

Abstract
The polarized trafficking of axonal and dendritic proteins is essential for the structure and function of neurons. Cyclin-dependent kinase 5 (CDK-5) and its activator CDKA-1/p35 regulate diverse aspects of nervous system development and function. Here, we show that CDK-5 and CDKA-1/p35 are required for the polarized distribution of neuropeptide-containing dense-core vesicles (DCVs) in Caenorhabditis elegans cholinergic motor neurons. In cdk-5 or cdka-1/p35 mutants, the predominantly axonal localization of DCVs containing INS-22 neuropeptides was disrupted and DCVs accumulated in dendrites. Time-lapse microscopy in DB class motor neurons revealed decreased trafficking of DCVs in axons and increased trafficking and accumulation of DCVs in cdk-5 mutant dendrites. The polarized distribution of several axonal and dendritic markers, including synaptic vesicles, was unaltered in cdk-5 mutant DB neurons. We found that microtubule polarity is plus-end out in axons and predominantly minus-end out in dendrites of DB neurons. Surprisingly, cdk-5 mutants had increased amounts of plus-end-out microtubules in dendrites, suggesting that CDK-5 regulates microtubule orientation. However, these changes in microtubule polarity are not responsible for the increased trafficking of DCVs into dendrites. Genetic analysis of cdk-5 and the plus-end-directed axonal DCV motor unc-104/KIF1A suggest that increased trafficking of UNC-104 into dendrites cannot explain the dendritic DCV accumulation. Instead, we found that mutations in the minus-end-directed motor cytoplasmic dynein, completely block the increased DCVs observed in cdk-5 mutant dendrites without affecting microtubule polarity. We propose a model in which CDK-5 regulates DCV polarity by both promoting DCV trafficking in axons and preventing dynein-dependent DCV trafficking into dendrites.
Introduction
Neurons are highly polarized cells with molecularly and functionally distinct dendritic and axonal compartments that ensure the directional flow of information. The establishment and maintenance of neuronal polarity requires strict mechanisms for the polarized sorting of presynaptic and postsynaptic proteins to axons and dendrites, respectively.
Cyclin-dependent kinase 5 (CDK-5) and its cyclin-like activator CDKA-1/p35 regulate diverse cellular functions in the nervous system, such as cell migration, axon outgrowth, and synaptic transmission (Dhavan and Tsai, 2001; Cheung et al., 2006). In particular, CDK-5 regulates the transport of membrane-bound organelles in cultured squid and rodent axons (Ratner et al., 1998; Morfini et al., 2004; Pandey and Smith, 2011) and was recently shown to promote polarized trafficking in Caenorhabditis elegans (Ou et al., 2010). Ou et al. (2010) showed that CDK-5 and another cyclin-dependent kinase, PCT-1, regulate the polarized trafficking of synaptic vesicles (SVs) in cholinergic motor neuron axons by inhibiting retrograde trafficking by the minus-end-directed microtubule motor dynein. Interestingly, this polarity mechanism was cell type specific, because these kinases are required for the polarized trafficking of SVs in DA class motor neurons but, surprisingly, not in related DB class motor neurons (Ou et al., 2010). This research raises several interesting questions regarding whether CDK-5 has a broader role in regulating neuronal polarity: (1) Does CDK-5 affect the polarized trafficking of other cargo, such as secreted cargo? (2) Does CDK-5 regulate fundamental aspects of polarity such as microtubule orientation? and (3) Does CDK-5 regulate any aspect of neuronal polarity in DB motor neurons?
