

Abstract
Heart disease remains the leading cause of death and disability in the Western world. Current therapies aim at treating the symptoms rather than the subcellular mechanisms, underlying the etiology and pathological remodeling in heart failure. A universal characteristic, contributing to the decreased contractile performance in human and experimental failing hearts, is impaired calcium sequestration into the sarcoplasmic reticulum (SR). SR calcium uptake is mediated by a Ca2+-ATPase (SERCA2), whose activity is reversibly regulated by phospholamban (PLN). Dephosphorylated PLN is an inhibitor of SERCA and phosphorylation of PLN relieves this inhibition. However, the initial simple view of a PLN/SERCA regulatory complex has been modified by our recent identification of SUMO, S100 and the histidine rich Ca-binding protein as regulators of SERCA activity. In addition, PLN activity is regulated by two phosphoproteins, the inhibitor-1 of protein phosphatase 1 and the small heat shock protein 20, which impact the overall SERCA-mediated Ca-transport. This review will highlight the regulatory mechanisms of cardiac contractility by the multimeric SERCA/PLN-ensemble and the potential for new therapeutic avenues targeting this complex by using small molecules and gene transfer methods.
Keywords: heart failure, contractility, sarcoplasmic reticulum, calcium
An important role of Ca2+ in muscle contraction was first indicated a century ago by Ringer (1883), who demonstrated that the frog’s heart would not contract in the absence of extracellular Ca2+. Since then, it has been shown that Ca2+ is a physiological regulator of contraction, energetics, cell survival and other processes in muscle. Aberrant Ca-homeostasis is a universal characteristic of human and experimental heart failure and studies over the years by many laboratories which have the understanding that the subcellular mechanisms underlying regulation of Ca-handling in the normal and diseased heart. The precipitating events that lead to single cardiac myocyte dysfunction usually begin with an injury to the ventricle (myocardial infarction, ischemia, infection, valvular disease, familial, idiopathic), which activates the renin angiotensin and sympathetic nervous system along with cytokines. These in turn can cause direct damage to the individual cardiac myocytes resulting in contractile dysfunction and abnormal calcium cycling and eventual apoptosis and death (Figure 1). This review will focus on the regulation of Ca-cycling and contractility by the sarcoplasmic reticulum (SR) Ca2+-ATPase/Phospholamban (SERCA/PLN) complex and their interacting partners under physiological and pathophysiological conditions in the heart. The role of each Ca-cycling protein in this multimeric complex, which contributes to the pathophysiology of heart failure, and novel therapeutic strategies aimed at improving calcium homeostasis, will be discussed.
Figure 1. The SERCA2a/PLN regulatory complex in cardiac calcium cycling and survival.
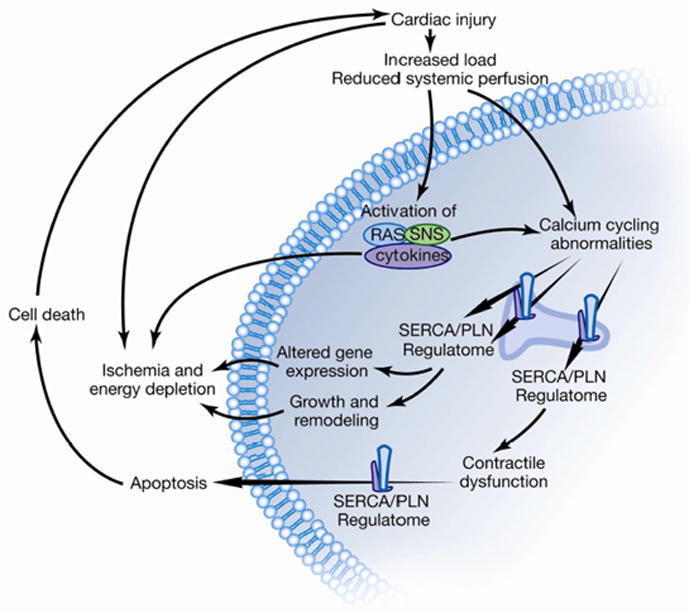
Injury to the ventricle such as myocardial infarction, ischemia, infection, valvular disease, familial, idiopathic activates the renin angiotensin and sympathetic nervous system along with cytokines. These in turn can cause direct damage to the individual cardiac myocytes resulting in contractile dysfunction and abnormal calcium cycling and eventual apoptosis and death. Targeting the SERCA2a/PLN proteome may have beneficial effects in abrogating the damage done to the individual cardiomyocytes.
SR Ca2+-ATPase and Ca2+ Homeostasis
There are three isoforms of the SR or endoplasmic reticulum (ER) Ca2+-ATPase (SERCA2). SERCA2 is the predominant variant of all SERCA isoforms and phylogenetically the oldest. Three different splice transcripts have been reported so far, SERCA2a, SERCA2b, and SERCA2c, which only differ at the C-terminus. SERCA2a is expressed in cardiac muscle and slow twitch skeletal muscle; SERCA2b is present in adult smooth muscle and nonmuscle tissues; and SERCA2c is found in cardiac muscle as well as non-muscle tissue including epithelial, mesenchymal, and hematopoietic cells. The proposed general model of the enzyme has three cytoplasmic domains joined to a set of 10 transmembrane helices by a narrow extramembrane pentahelical stalk 1. In heart, SERCA2a activity controls both the rate of cytosolic Ca2+ removal and the degree of SR Ca2+ load, representing a fundamental determinant of both cardiac relaxation and contraction.
Studies in genetically altered models have defined the functional role of the SERCA pump in Ca2+ homeostasis and cardiac physiology. Transgenic mice overexpressing SERCA2a by 1.2-or 1.5-fold exhibited increased SR Ca2+ transport and enhanced rates of cardiac contractility and relaxation 2,3,4. No cardiac pathology was observed in these animals, suggesting that SERCA2a overexpression can be tolerated by the heart. On the other hand, absence of the SERCA2 gene is lethal, with homozygous null (SERCA2 −/−) mice dying early in development 5. Heterozygous (SERCA2 +/−) mice are viable, showing 35% decrease in SERCA2 protein levels as a result of the loss of one copy of the SERCA2 allele and exhibited decreased myocyte contractility and SR Ca2+ load. Although no cardiac pathology was exhibited under basal conditions, reduction in SERCA2 levels in combination with an increased hemodynamic load resulted in an accelerated pathway to heart failure 6. These mice show impaired intracellular Ca2+ homeostasis and decreased rates of cardiac contractile function, indicating the requirement for two functional copies of the SERCA2 gene for effective SR Ca2+ cycling and cardiac function 5,7. More recently, an inducible cardiomyocyte-specific excision of SERCA2 resulted in severe reduction in both systolic and diastolic function and high mortality 8 weeks following gene excision.
SERCA2a Binding Partners
While experimental evidence suggests that proteins involved in SR Ca2+ release, such as the ryanodine receptor, function as part of a macromolecular complex, the existence of such a protein complex in the regulation of SR Ca2+ uptake has only recently begun to emerge (Figure 2). In particular, SERCA2a has been found to interact with proteins of the SR lumen, such as histidine rich calcium binding protein (HRC) 8 and calreticulin 9, while its cytosolic region has been shown to bind to S100A 10. Furthermore, PLN and sarcolipin (SLN) have been found to bind to the cytosolic and/or transmembrane domains of SERCA2a, with accumulating evidence suggesting that these interactions lead to inhibition of the pump’s affinity for Ca2+ 11,12. PLN has proven to be a major regulator of SERCA2a activity and so far, it is the only SERCA2a-associated protein directly involved in cardiac disease development, including heart failure.
Figure 2. Regulation of SR Ca-transport by a multimeric protein complex.
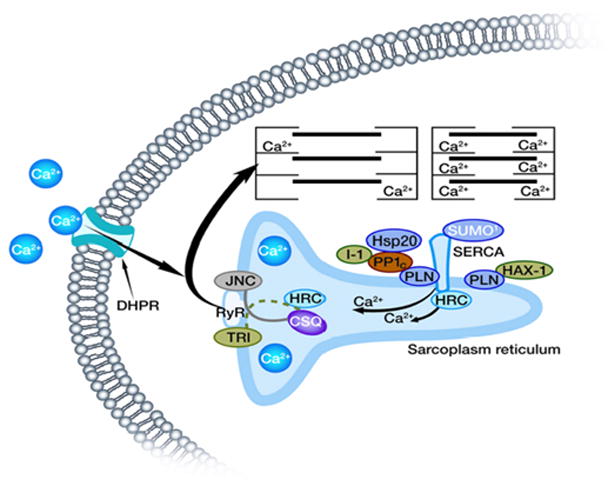
SERCA2a activity is regulated by its reversible inhibitor PLN, SUMO and the histidine rich Ca-binding protein (HRC). Phosphorylation of PLN is mediated by cAMP-dependent or Ca-CAM-dependent PKs and dephosphorylation occurs by protein phosphatase 1 (PP1). The activity of PP1 is regulated by inhibitor-1 (I-1).
The Histidine-Rich Calcium Binding Protein
The histidine-rich calcium binding protein (HRC), similarly to calsequestrin, is a low-affinity, high-capacity Ca-binding protein located in the cardiac SR lumen 8. Interestingly, recent studies have revealed that HRC may have a complex role in the cardiomyocyte and may mediate a cross-talk between SR Ca-uptake and release, as reviewed by Arvanitis et. al. 13. Indeed, HRC is a regulator of both SERCA2a activity, through its direct binding interaction, and RyR function through its binding to triadin. These interactions may be regulated in a Ca-dependent manner during the cardiac cycle 8. The role of this protein in heart function has been elucidated through adenoviral expression studies and transgenic models. Acute HRC overexpression in cardiomyocytes was associated with decreases in Ca2+-induced Ca2+-release (CICR) and the Ca2+-decay rate, resulting in depressed contractility 14. Importantly, a modest increase of HRC expression had a more pronounced effect on Ca2+ transients and cardiomyocyte contractility than a 20-fold overexpression of calsequestrin 15. Accordingly, transgenic overexpression of HRC in the heart 16 depressed SR Ca2+ uptake rates, providing further support for the inhibitory role of HRC on SERCA2a and intracellular Ca2+ cycling. In addition, chronic overexpression of HRC in the heart, compromised the heart’s response to stress, and eventually progressed to hypertrophy and heart failure upon aging 16. Thus, increases in the apparent stoichiometry of HRC/SERCA2, as observed in human failing hearts, may contribute to the depressed SR Ca2+ uptake and impaired cardiomyocytes Ca2+ cycling 14,16. Paradoxically, overexpression of HRC conferred a cardioprotective effect against an ischemic insult, which was at least partially attributed to reduced mitochondrial Ca load and attenuated apoptotic and necrotic injuries. Furthermore, HRC null mice exhibited normal basal cardiac function but they were more susceptible to isoproterenol-induced hypertrophy. Importantly, ablation of HRC impaired weight gain, possibly due to alterations in cell metabolism 17.
Recently, a human HRC variant (S96A) was identified, associated with life-threatening ventricular arrhythmias in dilated cardiomyopathy patients 18. The substitution of Ala in position 96 of the HRC alters its regulatory effects resulting in overall disturbed SR Ca-homeostasis, as evidenced by reduced Ca transient amplitude and prolongation of Ca decay time 19. Importantly, acute expression of the mutant HRC in failing cardiomyocytes was associated with aberrant Ca transients and arrhythmogenesis 19, consistent with the phenotype of human carriers.
Collectively, these studies have established a critical role of HRC in maintaining Ca homeostasis in the SR by regulating Ca storage, release and uptake.
PLN Regulation of SERCA2
In the early 1970s, it was suggested that the effects of various catecholamines on cardiac function may be partly attributed to phosphorylation of the SR by cAMP-dependent protein kinase(s). In 1972, two independent groups reported the ability of cyclic AMP-dependent protein kinases to phosphorylate the cardiac SR 20,21. It soon became clear that the substrate for protein kinase A (PKA) was not the SR Ca2+-ATPase but a low molecular weight protein. At the annual picnic of Arnie’s Katz’s lab in the summer of 1973, his wife Phyllis (a classicist) suggested that the new phosphoprotein be named phospholamban from the Greek root words phosphate and “lambano”, which mean “to receive phosphate.” 22. PLN is a small protein, comprising 52 amino acid residues. Indeed, subsequent studies confirmed that PLN is a critical mediator of the β-adrenergic stimulatory effects and changes in its phosphorylation were associated with functional alterations of the cardiac SR and contractility.
Ca2+-CaM-dependent PKs have also been shown to phosphorylate PLN and this occurs independently of PKA phosphorylation 23,24. Phosphorylation by cAMP-dependent PK occurs on Ser 16, whereas Ca2+-CaM-dependent PK catalyzes exclusively the phosphorylation of Thr 17 23,24. Dephosphorylated PLN exerts an inhibitory effect on SERCA2a and phosphorylation by either kinase was shown to result in stimulation of the SR Ca2+-ATPase activity and the initial rates of SR Ca2+ transport by relieving inhibition on the SR Ca-uptake pump (Figure 3). Stimulation was associated with an increase in the apparent affinity of the SR Ca2+-ATPase for Ca2+ (KCa).
Figure 3. Modeling of PLN.
