cardio facial syndrome (22q11.2 deletion) J. Autism Dev. Disord. 2007;37:1776–1786. doi: 10.1007/s10803-006-0308-6. [DOI] [PubMed] [Google Scholar]
- 21.Kates W.R., Antshel K.M., Fremont W.P., Shprintzen R.J., Strunge L.A., Burnette C.P., Higgins A.M. Comparing phenotypes in patients with idiopathic autism to patients with velocardiofacial syndrome (22q11 DS) with and without autism. Am. J. Med. Genet. A. 2007;143A:2642–2650. doi: 10.1002/ajmg.a.32012. [DOI] [PubMed] [Google Scholar]
- 22.Niklasson L., Rasmussen P., Oskarsdottir S., Gillberg C. Autism, ADHD, mental retardation and behavior problems in 100 individuals with 22q11 deletion syndrome. Res. Dev. Disabil. 2009;30:763–773. doi: 10.1016/j.ridd.2008.10.007. [DOI] [PubMed] [Google Scholar]
- 23.Chow E.W., Watson M., Young D.A., Bassett A.S. Neurocognitive profile in 22q11 deletion syndrome and schizophrenia. Schizophr. Res. 2006;87:270–278. doi: 10.1016/j.schres.2006.04.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Sebat J., Lakshmi B., Malhotra D., Troge J., Lese-Martin C., Walsh T., Yamrom B., Yoon S., Krasnitz A., Kendall J., et al. Strong association of de novo copy number mutations with autism. Science. 2007;316:445–449. doi: 10.1126/science.1138659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Szatmari P., Paterson A.D., Zwaigenbaum L., Roberts W., Brian J., Liu X.Q., Vincent J.B., Skaug J.L., Thompson A.P., Senman L., et al. Mapping autism risk loci using genetic linkage and chromosomal rearrangements. Nat. Genet. 2007;39:319–328. doi: 10.1038/ng1985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Marshall C.R., Noor A., Vincent J.B., Lionel A.C., Feuk L., Skaug J., Shago M., Moessner R., Pinto D., Ren Y., et al. Structural variation of chromosomes in autism spectrum disorder. Am. J. Hum. Genet. 2008;82:477–488. doi: 10.1016/j.ajhg.2007.12.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Christian S.L., Brune C.W., Sudi J., Kumar R.A., Liu S., Karamohamed S., Badner J.A., Matsui S., Conroy J., McQuaid D., et al. Novel submicroscopic chromosomal abnormalities detected in autism spectrum disorder. Biol. Psychiatry. 2008;63:1111–1117. doi: 10.1016/j.biopsych.2008.01.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Cai G., Edelmann L., Goldsmith J.E., Cohen N., Nakamine A., Reichert J.G., Hoffman E.J., Zurawiecki D.M., Silverman J.M., Hollander E., et al. Multiplex ligation-dependent probe amplification for genetic screening in autism spectrum disorders: efficient identification of known microduplications and identification of a novel microduplication in ASMT. BMC Med. Genomics. 2008;1:50. doi: 10.1186/1755-8794-1-50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Bucan M., Abrahams B.S., Wang K., Glessner J.T., Herman E.I., Sonnenblick L.I., Alvarez Retuerto A.I., Imielinski M., Hadley D., Bradfield J.P., et al. Genome-wide analyses of exonic copy number variants in a family-based study point to novel autism susceptibility genes. PLoS Genet. 2009;5:e1000536. doi: 10.1371/journal.pgen.1000536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Pinto D., Pagnamenta A.T., Klei L., Anney R., Merico D., Regan R., Conroy J., Magalhaes T.R., Correia C., Abrahams B.S., et al. Functional impact of global rare copy number variation in autism spectrum disorders. Nature. 2010;466:368–372. doi: 10.1038/nature09146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Itsara A., Wu H., Smith J.D., Nickerson D.A., Romieu I., London S.J., Eichler E.E. De novo rates and selection of large copy number variation. Genome Res. 2010;20:1469–1481. doi: 10.1101/gr.107680.