We addressed these questions by investigating the role of CDK-5 in regulating the polarized trafficking of neuropeptides in motor neurons in C. elegans. Much research has focused on the molecular mechanisms involved in the polarized trafficking of transmembrane proteins to axons and dendrites, and SV precursors to presynaptic sites (Horton and Ehlers, 2003; Ou and Shen, 2011). Less is known about the polarized trafficking mechanisms by which neurons target secreted cargo, like neuropeptide-filled dense-core vesicles (DCVs), to their sites of release. DCVs carry diverse cargo such as neuropeptides, neurotrophins, and peptide hormones, and are involved in modulating synaptic transmission, plasticity, and behavior (Li and Kim, 2008; Lessmann and Brigadski, 2009). Unlike SVs, which are recycled and refilled with neurotransmitter at the presynaptic terminal, DCVs are packaged with their protein cargo at the trans-Golgi network in the cell body, undergo a complex biogenesis process, and must be continuously transported to their sites of release (Borgonovo et al., 2006; Dikeakos and Reudelhuber, 2007; Edwards et al., 2009; Sumakovic et al., 2009). In addition, depending on their cargo, DCVs can be targeted to release sites in axons and/or dendrites (Fisher et al., 1988; Landry et al., 2003; Lessmann and Brigadski, 2009). However, little is known about the mechanisms that control DCV targeting to either subcellular compartment.
Here, we show that CDK-5 is required for the polarized trafficking of DCVs in DA and DB motor neurons, and propose a model in which CDK-5 both promotes DCV trafficking in axons and prevents dynein-dependent DCV trafficking into dendrites.
Materials and Methods
Strains.
Strains were maintained on OP50 Escherichia coli at 20°C as described by Brenner et al. (1974). The following strains were used in this study: N2 Bristol, nuIs195(Punc-129::ins-22::venus), ceIs72(Punc-129::ida-1::gfp), nuIs165(Punc-129::unc-10::gfp), nuIs174(Punc-129::gfp::rab-3), pzEx77(Punc-17::cdk-5), pzEx107(Punc-17::cdk-5(D144N)), pzEx114(Punc-129::cdka-1), pzEx140(Punc-129::cdk-5), pzEx133(Punc-129::fbn-1::gfp), pzEx156(Punc-129::ebp-1::gfp), pzEx220(Punc-129::gfp::dli-1), yuEx46(Punc-129::unc-9::gfp; Punc-129::mCherry) (gift from Lars Dreier, University of California Los Angeles, Los Angeles, CA), cdk-5(gm336), cdk-5(ok626), cdka-1(gm335), unc-10(e102), unc-104(e1265), dhc-1(js319), and dli-1(ku266). The cdk-5 allele gm336 is a predicted null mutation and consists of a 760 bp deletion that eliminates the start codon as previously described (Juo et al., 2007). The ok626 allele is also a predicted null mutation that consists of a 1.6 kb deletion that eliminates the cdk-5 start codon but also deletes part of a neighboring gene T27E9.4.
Constructs, transgenes, and germline transformation.
Plasmids were generated using standard cloning techniques, and details are available upon request. Punc-129::cdk-5 (FJ#55) and Punc-129::cdka-1 (FJ#56) were generated by subcloning cdk-5 from pV6::cdk-5 (KP#1414) and cdka-1 from pV6::cdka-1 (KP#1413) into the Punc-129 expression vector KP#1271. The unc-129 promoter contains a 2644 bp fragment upstream of the unc-129 start codon that was amplified by PCR using the following primers (ODS276, 5′-GGGGGGGCATGCGGAAACATGATATCGACGGAC-3′, and ODS277, 5′-GGGGGGGGATCCcttgcttgctcttccaattttcc-3′) (Sieburth et al., 2005). Wild-type and kinase-dead cdk-5 were subcloned from pV6::cdk-5 (KP#1414) and pV6::cdk-5(D144N) (KP#1415), respectively, and inserted under the control of the unc-17 promoter. The unc-17 promoter consists of a 3213 bp fragment of the unc-17 5′-UTR that was amplified with the following PCR primers (UNC17-FWD, 5′-TTCACATCCCCCGAAATTTCC-3′, and UNC17-REV, 5′-GACTTTTAATTTATAAAATCACATTTTG-3′).
