

Abstract
Gene silencing using RNA interference (RNAi) has become a prominent biological tool for gene annotation, pathway analysis, and target discovery in mammalian cells. High-throughput screens conducted using whole-genome siRNA libraries have uncovered rich sets of new genes involved in a variety of biological processes and cellular models of disease. However, high-throughput RNAi screening is not yet a mainstream tool in life science research because current screening platforms are expensive and onerous. Miniaturizing the RNAi screening platform to reduce cost and increase throughput will enable its widespread use and harness its potential for rapid genome annotation. With this aim, we have combined semi-conductor microfabrication and nanolitre dispensing techniques to develop miniaturized electroporation-ready microwell arrays loaded with siRNA molecules in which multiplexed gene knockdown can be achieved. Arrays of microwells are created using high-aspect ratio biocompatible photoresists on optically transparent and conductive Indium-Tin Oxide (ITO) substrates with integrated micro-electrodes to enable in situ electroporation. Non-contact inkjet microarraying allows precise dispensing of nanolitre volumes into the microwell structures. We have achieved parallel electroporation of multiple mammalian cells cultured in these microwell arrays and observed efficient knockdown of genes with surface-bound, printed siRNAs. Further integration of microfabrication and non-contact nanolitre dispensing techniques described here may enable single-substrate whole-genome siRNA screening in mammalian cells.
Introduction
Gene knockdown using RNA interference (RNAi) has emerged as a robust, rapid method to annotate the mammalian genome.1,2 Genome-size libraries of RNAi molecules (typically siRNAs) have been screened to identify new genes that modulate specific pathways of interest such as apoptosis, mitosis, endocytosis, and host-pathogen interactions.3–10 These genome-wide screens have shown the enormous potential of RNAi screening technology to annotate thousands of genes of unknown function and rapidly identify novel molecular components that may be altered in disease.11,12 However, the cost and logistics of current RNAi screening technology have limited its use. Present RNAi screening platforms for mammalian cells (96 or 384 well format) rely on the use of expensive robotic equipment and costly reagents (e.g., siRNA libraries, transfection and readout reagents). The investment required per screen also limits the ability to conduct experiments with multiple replicates to enhance data quality. To capitalize on the benefit of rapid gene discovery and pathway analysis that RNAi screening can provide, it is important to develop methodology that can overcome these limitations.
One way to increase the use of HTS RNAi screening in mammalian cells would be to miniaturize the process.13 An efficient miniaturized RNAi screening platform could dramatically lower reagent costs and do away with the need for robotics, important steps to spread the use of genome-wide RNAi screening. A decade ago, cell-based microarrays were described as a novel method for performing high-throughput genomic screens: cDNAs and siRNAs microarrayed on glass slides were chemically ‘reverse transfected’ into a lawn of overlaying cells.14,15 This methodology has been expanded beyond chemical-mediated transfection to deliver nucleic acids into mammalian cells using viruses, electroporation, and less conventional methods such as nano-wires.16–19 However, few genome-wide screens in mammalian cells have been performed to date using this miniaturized format. There are at least three potential reasons as to why this has been the case. First, cell-based microarrays lack an efficient and simple method to transfect multiple cell types that can scale to whole genome levels (i.e. chemical reagents do not work for many cell types and genome-size libraries of infectious particles are a great challenge to generate). Second, because cells grow as a continuous lawn, lack of segregation of microscale cultures over the course of the experiment can confound phenotypic analysis. Third, assay readout and analysis is complicated by the lack of features to accurately identify thousands of individual microscale cultures during imaging.
We are developing a miniaturized RNAi screening platform that seeks to resolve these issues. In our scheme, nucleic acids will be delivered by electroporation, as this is a highly efficient method to transfect a wide variety of mammalian cells, including primary cells. In addition, because electroporation is a physical phenomenon that works with naked nucleic acids, this approach simplifies liquid handling of RNAi libraries (no need for complexion or packaging) and reduces reagent costs (no chemicals required). To spatially isolate cell populations, we will incorporate microfabrication techniques to create microwell arrays that segregate microscale cell cultures. These microwells will provide visual markers to distinguish cell populations during assay imaging and can also serve to minimize cell migration. Finally, to insure compatibility with existing readout methods (primarily high-content microscopy and fluorescence-based assays), we will use Indium-Tin Oxide (ITO) as the substrate material. Transparent and conductive, ITO will enable imaging of cell cultures and also act as the cathode during electroporation.
We have previously shown that simple microwell arrays on ITO-coated glass slides can be used to electroporate unbound naked siRNAs.20 Here, we have extended those proof-of-principle experiments and used microfabrication techniques to create electroporation-ready microwell arrays. These microwell arrays are created with biocompatible SU-8 and use an integrated electrode grid layer to distribute the voltage to the entire substrate and insure uniform electroporation. We have used non-contact inkjet microarrayers to precisely load multiple siRNA species into array microwells that are chemically modified to enhance retention of siRNA molecules prior to electroporation. Finally, we show efficient in situ electroporation of microwell array-bound siRNAs in a multiplexed fashion to obtain clear knockdown of endogenous genes in several cell types. These electroporation-ready microwell arrays may be scaled to provide the basis of a next-generation RNAi screening platform for mammalian cells.