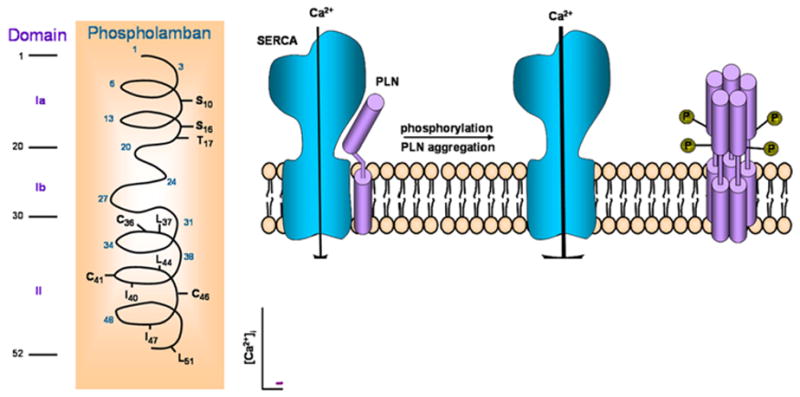
There are three domains: cytosolic domain Ia, containing the phosphorylated Ser16 and Thr17 sites, cytosolic domain Ib and transmembrane domain II, containing AA that are important in pentamer stabilization and functional regulation of SERCA2a. The PLN monomer interacts with SERCA2a and inhibits its activity, while PLN phosphorylation leads to pentameric assembly and relief of the PLN inhibitory effects.
In vitro, PLN is also phosphorylated by two additional PKs: PK-C and a cGMP-dependent PK. Protein kinase C (Ca2+/phospholipids–dependent PK) phosphorylates the protein at a site distinct (Ser 10) from those phosphorylated by either cAMP-dependent PK or Ca2+-CaM–dependent PK, while cGMP-kinase phosphorylates PLN at Ser 16. Importantly, cardiac SR appears to contain an intrinsic protein phosphatase activity, which can dephosphorylate both cAMP-dependent and Ca2+/CAM-dependent sites on PLN 25.
On the basis of these observations, it was initially proposed that phosphorylated PLN acts as a stimulator of the cardiac SR Ca2+-ATPase (SERCA2) activity. However, in the late 1980s, there was a significant breakthrough demonstrating that dephosphorylated PLN is actually an inhibitor of cardiac SERCA2 and that phosphorylation relieves this inhibition, giving the appearance of phosphorylation-induced stimulation 26,27. This finding, together with the identification of a cardiac SR–associated protein phosphatase that can dephosphorylate PLN 25, has led to our current understanding of PLN as a reversible inhibitor of the cardiac SR Ca2+ ATPase activity.
Mechanisms of PLN Regulation
PLN is in dynamic equilibrium between monomeric and oligomeric states. In the dephosphorylated form, a substantial fraction of PLN monomers exists and this has been proposed to be the active species of PLN that binds SERCA2 and inhibits it. Upon phosphorylation, PLN appears to form mainly pentamers, which is due to changes in the isoelectric point (from 10 to 6.7) of the protein 28 and this oligomer has been suggested to be inactive or at least less active than the monomeric unit (Figure 3).
The complete amino acid sequence of PLN has been determined for various tissues and species from fish to humans, where it has been shown to exhibit a high degree of homology further supporting the notion that PLN is a critical regulator of cardiac function. There is currently no evidence for the existence of any isoforms for this protein and the PLN gene has been mapped to human chromosome 6 29. The calculated molecular weight of PLN is 6080 Da, and the protein has been proposed to contain two major domains: a hydrophilic domain (domain I) with three unique phosphorylatable sites (Ser 10, Ser 16 and Thr 17), and a hydrophobic C-terminal domain, anchored into the SR membrane. The hydrophilic domain (amino acids 1–30) has been further divided into two sub domains: domain Ia (amino acids 1–20) and Ib (amino acids 21–30). Domain Ia has a net positive charge in the dephosphorylated form and consists of an α-helix followed by a Pro residue at position 21 (stalk region). Domain Ib has been suggested to be relatively unstructured 1,30. The hydrophobic domain (II; amino acids 31–52) forms an α-helix in the SR membrane (Figure 3).
PLN migrates as a 24- to 28-kDa pentamer on SDS gels and dissociates into dimers and monomers upon boiling in SDS prior to electrophoresis. Site-specific mutagenesis experiments identified Cys (Cys 36, Cys 41, and Cys 46), Leu (Leu 37, Leu 44, and Leu 51) and Ile (Ile 40 and Ile 47) residues in the hydrophobic transmembrane domain as essential amino acids for PLN pentamer formation 30. The leucine and isoleucine amino acids are suggested to form five zippers in the membrane that stabilize the pentameric form of the protein with a central pore, defined by the surface of the hydrophobic amino acids 28.
Cyclic AMP-dependent phosphorylation of PLN reverses its inhibitory effect on the Ca2+ pump. The inhibitory role of PLN on SR and cardiac function has been directly confirmed using transgenic animal models. Overexpression of the protein (PLN-overexpressing mice) was associated with inhibition of SR Ca2+ transport, Ca2+ transient, and depression of basal left ventricular function 31. On the other hand, partial (PLN-heterozygous mice) or complete ablation of the protein (PLN-deficient mice) in mouse models was associated with increases in SR Ca2+ transport and cardiac function 32,32, 33,34. Actually, a close linear correlation between the levels of PLN and contractile parameters in PLN overexpressors, WTs, PLN-heterozygous and PLN-homozygous knockout hearts was observed 35, indicating that PLN is a prominent regulator of myocardial contractility. These findings suggest that changes in the level of this protein may result in parallel changes in SR function and cardiac contractility.
The region of PLN interacting with the Ca2+-ATPase may involve amino acids 2–18. This association is disrupted by phosphorylation of Ser 10, Ser 16 or Thr 17 (phosphorylated by protein kinase C, cAMP-dependent, and Ca2+-calmodulin dependent protein kinase, respectively) in PLN, because the positive charges of the PLN cytosolic domain are partially neutralized by the phosphate moiety in this vicinity. Phosphorylation of PLN by the cAMP-dependent protein kinase at Ser 16 is associated with local unwinding of the α-helix at position 12–16 resulting in conformational changes in the recognition unit of the protein 36.
Interestingly, PLN peptides, corresponding to the hydrophobic membrane-spanning domain, also affect Ca2+-ATPase activity by lowering its affinity for Ca2+ 30. The importance of the membrane-spanning region of PLN in inhibiting SR Ca2+-ATPase activity was demonstrated through mutagenesis studies 30. It was shown that substitution of the pentamer-stabilizing residues (Leu 37, Leu 44, Leu 51, Ile 40, and Ile 47) in the membrane-spanning region (domain II) by Ala resulted in monomeric mutants, which were more effective inhibitors of the SR Ca2+-ATPase activity than wild-type PLN. These PLN monomeric mutants were called “supershifters” because they decreased the apparent affinity of SR Ca2+-ATPase to a greater extent than wild-type PLN. Thus, it was proposed that monomeric PLN is the active form, which is involved in the interaction with SR Ca2+-ATPase. Furthermore, scanning alanine-mutagenesis studies have identified the amino acid residues in the transmembrane domain of PLN (Leu 31, Asn 34, Phe 35, Ile 38, Leu 42, Ile 48, Val 49, and Leu 52), which are associated with loss of function 30. These amino acids are located on the exterior face of each helix in the pentameric assembly of PLN (opposite from the pentamer-stabilizing face). The importance of these transmembrane domain residues of PLN to SR Ca2+ -ATPase function has been demonstrated in vivo using several transgenic models. N27A PLN can act as a superinhibitor and N27A hearts have depressed Ca2+ -ATPase activity and cardiac function, which is not recovered completely by β-adrenergic stimulation, and the hearts progress to failure 37,38. Mice expressing the V49G variant in the PLN transmembrane domain also exhibited diminished cardiac function and hypertrophy which progressed to dilated cardiomyopathy 39. Moreover, L37A and I40A PLN transgenic mice showed similar decreased function and pathology 40.
As indicated above, the PLN monomer has been proposed to be the active species for interaction with the SR Ca2+-ATPase and the pentamers are regarded as functionally inactive forms of PLN 30. Phosphorylation of PLN monomers promotes association into inactive pentamers. Thus, two important steps for SR Ca2+-ATPase inhibition have been suggested: (1) dissociation of monomeric PLN from dephosphorylated pentamers; and (2) binding of PLN monomers to the SR Ca2+-ATPase. There are at least two interaction sites between PLN and the SR Ca2+-ATPase: one in the cytoplasmic domains of the two proteins and another one within the transmembrane sequences.
In Vivo Phosphorylation of PLN and Regulation of Contractility
The stimulatory effects of β-adrenergic agonists in the heart led scientists to initially explore cAMP-induced phosphorylation of contractile proteins, identifying troponin I as a cAMP-PK substrate in 1975 41. It was not until 1982 that the first in vivo evidence of PLN phosphorylation was presented in vivo 42. In these experiments, the ATP pool was labeled with [32P] orthophosphate. Microsomal fractions enriched in SR were prepared from hearts freeze-clamped during stimulation with different agonists (catecholamines, forskolin, phosphodiesterase inhibitors, phorbol esters) and analyzed by gel electrophoresis and autoradiography for 32P incorporation. β-adrenergic agonist (isoproterenol) stimulation of the perfused hearts produced an increase in 32P incorporation into PLN 42,43. The stimulation of 32P incorporation into PLN was associated with an increased rate of Ca2+ uptake into SR membrane vesicles and an increased SR Ca2+-ATPase activity 43,44.
These biochemical changes were associated with increases in left ventricular functional parameters (contractility and relaxation). The in vivo phosphorylation of PLN was specific only for inotropic agents that increased the cAMP content of the myocardium (β-adrenergic agonists, forskolin, and phosphodiesterase inhibitors). On the other hand, positive inotropic interventions which increased the intracellular Ca2+ level by cAMP-independent mechanisms (α-adrenergic agonists, ouabain, and elevated [Ca2+]) did not stimulate PLN phosphorylation or relaxation. In addition although protein kinase C (PKC) and cGMP-dependent PK (PKG) were shown to phosphorylate PLN in vitro, stimuli that activate PK-C or elevate the cGMP levels did not increase PLN phosphorylation in beating guinea pig hearts 45,46. Thus, the physiological relevance of PK-C and PK-G dependent PLN-phosphorylation is not clear at present. Importantly, phosphorylation of Ser 16 correlated most closely with changes in cardiac function in beating hearts compared to PKA-phosphorylation of troponin I 47. Based on these results and findings in transgenic animals 48, it has been proposed that: 1) prevention of Ser 16 phosphorylation (Ser 16 → Ala mutation) results in attenuation of the β-adrenergic response in mammalian hearts; and 2) that phosphorylation of Ser 16 is a prerequisite for Thr 17 phosphorylation. Indeed in mice containing an alanine substitution for Ser 16, there were diminished responses to the β-adrenergic stimulation. Also, there was no phosphorylation at Thr 17 in these animals 48. Conversely, substituting alanine for Thr 17 did not interfere with phosphorylation of Ser 16, and hearts were responsive to β-adrenergic stimulation 49. Moreover, overexpression of a non-phosphorylatable form of PLN (both Ser 16 and Thr 17 sites mutated to Ala) resulted in maximum inhibition of the SR Ca2+ ATPase calcium affinity 50. It should be noted that Thr 17 has been shown to be phosphorylated independently of Ser 16 under conditions which activate CaMKII and suppress phosphatase activity such as increased stimulation frequency of the heart, elevated intracellular Ca2+, ischemia-reperfusion injury, and acidosis 23,51.
The functional alterations in the SR Ca2+-ATPase activity may explain, at least partly, the activating and relaxing effects of β-adrenergic agents in cardiac muscle. The cAMP-dependent phosphorylation of PLN under either in vitro or in vivo conditions increases the rate of SR Ca2+ transport and SR Ca2+-ATPase activity. Such an increase in Ca2+ transport is expected to contribute primarily to the relaxing effects of catecholamines. The increased phosphorylation of PLN and the increased Ca2+ levels accumulated by the SR would lead to the availability of higher levels of Ca2+ to be subsequently released for binding to the contractile proteins. The critical and prominent role of PLN in the mediation of β-adrenergic functional responses was also confirmed in transgenic animal studies. Cardiac myocytes or work-performing heart preparations from PLN -deficient mice exhibited largely attenuated responses to β-adrenergic agonist stimulation 33,52, indicating that PLN is a key phosphoprotein in the heart’s responses to β-adrenergic agonists.
Besides PLN, increases in the phosphorylation of other myocardial phosphoproteins, including the L-type Ca-channel and phospholemman in the outer cell membrane, troponin I and C-protein in the myofilaments, and the SR Ca2+-release channel (ryanodine receptor) have been also shown to contribute to the stimulatory effects of β-adrenergic agonists in the heart.
PLN Human Mutations
In recent years, several human mutations in the PLN gene have been identified, and additional insights into PLN regulation of SERCA2 have been obtained. Two of these mutations (R9C and R14del) are associated with increases in PLN inhibition of the Ca-affinity of SERCA2 53,54. Their mechanisms appear to be partially due to decreases in cAMP-dependent protein kinase (PKA) phosphorylation of wild type PLN, preventing relief of the PLN inhibitory effects. The chronic inhibition of SERCA by mutant PLN led to dilated cardiomyopathy and premature death in families of carriers of R9C or R14del-PLN 53,54. These findings were also observed in mouse models carrying the R9C or R14del-PLN mutations 53,54. Interestingly, in the absence of endogenous PLN, the R14del mutant fails to co-localize with SERCA2a, resulting in lack of inhibition of SR Ca-uptake and enhanced contractility in the mouse 54. The mutant PLN is misrouted to the sarcolemma where it interacts with Na/K-ATPase, leading to cardiac remodeling despite the enhanced contractility 54. In addition, two more substitutions at Arg9 were identified in a heart failure cohort, namely R9L and R9H55. A loss of function human PLN mutation (Leu 39 stop) was also found to result in dilated cardiomyopathy and premature death in the homozygous state 56. The Leu 39 stop mutation produces a truncated PLN protein that when introduced in HEK 293 cells resulted in an unstable protein. The low levels of L39stop-PLN detected were misrouted to other membranes, leaving SERCA2a activity in the unhibited state. Finally, mutations have also been identified in the promoter region of the PLN gene, which are associated with increased 57,58 or decreased promoter activity 59, presumably leading to alterations in PLN levels. The cardiac phenotypes associated with these naturally occurring mutations in PLN have revealed an important difference between human and mice. Mutations that are deleterious for humans are not necessarily so for mice. This may be due to the differences in cardiac reserve and regulation of Ca-balance in the cardiomyocytes of the two species. The mouse heart beats at nearly its maximal rate while the human heart has a large cardiac reserve, allowing it to increase its rate by 2–3 fold. PLN is a major regulator of SR Ca-content during increases in heart rate and defective PLN regulation would compromise the cardiac reserve. In addition, several differences underlie Ca-cycling regulation in the two species, such as differences in myosin heavy chain isoforms and the role of Na-Ca-exchanger on a beat-to-beat basis. Thus, a fine balance by PLN regulation is of greater importance in humans than in mice.