The fbn-1 and ebp-1 open reading frame were obtained by RT-PCR from wild-type cDNA and subcloned under the control of the unc-129 promoter. NotI sites were introduced at the C termini of both genes and used to insert GFP to create Punc-129::FBN-1::GFP (FJ#57) and Punc-129::ebp-1::GFP (FJ#62). GFP::DLI-1 was subcloned from Pjkk-1::GFP::DLI-1 (gift from Naoki Hisamoto, Nagoya University, Nagoya, Japan) and inserted under the control of the unc-129 promoter to create Punc-129::GFP::DLI-1 (FJ#68). Transgenic strains were generated by microinjection of various plasmids at the following concentrations: 50 ng/μl for Punc-17::cdk-5(wt), Punc-17::cdk-5(D144N), Punc-129::cdk-5, and Punc-129::cdka-1; 25 ng/μl for Punc-129::fbn-1::gfp and Punc-129::gfp::dli-1; and 1 ng/μl for Punc-129::ebp-1::gfp. Plasmids were injected with various co-injection markers: Pttx-3::dsred, Pmyo-2::nls::gfp, or Pmyo-2::nls::mcherry.
Fluorescent microscopy and quantification.
All imaging was performed using a Zeiss M1 AxioImager microscope. For all experiments, except for time-lapse imaging, young adult hermaphrodites were immobilized with 30 mg/ml 2,3-butanedione monoxamine (Sigma-Aldrich) for 5–7 min and mounted on 2% agarose pads before imaging. Images were taken using a Zeiss 100× PlanApo objective (NA 1.4) and an Orca-ER (Hamamatsu) CCD camera. Maximum-intensity projections of Z-series stacks and line scans of fluorescent puncta were obtained using MetaMorph (version 7.1) software (Molecular Devices). For quantitative analyses of fluorescent ventral and dorsal nerve cord puncta, maximum-intensity projections of Z series stacks (total depth, 1 μm) were made. Exposure settings and gain were set to fill the 12 bit dynamic range without saturation and were constant for all images of a given fluorescent marker. FluoSphere yellow-green fluorescent beads (Invitrogen) were imaged daily to correct for day-to-day variation in microscope light bulb intensity. All images (unless stated otherwise) were taken from animals oriented with the dorsal or ventral nerve cord up, and laterally oriented animals were excluded. Images containing mobile INS-22::Venus puncta were excluded from analysis.
The unc-129 promoter drives expression of genes in a subset of DA motorneurons, which have processes projecting toward the head, and a subset of DB neurons, which project toward the tail (see Fig. 1). DA6 is the most posterior DA neuron with visible expression of fluorescent markers driven by the unc-129 promoter; thus, all fluorescent signal posterior to the DA6 neuron is derived only from DB neurons. To isolate only DB axons in the dorsal nerve cord (DNC), we drew line scans along the DNC posterior to the site of DA6 commissure entry. To isolate only DB dendrites in the ventral nerve cord (VNC), we drew line scans posterior to the DA6 cell body. In our analysis of microtubule orientation in the DB axons (see Fig. 4), we find that >95% of microtubules in this area of the DNC are oriented in the same direction, suggesting that this section of the nerve cord is not contaminated by fluorescent signal from DA neurons. To image INS-22::Venus primarily in DA neurons, we imaged anterior to the DA3 cell body for the VNC and anterior to the site of DA3 commissure entry into the DNC (see Fig. 1, DA-rich region indicated). In most animals, there was no expression of fluorescent markers in any DB neurons in this region; however, in ∼20% of wild-type animals, there was weak expression of INS-22::Venus in the DB3 cell body, suggesting that the vast majority of fluorescent signal in this section of the nerve cord is derived from DA neurons, with a small amount of signal from DB neurons.
Figure 1.

The unc-129 promoter controls expression in a subset of DA and DB motor neurons. A, Schematic diagram of DA and DB motor neurons under the control of the unc-129 promoter used in this study. DA and DB neurons have cell bodies located on the ventral side with dendrites in the VNC and axons in the DNC. DA neurons (red) project their processes toward the anterior, whereas DB neurons (blue) project their processes toward the posterior. The DA-rich and DB-only regions of the VNC and DNC imaged throughout this study are indicated. B, A photomontage of the VNC of transgenic animals expressing soluble mCherry under the control of the unc-129 promoter. Specific DA and DB neuron cell bodies and processes expressing mCherry are shown. Neuron identities were determined based on the position of their cell bodies and the projections of their commissures. Strong mCherry expression was observed in DA1–6 and DB4–7, whereas occasional, weak expression was seen in DB3, whose cell body is located just anterior to DA2 (see A). To isolate fluorescent signals derived only from DB neurons, neuronal processes were imaged in regions of the VNC and DNC posterior to the DA6 cell body (DB-only region). To enrich for fluorescent signals derived from DA neurons, processes were imaged in regions of the VNC and DNC anterior to the DA3 cell body (DA-rich region). The animals are oriented with anterior to the left and dorsal to the top.