Materials and methods
Microwell array fabrication
Indium Tin Oxide deposited polished slides (CG-41IN-S107, Delta Technologies, MN) were cleaned for 3 min with deionized water, methanol, and chloroform successively in a sonicated water bath. The slides were then air dried and dehydrated at 200 °C overnight. Slides were treated in oxygen plasma for 5 min (PEIIB, Technics) and a layer of positive photoresist was spin-coated at 500 rpm for 15 s and subsequently at 2500 rpm for 40 s. The slides were then pre-baked at 100 °C for 20 min and allowed to cool for 15 min. Photolithography was performed with a first mask (aligned to the edge of the slide) by exposing for 20 s to 365 nm UV radiation (MA6/BA6, Suss Microtech, Germany) at 7.5 W cm−2. Slides were then developed for 6 min in MF319 developer, rinsed with deionized water and nitrogen dried. Post-baking of the slides was done at 100 °C for 10 min and a second plasma etch was carried out to remove photoresist residue in developed areas. Metal deposition of Ti/Au (20 nm/150 nm) was performed by sputtering in a vacuum at 2.8e−6 Torr (Discovery 18, Denton Vacuum LLC, NJ). Lift-off was carried out overnight in acetone to obtain the electrode grid pattern on the ITO. Thereafter, the slides were dehydrated at 100 °C for 30 min, cooled to room temperature and oxygen plasma treated to remove residual solvents. Slides were then spin-coated with SU-8-2050 (Microchem, MA) for 15 s at 500 rpm and 40 s at 4000 rpm. A soft-bake step was performed at 60 °C for 2 min followed by 100 °C for 7 min. The slides were aligned and photolithography was done with a second mask using exposure to UV for 60 s (MA6, Suss Microtech, Germany). A post-exposure bake was performed at 60 °C for 3 min and 100 °C for 8 min. The slides were allowed to cool for 15 min and developed (SU-8 Developer, Microchem, MA) for 6 min. After a 1 min isopropanol wash, the slides were nitrogen dried and hard baked at 160 °C for 3 h in gradual steps of five intermediate temperatures.
Substrate surface modification
Microfabricated microwell array slides were treated with oxygen plasma for 6 min (Plasma Prep II, SPI Supplies, PA) at 150–200 mTorr. The slides were then immediately immersed in 2.5% 3-Amino Propyl Triethoxy Silane (APTES, Sigma Aldrich) in toluene at room temperature for 6 h. Subsequently, the slides were washed with fresh toluene, ethanol, and dried in nitrogen. Presence of amine groups on the microwell array ITO surface was verified by pipetting 400 nM Oregon-green NHS ester pH 9 in bicarbonate buffer (Invitrogen, CA), on a corner of all slides. The ester groups were allowed to react with the surface amine groups for 30 min at room temperature. The slides were then washed with water and nitrogen dried. Presence of a fluorescent spot under an inverted microscope (Nikon Eclipse, TE-2000U) indicated successful amine-silanization of the surface. This simple surface modification was sufficient to ensure strong cell adherence throughout the timeline of experiments. This could be due to the fact that the cell types used can proliferate in unmodified ITO of the kind used in the microfabrication of our substrates (typical surface roughness of <0.075 micrometre/20 mm, peak-to-peak).
Electroporation setup
Electroporation experiments were carried out in a custom setup (Figure S3) using a square wave electroporator (ECM 830 Electroporation System, BTX, MA). Microfabricated slides (consisting of eight columns of two 81 microwell arrays) were diced into eight pieces using a diamond scribe, such that each piece held two microwell arrays. Four pieces were housed in the custom setup and the exposed Ti/Au of each microwell array was clipped using copper connectors coupled to the cathode terminal of the electroporator. Appropriately-sized aluminum pieces serving as the anode were placed on top of the microwell arrays, separated by glass spacers. The effective inter-electrode distance was set at 300 μm. For electroporation, 16 μL of cell suspension in buffer (see below) was pipetted on top of the microwell arrays prior to placing the anode. An electroporation pulse was applied to each of the diced pieces housed in the custom setup such that both microwell arrays on the same piece were electroporated simultaneously.
Inkjet loading of microwell arrays
Lyophilized siRNA molecules targeting RPS27a, GFP, and a non-targeting control (siRPS27a, siGFP, and siControl; Qiagen, CA) were reconstituted to a concentration of 200 μM in PBS and stored at −20 °C. A piezoelectric inkjet printer (Rainmaker, Engineering Arts, AZ) was used to dispense 8 nL of siRNA solutions (50 μM in PBS) into the microwells according to the desired pattern. Fluorescence-conjugated control siRNAs (Alexa-488 and Rhodamine) were mixed in (1 μM) with each unlabeled siRNA to visually verify the quality of siRNA printing. A ProScanArray HT confocal laser slide scanner (Perkin Elmer, MA) was used to scan each slide and confirm accurate loading of siRNAs. Printed microwell arrays were desiccated in a bench-top vacuum container until use.