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Sanders S.J., Ercan-Sencicek A.G., Hus V., Luo R., Murtha M.T., Moreno-De-Luca D., Chu S.H., Moreau M.P., Gupta A.R., Thomson S.A., et al. Multiple recurrent de novo CNVs, including duplications of the 7q11.23 Williams syndrome region, are strongly associated with autism. Neuron. 2011;70:863–885. doi: 10.1016/j.neuron.2011.05.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Guilmatre A., Dubourg C., Mosca A.L., Legallic S., Goldenberg A., Drouin-Garraud V., Layet V., Rosier A., Briault S., Bonnet-Brilhault F., et al. Recurrent rearrangements in synaptic and neurodevelopmental genes and shared biologic pathways in schizophrenia, autism, and mental retardation. Arch. Gen. Psychiatry. 2009;66:947–956. doi: 10.1001/archgenpsychiatry.2009.80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.International Schizophrenia Consortium. Rare chromosomal deletions and duplications increase risk of schizophrenia. Nature. 2008;455:237–241. doi: 10.1038/nature07239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Stefansson H., Rujescu D., Cichon S., Pietilainen O.P., Ingason A., Steinberg S., Fossdal R., Sigurdsson E., Sigmundsson T., Buizer-Voskamp J.E., et al. Large recurrent microdeletions associated with schizophrenia. Nature. 2008;455:232–236. doi: 10.1038/nature07229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Xu B., Roos J.L., Levy S., van Rensburg E.J., Gogos J.A., Karayiorgou M. Strong association of de novo copy number mutations with sporadic schizophrenia. Nat. Genet. 2008;40:880–885. doi: 10.1038/ng.162. [DOI] [PubMed] [Google Scholar]
- 37.Kirov G., Grozeva D., Norton N., Ivanov D., Mantripragada K.K., Holmans P., Craddock N., Owen M.J., O'Donovan M.C. Support for the involvement of large copy number variants in the pathogenesis of schizophrenia. Hum. Mol. Genet. 2009;18:1497–1503. doi: 10.1093/hmg/ddp043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Need A.C., Attix D.K., McEvoy J.M., Cirulli E.T., Linney K.L., Hunt P., Ge D., Heinzen E.L., Maia J.M., Shianna K.V., et al. A genome-wide study of common SNPs and CNVs in cognitive performance in the CANTAB. Hum. Mol. Genet. 2009;18:4650–4661. doi: 10.1093/hmg/ddp413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Bassett A.S., Costain G., Fung W.L., Russell K.J., Pierce L., Kapadia R., Carter R.F., Chow E.W., Forsythe P.J. Clinically detectable copy number variations in a Canadian catchment population of schizophrenia. J. Psychiatr. Res. 2010;45:1005–1009. doi: 10.1016/j.jpsychires.2010.06.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Levinson D.F., Duan J., Oh S., Wang K., Sanders A.R., Shi J., Zhang N., Mowry B.J., Olincy A., Amin F., et al. Copy number variants in schizophrenia: confirmation of five previous findings and new evidence for 3q29 microdeletions and VIPR2 duplications. Am. J. Psychiatry. 2011;168:302–316. doi: 10.1176/appi.ajp.2010.10060876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Buizer-Voskamp J.E., Muntjewerff J.W., Strengman E., Sabatti C., Stefansson H., Vorstman J.A., Ophoff R.A. Genome-wide analysis shows increased frequency of copy number variation deletions in Dutch schizophrenia patients. Biol. Psychiatry. 2011;70:655–662. doi: 10.1016/j.biopsych.2011.02.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Grozeva D., Conrad D.F., Barnes C.P., Hurles M., Owen M.J., O'Donovan M.C., Craddock N., Kirov G. Independent estimation of the frequency of rare CNVs in the UK population confirms their role in schizophrenia. Schizophr. Res. 2012;135:1–7. doi: 10.1016/j.schres.2011.11.