Figure 4.

CDK-5 regulates the orientation of microtubules in DB dendrites. A, Schematic diagram of a DB motor neuron (top panel). The boxed region denotes that the axon was imaged for data presented in A and B. Representative kymographs generated from a 25 s movie of EBP-1::GFP movement in the axon of wild-type and cdk-5(gm336) mutant animals (bottom panels). B, Quantification of percentage of plus-end- and minus-end-out microtubules in wild-type (n = 76 puncta) and cdk-5 (n = 93 puncta) mutant animals based on analysis of EBP-1::GFP movement in DB axons. C, Schematic diagram of a DB motor neuron (top panel). The boxed region denotes that the dendrite was imaged for data presented in C and D. Representative kymographs generated from a 25 s movie of EBP-1::GFP movement in the dendrite of wild-type and cdk-5(gm336) mutant animals (bottom panels). D, Quantification of percentage of plus-end- and minus-end-out microtubules in wild-type (n = 107 puncta) and cdk-5 (n = 195 puncta) mutant animals based on analysis of EBP-1::GFP movement in DB dendrites. Values that differ significantly from wild type (χ2 test, Yates' correction) are denoted on the graphs (**p < 0.001).
Line scans of nerve cord puncta were generated using MetaMorph (version 6.0) and were then analyzed in Igor Pro (version 5) using custom-written software as previously described (Burbea et al., 2002). Puncta intensity and density were calculated for each image. Puncta intensity is the fractional increase in peak fluorescence of each puncta over fluorescent bead intensity for that date. Puncta density is the average number of puncta per 10 μm of nerve cord.
For DB6 cell body imaging, lateral and ventral up animals were imaged, and Z stacks were taken to a depth of 2 μm. Average fluorescence of three fluorescent patches per cell body was measured using MetaMorph software, and then corrected for daily bead values and analyzed in Microsoft Excel.
For GFP::DLI-1 imaging, lateral and ventral up animals were imaged, and Z stacks were taken to a depth of 1 μm. The fluorescence of the most posterior GFP::DLI-1 punctum in the VNC was measured using MetaMorph software, and then corrected for daily bead values and analyzed in Microsoft Excel.
Changes in puncta intensity and density were analyzed for statistical significance using Student's t test for two genotype comparisons and the Tukey–Kramer test for multiple genotype comparisons.
Time-lapse microscopy.
For time-lapse microscopy of INS-22::Venus, young adult worms were paralyzed in 1.5 mm Levamisol (Sigma-Aldrich) dissolved in M9 buffer for 6–7 min. Animals were mounted on 2% agarose pads containing 1.5 mm Levamisol. Time-lapse images were taken at 4 Hz speed for 20 s and saved as a Z series. For dendritic INS-22::Venus, movies were taken of DB6 dendrites in a region of the VNC between the DA6 and DB7 cell bodies (see Fig. 1). Because DB processes project posteriorly, all movements toward the tail were designated anterograde, and all movements toward the head were designated retrograde. For axon commissure time-lapse imaging, movies were taken of DB6 commissures in ventral-up and ventrolaterally oriented animals, with line scans drawn from the DB6 cell body to the last in-focus INS-22::Venus punctum. These scans were used to generate kymographs in MetaMorph (version 7.1). INS-22::Venus puncta direction of movement and velocity were calculated by tracing these kymographs. Each mobile punctum was traced during its longest uninterrupted period of movement. Puncta were defined as mobile if they moved distances greater than twice their own width and at velocities >0.1 μm/s. Puncta that changed direction were traced for both directions for analysis of puncta velocity and direction of movement, but were only counted once for analysis of total puncta. Kymograph traces were compiled for analysis in Microsoft Excel. Average anterograde and retrograde velocity, and the number of stationary and anterogradely and retrogradely moving puncta were calculated for each kymograph (i.e., worm), and these data were compiled for each genotype and analyzed in Microsoft Excel. For two genotype comparisons, Student's t tests were used to detect differences between genotypes. For greater than two genotype comparisons, the Tukey–Kramer test was used to detect differences between genotypes. The Kolmogorov–Smirnov (KS) test was used for puncta velocity comparisons.