Cell culture and preparation
Immortalized mammalian cells (HeLa and HEK 293) were cultured in 75 cm2 flasks and split every third day using standard protocols. On the day of experimentation, cells at 70–80% confluence were washed with PBS, treated with 1 mL Accutase (Invitrogen, CA) and incubated for 5 min at 37 °C. Thereafter, 9 mL of DMEM supplemented with 10% Fetal Bovine Serum was added to the flask. Cells were centrifuged at 1200 rpm for 3 min, washed, and resuspended in Opti-MEM to the desired cell density (see below). Thioglycollate-elicited peritoneal GFP-expressing primary mouse macrophages were prepared from 3 month-old male C57BL/6-Tg(UBC-GFP)30Scha/J transgenic mice (Jackson Labs, ME) as described in Molteni et al.21
Electroporation of cells in microwell arrays and phenotypic analysis
Cells were diluted in Opti-MEM to a final density of 1800 cells μL−1 for HeLa or HEK 293 cells and 5000 cells μL−1 for primary mouse macrophages. In experiments where siRNAs were suspended in electroporation buffer, siRNAs (siRPS27a, siGFP or siControl) were used at a final concentration of 5 μM in the cell suspension to be electroporated. In experiments with siRNAs bound to the surface of the microwell array, no siRNAs were added to the suspended cells. Cells (16 μL) were pipetted on top of the microwell array and sandwiched between the ITO cathode and the overlaying aluminum anode separated by the spacer. The media covered the entire microwell array so that cathode and anode were in contact. At 1 min post-seeding, when cells had dropped to the bottom of the microwells, they were electro-porated using appropriate conditions (30 V amplitude/0.5 msec pulse-width for HeLa and HEK 293 cells; 40 V amplitude/1 msec pulse-width for primary mouse macrophages). Cells were allowed 10 min to recover and attach to the ITO surface prior to removal of the microwell array from the electroporation setup and incubation in cell culture media inside standard 6-well dishes. Imaging was performed on an inverted fluorescence microscope (Nikon TE-2000U) over 2–3 days for experiments with HeLa and HEK 293 cells, and up to a week for primary mouse macrophages. To determine average numbers of cells contained within the microwells (2 days post-electroporation), HeLa cell nuclei were stained with Hoechst (Invitrogen, CA) and counted using ImageJ (Open Source; http://rsbweb.nih.gov/ij/). Viable cells were identified by Calcein AM staining (Invitrogen, CA) and exclusion of Propidium Iodide (example in Figure S5).
Finite Element Modeling (FEM) of electric field
To understand the distribution of electric fields within individual 400 × 400 × 40 μm microwells on SU-8-patterned ITO slides, FEM simulations were performed in FEMLAB (Comsol, CA) using the AC/DC Module. The geometry was adapted from actual fabrication parameters, with boundary conditions as shown in Figure S7A. The ITO surface was modeled with a thickness of 200 nm and material property values of electrical conductivity of 3.75e6 S m−1 and surface resistivity of 4 Ω sq−1. The embedded gold transmission lines were modeled using values of electrical conductivity of 4.1e7 S m−1 and resistivity of 0.122 Ω sq−1. The microwell boundary was modeled using values for SU-8 electrical permittivity 4.8 and electrical conductivity 1e−7 S m−1. The distance between the anode and cathode was modeled at 300 μm with media of conductivity 1.6 S m−1 in between the electrodes. The anode was modeled as a positive contact (voltage 30 V), and the ITO surface served as cathode (voltage 0 V). To evaluate the field intensity surrounding cells within the micro-wells, we used twenty-four 10 × 10 μm spherical particles as cell mimics, with membrane permittivity of 9 and conductivity 1e−8 S m−1, and cytoplasm permittivity of 70 and electrical conductivity of 1.6 S m−1. After meshing and solving under stationary conditions, the electric field intensity along the Y axis (perpendicular to the ITO surface) was plotted and used for analysis.
Results
Microfabrication of biocompatible electroporation-ready microwell arrays
A general scheme of the approach we envision to develop a next-generation miniaturized RNAi screening platform for mammalian cells is shown in Fig. 1. Central to our methodology is the development of an electroporation-ready transparent microwell array that can be robotically loaded with multiple siRNA species, onto which cells would be seeded for electroporation, incubation and subsequent phenotype assessment. To create such an electroporation-compatible substrate, a microwell array comprised of an underlying titanium/gold electrode grid and SU-8 microstructures was patterned on ITO-coated glass slides using a two-mask cleanroom process (Figure S1). ITO-coated slides of standard dimensions (1 × 3 in) were designed to yield 16 individual microwell arrays (each containing 81 microwells) adequately spaced for dicing (Fig. 2A and S3A). Given the limited conductivity of ITO, the electrode grid is used to distribute the electric field intensity evenly across the entire microwell array during application of the electroporation pulse. The dark background of the electrode grid also serves as an opaque marker that allows automated registration of each microwell during assay and imaging. The electrode grid was completely insulated by the overlaying SU-8 photoresist, which formed the physical microwell layer (35 ± 5 micron thickness). De-lamination of SU-8 microwells during cell culture was evident if a hard-bake step was not conducted after SU-8 development (data not shown). Hence, a ramped hard-bake protocol was implemented to ensure low stress and minimal SU-8 cracking during the final cure step; this modification guaranteed structural stability for up to 7 days in culture.
Fig. 1.

Schematic of the electroporation protocol in miniaturized electroporation-ready microwell arrays. Left column – top view of microwell array; Center column – cross-sectional view of a single microwell; Right column – magnified view of surface interactions. First row – microwell array consisting of an ITO-coated glass surface patterned with an electrode grid, which is further insulated by a pattern of thick photoresist (SU-8). The ITO layer is amine-functionalized to generate a positively charged surface. Second row – registered and multiplexed loading of nucleic acids (siRNA) into the microwell array. Negatively charged siRNAs electrostatically bind the positive surface. Third row – cell seeding onto the microwell array in electroporation buffer by sandwiching the plated volume at a precise distance from the bottom ITO surface. Cells attach to the bottom of the microwells and are in proximity to the bound siRNA. Fourth row – using the top sandwich electrode as an anode and the bottom ITO layer as the cathode, an electroporation pulse is applied to all microwells simultaneously. Current flows from the patterned electrode grid into the ITO layer and through the electroporation buffer. The microwell array material (SU-8) insulates the grid and prevents direct current flow into the buffer. Fifth row – cells are allowed to recover and strengthen attachment post-electroporation. Sixth row – the microwell array is incubated for the period of the assay and imaged for cellular phenotypes.