HAX-1 Regulation of PLN/SERCA Activity
The HS-1 associated protein X-1 (HAX-1), which has a MW of ~35 kDa is ubiquitously expressed in mitochondria and was originally identified as an intracellular protein with anti-apoptotic function. Indeed, overexpression of HAX-1 protected cardiomyocytes against hypoxia/reoxygenation-induced apoptosis 60. Interestingly, more recent studies have revealed that HAX-1 also localizes to the SR and is an important regulator of Ca cycling. These effects are primarily mediated through its interaction with PLN and to a lesser extent through direct SERCA2a modulation (Figure 2). Indeed, HAX-1 has been shown to interact directly with PLN, with the minimal binding region of PLN to Hax-1 being amino acids 16–22, which includes both the Ser16 and Thr17 phosphorylation sites. This suggested that binding of HAX-1 to PLN may control the activity of PLN 61. Similarly to the SERCA2/PLN interaction, binding of HAX-1 to PLN was found to be diminished upon phosphorylation of PLN by cAMP-dependent protein kinase and increasing Ca2+ concentrations, indicating that HAX-1 may regulate the functional properties of PLN in the heart. Indeed, the PLN/HAX-1 interaction was shown to play a role in modulating Ca2+ cycling and cardiac contractility in vivo. Cardiac overexpression of HAX-1 decreased the affinity of SERCA2 for Ca and depressed myocyte calcium kinetics and mechanics. Accordingly, down-regulation of HAX-1 enhanced calcium cycling and contractility. The inhibitory effects of HAX-1 were abolished upon phosphorylation of PLN, consistent with the relief of the HAX-1/PLN interaction. The mechanism underlying the inhibitory effects of HAX-1 appeared to involve increased formation of PLN monomers, the active/inhibitory units of the calcium pump. Indeed ablation of PLN rescued the HAX-1 inhibitory effects on Ca-cycling and contractility in vivo 62 (Figure 4). Interestingly, HAX-1 fails to translocate to the SR in the absence of PLN, suggesting that the interaction of HAX-1 with PLN is imperative for its physiological function 63. It is important to note that HAX-1 also interacts directly with SERCA2a 64 but evidence from genetic models and adenovirally-infected cardiomyocytes suggest that the major effect of HAX-1 on SR calcium transport involves the Ca affinity of the pump and not the maximal velocity, as would be expected from direct SERCA modulation. Importantly, the HAX-1/PLN complex may regulate SR/ER and mitochondrial Ca2+ homeostasis, influencing the initiation of the apoptotic cell death signaling cascades. Indeed, expression studies in HEK cells showed that the anti-apoptotic effects of HAX-1, following hypoxia/reoxygenation-induced cell death, were enhanced in the presence of PLN 61. Similarly to Bcl-2, the beneficial effects of HAX-1 are associated with decreased SR calcium content leading to reduced mitochondria Ca load. Collectively, these findings indicate that HAX-1 represents a new regulator of PLN/SERCA activity, which may link Ca2+ handling and cell survival.
Figure 4. HAX-1 regulates PLN/SERCA activity.
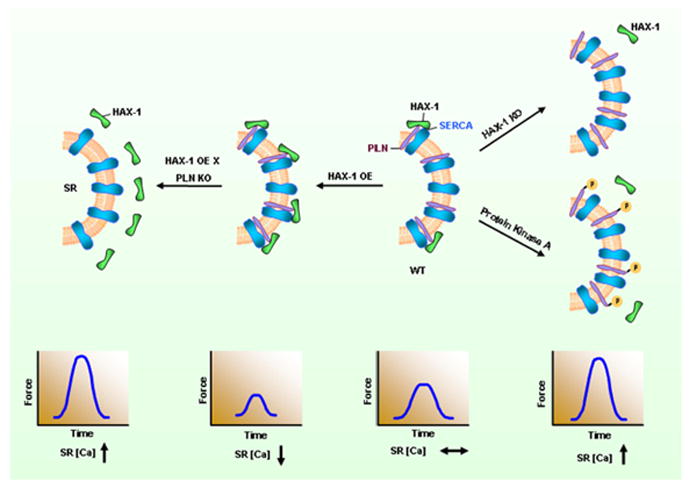
The anti-apoptotic protein HAX-1 interacts with PLN and enhances its inhibitory effects on SERCA2a and contractility. PKA phosphorylation of PLN or ablation of HAX-1 abolish the inhibition of HAX-1 on PLN/SERCA and Ca-cycling. Accordingly, overexpression of HAX-1 increases PLN inhibition and depresses contractility.
Inhibitor-1/Protein Phosphatase 1
Early studies indicated that the major phosphatase dephosphorylating PLN is PP1 65,66 and the activity of this enzyme is significantly increased in human failing hearts 67. These increases in PP1 may be one of the underlying factors in cardiac dysfunction and remodeling, as suggested by the generation of a transgenic model, expressing similar levels of PP1 as the human failing hearts 68. The transgenic hearts exhibited depressed basal contractile function and a blunted β-adrenergic response, which resulted in hypertrophy, heart failure and early death. PP1 is a holoenzyme comprised of its catalytic domain, which possesses its phosphatase activity, complexed with as many as 100 established or putative regulatory subunits 69. At the level of the cardiac SR, PP1 is regulated by its endogenous inhibitors, I-1 and I-2. Indeed, further insights into the role of this phosphatase were provided by studies of its endogenous inhibitors, I-1 and I-2. Cardiac-specific expression of a truncated and constitutively active form of I-2 (I-2*) depressed PP1 activity, enhanced contractile parameters and increased Ca2+ transient kinetics 70. These effects were associated with increased PLN phosphorylation at Ser16 but not at Thr17, suggesting a site-specific preference for PLN. Collectively, these results indicated that PP1 is a critical negative modulator of cardiac function.
Another endogenous regulator of PP1 is I-1, which gets activated upon phosphorylation at Thr35 by the cAMP-dependent kinase, PKA, resulting in attenuated PP1 activity 71,72,73. Indeed, I-1 knock-out (KO) mice exhibited depressed basal cardiac function in vivo. Furthermore β-adrenergic responses were blunted in isolated perfused hearts, associated with enhanced PP1 activity and decreased PLN phosphorylation 68. Accordingly, adenovirally-mediated overexpression of I-1 ex vivo enhanced contractility and increased PLN phosphorylation, upon β-adrenergic stimulation 74. Further studies in vivo showed that overexpression of a truncated (AA: 1–65) and constitutively active (T35D) form of I-1 (I-1c) resulted in decreases in the SR-associated PP1 activity, enhanced PLN-phosphorylation at both Ser16 and Thr17 and enhanced contractility both basally and after β-adrenergic stimulation 75. Phosphorylation of the RyR at Ser2808 and TnI phosphorylation at Ser22/Ser23 were unaffected. These results were confirmed by studies in an inducible transgenic model, which allowed for expression of I-1c in the adult heart 76. Collectively, these experiments suggested that I-1 may be acting as a molecular inotrope by suppressing PP1 activity and allowing for unopposed increases in the phosphorylation of PLN, which amplifies the β-agonist response (Figure 5). However, other studies have indicated opposite findings on the beneficial72 effects of increased I-1 function in the heart 77. Inducible I-1c expression on an I-1 deficient background in the adult heart was associated with enhanced function, but these hearts underwent remodeling through the aging process and they exhibited increased susceptibility to arrhythmias upon prolonged β-agonist stimulation 77. These findings may be due to increased pSer2815-RyR and potential diastolic Ca-leak from the SR 78,79. The reason for this apparent discrepancy between labs on the regulatory role of I-1 in vivo may be due to differences in overexpression levels, genetic background and/or environmental conditions.
Figure 5. Beta adrenergic agonist stimulation and protein phosphatase 1 in regulation of the PLN/SERCA activity.
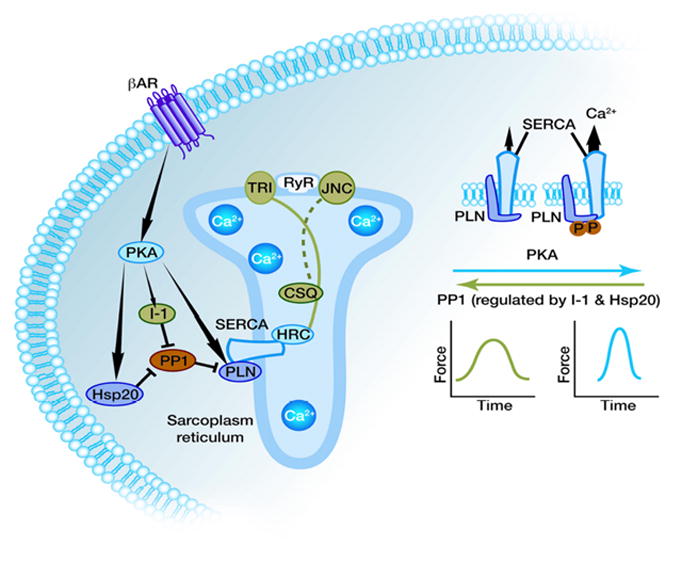
PKA activation results in increased phosphorylation of PLN, Inhibitor-1 and Hsp20 amplifying the stimulatory effects b-AR stimulation on SR Ca-transport and contractility.
Interestingly, there are additional phosphorylation sites on I-1: Ser67 and Thr75 that are phosphorylated by PKC. Studies in genetically-altered mouse models indicated that phosphorylation of I-1 at Ser67 by PKC-α was associated with increased PP1 activity and depressed contractility in vivo80. Further studies in isolated cardiomyocytes showed that phosphorylation at these sites was associated with enhanced PP1 activity, decreased PLN phosphorylation and depressed contractile function 81,82. Interestingly, these studies also showed that activation of the cAMP pathway was not able to fully reverse the depressed contractility, which was attributed to the inability of Thr35 to be phosphorylated efficiently in the PKC phosphorylation site mutants. Overall, these data suggest that I-1 may be an important mediator of the crosstalk between the PKA and PKC pathways in the heart. In addition, the cAMP and Ca2+ signaling pathways are also integrated at the level of I-1, as Thr35 is dephosphorylated by PP2A and PP2B, relieving PP1 inhibition and restoring cardiac function 83. Thus, I-1 appears to be a more complex regulator than previously thought and may modulate PP1’s activity according to differential cellular conditions, which activate these signaling pathways.
More recently, a human polymorphism has been identified in the human I-1 gene (G147D), which was associated with blunted β-adrenergic responses and depressed PLN phosphorylation in isolated cardiomyocytes 84, suggesting that this transversion may contribute to decreased SR Ca2+ cycling and functional deterioration in the failing heart. Thus, restoration of proper PP1 activity by I-1 may be a novel therapeutic strategy to rectify the disturbed Ca2+ homeostasis and Ca2+ cycling in the failing heart.
Heat Shock Protein 20
It is now widely acknowledged that the induction of heat shock proteins is cytoprotective, which is attributed at least partly, to chaperone activities of these “stress proteins”. Of particular interest in this family is a protein of ~20 kDa, namely Hsp20 or HspB6. This is the only small heat shock protein that has the consensus motif (RRAS) for protein kinase A/protein kinase G (PKA/PKG)-dependent phosphorylation at its Ser16 site 85. This suggested that Hsp20 may be subject to neurohormonal control via the β-adrenergic cascade in the heart. Indeed, it has been shown that Hsp20 expression and phosphorylation at Ser 16 is increased upon sustained β-adrenergic signaling in cardiomyocytes 86. In a congestive heart failure (CHF) model, both the expression of phosphorylated and unphosphorylated forms of Hsp20 were found to be significantly increased compared with the normal group 86. Several studies have focused on elucidating the role of Hsp20 and its phosphorylation on cardiac contractility and cardioprotection. Interestingly, it has been suggested that Hsp20 regulates myocardial contraction via regulation of the PP1/PLN axis 87. In vitro studies in cardiomyocytes, using phospho-peptide analogs 88 or adenoviruses, indicated that Hsp20 was associated with significant increases in mechanical and Ca-kinetic parameters 85,88 Importantly, cardiac overexpression of Hsp20 significantly enhanced contractile parameters and Ca-kinetics 87. Conversely, the knockdown of Hsp20 by anti-sense RNA or microRNA-320 was associated with depressed contractility 89. The underlying mechanisms appear to involve direct binding of Hsp20 to type 1 phosphatase (PP1) and inhibition of PP1 activity, associated with enhanced phosphorylation of PLN and SR Ca-cycling (Figure 5). This hypothesis was supported by the observation that PP1 activity was reduced in Hsp20 transgenic mice. Furthermore, the inotropic effects of Hsp20 were abrogated in adenovirally-infected cardiomyocytes expressing a non-phosphorylatable form of PLN (S16A/T17A), further supporting the notion that the effects of Hsp20 on contractility are mediated through modulation of the PP1/PLN axis.