Kymograph analysis using MetaMorph software was also used to measure the fluorescence intensity of mobile INS-22::Venus puncta in dendrites. The maximum fluorescence intensity of each mobile punctum was measured at the t = 0 time point of each kymograph, to minimize the effects of photobleaching. Maximum intensity measurements were analyzed using Microsoft Excel and IgorPro (version 5). Occasionally, mobile INS-22::Venus puncta were observed to undergo a splitting event, in which a single punctum divides into two separate puncta during the time-lapse movie. While these events are rare, it suggests that a mobile punctum may represent more than one DCV. For this reason, splitting puncta were excluded from mobile INS-22::Venus fluorescence intensity analyses to enrich for puncta representing single DCVs.
For time-lapse microscopy of EBP-1 movement, animals were paralyzed in 2 mm Levamisol dissolved in M9 for 8–9 min and mounted on a 2% agarose pad containing 2 mm Levamisol. In the VNC, line scans were drawn in DB6 or DB7 dendrites from the DA6 or DB7 cell body toward the tail. In the DNC, line scans were drawn toward the tail in DB6 axons immediately posterior to the site of DA6 commissure entry into the nerve cord (DB-only region is indicated on Fig. 1). Time-lapse images were taken at a speed of 4 Hz for 25 s. EBP-1::GFP direction of movement and velocity were calculated by tracing puncta in these kymographs. Only puncta that could be followed for 2 s or more were included. Occasionally, oscillating puncta were visible, consistent with reports from Stepanova et al. (2003), and these puncta were excluded from analysis. Direction of movement and velocity of EBP-1::GFP puncta were analyzed using Microsoft Excel. Changes in percentages of plus-end- and minus-end-out microtubules were compared across genotypes using χ2 analysis, with a Yates correction used for two genotype comparisons (i.e., 2 × 2 contingency tables).
Results
CDK-5 regulates the polarized distribution of DCVs in DB motor neurons
We investigated whether CDK-5 regulates the polarized trafficking of neuropeptide-containing DCVs in DA and DB cholinergic motor neurons in C. elegans. Each DA/DB neuron has a simple polarized morphology consisting of a cell body and a single dendrite in the VNC, where it receives inputs, and a single axon in the DNC, where it makes synapses onto muscle and GABAergic motor neurons (Figs. 1, 2A,D) (White et al., 1986). DA class motor neurons extend their processes toward the anterior and are involved in backward locomotion, whereas DB class motor neurons extend their processes toward the posterior and are involved in forward locomotion (Chalfie et al., 1985; White et al., 1986; Haspel et al., 2010). We used the unc-129 promoter to drive expression of fluorescent markers in a subset of DA and DB motor neurons (Fig. 1A) (Sieburth et al., 2005, 2007). Figure 1B shows an image of the VNC illustrating specific DA and DB cell bodies and processes that express soluble mCherry under the control of the unc-129 promoter. In this study, we imaged specific regions of the VNC (dendrites) and DNC (axons) to separate fluorescent signals from either DA or DB motor neurons (Fig. 1).
Figure 2.