Fig. 2.
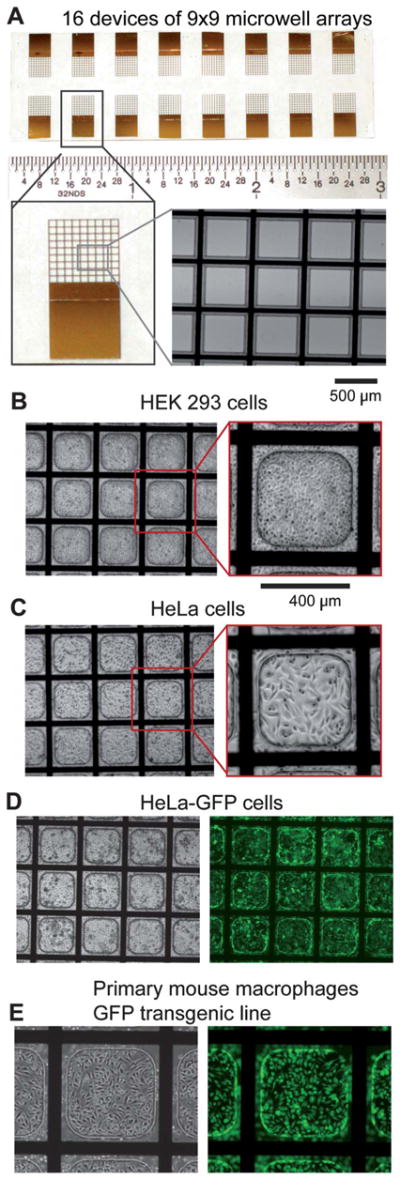
Microwell array design and biocompatibility. A) Top-Image of a complete, fabricated microwell array on ITO-coated glass slides. Sixteen devices consisting of a 9 × 9 microwell array each are spaced for dicing and individual experimentation. Lower left-Image of a single device. Lower right-Magnified image of a microwell array where the opaque (black) areas are the patterned titanium/gold electrode grid and the insulating microwell material surrounding the grid made of SU-8. B) Image of HEK 293 cells, C) HeLa cells, and D) HeLa-GFP cells cultured in microwell arrays 48 h post-seeding. E) Primary mouse macrophages derived from a GFP transgenic mouse line cultured and imaged in phase (left) and fluorescence (right) in a microwell array.
After fabrication, the microwell arrays were amine-silanized to create a positively charged layer on the ITO surface to enhance electrostatic binding of siRNAs and facilitate release during electroporation (Figure S2A). Conversion of surface hydroxyl groups into functional amine groups was verified on spare edge areas by reacting a fluorophore-conjugated NHS-ester molecule or by dispensing negatively charged fluorescent siRNAs onto the substrate (Figure S2B). Both of these stains were easily detected on amine-functionalized ITO surfaces after extensive washing (Figure S2C). No significant siRNA attachment was observed in the absence of surface modification. SU-8 surface hydrophobicity decreased after oxygen plasma treatment, but seemed to increase slightly after amine-functionalization. Oxygen plasma treatment and surface functionalization also reduced bubble formation during cell culture (data not shown). To assess the biocompatibility of the materials used in the fabrication of these electroporation-ready microwell arrays, HEK293, HeLa, HeLa-GFP cells, and primary mouse macrophages were cultured on these substrates for 2 to 7 days and morphology and growth monitored (Fig. 2). Cells seeded within the microwells were healthy and proliferated normally. With the current dimensions of the microwells (400 × 400 micron, curved corners), when 80 cells per microwell are seeded, 255 ± 18 HeLa cells can be found 48 h post-seeding, a sufficient number for conclusive phenotypic analysis (Figure S3).