Furthermore, increases in Hsp20 levels are associated with cardioprotection. Indeed, overexpression of Hsp20 in cardiomyocytes is protective against Iso-induced apoptosis 90 and simulated I/R injury 91. In vivo studies in Hsp20 transgenic mice showed that these hearts are resistant to chronic beta-agonist-induced cardiac remodeling, interstitial fibrosis and apoptosis. Contractility was also preserved in hearts with increased Hsp20 levels. Furthermore, Hsp20-transgenic hearts, subjected to global no-flow ischemia/reperfusion, exhibited improved recovery of contractile performance with reduced necrosis and apoptosis 92. Moreover, the infarct region-to-risk region ratio was significantly reduced in Hsp20-hearts, compared to wild-type hearts. Interestingly, the levels of Hsp20 phosphorylation were significantly increased in post- ischemic/reperfused transgenic hearts, suggesting that Hsp20 phosphorylation may play an important role in cardioprotection against ischemic injury. Indeed overexpression of a S16A-Hsp20 mutant abrogated the protective effects of Hsp20 in the heart and increased infarct size as well as apoptosis 86. The beneficial effects of Hsp20 in cardioprotection were further demonstrated in response to Doxorubicin-induced cardiotoxicity. The mechanism underlying these cardioprotective effects is associated with reduced apoptosis through the Bax/BCl-2 and Akt pathways. Furthermore, a human mutant with P20L substitution was identified, which was associated with diminished phosphorylation at Ser16 and complete abrogation of the Hsp20 cardioprotective effects against ischemia/reperfusion 91. Since PP1 and PLN have also been implicated in regulation of the apoptotic pathways, it is intriguing to speculate that the anti-apoptotic, cardioprotective effects of Hsp20 may also been mediated, at least partly, through PP1 and PLN. Together, these results support the beneficial effects of Hsp20 and its phosphorylation in at multiple levels by modulating cardiac contractility and survival.
S100A1
The S100A1 contains two EF-hand calcium-binding motifs 93 and is highly prevalent in cardiac cells, localizing at the SR, the myofilaments, and the mitochondria 94. S100A1 exerts its biological function by its interaction with various target proteins to regulate cellular processes. Previous reports suggested that S100A1 interacts with SERCA2a and PLN 10, as well as with RyR 95. Therefore, it seems that in response to intra-SR Ca2+ signals, S100A1 affects SR Ca2+ uptake and/or release. Indeed, it has been shown that diminished cardiac S100A1 protein levels are characteristic of end-stage failing human hearts 96. S100A1 may be affecting SERCA and RyR due to its potent molecular chaperone character 97, functioning predominantly as a Ca2+-sensor rather than a Ca2+-storage protein 98. In addition, S100A1 has been localized within the mitochondria and the myofilaments but their roles within these cellular structures have not been well defined yet. S100A1 has been shown to be a promising target for cardiac gene therapy 99. S100A1 gene transfer in isolated cardiomyoctes resulted in enhanced contraction and calcium transients. In rodent models of heart failure, AAV6-S100A1 gene therapy was able to improve cardiac function and to reverse left ventricular remodeling in the long term (8 weeks after gene therapy) 99 in a rat model of heart failure. In the same way, cardiac hypertrophy was reduced and cardiomyocyte Ca2+ cycling maintained. Furthermore, to compare current pharmacological interventions, S100A1 gene therapy was tested against chronic β-AR blockade by metoprolol and as a combination therapy. S100A1 therapy alone resulted in superior cardiac performance in comparison to metoprolol and combination therapy revealed synergistic effects of β-AR-blockage and restoration of S100A1 protein levels 99. In the latest study by Pleger et al., MI was induced by balloon occlusion of the left circumflex coronary artery for two hours 100. AAV9-S100A1 cardiac-restricted gene therapy was performed two weeks later via the anterior vein into the left ventricular remote myocardium 100. After 14 weeks, gene transfer resulted in cardiac restricted gene expression without any signs for extra-cardiac expression analyzed by AAV-luciferase expression. Cardiac function was severely impaired after MI and hearts showed signs of left ventricular remodeling. In comparison, AAV9-S100A1 treatment prevented cardiac deterioration and reversed ventricular remodeling by re-establishing S100A1 expression level. Furthermore, S100A1 treatment was able to reconstitute cytosolic and SR Ca2+ cycling as well as energy homeostasis in isolated cardiomyocytes 100.
SUMO1
The levels and activity of SERCA2a in cardiomyocytes can also be modulated in parallel with the levels of a cytoplasmic protein, small ubiquitin-like modifier type 1 (SUMO1) 101. SUMOs are a family of peptides that alter the function of other proteins in cells through a post-translational modification described as sumoylation. Sumoylation is involved in the modulation of various cellular processes such as transport from the nucleus to the cytosol, transcription, and protein stabilization and degradation through the process of reversible covalent linking of SUMO to the target protein 101. The activities of many important intracellular proteins are modified in this way, including steroid receptors, proto-oncogenes, tumor suppressors, and cardiac transcription factors.
Kho et al. found that sumoylation appeared to prolong the lifetime of SERCA2a in the cell as well as increase the intrinsic activity of SERCA2a ATPase 102. The authors also found that SERCA2a and SUMO1 levels were both reduced in mouse and pig models of heart failure and in cardiomyocytes isolated from failing human ventricles 102. To determine whether reduced SUMO1 levels are responsible for reduced SERCA2a protein levels and reduced cardiac function, SUMO1 was increased by gene transfer in a mouse model of heart failure (induced by thoracic aortic constriction). Increasing SUMO1 levels led to a restoration of SERCA2a levels, improved hemodynamic performance, and reduced mortality among the animals with heart failure. Reduction of SUMO1 expression using a short hairpin RNA approach reduced SERCA2a levels and adversely affected the pump function of the mouse hearts. The link to the sodium–calcium exchanger was recapitulated in these experiments, since sodium–calcium exchanger levels were higher after SUMO1 down-regulation, again suggesting that the expression of these two key calcium-handling proteins is related. A key additional finding was that reduced cardiac function due to SERCA2a down-regulation could not be improved by up-regulation of SUMO1 without its stimulatory effect on SERCA2a levels. This is an important observation, because sumoylation leads to translational modification of a number of intracellular proteins that might have changed cardiac function independently of SERCA2a.
Kho and colleagues 102 report a SUMO-induced modification of a cardiac protein and have shown a new mechanism for modulation of SERCA2a activity and cardiac-pump function. As our understanding of SUMOylation increases, targeting it for therapeutic benefits to the heart may be possible.
The SERCA/PLN Regulatory Complex in Heart Failure
In human and experimental heart failure both the level and the activity of the SR Ca2+ pump are decreased contributing to the deteriorated cardiac function 103,104. Furthermore, the protein level of PLN remains unaltered and this results in an increased fraction of SERCA in the inhibited form by PLN 13. In addition, the phosphorylated PLN levels are decreased further compounding reduced SR Ca2+ ATPase activity 104. This diminished phosphorylation of PLN can be attributed to an attenuation of the beta-adrenergic cascade due to receptor desensitization, receptor downregulation and uncoupling, which occurs during disease progression 105,106. Importantly, it has been recognized that activation of phosphatases may also contribute to the dephosphorylation of PLN and depressed cardiac function 68,107,108
Furthermore, decreases in the protein levels of HAX-1 and inhibitor-1 as well as PKA-phosphorylation of I-1 have been observed in failing hearts, which are expected to further contribute to the depressed SR Ca-cycling homeostasis. On the other hand, the levels of HRC 16 and Hsp20 as well as PKA-phosphorylation of Hsp20 86 are significantly increased in heart failure, suggesting important compensatory responses for the deteriorated cardiac function. Thus, complex regulatory mechanisms underlie the function of the SERCA/PLN modulators under physiological and pathophysiological conditions.
The SR Ca-Transport Interactome (“Assemble”) as Target in Heart Failure
There is a need for innovative and targeted therapy to improve the impaired contractile function and halt remodeling in heart failure. Reversing the contractile failure of cardiac myocytes with the use of standard pharmacological inotropic agents has been controversial, most likely due to the pleiotropic intracellular effects of the targeted molecules 109. As such, it has been suggested that a targeted approach may be more effective in alleviating the depressed function and progression of remodeling.
Given the central importance of the SR Ca2+ ATPase to proper SR Ca2+ cycling, excitation-contraction coupling, and thus cardiac function, SERCA2 has been a focus of potential gene-targeted therapy for heart failure, especially since pump activity is depressed in failing hearts. SERCA2a gene-transfer in animal models and human failing cardiomyocytes has shown beneficial effects, including improved contractility and energetics as well as prevention of arrhythmias and hypertrophy. For example, adenoviral delivery of SERCA2a to human failing cardiomyocytes rescued the depressed contractility and Ca2+ transients 110. Adenovirus-mediated gene-transfer also improved function and survival in rat failing hearts. Additionally SERCA2a delivery suppressed arrhythmias as well as infarct size in a rat model of ischemia/reperfusion injury 110. These beneficial effects in heart failure have also been demonstrated in multiple models of heart failure in large animals 110. Along with the demonstration of improved myocardial mechanical function, the overexpression of SERCA2a had multiple effects including improved myocardial energetics, endothelial function and anti-arrhythmic effects. There has been a well documented decrease in creatine kinase activity in failing hearts resulting in a decrease in the supply of ATP to essential areas in cardiac cells. The decrease in SERCA2a-mediated Ca2+ regulation in the failing heart increases the energy cost for Ca2+ regulation, together resulting in inefficient energy use 111. Sakata et al investigated the effects of SERCA2a overexpression on mechanoenergetics in rats. In these studies they measured the oxygen cost of left ventricular (LV) contractility per beat for Ca2+ handling in excitation–contraction coupling per unit change in LV contractility. In diseased hearts from diabetic and aortic-banded rats, the oxygen cost of LV contractility was increased; SERCA2a overexpression restored the increased oxygen cost of LV contractility to the normal level, indicating an improvement in energy utilization (Figure 6). Of note, chronic stimulation of the myocardium by [beta]-adrenergic agents increases mortality, probably due to enhanced mechanical function without the improvement of energy utilization, leading to increase total energy consumption. This improvement in energy use could, therefore, hold great significance for future clinical trials of SERCA2a gene therapy for congestive HF. This clear difference mechanoenergetic efficacy could directly influence patient survival. In fact, Del Monte and coworkers showed improved survival in rats after SERCA2a overexpression, coupled with the improvement in energy consumption. The effects of SERCA2a gene transfer seem to reverse the adverse remodeling in the cardiac cells with resulting improvements in energetics. The effects of SERCA2a on energetics cannot be viewed as an acute effect, such as the infusion of dobutamine but has to be considered in terms of a chronic effect on overall cardiac remodeling and reduction in ventricular size. Recently Lyon et al have shown that SERCA2a gene transfer does in fact reverse abnormalities of sarcolemmal structure in heart failure with reappearance of z-grooves and T-tubules in reverse remodeled hearts 112.
Figure 6. Strategies of improving calcium handling and SR Ca-content in heart failure.
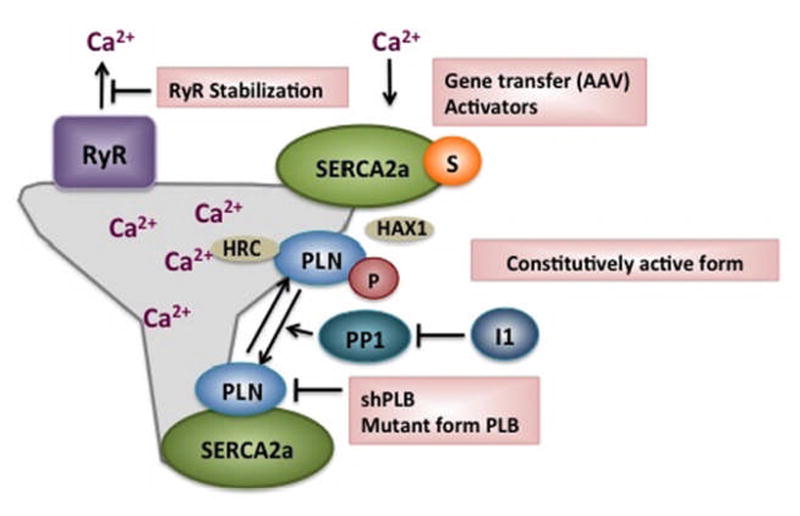
These strategies have focused pharmacologically on inhibiting the Na/K ATPase which results in increased intracellular Ca2+ and more recently on stabilizing the Ryanodine Receptor in resulting in decreased SR Ca2+ leak. Gene editing techniques have focused on enhancing SERCA2a’s activity by either increasing the level of SERCA2a or altering the expression of its partners.
Furthermore, overexpression of SERCA2a increases coronary blood flow and in doing so improves cardiac function 113. One of the reasons for this improvement has been shown to be the increased production of NO caused by SERCA2a overexpression through an increase in eNOS levels and phosphorylation. In a recent study, Hadri et al demonstrated that eNOS expression was increased in SERCA2a-overexpressing human coronary endothelial cells as a result of SERCA2a-mediated activation of the eNOS promoter. SERCA2a appears to play a role in the induction of eNOS transcription via an element in the promoter region from bp −5,039 to −4,861. This region was shown to contain a 269-bp activator element acting as an enhancer of transcription. The enhancer contains myeloid zinc finger–like, AP-2, Sp1, and Ets binding sites and is important for the endothelial specificity of eNOS promoter 113. This additional effect of overexpression of SERCA2a might have a synergetic effect in the prevention of cardiovascular disease.