CDK-5 regulates the polarized distribution of DCVs in DB motor neurons. A, Schematic diagram of a DB motor neuron with axon in the DNC and dendrite in the VNC (top panel). This diagram is oriented with the anterior of the animal to the left for this and all subsequent figures. The boxed region denotes that the axon was imaged for data presented in A–C and G–I. Representative images of INS-22::Venus in DB axons of young adult wild-type, cdk-5(ok626), cdk-5(gm336), Punc-129::cdk-5;cdk-5(gm336) rescue, cdka-1(gm335), and Punc-129::cdka-1;cdka-1(gm335) rescue animals (bottom panels). B, C, Quantification of INS-22::Venus puncta intensity (B) and density (C) in axons of wild-type (n = 30), cdk-5(ok626) (n = 25), cdk-5(gm336) (n = 35), cdk-5 rescue (n = 19), cdka-1(gm335) (n = 23), and cdka-1 rescue (n = 20) animals. D, Schematic diagram of a DB motor neuron (top panel). The boxed region denotes that the dendrite was imaged for data presented in D–F and J–L. Representative images of INS-22::Venus in DB dendrites of wild-type, cdk-5(ok626), cdk-5(gm336), Punc-129::cdk-5;cdk-5(gm336) rescue, cdka-1(gm335), and Punc-129::cdka-1;cdka-1(gm335) rescue animals are shown. For this and all other dendrite images, the white asterisk (*) indicates the position of a motor neuron cell body. E, F, Quantification of INS-22::Venus puncta intensity (E) and density (F) in dendrites of wild-type (n = 27), cdk-5(ok626) (n = 19), cdk-5(gm336) (n = 19), cdk-5 rescue (n = 19), cdka-1(gm335) (n = 15), and cdka-1 rescue (n = 18) animals. G, Representative images of IDA-1::GFP in DB axons of young adult wild-type and cdk-5(gm336) mutant animals. H, I, Quantification of IDA-1::GFP puncta intensity (H) and density (I) in axons of wild-type (n = 27) and cdk-5(gm336) (n = 25) mutant animals. J, Representative images of IDA-1::GFP in DB dendrites of wild-type and cdk-5(gm336) mutant animals. K, L, Quantification of IDA-1::GFP puncta intensity (K) and density (L) in dendrites of wild-type (n = 23) and cdk-5(gm336) (n = 25) mutant animals. For this and all subsequent figures, error bars denote SEM. Values that differ significantly [Tukey–Kramer (B, C, E, F) and Student's t test (H, I, K, L)] from wild type (marked by asterisks above each bar) or from other genotypes (comparisons marked by brackets) are denoted on the graphs (#p < 0.05; *p < 0.01; **p < 0.001), and values that do not differ significantly (p > 0.05) are denoted by n.s.
To visualize DCVs in DA and DB motor neurons, we expressed a Venus-tagged neuropeptide, insulin-like protein 22 (INS-22::Venus), under the control of the unc-129 promoter (Sieburth et al., 2005, 2007). We chose the neuropeptide ins-22 because it is expressed in ventral cord motor neurons and has been implicated in regulating synaptic transmission at the C. elegans neuromuscular junction (Pierce et al., 2001; Sieburth et al., 2005). In addition, INS-22::Venus has been well characterized as a DCV marker (Sieburth et al., 2005, 2007; Ch'ng et al., 2008; Edwards et al., 2009). In wild-type animals, INS-22::Venus has a highly polarized subcellular distribution where it is largely excluded from the motor neuron dendrites and is localized in a punctate manner in the motor neuron axons (Fig. 2A–F) (Sieburth et al., 2007). INS-22::Venus colocalizes with and adjacent to synaptobrevin in the axon (Sieburth et al., 2007), consistent with electron microscopy findings that indicate that DCVs are broadly distributed at presynaptic sites and only slightly enriched at the active zone (Hammarlund et al., 2008). Trafficking of INS-22::Venus to the axon requires the anterograde kinesin motor UNC-104/KIF1A, as has been shown for several other DCV markers and endogenous neuropeptides (Jacob and Kaplan, 2003; Zahn et al., 2004; Sieburth et al., 2005, 2007; Barkus et al., 2008).