Electroporation of siRNAs suspended in buffer in microwell arrays
To assess whether electroporation of siRNAs into mammalian cells is efficient in these microwell arrays (i.e. they are capable of delivering a reliable pulse), we first tested the microfabricated substrates using siRNAs suspended in buffer. Multiple parameters were considered to optimize cell electroporation, including anode distance, cell-seeding time prior to electroporation, cell adhesion time post-electroporation, electroporation buffer composition, siRNA concentration, and electroporation pulse parameters. The apparatus and scheme used to electroporate in parallel these 81 microwell arrays are shown in Figure S4. Optimization of electroporation parameters was initially conducted using Propidium Iodide as an instant marker of cell permeabilization. With a separation of 300 microns between the anode and the microwell array, greater than 95% of dispensed cells were seeded in the microwells within 1 min, after which an electroporation pulse was delivered. For the cell types tested (HeLa, HEK 293, and primary mouse macrophages), 10–12 min was sufficient for cell adhesion post-electroporation so that subsequent exchange of electroporation buffer with culture media involved minimal loss of attached cells. While the electroporation buffer overflows individual microwells (covering the entire anode-cathode space), this does not create significant cross-contamination, as the siRNAs are bound to the surface and remain so prior to electroporation. Two siRNAs targeting known proteins were used to generate knockdown phenotypes and multiple labels were used to assess cell viability (Figure S5). First, a siRNA targeting the Ribosomal Protein Subunit 27a (siRPS27a) was used to induce rapid cell death due to interference with ribosomal assembly and protein translation.22 Second, a siRNA targeting green fluorescent protein (siGFP) was used in HeLa cells stably expressing GFP; functional knockdown was measured as a decrease in fluorescence intensity. Considerable cell death (>95%) was observed in HeLa and HEK 293 cells electroporated with siRPS27a in microwell arrays at 48 h post-electroporation (Figure S6A–C). In contrast, no significant cell death was seen when cells were electroporated with control siRNAs, or in the absence of an electroporation pulse. Cell viability was assessed using Calcein AM (viable cells staining positive) and exclusion of Propidium Iodide (Figure S5). Similarly, >90% of primary mouse macrophages electroporated with siRPS27a died, though the phenotype manifested slower kinetics than in rapidly growing cells (5 days vs. 48 h). Electroporation of siGFP into GFP-expressing primary mouse macrophages (derived from a transgenic line that expresses GFP ubiquitously) led to a reduction of GFP total fluorescence intensity of greater than 60% within the microwells compared to control (Figure S6D). Electroporation of a plasmid encoding GFP into HEK 293 cells resulted in a significant proportion of cells (>40%) that expressed high levels of GFP protein (Figure S6E). Finite Element Modeling of the electric field surrounding the cells required to effect electroporation indicates that the Ex component varied between ± 50 V cm−1 distance of 10 μm and ± 20 V cm−1 at 5 μm from the ITO surface (Ex average 35 V cm−1), while Ey values were between 1100–1470 V cm−1 (Ey average 1270 V cm−1) (Figure S7). Together, these results indicate that our micro-fabricated microwell arrays constitute an efficient substrate to electroporate multiple mammalian cell types with various nucleic acids.
Electroporation of siRNAs bound to the surface of microwell arrays
Having shown that the microfabricated substrates can be used as efficient electroporation devices, we tested the ability of these electroporation-ready microwell arrays to bind and retain siR-NAs. Since our goal is to eventually use this technology to deliver thousands of different siRNA molecules in parallel (i.e. one siRNA species per microwell), it is important that individual siRNA molecules do not diffuse readily to neighboring micro-wells during buffer exposure, cell seeding, and electroporation. To maximize siRNA binding and to prevent the possibility of diffusion of siRNAs from the surface upon exposure to electroporation buffer, the ITO surface of the microwell arrays was modified to have functional amine groups as described above. To test whether this modification would enable sufficient electrostatic binding between the negatively charged siRNA molecules and the positively charged amine-functionalized ITO surface to prevent siRNA diffusion during buffer manipulations, functional and control siRNAs were pipetted and desiccated on top of full amine-functionalized microwell arrays. Overnight desiccation did not affect siRNA stability as evidenced by highly efficient (>95%) cell death post-electroporation in microwell arrays coated with siRPS27a (Fig. 3A). To evaluate whether gene knockdown can be restricted to specific areas of the microwell array and confirm that siRNA diffusion from ITO-modified surfaces is not a problem, half and quarter arrays were coated with siRPS27a or siControl siRNAs. Upon electroporation, cell death was observed only in the siRPS27a-coated areas of these arrays (Fig. 3B,C). No cell death was observed without the application of an electroporation pulse. Moreover, spatially confined knockdown of multiple genes (siRPS27a, siGFP, and siControl) was achieved in a single microwell array using the four quadrants (Figure S8). These results indicate that efficient, restricted gene knockdown can be achieved using surface-bound siRNAs in these electroporation-ready microwell arrays.
Fig. 3.

Electroporation of functional siRNAs bound within microwell arrays and evaluation of cellular phenotypes. A) Left-Schematic showing that the full area microwell arrays were coated with either siRPS27a or siControl. Right-Death phenotypes were observed only in the siRPS27a-coated microwells and only under electroporation conditions. No death was observed in siRPS27a-coated microwells without electroporation. B) Left-Schematic showing half areas of the microwell array coated with siRPS27a or siControl. Right-Death phenotypes were observed only on the siRPS27a-coated half. C) Left-Schematic showing quadrant areas of the microwell array coated with siRPS27a or siControl. Right-Death phenotypes were observed only in quadrants containing siRPS27a. Experiments were performed with HeLa cells and phenotypes assessed 48 h post-electroporation.
Registered loading of siRNA into microwell arrays
Next, we tested existing liquid handling technologies to load the electroporation-ready substrates with multiple siRNAs each restricted to an individual microwell. Accurate alignment of the dispensing head with individual microwells and precise dispensing of small volumes (below 10 nL given microwell dimensions) are required to prevent sample merging and spill-over in this small format. For this purpose, contact pin-based microarrayers proved to have low spotting accuracy and limited control over alignment, which resulted in inefficient siRNA loading into the microwells (data not shown). However, an alternative approach using non-contact piezoelectric inkjet printing yielded precise, reproducible spotting (>99% on-target) with complete alignment accuracy over the microwell arrays. No overflow of piezoelectric inkjet printed Alexa-fluor-488 and Cy5 conjugated siRNAs dispensed in an alternating pattern was observed into adjoining empty microwells (Fig. 4 A,B). The non-contact arrayer could dispense different siRNAs accurately in various design patterns across all 16 microwell arrays present on a microscope-slide size substrate (Fig. 4 C,D).