The antiarrhythmic effects of SERCA2a overexpression were demonstrated in acute ischemia-reperfusion 114 and in heart failure 114,115,116. In chronic heart failure after myocardial infarction in rats, the overexpression of SERCA2a was associated with a reduction of spontaneous and provoked ventricular arrhythmia along with a reduction in calcium leak from the SR 116. The latter findings support the clinical safety of SERCA2a gene therapy in the arrhythmia-prone heart failure population.
In other recent studies, SERCA2a gene transfer has been shown to decrease alternans, a substrate of ventricular arrhythmias both in vitro and in vivo117. The improved contractile performance, along with improved myocardial energetics, endothelial function and coronary flow has resulted in improved survival of HF animals following gene transfer of SERCA2a. In porcine and ovine models of HF, gene transfer of SERCA2a with AAV vectors carrying SERCA2a resulted in a decrease in ventricular volumes, enhanced ejection fraction and de-activation of HF biomarkers 118,119. Most importantly, these studies showed that there was a gene-dose-effect whereby an increase in the concentration of vectors delivered resulted In higher gene expression of SERCA2a and improved performance in the HF animals.
Based on the extensive evidence that SERCA2a induces beneficial effects in multiple models of heart failure, the first clinical trial of gene therapy in patients with HF was launched in the United States in 2007 -CUPID (Calcium Up-Regulation by Percutaneous Administration of Gene Therapy in Cardiac Disease) 120,121. Patients treated with AAV1.SERCA2a demonstrated improvement or stabilization in New York Heart Association (NYHA) class, MLWHFQ (Minnesota Living With Heart Failure Questionnaire), six-minute walk test (6MWT), maximal Oxygen consumption during exercise (VO2 max), N-Terminal pro-hormone of Brain Natriuretic Peptide levels (NT-proBNP), and LV end-systolic volumes 122. Significant increases in time to adjudicated CV events, and a decreased frequency of CV events per patient were observed in all patients receiving AAV1.SERCA2a. No increases in adverse events, disease-related events, laboratory abnormalities or arrhythmias were observed in AAV1.SERCA2a treated patients compared to placebo. Additionally, after 12 months of receiving a single infusion of AAV1.SERCA2a, patients treated with the highest dose versus placebo had an 88 percent risk reduction of major cardiovascular events such as death, need for left ventricular assist device (LVAD) or cardiac transplant, episodes of worsening of heart failure, and number of heart failure-related hospitalizations 122. These positive results have persisted at 24 months. Furthermore, there were no changes in arrhythmias as recorded by the implantable cardioverter defibrillators (ICDs) that were required in all patients.
Targeting PP1/PLN
Given the aberrant regulation of type 1 phosphatase activity in cardiac pathology, several groups have investigated the potential of phosphatase inhibition as a therapeutic approach to enhance cardiac function in heart failure. Specifically, studies have examined the inhibition of PP1 through its regulators, inhibitor-2 and inhibitor-1. Gene delivery of I-2 in a cardiomyopathic heart failure hamster model 123, during the transition from moderate to severe dysfunction, resulted in prevention of heart failure progression and prolonged survival. These beneficial effects were associated with decreased PP1 activity and increased PLN phosphorylation at Ser16 without any changes in the phosphorylation of RyR. In contrast to the acute beneficial effects, cardiac-specific expression of the constitutively active I-2 (I-2*) was associated with heart failure and depressed PLN phosphorylation at Ser16 after pressure overload induced by aortic constriction. The apparent discrepancies may be related to confounding, compensatory effects of ‘chronic’ inhibition of PP1 in the transgenic mice.
I-1-targeted interventions also exhibited therapeutic promise in heart failure. I-1 ablation reduced the incidence of isoprenaline-induced arrhythmias, hypertrophy and death, possibly due to negation of the adverse effects associated with the PKC sites 124. Adenoviral-mediated expression of the constitutively active (T35D) and truncated form of I-1 (I-1c), which lacks the detrimental PKC-phosphorylation sites, enhanced contractile parameters and Ca kinetics of human failing cardiomyocytes 68. In vivo, overexpression of I-1c in the heart also enhanced Ca-cycling and contractile parameters. Notably, upon trans-aortic constriction, I-1c hearts maintained the enhanced cardiac performance and these mice exhibited an attenuated progression to heart failure, characterized by a diminished extent of cardiac hypertrophy and fibrosis, with no decompensation. Furthermore, these mice maintained their enhanced cardiac function upon chronic isoproterenol-stimulation, associated with cardiac remodeling 75.
More recently, it has been reported that inducible expression of I-1c in the adult heart protects against ischemia/reperfusion (I/R) injury 76. In particular, expression of I-1c ameliorated contractile dysfunction and attenuated cellular damage post-I/R. However, other studies 124 indicated detrimental effects of prolonged I-1c expression in the heart, associated with enhanced RyR phosphorylation at the CaMKII-site, S2815. Nevertheless, gene delivery of I-1c in pre-existing heart failure in a rat model of pressure overload restored contractility to non-failing levels. Of special interest is the fact that these beneficial effects were mediated by enhanced phosphorylation of PLN, while the PKA-phosphorylation levels of the RyR were unchanged. Interestingly, a recent study by Kawashima and colleagues in 2009 125 showed that specific inhibition of PP1 by I-1 stimulated SR Ca-uptake but had no effect on Ca-release, suggesting that I-1 may be preferentially regulating PLN and not the RyR. The beneficial effects of I-1c in rodent models have been confirmed in large animal models of heart failure whereby acute and chronic gene transfer of I-1c by viral gene transfer induced both short-term and long-term beneficial hemodynamic effects. In porcine ischemic models of heart failure, AAV9.I1c induced long term hemodynamic benefits and improved cardiac remodeling (Figure 6). Future studies may further address the beneficial effects of PP1 inhibition through I-1c in functional recovery and remodeling in heart failure.
CONCLUSION AND PERSPECTIVE
In summary, several lines of evidence indicate that the SERCA/PLN regulatory complex is of paramount importance in proper cardiac Ca-cycling and function. In addition, it has recently become apparent that this fine-tuned regulation by SERCA/PLN represents a nodal point in the interaction of several protein partners that mediate a fine cross-talk between Ca and several signaling pathways, underlying cardiac performance and cell death. Indeed, disturbances in the regulatory function of SERCA/PN have been implicated as important contributors to the depressed cardiac function and remodeling in failing hearts. As such, targeting SERCA, PLN or the PLN/PP1 through I-1 or Hsp20 appears beneficial in alleviating the detrimental effects of heart failure, through specific increases in SR Ca-transport. Notably, the benefits of targeting SERCA have been shown in a recent clinical trial and the benefits of the other regulatory partners are at various stages of development. These new paradigms have led to experimental trials in enhancing SERCA2a uptake. There is now intensive scrutiny of the various proteins that lead to calcium cycling abnormalities and this focus will ultimately lead to novel treatment modalities in heart failure.
Acknowledgments
The authors would like to thank Dr. P. Nicolaou for useful suggestions in the preparation of this article.
Sources of Funding
This study was supported by National Institutes of Health Grants HL-26057, HL-64018, HL100396, HL088434, HL093183, HL080498, HL083156, and HL100396 and NIH/NHLBI Contract HHSN268201000045C.
Non standard abbreviations and acronyms
- Ad
Adenovirus
- AAV
Adeno-Associated Virus
- HAX-1
HS-1-associated protein X-1
- HRC
Histidine Rich Calcium Binding Protein
- Hsp20
Heat shock protein 20
- I-1
Inhibitor-1
- I/R
Ischemia/reperfusion
- KO
Knockout
- MLWHFQ
Minnesota Living with Heart Failure Questionnaire
- NYHA
New York Heart Association
- OE
Over-expression
- PLN
Phospholamban
- PP1c
Protein Phosphatase 1 catalytic subunit
- RyR
Ryanodine Receptor
- SERCA
Sarcoplasmic/endoplasmic reticulum calcium ATPase
- SUMO1
Small ubiquitin-like modifier type 1
- VO2max
Maximal oxygen consumption
- WT
Wild-type
Footnotes
Disclosures
Dr. Kranias is a scientific founder of Nanocor. Dr. Hajjar is scientific founder of Celladon.
References
- 1.MacLennan DH, Abu-Abed M, Kang C. Structure-function relationships in Ca2+ cycling proteins. J Mol Cell Cardiol. 2002;34:897–918. doi: 10.1006/jmcc.2002.2031. [DOI] [PubMed] [Google Scholar]
- 2.He H, Giordano FJ, Hilal-Dandan R, Choi DJ, Rockman HA, McDonough PM, Bluhm WF, Meyer M, Sayen MR, Swanson E, Dillmann WH. Overexpression of the rat sarcoplasmic reticulum Ca2+ ATPase gene in the heart of transgenic mice accelerates calcium transients and cardiac relaxation. J Clin Invest. 1997;100:380–9. doi: 10.1172/JCI119544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Baker DL, Hashimoto K, Grupp IL, Ji Y, Reed T, Loukianov E, Grupp G, Bhagwhat A, Hoit B, Walsh R, Marban E, Periasamy M. Targeted overexpression of the sarcoplasmic reticulum Ca2+-ATPase increases cardiac contractility in transgenic mouse hearts. Circ Res. 1998;83:1205–14. doi: 10.1161/01.res.83.12.1205. [DOI] [PubMed] [Google Scholar]
- 4.Vetter R, Rehfeld U, Reissfelder C, Weiss W, Wagner KD, Gunther J, Hammes A, Tschope C, Dillmann W, Paul M. Transgenic overexpression of the sarcoplasmic reticulum Ca2+ATPase improves reticular Ca2+ handling in normal and diabetic rat hearts. Faseb J. 2002;16:1657–9. doi: 10.1096/fj.01-1019fje. [DOI] [PubMed] [Google Scholar]
- 5.Periasamy M, Reed TD, Liu LH, Ji Y, Loukianov E, Paul RJ, Nieman ML, Riddle T, Duffy JJ, Doetschman T, Lorenz JN, Shull GE. Impaired cardiac performance in heterozygous mice with a null mutation in the sacro(endo)plasmic reticulum Ca2+-ATPase isoform 2 (SERCA2) gene. J Biol Chem. 1999;274:2556–62. doi: 10.1074/jbc.274.4.2556. [DOI] [PubMed] [Google Scholar]
- 6.Schultz Jo el J, Glascock BJ, Witt SA, Nieman ML, Nattamai KJ, Liu LH, Lorenz JN, Shull GE, Kimball TR, Periasamy M. Accelerated onset of heart failure in mice during pressure overload with chronically decreased SERCA2 calcium pump activity. Am J Physiol Heart Circ Physiol. 2004;286:H1146–53. doi: 10.1152/ajpheart.00720.2003. [DOI] [PubMed] [Google Scholar]
- 7.Ji Y, Lalli MJ, Babu GJ, Xu Y, Kirkpatrick DL, Liu LH, Chiamvimonvat N, Walsh RA, Shull GE, Periasamy M. Disruption of a single copy of the SERCA2 gene results in altered Ca2+ homeostasis and cardiomyocyte function. J Biol Chem. 2000;275:38073–80. doi: 10.1074/jbc.M004804200. [DOI] [PubMed] [Google Scholar]
- 8.Arvanitis DA, Vafiadaki E, Fan GC, Mitton BA, Gregory KN, Del Monte F, Kontrogianni-Konstantopoulos A, Sanoudou D, Kranias EG. Histidine-rich ca-binding protein interacts with sarcoplasmic reticulum ca-ATPase. Am J Physiol Heart Circ Physiol. 2007;293:H1581–9. doi: 10.1152/ajpheart.00278.2007. [DOI] [PubMed] [Google Scholar]