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Hiroi N., Zhu H., Lee M., Funke B., Arai M., Itokawa M., Kucherlapati R., Morrow B., Sawamura T., Agatsuma S. A 200-kb region of human chromosome 22q11.2 confers antipsychotic-responsive behavioral abnormalities in mice. Proc. Natl Acad. Sci. USA. 2005;102:19132–19137. doi: 10.1073/pnas.0509635102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Paylor R., Glaser B., Mupo A., Ataliotis P., Spencer C., Sobotka A., Sparks C., Choi C.H., Oghalai J., Curran S., et al. Tbx1 haploinsufficiency is linked to behavioral disorders in mice and humans: implications for 22q11 deletion syndrome. Proc. Natl Acad. Sci. USA. 2006;103:7729–7734. doi: 10.1073/pnas.0600206103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Suzuki G., Harper K.M., Hiramoto T., Sawamura T., Lee M., Kang G., Tanigaki K., Buell M., Geyer M.A., Trimble W.S., et al. Sept5 deficiency exerts pleiotropic influence on affective behaviors and cognitive functions in mice. Hum. Mol. Genet. 2009;18:1652–1660. doi: 10.1093/hmg/ddp086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Bartsch I., Sandrock K., Lanza F., Nurden P., Hainmann I., Pavlova A., Greinacher A., Tacke U., Barth M., Busse A., et al. Deletion of human GP1BB and SEPT5 is associated with Bernard-Soulier syndrome, platelet secretion defect, polymicrogyria, and developmental delay. Thromb. Haemost. 2011;106:475–483. doi: 10.1160/TH11-05-0305. [DOI] [PubMed] [Google Scholar]
- 47.Caltagarone J., Rhodes J., Honer W.G., Bowser R. Localization of a novel septin protein, hCDCrel-1, in neurons of human brain. Neuroreport. 1998;9:2907–2912. doi: 10.1097/00001756-199808240-00042. [DOI] [PubMed] [Google Scholar]
- 48.Kinoshita A., Noda M., Kinoshita M. Differential localization of septins in the mouse brain. J. Comp. Neurol. 2000;428:223–239. doi: 10.1002/1096-9861(20001211)428:2<223::aid-cne3>3.0.co;2-m. [DOI] [PubMed] [Google Scholar]
- 49.Michalon A., Koshibu K., Baumgartel K., Spirig D.H., Mansuy I.M. Inducible and neuron-specific gene expression in the adult mouse brain with the rtTA2S-M2 system. Genesis. 2005;43:205–212. doi: 10.1002/gene.20175. [DOI] [PubMed] [Google Scholar]
- 50.Beaton E.A., Simon T.J. How might stress contribute to increased risk for schizophrenia in children with chromosome 22q11.2 deletion syndrome? J. Neurodev. Disord. 2011;3:68–75. doi: 10.1007/s11689-010-9069-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Brain P. What does individual housing mean to a mouse? Life Sci. 1975;16:187–200. doi: 10.1016/0024-3205(75)90017-x. [DOI] [PubMed] [Google Scholar]
- 52.Voikar V., Polus A., Vasar E., Rauvala H. Long-term individual housing in C57BL/6J and DBA/2 mice: assessment of behavioral consequences. Genes Brain Behav. 2005;4:240–252. doi: 10.1111/j.1601-183X.2004.00106.x. [DOI] [PubMed] [Google Scholar]
- 53.Terranova M.L., Laviola G., Alleva E. Ontogeny of amicable social behavior in the mouse: gender differences and ongoing isolation outcomes. Dev. Psychobiol. 1993;26:467–481. doi: 10.1002/dev.420260805. [DOI] [PubMed] [Google Scholar]
- 54.Panksepp J.B., Jochman K.A., Kim J.U., Koy J.J., Wilson E.D., Chen Q., Wilson C.R., Lahvis G.P. Affiliative behavior, ultrasonic communication and social reward are influenced by genetic variation in adolescent mice. PLoS One. 2007;2:e351. doi: 10.1371/journal.pone.0000351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Abramov U., Raud S., Koks S., Innos J., Kurrikoff K., Matsui T., Vasar E. Targeted mutation of CCK(2) receptor gene antagonises behavioural changes induced by social isolation in female, but not in male mice. Behav. Brain Res. 2004;155:1–11. doi: 10.1016/j.bbr.2004.03.027. [DOI] [PubMed] [Google Scholar]