Fig. 4.
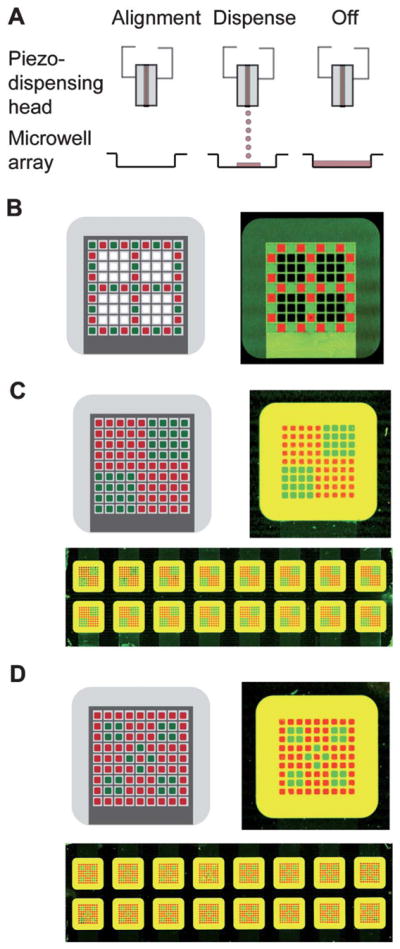
Registered loading of siRNAs into microwell arrays using a piezo-based inkjet printer. A) Schematic showing alignment of piezo-dispense head and a microwell array prior to dispensing of siRNA. Nanolitre volumes (8 nL) are dispensed into each microwell, coating the entire bottom surface. B) Left-Alternating design pattern programmed into the inkjet microarrayer. Right-Scan of a single microwell array loaded with Alexa-fluor-488 (green) and Cy5 (red) conjugated siRNAs using the alternating pattern. C) Top left-Design pattern 1 comprising of a quadrant to be printed into the microwell array. Top right-Scanned image of a single microwell array loaded with the quadrant pattern. Bottom-Scanned image of an entire slide (16 devices) printed in the quadrant pattern. D) Top left-Design 2 consisting of a checkered pattern to be printed into the microwell array. Top right-Scanned image of a single microwell array loaded with the checkered pattern. Bottom-Scanned image of an entire slide (16 devices) printed with the checkered pattern. Concentrations of fluorescently labeled siRNAs loaded were 5 μM in (B) and 1 μM in (C) and (D).
Electroporation of siRNAs printed in the microwell arrays
Having found a robust and accurate method to load the electroporation-ready microwell arrays with multiple different siRNA species, we next sought to achieve highly resolved (microwell-to-microwell) gene knockdown using piezo-dispensed siRNAs in various patterns. Control and RPS27a siRNAs were printed on two different patterns in 9 × 9 microwell arrays, the arrays were seeded with cells, electroporation was performed, and phenotypes evaluated 48 h later. In the first pattern, siRPS27a and siControl were dispensed in opposing quadrants, while in the second pattern they were dispensed in a checkered design at the center of the microwell array (Fig. 5). Phenotypes obtained for the first pattern were spatially registered with the siRNA printing design. Adjoining microwells loaded with siRPS27a and siControl yielded the expected phenotypes: cell death was clearly limited to those wells containing siRPS27a (Fig. 5). Similar results were obtained with microwell arrays loaded with siGFP and siControl in this pattern (Figure S9). In the second more complex design, the phenotypes also corresponded to the siRNA printed on each specific microwell, though some minor phenotypic spillover seemed apparent in some adjoining microwells. This may be due to the very high concentration of siRNA used (50 μM); decreasing printed siRNA concentration should minimize this issue. These results indicate that effective and restricted gene knockdown can be achieved with piezo-dispensed siRNAs in these electroporation-ready microwell arrays.
Fig. 5.

In situ electroporation of functional siRNAs printed onto microwells arrays. A) Top left-Electroporation and assessment of phenotypes using Design pattern 1 with HeLa cells within microwell arrays loaded with siControl (red areas) and siRPS27a (green areas). Quadrant areas are depicted by Q1 & Q4 (for siControl) and Q2 & Q3 (for siRPS27a). Areas Q12 and Q34 consist of bordering siControl and siRPS27a microwells. Bottom-Phase contrast images of all areas taken 48 h post-electroporation. Top right-Magnified image of area Q12 where death phenotypes are observed only in microwells printed with siRPS27a. B) Top left-Electroporation and assessment of phenotypes using Design pattern 2 with HeLa cells within microwell arrays loaded with siControl (red areas) and siRPS27a (green areas). Areas A1, A2 and A3 were chosen to assess phenotypes. Bottom-Phase images of areas A1, A2, and A3 at 48 h post-electroporation. Top right-Magnified image of area A2 with checkered death phenotypes obtained in siRPS27a-printed microwells.