- 9.Ihara Y, Kageyama K, Kondo T. Overexpression of calreticulin sensitizes SERCA2a to oxidative stress. Biochem Biophys Res Commun. 2005;392:1343–9. doi: 10.1016/j.bbrc.2005.02.112. [DOI] [PubMed] [Google Scholar]
- 10.Kiewitz R, Acklin C, Schafer BW, Maco B, Uhrik B, Wuytack F, Erne P, Heizmann CW. Ca2+-dependent interaction of S100A1 with the sarcoplasmic reticulum Ca2+-ATPase2a and phospholamban in the human heart. Biochem Biophys Res Commun. 2003;306:550–7. doi: 10.1016/s0006-291x(03)00987-2. [DOI] [PubMed] [Google Scholar]
- 11.Asahi M, Nakayama H, Tada M, Otsu K. Regulation of sarco(endo)plasmic reticulum Ca2+ adenosine triphosphatase by phospholamban and sarcolipin: Implication for cardiac hypertrophy and failure. Trends Cardiovasc Med. 2003;13:152–7. doi: 10.1016/s1050-1738(03)00037-9. [DOI] [PubMed] [Google Scholar]
- 12.Bhupathy P, Babu GJ, Periasamy M. Sarcolipin and phospholamban as regulators of cardiac sarcoplasmic reticulum Ca2+ ATPase. J Mol Cell Cardiol. 2007;42:903–11. doi: 10.1016/j.yjmcc.2007.03.738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Arvanitis DA, Vafiadaki E, Sanoudou D, Kranias EG. Histidine-rich calcium binding protein: The new regulator of sarcoplasmic reticulum calcium cycling. J Mol Cell Cardiol. 2010;50:43–49. doi: 10.1016/j.yjmcc.2010.08.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Fan GC, Gregory KN, Zhao W, Park WJ, Kranias EG. Regulation of myocardial function by histidine-rich, calcium-binding protein. Am J Physiol Heart Circ Physiol. 2004;287:H1705–11. doi: 10.1152/ajpheart.01211.2003. [DOI] [PubMed] [Google Scholar]
- 15.Sato Y, Ferguson DG, Sako H, Dorn GW, 2nd, Kadambi VJ, Yatani A, Hoit BD, Walsh RA, Kranias EG. Cardiac-specific overexpression of mouse cardiac calsequestrin is associated with depressed cardiovascular function and hypertrophy in transgenic mice. J Biol Chem. 1998;273:28470–7. doi: 10.1074/jbc.273.43.28470. [DOI] [PubMed] [Google Scholar]
- 16.Gregory KN, Ginsburg KS, Bodi I, Hahn H, Marreez YM, Song Q, Padmanabhan PA, Mitton BA, Waggoner JR, Del Monte F, Park WJ, Dorn GW, 2nd, Bers DM, Kranias EG. Histidine-rich ca binding protein: A regulator of sarcoplasmic reticulum calcium sequestration and cardiac function. J Mol Cell Cardiol. 2006;40:653–65. doi: 10.1016/j.yjmcc.2006.02.003. [DOI] [PubMed] [Google Scholar]
- 17.Jaehnig EJ, Heidt AB, Greene SB, Cornelissen I, Black BL. Increased susceptibility to isoproterenol-induced cardiac hypertrophy and impaired weight gain in mice lacking the histidine-rich calcium-binding protein. Mol Cell Biol. 2006;26:9315–9326. doi: 10.1128/MCB.00482-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Arvanitis DA, Sanoudou D, Kolokathis F, Vafiadaki E, Papalouka V, Kontrogianni-Konstantopoulos A, Theodorakis GN, Paraskevaidis IA, Adamopoulos S, Dorn GW, 2nd, Kremastinos DT, Kranias EG. The Ser96Ala variant in histidine-rich calcium-binding protein is associated with life-threatening ventricular arrhythmias in idiopathic dilated cardiomyopathy. Eur Heart J. 2008;29:2514–25. doi: 10.1093/eurheartj/ehn328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Han P, Cai W, Wang Y, Lam CK, Arvanitis DA, Singh V, Chen S, Zhang H, Zhang R, Cheng H, Kranias EG. Catecholaminergic induced arrhythmias in failing cardiomyocytes associated with human HRCS96A variant overexpression. Am J Phys Heart Circ Phys. 2011;301:H1588–1595. doi: 10.1152/ajpheart.01153.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kirchberger MA, Tada M, Repke DI, Katz AM. Cyclic adenosine 3′, 5′-monophosphate-dependent protein kinase stimulation of calcium uptake by canine cardiac microsomes. J Mol Cell Cardiology. 1972;4:673–680. doi: 10.1016/0022-2828(72)90120-4. [DOI] [PubMed] [Google Scholar]
- 21.Wollenberger A. Nucleotides and the regulation of heart beat. Abstracts of the Fifth International Congress of Pharmacology; 1972. pp. 231–233. [Google Scholar]
- 22.Katz AM. Discovery of phospholamban: A personal history. in: Cardiac sarcoplasmic reticulum function and regulation of contractility. Annal N Y Acad Sci. 1998;853:9–19. doi: 10.1111/j.1749-6632.1998.tb08252.x. [DOI] [PubMed] [Google Scholar]
- 23.Mattiazzi A, Mundina-Weilenmann C, Guoxiang C, Vittone L, Kranias EG. Role of phospholamban phosphorylation on Thr17 in cardiac physiological and pathological conditions. Cardiovasc Res. 2005;68:366–375. doi: 10.1016/j.cardiores.2005.08.010. [DOI] [PubMed] [Google Scholar]
- 24.Davis BA, Edes I, Gupta RC, Young EF, Kim HW, Steenaart NA, Szymanska G, Kranias EG. The role of phospholamban in the regulation of calcium transport by cardiac sarcoplasmic reticulum. Molecular and Cellular Biochemistry. 1990;99:83–88. doi: 10.1007/BF00230337. [DOI] [PubMed] [Google Scholar]
- 25.Kranias EG. Regulation of calcium transport by protein phosphotase activity associated with cardiac scrcoplasmic reticulum. J Biol Chem. 1985;260:11006–11010. [PubMed] [Google Scholar]
- 26.James P, Inui M, Tada M, Chiesi M, Carafoli E. Nature and site of phospholamban regulation of the Ca2+ pump of sarcoplasmic reticulum. Nature. 1989;342:90–92. doi: 10.1038/342090a0. [DOI] [PubMed] [Google Scholar]
- 27.Kim HW, Steenaart NA, Ferguson DG, Kranias EG. Functional reconstitution of the cardiac sarcoplasmic reticulum Ca2+-ATPase with phospholamban in phospholipid vesicles. J Biol Chem. 1990;265:1702–1709. [PubMed] [Google Scholar]
- 28.Simmerman HK, Jones L. Phospholamban: Protein structure, mechanism of action, and role in cardiac function. Physiol Rev. 1998;78:921–947. doi: 10.1152/physrev.1998.78.4.921. [DOI] [PubMed] [Google Scholar]
- 29.Fujii J, Zarain-Herzberg A, Willard HF, Tada M, MacLennan DH. Structure of the rabbit phospholamban gene, cloning of the human cDNA, and assignment of the gene to human chromosome 6. J Biol Chem. 1991;266:11669–11675. [PubMed] [Google Scholar]
- 30.MacLennan DH, Kimura Y, Toyofuku T. Sites of regulatory interaction between calcium ATPases and phospholamban. Ann N Y Acad Sci. 1998;853:31–42. doi: 10.1111/j.1749-6632.1998.tb08254.x. [DOI] [PubMed] [Google Scholar]
- 31.Kadambi VJ, Ponniah S, Harrer JM, Hoit BD, Dorn GW, II, Walsh RA, Kranias EG. Cardiac-specific overexpression of phospholamban alters calcium kinetics and resultant cardiomyocyte mechanics in transgenic mice. J Clin Invest. 1996;97:533–539. doi: 10.1172/JCI118446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Luo W, Grupp IL, Harrer J, Ponniah S, Grupp G, Duffy JJ, Doetschman T, Kranias EG. Targeted ablation of the phospholamban gene is associated with markedly enhanced myocardial contractility and loss of β-agonist stimulation. Circ Res. 1994;75:401–409. doi: 10.1161/01.res.75.3.401. [DOI] [PubMed] [Google Scholar]
- 33.Luo W, Wolska BM, Grupp IL, Harrer JM, Haghighi K, Ferguson DG, Slack JP, Grupp G, Doetschman T, Solaro RJ, Kranias EG. Phospholamban gene dosage effects in the mammalian heart. Circ Res. 1996;78:839–847. doi: 10.1161/01.res.78.5.839. [DOI] [PubMed] [Google Scholar]
- 34.Hoit BD, Khoury SF, Kranias EG, Ball N, Walsh RA. In vivo echocardiographic detection of enhanced left ventricular function in gene-targeted mice with phospholamban deficiency. Circ Res. 1995;77:632–637. doi: 10.1161/01.res.77.3.632. [DOI] [PubMed] [Google Scholar]
- 35.Lorenz JN, Kranias EG. Regulatory effects of phospholamban on cardiac function in intact mice. Am J Physiol. 1997;273:H2826–H2831. doi: 10.1152/ajpheart.1997.273.6.H2826. [DOI] [PubMed] [Google Scholar]
- 36.Mortishire-Smith RJ, Pitzenberger SM, Burke CJ, Middaugh CR, Garsky VM, Johnson RG. Solution structure of the cytoplasmic domain of phopholamban: Phosphorylation leads to a local perturbation in secondary structure. Biochemistry. 1995;34:7603–7613. doi: 10.1021/bi00023a006. [DOI] [PubMed] [Google Scholar]
- 37.Zhai J, Schmidt AG, Hoit BD, Kimura Y, MacLennan DH, Kranias EG. Cardiac-specific overexpression of a superinhibitory pentameric phospholamban mutant enhances inhibition of cardiac function in vivo. J Biol Chem. 2000;275:10538–10544. doi: 10.1074/jbc.275.14.10538. [DOI] [PubMed] [Google Scholar]
- 38.Schmidt AG, Zhai J, Carr AN, Gerst MJ, Lorenz JN, Pollesello P, Annila A, Hoit BD, Kranias EG. Structural and functional implications of the phospholamban hinge domain: Impaired SR Ca2+ uptake as a primary cause of heart failure. Cardiovasc Res. 2002;56:248–259. doi: 10.1016/s0008-6363(02)00541-2. [DOI] [PubMed] [Google Scholar]
- 39.Haghighi K, Schmidt AG, Hoit BD, Brittsan AG, Yatan A, Lester JW, Zhai J, Kimura Y, Dorn GW, II, MacLennan DH, Kranias EG. Superinhibition of sarcoplasmic reticulum function by phospholamban induces cardiac contractile failure. J Biol Chem. 2001;276:24145–24152. doi: 10.1074/jbc.M102403200. [DOI] [PubMed] [Google Scholar]
- 40.Zvaritch E, Backx PH, Jirik F, Kimura Y, de Leon S, Schmidt AG, Hoit BD, Lester JW, Kranias EG, MacLennan DH. The transgenic expression of highly inhibitory monomeric forms of phospholamban in mouse heart impairs cardiac contractility. J Biol Chem. 2000;275:14985–14991. doi: 10.1074/jbc.275.20.14985. [DOI] [PubMed] [Google Scholar]
- 41.England PJ. Correlation between contractions and phosphorylation of the inhibitory subunit of troponin in perfused rat heart. FEBS Lett. 1975;50:57–60. doi: 10.1016/0014-5793(75)81040-4. [DOI] [PubMed] [Google Scholar]
- 42.Kranias EG, Solaro RJ. Phosphorylation of troponin I and phospholamban during catecholamine stimulation of rabbit heart. Nature. 1982;298:182–184. doi: 10.1038/298182a0. [DOI] [PubMed] [Google Scholar]
- 43.Lindemann JP, Jones LR, Hathaway DR, Henry BG, Watanabe AM. β-Adrenergic stimulation of phospholamban phosphorylation and Ca2+-ATPase activity in guinea pig ventricles. J Biol Chem. 1983;258:464–471. [PubMed] [Google Scholar]
- 44.Kranias EG, Garvey JL, Srivastava RD, Solaro RJ. Phosphorylation and functional modifications of sarcoplasmic reticulum and myofibrils in isolated rabbit hearts stimulated with isoprenaline. Biochem J. 1985;226:113–121. doi: 10.1042/bj2260113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Edes I, Kranias EG. Phospholamban and troponin I are substrates for protein kinase C in vitro but not in intact beating guinea pig hearts. Circ Res. 1990;67:394–400. doi: 10.1161/01.res.67.2.394. [DOI] [PubMed] [Google Scholar]
- 46.Huggins JP, Cook EA, Piggott JR, Mattinsley TJ, England PJ. Phospholamban is a good substrate for cyclic GMP-dependent protein kinase in vitro, but not in intact cardiac or smooth muscle. Biochem J. 1989;260:829–835. doi: 10.1042/bj2600829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Talosi L, Edes I, Kranias EG. Intracellular mechanisms mediating reversal of β-adrenergic stimulation in intact beating hearts. Am J Physiol. 1993;264:H791–H797. doi: 10.1152/ajpheart.1993.264.3.H791. [DOI] [PubMed] [Google Scholar]
- 48.Luo W, Chu G, Sato Y, Zhou Z, Kadambi VJ, Kranias EG. Transgenic approaches to define the functional role of dual site phospholamban phosphorylation. J Biol Chem. 1998;273:4734–4739. doi: 10.1074/jbc.273.8.4734. [DOI] [PubMed] [Google Scholar]
- 49.Chu G, Lester JW, Young KB, Luo W, Zhai J, Kranias EG. A single site (Ser16) phosphorylation in phospholamban is sufficient in mediating its maximal cardiac responses to β-agonists. J Biol Chem. 2000;275:38938–38943. doi: 10.1074/jbc.M004079200. [DOI] [PubMed] [Google Scholar]
- 50.Brittsan AG, Carr AN, Schmidt AG, Schmidt AG, Kranias EG. Maximal inhibition of SERCA2 Ca2+ affinity by phospholamban in transgenic hearts overexpressing a non-phosphorylatable form of phospholamban. J Biol Chem. 2000;275:12129–12135. doi: 10.1074/jbc.275.16.12129. [DOI] [PubMed] [Google Scholar]