To investigate whether CDK-5 regulates the polarized distribution of DCV cargo in C. elegans, we analyzed the distribution of INS-22::Venus-containing DCVs in DA and DB motor neurons in cdk-5(gm336) and cdk-5(ok626) mutant animals using quantitative fluorescence microscopy. Both cdk-5(ok626) and cdk-5(gm336) alleles are predicted null mutations (Juo et al., 2007). We first imaged INS-22::Venus fluorescence in a posterior region of the dorsal nerve cord where it is only expressed in DB axons (Fig. 1A) and quantified the density and fluorescence intensities of INS-22::Venus puncta using custom-written software (see Materials and Methods) (Burbea et al., 2002). INS-22::Venus puncta fluorescence intensities were decreased by ∼35–40% (p < 0.001) in the motor neuron axons of cdk-5(ok626) and cdk-5(gm336) mutant animals compared with wild-type controls (Fig. 2A,B). We also analyzed the distribution of INS-22::Venus in DB motor neuron axons of animals with loss-of-function mutations in the activator of CDK-5, CDKA-1/p35. Similarly, we found that INS-22::Venus puncta fluorescence intensities decreased by ∼33% (p < 0.001) in cdka-1(gm335) loss-of-function mutants compared with wild-type controls (Fig. 2A,B). There was no significant change in the density of INS-22::Venus puncta in DB motor neuron axons of cdk-5 or cdka-1 mutant animals (Fig. 2C). These results suggest that the abundance of INS-22-containing DCVs in the axons of DB motor neurons is regulated by CDK-5 and its activator CDKA-1/p35.
To determine whether the loss of INS-22::Venus fluorescence in the axon was caused by alterations in neuropeptide trafficking in cdk-5 mutants, we examined DB motor neuron cell bodies and dendrites for INS-22::Venus accumulation. We found no change in the abundance of INS-22::Venus in DB motor neuron cell bodies of cdk-5(gm336) mutants compared with controls (INS-22::Venus fluorescence [normalized (norm.)] ± SEM: WT, 1.00 ± 0.04; cdk-5, 1.02 ± 0.05; p > 0.05) (see Fig. 6B,C); however, in the dendrites, we found a significant increase (p < 0.001) in INS-22::Venus puncta density in cdk-5(ok626), cdk-5(gm336), and cdka-1(gm335) mutants (Fig. 2D,F). Furthermore, analysis of a DCV membrane marker, GFP-tagged IDA-1/IA-2 (insulinoma-associated protein 2) (Solimena et al., 1996; Cai et al., 2004; Zhou et al., 2007; Edwards et al., 2009), in wild-type and cdk-5 mutants revealed similar defects in the polarized distribution of DCVs in DB motor neurons (Fig. 2G–L). These results indicate that CDK-5 and its activator CDKA-1/p35 regulate the polarized distribution of DCVs in DB motor neurons.
Figure 6.

UNC-104/Kif1A is not required for the increase in dendritic DCVs in cdk-5 mutants. A, Schematic diagram of a DB motor neuron (top panel). The boxed region denotes that the axon was imaged for data presented in A. Representative images of INS-22::Venus in DB axons of wild-type and unc-104(e1265) mutant animals (bottom panels). B, Schematic diagram of a DB motor neuron (top panel). The boxed region denotes that the cell body was imaged for data presented in B and C. Representative images of INS-22::Venus in the DB6 motor neuron cell body of wild-type, cdk-5(gm336), unc-104(e1265), and cdk-5;unc-104 double-mutant animals (bottom panels). C, Quantification of INS-22::Venus fluorescence intensity in DB cell bodies of wild-type (n = 24), cdk-5 (n = 14), unc-104 (n = 11), and cdk-5;unc-104 (n = 14) double-mutant animals. D, Schematic diagram of a DB motor neuron (top panel). The boxed region denotes that the dendrite was imaged for data presented in D–F. Representative images of INS-22::Venus in DB dendrites of wild-type, cdk-5(gm336), unc-104(e1256), and cdk-5;unc-104 double-mutant animals (bottom panels). E, F, Quantification of INS-22::Venus puncta intensity (E) and density (F) in DB dendrites of wild-type (n = 27), cdk-5 (n = 19), unc-104 (n = 18), and cdk-5;unc-104 (n = 18) animals. Values that differ significantly (Tukey–Kramer test) from wild type (marked by asterisks above each bar) or from other genotypes (comparisons marked by brackets) are denoted on the graphs (**p < 0.001; n.s., p > 0.05).