Discussion
In this report we describe the creation of electroporation-ready microwell arrays that can be used to introduce multiple surface-bound siRNA species in a parallel fashion. In situ electroporation of bound siRNAs on modified ITO substrates is restricted and highly efficient: expected phenotypes are observed only on microwells loaded with phenotype-inducing siRNAs. The charge of the ITO surface is altered by the presence of positively charged amine groups that electrostatically bind siR-NAs and prevent siRNA diffusion during cell plating. This binding is reversible and the siRNAs remain fully functional upon electroporation. These results indicate that manufacture of siRNA-loaded substrates will be relatively simple, for siRNAs do not need to be complexed with chemical transfection reagents, thus making transfer from stock plates a one-step process. The concentration of printed siRNA used in these experiments was 50 μM, significantly greater than that usually used in standard electroporation cuvettes. However, we have not extensively explored ITO surface-siRNA interactions in this work, so it is very possible that further surface chemistry modifications may reduce the concentration of printed siRNA required to achieve functional gene knockdown. Nonetheless, it should be noted that even at these concentrations, because so little siRNA is used (8 nL) this miniaturized RNAi screening platform offers dramatic savings over current screening methodology, particularly when additional costs (assay reagents, plates, media, instrumentation) are taken into account.
Microfabrication of the microwell arrays using high-aspect ratio photo-polymers such as SU-8 provides high-quality spatially identifiable regions for microscale cell cultures. SU-8 serves as a biocompatible and stable material for culturing multiple cell types (HeLa, HEK 293, primary mouse macrophages) for up to a week, sufficient time to conduct most screens. In addition, SU-8 processing is compatible with whole-wafer fabrication and flexible for scaling microwell array size. Incorporation of the underlying patterned conductive grid made of Ti/Au provides a convenient way to ensure uniform electric field throughout the array and compensate for the insufficient conductivity of ITO.20 Together, the opaque Ti/Au layer and the SU-8 provide clear markers for image acquisition and analysis. Incorporation of such markers may be critical in miniaturized RNAi screening platforms to prevent confounding of results, particularly as arrays grow to include thousands of siRNA species. Furthermore, SU-8 can be functionally modified to further modulate cell migration between neighboring microwells on the array.23
Microarray printing of nucleic acids on flat substrates (without microstructure features) is well established for both contact and non-contact printers. However, swift, targeted microarraying to microstructured regions on the substrate is challenging because the print head and substrate features must be precisely aligned over the entire substrate. Contact printers can be aligned with microwells on ITO slides as long as there is sufficient clearance between the print head and the microwell edges.20 In our microfabricated microwell arrays, contact printing failed to coat the entire microwell surface evenly, and larger pins would collide with the microwell profiles. In contrast, non-contact inkjet technology provided precision targeting and loading of siRNA molecules from stock plates into the microwells in any desired pattern. Given the high tolerance of error in positioning (due to the non-contact nature of dispensing), siRNAs were coated over the entire microwell bottom. Non-contact inkjet dispensing of nucleic acids is a mature technology and its integration for quickly dispensing libraries of siRNAs into microwells to manufacture genome-scale arrays will be relatively straightforward.
To our knowledge, this is the first report of in situ electroporation of mammalian cells with surface-bound siRNAs immediately after cell seeding. There are several potential advantages to this approach over past reports of culturing cells for a day prior to electroporation.19 Electroporation may be more efficient because when first seeded, cells have a relatively uniform shape and size. In addition, because siRNAs are bound to the ITO surface as a layer that can potentially be degraded by enzymes present in cell culture media, it may be preferable to minimize their exposure to media prior to electroporation. An important aspect of the electroporation setup is the small distance between the ITO cathode and anode (300 μm), which seems to afford two benefits. First, the cells drop down to the bottom of the microwells quickly, which provides the opportunity to electroporate shortly thereafter and thus minimize any potential inter-well siRNA diffusion. Second, the small electrode distance seems to enable more uniform cell distribution, at least in these small 9 × 9 microwell arrays. The cells remain attached to the bottom of the microwells post-electroporation, and the array can be replenished with cell culture media. Using these electroporation-ready microwell arrays, clear phenotypes were observed upon electroporation of printed siRNAs targeting the RPS27a and GFP genes. Phenotypes were restricted to the corresponding microwells, with little or no contamination of phenotype between microwells. Future improvements of micro-well array design and cell handling methods (e.g., incorporation of microfluidics24–26) may completely eliminate the potential of phenotypic contamination.
In summary, we have created electroporation-ready microwell arrays that are automatically loaded with multiple siRNA species that can be electroporated in parallel into multiple mammalian cell types to effect gene knockdown. We have observed efficient and spatially restricted gene knockdown in these substrates. Because up to 20,670 microwells of the dimensions we have used can be accommodated in a single substrate the size of a 384-well plate, these results suggest that larger versions of these electroporation-ready microwell arrays may form the basis of a next-generation miniaturized HTS RNAi screening platform for mammalian cells.
Supplementary Material
Acknowledgments
We thank David Partida, Peter Kahn, and M.G. Finn for advice and technical assistance. This work was supported by NIH (5R01GM88597) and by the McDonalds Center for Obesity and Type 2 Diabetes. E.S. is the recipient of a Career Development Award from the American Diabetes Association.