- 51.Zhao W, Uehara Y, Chu G, Song Q, Qian J, Young K, Kranias EG. Threonine-17 phosphorylation of phospholamban: A key determinant of frequency-dependent increase of cardiac contractility. J Mol Cell Cardiol. 2004;37:607–612. doi: 10.1016/j.yjmcc.2004.05.013. [DOI] [PubMed] [Google Scholar]
- 52.Wolska BM, Stojanovic MO, Luo W, Kranias EG, Solaro RJ. Effect of ablation of phospholamban on dynamics of cardiac myocyte contraction and intracellular Ca2+ Am J Physiol. 1996;271:C391–C397. doi: 10.1152/ajpcell.1996.271.1.C391. [DOI] [PubMed] [Google Scholar]
- 53.Schmitt JP, Kamisago M, Asahi M, Li GH, Ahmad F, Mende U, Kranias EG, MacLennan DH, Seidman JG, Seidman CE. Dilated cardiomyopathy and heart failure caused by a mutation in phospholamban. Science. 2003;299:1410–1413. doi: 10.1126/science.1081578. [DOI] [PubMed] [Google Scholar]
- 54.Haghighi K, Pritchard T, Bossuyt J, Waggoner JR, Yuan Q, Fan GC, Osinska H, Anjak A, Rubinstein J, Robbins J, Bers DM, Kranias EG. The human phospholamban Arg14-deletion mutant localizes to plasma membrane and interacts with the Na/K-ATPase. J Mol Cell Cardiol. 2011 doi: 10.1016/j.yjmcc.2011.11.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Medeiros A, Biagi DG, Sobreira TJ, de Oliveira PS, Negrão CE, Mansur AJ, Krieger JE, Brum PC, Pereira AC. Mutations in the human phospholamban gene in patients with heart failure. Am Heart J. 2011;162:1088–1095. doi: 10.1016/j.ahj.2011.07.028. [DOI] [PubMed] [Google Scholar]
- 56.Haghighi K, Kolokathis F, Pater L, Lynch RA, Asahi M, Gramolini AO, Fan GC, Tsiapras D, Hahn HS, Adamopoulos S, Liggett SB, Dorn GW, 2nd, MacLennan DH, Kremastinos DT, Kranias EG. Human phospholamban null results in lethal dilated cardiomyopathy revealing a critical difference between mouse and man. J Clin Invest. 2003;111:869–876. doi: 10.1172/JCI17892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Minamisawa S, Sato Y, Tatsuguchi Y, Fujino T, Imamura S, Uetsuka Y, Nakazawa M, Matsuoka R. Mutation of the phospholamban promoter associated with hypertrophic cardiomyopathy. Biochem Biophys Res Commun. 2003;304:1–4. doi: 10.1016/s0006-291x(03)00526-6. [DOI] [PubMed] [Google Scholar]
- 58.Haghighi K, Chen G, Sato Y, Fan GC, He S, Kolokathis F, Pater L, Paraskevaidis I, Jones WK, Dorn GW, 2nd, Kremastinos DT, Kranias EG. A human phospholamban promoter polymorphism in dilated cardiomyopathy alters transcriptional regulation by glucocorticoids. Hum Mutat. 2008;29:640–7. doi: 10.1002/humu.20692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Medin M, Hermida-Prieto M, Monserrat L, Laredo R, Rodriguez-Rey JC, Fernandez X, Castro-Beiras A. Mutational screening of phospholamban gene in hypertrophic and idiopathic dilated cardiomyopathy and functional study of the PLN -42 C>G mutation. Eur J Heart Fail. 2007;9:37–43. doi: 10.1016/j.ejheart.2006.04.007. [DOI] [PubMed] [Google Scholar]
- 60.Han Y, Chen YS, Liu Z, Bodyak N, Rigor D, Bisping E, Pu WT, Kang PM. Overexpression of HAX-1 protects cardiac myocytes from apoptosis through caspase-9 inhibition. Circ Res. 2006;99:415–23. doi: 10.1161/01.RES.0000237387.05259.a5. [DOI] [PubMed] [Google Scholar]
- 61.Vafiadaki E, Sanoudou D, Arvanitis DA, Catino DH, Kranias EG, Kontrogianni-Konstantopoulos A. Phospholamban interacts with HAX-1, a mitochondrial protein with anti-apoptotic function. J Mol Biol. 2007;367:65–79. doi: 10.1016/j.jmb.2006.10.057. [DOI] [PubMed] [Google Scholar]
- 62.Zhao W, Waggoner JR, Zhang ZG, Lam CK, Han P, Qian J, Schroder PM, Mitton B, Kontrogianni-Konstantopoulos A, Robia SL, Kranias EG. The anti-apoptotic protein HAX-1 is a novel regulator of cardiac function. Proc Natl Acad Sci. 2009;106:20776–20781. doi: 10.1073/pnas.0906998106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Yap SV, Vafiadaki E, Strong J, Kontrogianni-Konstantopoulos A. HAX-1: A multifaceted antiapoptotic protein localizing in the mitochondria and the sarcoplasmic reticulum of striated muscle cells. J Mol Cell Cardiol. 2010;48:1266–79. doi: 10.1016/j.yjmcc.2009.10.028. [DOI] [PubMed] [Google Scholar]
- 64.Vafiadaki E, Arvanitis DA, Pagakis SN, Papalouka V, Sanoudou D, Kontrogianni-Konstantopoulos A, Kranias EG. The anti-apoptotic protein HAX-1 interacts with SERCA2 and regulates its protein levels to promote cell survival. Mol Biol Cell. 2009;20:306–18. doi: 10.1091/mbc.E08-06-0587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.MacDougall LK, Jones LR, Cohen P. Identification of the major protein phosphatases in mammalian cardiac muscle which dephosphorylate phospholamban. Eur J Biochem. 1991;196:725–34. doi: 10.1111/j.1432-1033.1991.tb15871.x. [DOI] [PubMed] [Google Scholar]
- 66.Steenaart NA, Ganim JR, Di Salvo J, Kranias EG. The phospholamban phosphatase associated with cardiac sarcoplasmic reticulum is a type 1 enzyme. Arch Biochem Biophys. 1992;293:17–24. doi: 10.1016/0003-9861(92)90359-5. [DOI] [PubMed] [Google Scholar]
- 67.Neumann J, Eschenhagen T, Jones LR, Linck B, Schmitz W, Scholz H, Zimmermann N. Increased expression of cardiac phosphatases in patients with end-stage heart failure. J Mol Cell Cardiol. 1997;29:265–72. doi: 10.1006/jmcc.1996.0271. [DOI] [PubMed] [Google Scholar]
- 68.Carr AN, Schmidt AG, Suzuki Y, del Monte F, Sato Y, Lanner C, Breeden K, Jing SL, Allen PB, Greengard P, Yatani A, Hoit BD, Grupp IL, Hajjar RJ, DePaoli-Roach AA, Kranias EG. Type 1 phosphatase, a negative regulator of cardiac function. Mol Cell Biol. 2002;22:4124–35. doi: 10.1128/MCB.22.12.4124-4135.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Ceulemans H, Bollen M. Functional diversity of protein phosphatase-1, a cellular economizer and reset button. Physiol Rev. 2004;84:1–39. doi: 10.1152/physrev.00013.2003. [DOI] [PubMed] [Google Scholar]
- 70.Kirchhefer U, Baba HA, Bokník P, Breeden KM, Mavila N, Brüchert N, Justus I, Matus M, Schmitz W, Depaoli-Roach AA, Neumann J. Enhanced cardiac function in mice overexpressing protein phosphatase inhibitor-2. Cardiovasc Res. 2005;68:98–108. doi: 10.1016/j.cardiores.2005.05.019. [DOI] [PubMed] [Google Scholar]
- 71.Neumann J, Gupta RC, Schmitz W, Scholz H, Nairn AC, Watanabe AM. Evidence for isoproterenol-induced phosphorylation of phosphatase inhibitor-1 in the intact heart. Circ Res. 1991;69:1450–7. doi: 10.1161/01.res.69.6.1450. [DOI] [PubMed] [Google Scholar]
- 72.Iyer RB, Koritz SB, Kirchberger MA. A regulation of the level of phosphorylated phospholamban by inhibitor-1 in rat heart preparations in vitro. Mol Cell Endocrinol. 1988;55:1–6. doi: 10.1016/0303-7207(88)90084-6. [DOI] [PubMed] [Google Scholar]
- 73.Gupta RC, Neumann J, Watanabe AM, Lesch M, Sabbah HN. Evidence for presence and hormonal regulation of protein phosphatase inhibitor-1 in ventricular cardiomyocyte. Am J Physiol. 1996;270:H1159–64. doi: 10.1152/ajpheart.1996.270.4.H1159. [DOI] [PubMed] [Google Scholar]
- 74.El-Armouche A, Rau T, Zolk O, Ditz D, Pamminger T, Zimmermann WH, Jäckel E, Harding SE, Boknik P, Neumann J, Eschenhagen T. Evidence for protein phosphatase inhibitor-1 playing an amplifier role in beta-adrenergic signaling in cardiac myocytes. FASEB J. 2003;1:437–9. doi: 10.1096/fj.02-0057fje. [DOI] [PubMed] [Google Scholar]
- 75.Pathak A, del Monte F, Zhao W, Schultz JE, Lorenz JN, Bodi I, Weiser D, Hahn H, Carr AN, Syed F, Mavila N, Jha L, Qian J, Marreez Y, Chen G, McGraw DW, Heist EK, Guerrero JL, DePaoli-Roach AA, Hajjar RJ, Krania EG. Enhancement of cardiac function and suppression of heart failure progression by inhibition of protein phosphatase 1. Circ Res. 2005;96:756–66. doi: 10.1161/01.RES.0000161256.85833.fa. [DOI] [PubMed] [Google Scholar]
- 76.Nicolaou P, Rodriguez P, Ren X, Zhou X, Qian J, Sadayappan S, Mitton B, Pathak A, Robbins J, Hajjar RJ, Jones K, Kranias EG. Inducible expression of active protein phosphatase-1 inhibitor-1 enhances basal cardiac function and protects against Ischemia/Reperfusion injury. Circ Res. 2009;104:1012–20. doi: 10.1161/CIRCRESAHA.108.189811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Wittköpper K, Fabritz L, Neef S, Ort KR, Grefe C, Unsöld B, Kirchhof P, Maier LS, Hasenfuss G, Dobrev D, Eschenhagen T, El-Armouche A. Constitutively active phosphatase inhibitor-1 improves cardiac contractility in young mice but is deleterious after catecholaminergic stress and with aging. J Clin Invest. 2010;120:617–26. doi: 10.1172/JCI40545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Wehrens XH, Lehnart SE, Reiken SR, Deng SX, Vest JA, Cervantes D, Coromilas J, Landry DW, Marks AR. Protection from cardiac arrhythmia through ryanodine receptor-stabilizing protein calstabin2. Science. 2004;304:292–6. doi: 10.1126/science.1094301. [DOI] [PubMed] [Google Scholar]
- 79.Yano M, Ono K, Ohkusa T, Suetsugu M, Kohno M, Hisaoka T, Kobayashi S, Hisamatsu Y, Yamamoto T, Kohno M, Noguchi N, Takasawa S, Okamoto H, Matsuzaki M. Altered stoichiometry of FKBP12.6 versus ryanodine receptor as a cause of abnormal ca(2+) leak through ryanodine receptor in heart failure. Circulation. 2000;102:2131–6. doi: 10.1161/01.cir.102.17.2131. [DOI] [PubMed] [Google Scholar]
- 80.Braz JC, Gregory K, Pathak A, Zhao W, Sahin B, Klevitsky R, Kimball TF, Lorenz JN, Nairn AC, Liggett SB, Bodi I, Wang S, Schwartz A, Lakatta EG, DePaoli-Roach AA, Robbins J, Hewett TE, Bibb JA, Westfall MV, Kranias EG, Molkentin JD. PKC-alpha regulates cardiac contractility and propensity toward heart failure. Nat Med. 2004;10:248–54. doi: 10.1038/nm1000. [DOI] [PubMed] [Google Scholar]
- 81.Rodriguez P, Mitton B, Waggoner JR, Kranias EG. Identification of a novel phosphorylation site in protein phosphatase inhibitor-1 as a negative regulator of cardiac function. J Biol Chem. 2006;281:38599–608. doi: 10.1074/jbc.M604139200. [DOI] [PubMed] [Google Scholar]
- 82.Rodriguez P, Mitton B, Nicolaou P, Chen G, Kranias EG. Phosphorylation of human inhibitor-1 at Ser67 and/or Thr75 attenuates stimulatory effects of protein kinase A signaling in cardiac myocytes. Am J Physiol Heart Circ Physiol. 2007;293:H762–9. doi: 10.1152/ajpheart.00104.2007. [DOI] [PubMed] [Google Scholar]
- 83.El-Armouche A, Bednorz A, Pamminger T, Ditz D, Didié M, Dobrev D, Eschenhagen T. Role of calcineurin and protein phosphatase-2A in the regulation of phosphatase inhibitor-1 in cardiac myocytes. Biochem Biophys Res Commun. 2006;346:700–6. doi: 10.1016/j.bbrc.2006.05.182. [DOI] [PubMed] [Google Scholar]
- 84.Chen G, Zhou X, Nicolaou P, Rodriguez P, Song G, Mitton B, Pathak A, Zachariah A, Fan GC, Dorn GW, 2nd, Kranias EG. A human polymorphism of protein phosphatase-1 inhibitor-1 is associated with attenuated contractile response of cardiomyocytes to {beta}-adrenergic stimulation. FASEB J. 2008;22:1790–6. doi: 10.1096/fj.07-097428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Chu G, Egnaczyk GF, Zhao W, Jo SH, Fan GC, Maggio JE, Xiao RP, Kranias EG. Phosphoproteome analysis of cardiomyocytes subjected to beta-adrenergic stimulation: Identification and characterization of a cardiac heat shock protein p20. Circ Res. 2004;94:184–93. doi: 10.1161/01.RES.0000107198.90218.21. [DOI] [PubMed] [Google Scholar]
- 86.Qian J, Ren X, Wang X, Zhang P, Jones WK, Molkentin JD, Fan GC, Kranias EG. Blockade of Hsp20 phosphorylation exacerbates cardiac ischemia/reperfusion injury by suppressed autophagy and increased cell death. Circ Res. 2009;105:1223–31. doi: 10.1161/CIRCRESAHA.109.200378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Qian J, Vafiadaki E, Florea S, Singh V, Song W, Lam CK, Yuan Q, Pritchard TJ, Rodriguez P, Wang Y, Haghighi K, Sanoudou D, Cai W, Wang HS, Fan GC, Kranias EG. Small heat shock protein 20 interacts with protein phosphatise-1 and enhances sarcoplasmic reticulum calcium cycling. Circ Res. 2011;108:1429–1438. doi: 10.1161/CIRCRESAHA.110.237644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Pipkin W, Johnson JA, Creazzo TL, Burch J, Komalavilas P, Brophy CM. Localization, macromolecular associations, and function of the small heat shock-related protein HSP20 in rat heart. Circulation. 2003;107:469–76. doi: 10.1161/01.cir.0000044386.27444.5a. [DOI] [PubMed] [Google Scholar]