- 56.Hiramoto T., Kang G., Suzuki G., Satoh Y., Kucherlapati R., Watanabe Y., Hiroi N. Tbx1: identification of a 22q11.2 gene as a risk factor for autism spectrum disorder in a mouse model. Hum. Mol. Genet. 2011;20:4775–4785. doi: 10.1093/hmg/ddr404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Flaherty L., Bolivar V. Congenic and consomic strains. In: Jones B.C., Mormede P., editors. Neurobehavioral Genetics. New York, NY: Taylor & Francis; 2007. pp. 115–127. [Google Scholar]
- 58.Rojas D.C., Smith J.A., Benkers T.L., Camou S.L., Reite M.L., Rogers S.J. Hippocampus and amygdala volumes in parents of children with autistic disorder. Am. J. Psychiatry. 2004;161:2038–2044. doi: 10.1176/appi.ajp.161.11.2038. [DOI] [PubMed] [Google Scholar]
- 59.Rojas D.C., Peterson E., Winterrowd E., Reite M.L., Rogers S.J., Tregellas J.R. Regional gray matter volumetric changes in autism associated with social and repetitive behavior symptoms. BMC Psychiatry. 2006;6:56. doi: 10.1186/1471-244X-6-56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Tan G.M., Arnone D., McIntosh A.M., Ebmeier K.P. Meta-analysis of magnetic resonance imaging studies in chromosome 22q11.2 deletion syndrome (velocardiofacial syndrome) Schizophr. Res. 2009;115:173–181. doi: 10.1016/j.schres.2009.09.010. [DOI] [PubMed] [Google Scholar]
- 61.Pinkham A.E., Hopfinger J.B., Pelphrey K.A., Piven J., Penn D.L. Neural bases for impaired social cognition in schizophrenia and autism spectrum disorders. Schizophr. Res. 2008;99:164–175. doi: 10.1016/j.schres.2007.10.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Andersson F., Glaser B., Spiridon M., Debbane M., Vuilleumier P., Eliez S. Impaired activation of face processing networks revealed by functional magnetic resonance imaging in 22q11.2 deletion syndrome. Biol. Psychiatry. 2008;63:49–57. doi: 10.1016/j.biopsych.2007.02.022. [DOI] [PubMed] [Google Scholar]
- 63.Winterer G., Weinberger D.R. Genes, dopamine and cortical signal-to-noise ratio in schizophrenia. Trends Neurosci. 2004;27:683–690. doi: 10.1016/j.tins.2004.08.002. [DOI] [PubMed] [Google Scholar]
- 64.Zoghbi H.Y. Postnatal neurodevelopmental disorders: meeting at the synapse. Science. 2003;302:826–830. doi: 10.1126/science.1089071. [DOI] [PubMed] [Google Scholar]
- 65.Kaufmann W.E., Moser H.W. Dendritic anomalies in disorders associated with mental retardation. Cereb. Cortex. 2000;10:981–991. doi: 10.1093/cercor/10.10.981. [DOI] [PubMed] [Google Scholar]
- 66.Beites C.L., Xie H., Bowser R., Trimble W.S. The septin CDCrel-1 binds syntaxin and inhibits exocytosis. Nat. Neurosci. 1999;2:434–439. doi: 10.1038/8100. [DOI] [PubMed] [Google Scholar]
- 67.Yang Y.M., Fedchyshyn M.J., Grande G., Aitoubah J., Tsang C.W., Xie H., Ackerley C.A., Trimble W.S., Wang L.Y. Septins regulate developmental switching from microdomain to nanodomain coupling of Ca(2+) influx to neurotransmitter release at a central synapse. Neuron. 2010;67:100–115. doi: 10.1016/j.neuron.2010.06.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Dong Z., Ferger B., Paterna J.C., Vogel D., Furler S., Osinde M., Feldon J., Bueler H. Dopamine-dependent neurodegeneration in rats induced by viral vector-mediated overexpression of the parkin target protein, CDCrel-1. Proc. Natl Acad. Sci. USA. 2003;100:12438–12443. doi: 10.1073/pnas.2132992100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Tada T., Simonetta A., Batterton M., Kinoshita M., Edbauer D., Sheng M. Role of Septin cytoskeleton in spine morphogenesis and dendrite development in neurons. Curr. Biol. 2007;17:1752–1758. doi: 10.1016/j.cub.2007.09.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Tsang C.W., Estey M.P., Diciccio J.E., Xie H., Patterson D., Trimble W.S. Characterization of presynaptic septin complexes in mammalian hippocampal neurons. Biol. Chem. 2011;392:739–749. doi: 10.1515/BC.2011.077. [DOI] [PubMed] [Google Scholar]