Footnotes
Electronic supplementary information (ESI) available. See DOI: 10.1039/c2lc20931d
References
- 1.Hannon GJ, Rossi JJ. Unlocking the potential of the human genome with RNA interference. Nature. 2004;431(7006):371–378. doi: 10.1038/nature02870. [DOI] [PubMed] [Google Scholar]
- 2.Elbashir SM, et al. Duplexes of 21-nucleotide RNAs mediate RNA interference in cultured mammalian cells. Nature. 2001;411(6836):494–498. doi: 10.1038/35078107. [DOI] [PubMed] [Google Scholar]
- 3.Aza-Blanc P, et al. Identification of modulators of TRAIL-induced apoptosis via RNAi-based phenotypic screening. Mol Cell. 2003;12(3):627–37. doi: 10.1016/s1097-2765(03)00348-4. [DOI] [PubMed] [Google Scholar]
- 4.Rines DR, et al. Whole genome functional analysis identifies novel components required for mitotic spindle integrity in human cells. GenomeBiology. 2008;9(2):R44. doi: 10.1186/gb-2008-9-2-r44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Bettencourt-Dias M, et al. Genome-wide survey of protein kinases required for cell cycle progression. Nature. 2004;432(7020):980–7. doi: 10.1038/nature03160. [DOI] [PubMed] [Google Scholar]
- 6.Perrimon N, Mathey-Prevot B. Applications of high-throughput RNA interference screens to problems in cell and developmental biology. Genetics. 2007;175(1):7–16. doi: 10.1534/genetics.106.069963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Konig R, et al. Human host factors required for influenza virus replication. Nature. 2010;463(7282):813–7. doi: 10.1038/nature08699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Konig R, et al. Global analysis of host-pathogen interactions that regulate early-stage HIV-1 replication. Cell. 2008;135(1):49–60. doi: 10.1016/j.cell.2008.07.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Pelkmans L, et al. Genome-wide analysis of human kinases in clathrin- and caveolae/raft-mediated endocytosis. Nature. 2005;436(7047):78–86. doi: 10.1038/nature03571. [DOI] [PubMed] [Google Scholar]
- 10.Kim J, et al. Functional genomic screen for modulators of ciliogenesis and cilium length. Nature. 2010;464(7291):1048–51. doi: 10.1038/nature08895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Moffat J, Sabatini DM. Building mammalian signalling pathways with RNAi screens. Nat Rev Mol Cell Biol. 2006;7(3):177–87. doi: 10.1038/nrm1860. [DOI] [PubMed] [Google Scholar]
- 12.Silva JM, et al. RNA interference microarrays: high-throughput loss-of-function genetics in mammalian cells. Proc Natl Acad Sci U S A. 2004;101(17):6548–52. doi: 10.1073/pnas.0400165101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Lehner B, Fraser AG. 5000 RNAi experiments on a chip. Nat Methods. 2004;1(2):103–4. doi: 10.1038/nmeth1104-103. [DOI] [PubMed] [Google Scholar]
- 14.Mousses S, et al. RNAi microarray analysis in cultured mammalian cells. Genome Res. 2003;13(10):2341–2347. doi: 10.1101/gr.1478703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Ziauddin J, Sabatini DM. Microarrays of cells expressing defined cDNAs. Nature. 2001;411(6833):107–110. doi: 10.1038/35075114. [DOI] [PubMed] [Google Scholar]
- 16.Bailey SN, et al. Microarrays of lentiviruses for gene function screens in immortalized and primary cells. Nat Methods. 2006;3(2):117–22. doi: 10.1038/nmeth848. [DOI] [PubMed] [Google Scholar]
- 17.Yamauchi F, Kato K, Iwata H. Spatially and temporally controlled gene transfer by electroporation into adherent cells on plasmid DNA-loaded electrodes. Nucleic Acids Res. 2004;32(22):e187. doi: 10.1093/nar/gnh176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Shalek AK, et al. Vertical silicon nanowires as a universal platform for delivering biomolecules into living cells. Proc Natl Acad Sci U S A. 2010;107(5):1870–1875. doi: 10.1073/pnas.0909350107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Fujimoto H, Kato K, Iwata H. Electroporation microarray for parallel transfer of small interfering RNA into mammalian cells. Anal Bioanal Chem. 2008;392(7):1309–1316. doi: 10.1007/s00216-008-2423-z. [DOI] [PubMed] [Google Scholar]
- 20.Jain T, et al. Highly parallel introduction of nucleic acids into mammalian cells grown in microwell arrays. Lab Chip. 2009;9(24):3557–66. doi: 10.1039/b913794g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Molteni V, et al. N-Acylthiadiazolines, a new class of liver X receptor agonists with selectivity for LXRbeta. J Med Chem. 2007;50(17):4255–9. doi: 10.1021/jm070453f. [DOI] [PubMed] [Google Scholar]
- 22.Wang YW, et al. In vitro and in vivo evidence of metallopanstimulin-1 in gastric cancer progression and tumorigenicity. Clin Cancer Res. 2006;12(16):4965–73. doi: 10.1158/1078-0432.CCR-05-2316. [DOI] [PubMed] [Google Scholar]
- 23.Wang Y, et al. Simple photografting method to chemically modify and micropattern the surface of SU-8 photoresist. Langmuir. 2006;22(6):2719–25. doi: 10.1021/la053188e. [DOI] [PubMed] [Google Scholar]
- 24.Fei ZZ, et al. Gene Transfection of mammalian cells using membrane sandwich electroporation. Anal Chem. 2007;79(15):5719–5722. doi: 10.1021/ac070482y. [DOI] [PubMed] [Google Scholar]
- 25.Geng T, et al. Flow-through electroporation based on constant voltage for large-volume transfection of cells. J Controlled Release. 2010;144(1):91–100. doi: 10.1016/j.jconrel.2010.01.030. [DOI] [PubMed] [Google Scholar]
- 26.Lee WG, Demirci U, Khademhosseini A. Microscale electroporation: challenges and perspectives for clinical applications. Integr Biol. 2009;1(3):242–251. doi: 10.1039/b819201d. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.