- 89.Ren XP, Wu J, Wang X, Sartor MA, Qian J, Jones K, Nicolaou P, Pritchard TJ, Fan GC. MicroRNA-320 is involved in the regulation of cardiac ischemia/reperfusion injury by targeting heat-shock protein 20. Circulation. 2009;119:2357–66. doi: 10.1161/CIRCULATIONAHA.108.814145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Fan GC, Chu G, Mitton B, Song Q, Yuan Q, Kranias EG. Small heat-shock protein Hsp20 phosphorylation inhibits beta-agonist-induced cardiac apoptosis. Circ Res. 2004;94:1474–82. doi: 10.1161/01.RES.0000129179.66631.00. [DOI] [PubMed] [Google Scholar]
- 91.Nicolaou P, Knöll R, Haghighi K, Fan GC, Dorn GW, 2nd, Hasenfub G, Kranias EG. Human mutation in the anti-apoptotic heat shock protein 20 abrogates its cardioprotective effects. J Biol Chem. 2008;283:33465–71. doi: 10.1074/jbc.M802307200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Fan GC, Ren X, Qian J, Yuan Q, Nicolaou P, Wang Y, Jones WK, Chu G, Kranias EG. Novel cardioprotective role of a small heat-shock protein, Hsp20, against ischemia/reperfusion injury. Circulation. 2005;111:1792–9. doi: 10.1161/01.CIR.0000160851.41872.C6. [DOI] [PubMed] [Google Scholar]
- 93.Rustandi RR, Baldisseri DM, Inman KG, Nizner P, Hamilton SM, Landar A, et al. Three-dimensional solution structure of the calcium-signaling protein apo-S100A1 as determined by NMR. Biochemistry. 2002;41:788–96. doi: 10.1021/bi0118308. [DOI] [PubMed] [Google Scholar]
- 94.Kato K, Kimura S. S100ao (alpha alpha) protein is mainly located in the heart and striated muscles. Biochim Biophys Acta. 1985;842:146–50. doi: 10.1016/0304-4165(85)90196-5. [DOI] [PubMed] [Google Scholar]
- 95.Treves S, Scutari E, Robert M, Groh S, Ottolia M, Prestipino G, Ronjat M, Zorzato F. Interaction of S100A1 with the Ca2+ release channel (ryanodine receptor) of skeletal muscle. Biochemistry. 1997;36:11496–503. doi: 10.1021/bi970160w. [DOI] [PubMed] [Google Scholar]
- 96.Remppis A, Greten T, Schafer BW, Hunziker P, Erne P, Katus HA, Heizmann CW. Altered expression of the ca(2+)-binding protein S100A1 in human cardiomyopathy. Biochim Biophys Acta. 1996;1313:253–7. doi: 10.1016/0167-4889(96)00097-3. [DOI] [PubMed] [Google Scholar]
- 97.Okada M, Hatakeyama T, Itoh H, Tokuta N, Tokumitsu H, Kobayashi R. S100A1 is a novel molecular chaperone and a member of the Hsp70/Hsp90 multichaperone complex. J Biol Chem. 2004;279:4221–33. doi: 10.1074/jbc.M309014200. [DOI] [PubMed] [Google Scholar]
- 98.Zimmer DB, Wright Sadosky P, Weber DJ. Molecular mechanisms of S100-target protein interactions. Microsc Res Tech. 2003;60:552–9. doi: 10.1002/jemt.10297. [DOI] [PubMed] [Google Scholar]
- 99.Pleger ST, Most P, Boucher M, Soltys S, Chuprun JK, Pleger W, Gao E, Dasgupta A, Rengo G, Remppis A, Katus HA, Eckhart AD, Rabinowitz JE, Koch WJ. Stable myocardial-specific AAV6-S100A1 gene therapy results in chronic functional heart failure rescue. Circulation. 2007;115:2506–2515. doi: 10.1161/CIRCULATIONAHA.106.671701. [DOI] [PubMed] [Google Scholar]
- 100.Pleger ST, Shan C, Ksienzyk J, Bekeredjian R, Boekstegers P, Hinkel R, Schinkel S, Leuchs B, Ludwig J, Qiu G, Weber C, Raake P, Koch WJ, Katus HA, Muller OJ, Most P. Cardiac AAV9-S100A1 gene therapy rescues post-ischemic heart failure in a preclinical large animal model. Sci Transl Med. 2011;3:92ra64. doi: 10.1126/scitranslmed.3002097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Schwartz RJ, Yeh ET. Weighing in on heart failure: The role of SERCA2a SUMOylation. Circ Res. 2012;110:198–199. doi: 10.1161/RES.0b013e318246f187. [DOI] [PubMed] [Google Scholar]
- 102.Kho C, Lee A, Jeong D, Oh JG, Chaanine AH, Kizana E, Park WJ, Hajjar RJ. SUMO1-dependent modulation of SERCA2a in heart failure. Nature. 2011;477:601–605. doi: 10.1038/nature10407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Limas CJ, Olivari MT, Goldenberg IF, Goldenberg IF, Levine TB, Benditt DG, Simon A. Calcium uptake by cardiac sarcoplasmic reticulum in human dilated cardiomyopathy. Cardiovasc Res. 1987;21:601–605. doi: 10.1093/cvr/21.8.601. [DOI] [PubMed] [Google Scholar]
- 104.Dash R, Frank KF, Carr AN, Moravec CS, Kranias EG. Gender influences on sarcoplasmic reticulum Ca2+-handling in failing human myocardium. J Mol Cell Cardiol. 2001;33:1345–53. doi: 10.1006/jmcc.2001.1394. [DOI] [PubMed] [Google Scholar]
- 105.Barki-Harrington L, Perrino C, Rockman HA. Network integration of the adrenergic system in cardiac hypertrophy. Cardiovasc Res. 2004;63:391–402. doi: 10.1016/j.cardiores.2004.03.011. [DOI] [PubMed] [Google Scholar]
- 106.Ferguson SS. Evolving concepts in G protein-coupled receptor endocytosis: The role in receptor desensitization and signaling. Pharmacol Rev. 2001;53:1–24. [PubMed] [Google Scholar]
- 107.Bibb JA, Nishi A, O’Callaghan JP, Ule J, Lan M, Snyder GL, Horiuchi A, Saito T, Hisanaga S, Czernik AJ, Nairn AC, Greengard P. Phosphorylation of protein phosphatase inhibitor-1 by Cdk5. J Biol Chem. 2001;276:14490–7. doi: 10.1074/jbc.M007197200. [DOI] [PubMed] [Google Scholar]
- 108.Park IK, DePaoli-Roach AA. Domains of phosphatase inhibitor-2 involved in the control of the ATP-mg-dependent protein phosphatase. J Biol Chem. 1994;269:28919–28. [PubMed] [Google Scholar]
- 109.Dorn GW, 2nd, Molkentin JD. Manipulating cardiac contractility in heart failure: Data from mice and men. Circulation. 2004;109:150–8. doi: 10.1161/01.CIR.0000111581.15521.F5. [DOI] [PubMed] [Google Scholar]
- 110.Lipskaia L, Chemaly ER, Hadri L, Lompre A, Hajjar RJ. Sarcoplasmic reticulum Ca2+ ATPase as a therapeutic target for heart failure. Expert Opin Biol Ther. 2010;10:29–41. doi: 10.1517/14712590903321462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Sakata S, Lebeche D, Sakata N, Sakata Y, Chemaly ER, Liang L, Tsuji T, Takewa Y, del Monte F, Peluso R, Zsebo K, Jeong D, Park WJ, Kawase Y, Hajjar RJ. Restoration of mechanical and energetic function in failing aortic-banded rat hearts by gene transfer of calcium cycling proteins. J Mol Cell Cardiol. 2007;42:852–61. doi: 10.1016/j.yjmcc.2007.01.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Lyon AR, Nikolaev VO, Miragoli M, Sikkel MB, Paur H, Benard L, Hulot JS, Kohlbrenner E, Hajjar RJ, Peters NS, Korchev YE, Macleod KT, Harding SE, Gorelik J. Plasticity of surface structures and β2-adrenergic receptor localization in failing ventricular cardiomyocytes during recovery from heart failure. Circ Heart Fail. 2012 doi: 10.1161/CIRCHEARTFAILURE.111.964692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Hadri L, Bobe R, Kawase Y, Ladage D, Ishikawa K, Atassi F, Lebeche D, Kranias EG, Leopold JA, Lompre AM, Lipskaia L, Hajjar RJ. SERCA2a gene transfer enhances eNOS expression and activity in endothelial cells. Mol Ther. 2010;18:1284–1292. doi: 10.1038/mt.2010.77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Prunier F, Kawase Y, Gianni D, Scapin C, Danik SB, Ellinor PT, Hajjar RJ, Del Monte F. Prevention of ventricular arrhythmias with sarcoplasmic reticulum Ca2+ ATPase pump overexpression in a porcine model of ischemia reperfusion. Circulation. 2008;118:614–624. doi: 10.1161/CIRCULATIONAHA.108.770883. [DOI] [PubMed] [Google Scholar]
- 115.del Monte F, Lebeche D, Guerrero JL, Tsuji T, Doye AA, Gwathmey JK, Hajjar RJ. Abrogation of ventricular arrhythmias in a model of ischemia and reperfusion by targeting myocardial calcium cycling. Proc Natl Acad Sci U S A. 2004;101:5622–5627. doi: 10.1073/pnas.0305778101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Lyon AR, Bannister ML, Collins T, Pearce E, Sepehripour AH, Dubb SS, Garcia E, O’Gara P, Liang L, Kohlbrenner E, Hajjar RJ, Peters NS, Poole-Wilson PA, Macleod KT, Harding SE. SERCA2a gene transfer decreases sarcoplasmic reticulum calcium leak and reduces ventricular arrhythmias in a model of chronic heart failure. Circ Arrhythm Electrophysiol. 2011;4:362–372. doi: 10.1161/CIRCEP.110.961615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Cutler MJ, Wan X, Laurita KR, Hajjar RJ, Rosenbaum DS. Targeted SERCA2a gene expression identifies molecular mechanism and therapeutic target for arrhythmogenic cardiac alternans. Circ Arrhythm Electrophysiol. 2009;2:686–694. doi: 10.1161/CIRCEP.109.863118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Kawase Y, Ly HQ, Prunier F, Lebeche D, Shi Y, Jin H, Hadri L, Yoneyama R, Hoshino K, Takewa Y, Sakata S, Peluso R, Zsebo K, Gwathmey JK, Tardif JC, Tanguay JF, Hajjar RJ. Reversal of cardiac dysfunction after long-term expression of SERCA2a by gene transfer in a pre-clinical model of heart failure. J Am Coll Cardiol. 2008;51:1112–1119. doi: 10.1016/j.jacc.2007.12.014. [DOI] [PubMed] [Google Scholar]
- 119.Byrne MJ, Power JM, Preovolos A, Mariani JA, Hajjar RJ, Kaye DM. Recirculating cardiac delivery of AAV2/1SERCA2a improves myocardial function in an experimental model of heart failure in large animals. Gene Ther. 2008;15:1550–1557. doi: 10.1038/gt.2008.120. [DOI] [PubMed] [Google Scholar]
- 120.Jaski BE, Jessup ML, Mancini DM, Cappola TP, Pauly DF, Greenberg B, Borow K, Dittrich H, Zsebo KM, Hajjar RJ. Calcium upregulation by percutaneous administration of gene therapy in cardiac disease (CUPID trial), a first-in-human phase 1/2 clinical trial. J Card Fail. 2009;15:171–181. doi: 10.1016/j.cardfail.2009.01.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Hajjar RJ, Zsebo K, Deckelbaum L, Thompson C, Rudy J, Yaroshinsky A, Ly H, Kawase Y, Wagner K, Borow K, Jaski B, London B, Greenberg B, Pauly DF, Patten R, Starling R, Mancini D, Jessup M. Design of a phase 1/2 trial of intracoronary administration of AAV1/SERCA2a in patients with heart failure. J Card Fail. 2008;14:355–367. doi: 10.1016/j.cardfail.2008.02.005. [DOI] [PubMed] [Google Scholar]
- 122.Jessup M, Greenberg B, Mancini D, Cappola T, Pauly DF, Jaski B, Yaroshinsky A, Zsebo KM, Dittrich H, Hajjar RJ. Calcium upregulation by percutaneous administration of gene therapy in cardiac disease (CUPID): A phase 2 trial of intracoronary gene therapy of sarcoplasmic reticulum Ca2+-ATPase in patients with advanced heart failure. Circulation. 2011;124:304–313. doi: 10.1161/CIRCULATIONAHA.111.022889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Yamada M, Ikeda Y, Yano M, Yoshimura K, Nishino S, Aoyama H, Wang L, Aoki H, Matsuzaki M. Inhibition of protein phosphatase 1 by inhibitor-2 gene delivery ameliorates heart failure progression in genetic cardiomyopathy. FASEB J. 2006;20:1197–9. doi: 10.1096/fj.05-5299fje. [DOI] [PubMed] [Google Scholar]
- 124.El-Armouche A, Wittköpper K, Degenhardt F, Weinberger F, Didié M, Melnychenko I, Grimm M, Peeck M, Zimmermann WH, Unsöld B, Hasenfuss G, Dobrev D, Eschenhagen T. Phosphatase inhibitor-1-deficient mice are protected from catecholamine-induced arrhythmias and myocardial hypertrophy. Cardiovasc Res. 2008;80:396–406. doi: 10.1093/cvr/cvn208. [DOI] [PubMed] [Google Scholar]
- 125.Kawashima H, Satoh H, Saotome M, Urushida T, Katoh H, Hayashi H. Protein phosphatase inhibitor-1 augments a protein kinase A-dependent increase in the Ca2+ loading of the sarcoplasmic reticulum without changing its Ca2+ release. Circ J. 2009;73:1133–40. doi: 10.1253/circj.cj-08-0871. [DOI] [PubMed] [Google Scholar]