- 71.Ramocki M.B., Zoghbi H.Y. Failure of neuronal homeostasis results in common neuropsychiatric phenotypes. Nature. 2008;455:912–918. doi: 10.1038/nature07457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Peng X.R., Jia Z., Zhang Y., Ware J., Trimble W.S. The septin CDCrel-1 is dispensable for normal development and neurotransmitter release. Mol. Cell. Biol. 2002;22:378–387. doi: 10.1128/MCB.22.1.378-387.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Suzuki G., Harper K.M., Hiramoto T., Funke B., Lee M., Kang G., Buell M., Geyer M.A., Kucherlapati R., Morrow B., et al. Over-expression of a human chromosome 22q11.2 segment including TXNRD2, COMT, and ARVCF developmentally affects incentive learning and working memory in mice. Hum. Mol. Genet. 2009;18:3914–3925. doi: 10.1093/hmg/ddp334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Zhu H., Lee M., Agatsuma S., Hiroi N. Pleiotropic impact of constitutive fosB inactivation on nicotine-induced behavioral alterations and stress-related traits in mice. Hum. Mol. Genet. 2007;16:820–836. doi: 10.1093/hmg/ddm027. [DOI] [PubMed] [Google Scholar]
- 75.Agatsuma S., Lee M., Zhu H., Chen K., Shih J.C., Seif I., Hiroi N. Monoamine oxidase A knockout mice exhibit impaired nicotine preference but normal responses to novel stimuli. Hum. Mol. Genet. 2006;15:2721–2731. doi: 10.1093/hmg/ddl206. [DOI] [PubMed] [Google Scholar]
- 76.McIlwain K.L., Merriweather M.Y., Yuva-Paylor L.A., Paylor R. The use of behavioral test batteries: effects of training history. Physiol. Behav. 2001;73:705–717. doi: 10.1016/s0031-9384(01)00528-5. [DOI] [PubMed] [Google Scholar]
- 77.Asada A., Takahashi J., Taniguchi M., Yamamoto H., Kimura T., Saito T., Hisanaga S. Neuronal expression of two isoforms of mouse Septin 5. J. Neurosci. Res. 2010;88:1309–1316. doi: 10.1002/jnr.22294. [DOI] [PubMed] [Google Scholar]
- 78.Nathanson J.L., Yanagawa Y., Obata K., Callaway E.M. Preferential labeling of inhibitory and excitatory cortical neurons by endogenous tropism of adeno-associated virus and lentivirus vectors. Neuroscience. 2009;161:441–450. doi: 10.1016/j.neuroscience.2009.03.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Follenzi A., Naldini L. HIV-based vectors. Preparation and use. Methods Mol. Med. 2002;69:259–274. [PubMed] [Google Scholar]
- 80.Hiroi N. Compartmental organization of calretinin in the rat striatum. Neurosci. Lett. 1995;197:223–226. doi: 10.1016/0304-3940(95)11942-p. [DOI] [PubMed] [Google Scholar]
- 81.Hiroi N., Graybiel A.M. Atypical and typical neuroleptic treatments induce distinct programs of transcription factor expression in the striatum. J. Comp. Neurol. 1996;374:70–83. doi: 10.1002/(SICI)1096-9861(19961007)374:1<70::AID-CNE5>3.0.CO;2-K. [DOI] [PubMed] [Google Scholar]
- 82.Xu M., Moratalla R., Gold L.H., Hiroi N., Koob G.F., Graybiel A.M., Tonegawa S. Dopamine D1 receptor mutant mice are deficient in striatal expression of dynorphin and in dopamine-mediated behavioral responses. Cell. 1994;79:729–742. doi: 10.1016/0092-8674(94)90557-6. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.