CDK-5 functions cell autonomously in motor neurons to regulate DCV polarity
To determine whether CDK-5 functions in motor neurons to regulate the polarized distribution of DCVs, we performed cell type-specific rescue experiments. We found that the changes in INS-22::Venus puncta fluorescence intensities and densities in DB axons and dendrites of cdk-5 mutants can be rescued by expression of wild-type cdk-5 cDNA under the control of a DA/DB motor neuron-specific promoter, Punc-129 (cdk-5 rescue) (Fig. 2A–F) (Sieburth et al., 2005). Likewise, the increase in INS-22::Venus puncta fluorescence intensity and density in DB dendrites of cdka-1 mutants can be rescued by expression of wild-type cdka-1 cDNA under the control of the unc-129 promoter (cdka-1 rescue) (Fig. 2D–F). Furthermore, expression of wild-type cdk-5 cDNA under the control of a cholinergic neuron-specific promoter, Punc-17 (cdk-5 wt rescue), also rescued the increased accumulation of INS-22::Venus in the dendrites of cdk-5(gm336) mutants, whereas a kinase-dead version of cdk-5 (cdk-5 k.d. rescue) did not rescue [average dendritic puncta density (per 10 μm) ± SEM: wild type, 1.47 ± 0.11; cdk-5(gm336), 3.41 ± 0.24; cdk-5 w.t. rescue, 1.19 ± 0.19; p < 0.001 vs cdk-5; cdk-5 k.d. rescue, 2.93 ± 0.24, p > 0.05 vs cdk-5]. Together, these data indicate that CDK-5 and CDKA-1/p35 function in cholinergic motor neurons to regulate DCV polarity in a kinase activity-dependent manner.
CDK-5 regulates the polarized distribution of DCVs in DA motor neurons
We next investigated whether CDK-5 regulates the polarized distribution of DCVs in DA class motor neurons. Because there is some overlap between DA and DB processes in the nerve cords, we maximized our DA signal by analyzing regions of the DNC and VNC where the vast majority of INS-22::Venus was in DA motor neuron axons and dendrites, respectively (Fig. 1) (see Materials and Methods). Although there was no change in the abundance of DCVs in cdk-5 mutant DA axons [average axonal puncta density (per 10 μm) ± SEM: wild type, 4.38 ± 0.16; cdk-5(gm336), 4.12 ± 0.21; p > 0.05; average axonal puncta intensity (norm.) ± SEM: wild type, 1.0 ± 0.07; cdk-5(gm336), 0.86 ± 0.05; p > 0.05], cdk-5 mutant dendrites had an increase in INS-22::Venus puncta density and intensity [average dendritic puncta density (per 10 μm) ± SEM: wild type, 2.50 ± 0.25; cdk-5(gm336), 3.44 ± 0.21; p < 0.001; average axonal puncta intensity (norm.) ± SEM: wild type, 1.0 ± 0.05; cdk-5(gm336), 2.31 ± 0.17; p < 0.001]. The DCV distribution defects observed in cdk-5 mutant DA dendrites were rescued by expression of cdk-5 cDNA under control of the unc-129 promoter [average dendritic puncta density (per 10 μm) ± SEM: wild type, 2.79 ± 0.24; cdk-5(gm336), 3.92 ± 0.21; cdk-5 w.t. rescue, 2.60 ± 0.34; p < 0.01 vs cdk-5; average dendritic puncta intensity (norm.) ± SEM: wild type, 1.0 ± 0.06; cdk-5(gm336), 2.01 ± 0.15; cdk-5 w.t. rescue, 1.40 ± 0.16; p < 0.001 vs cdk-5]. These data show that mutations in cdk-5 result in defects in the polarized distribution of INS-22::Venus-containing DCVs in both DB and DA motor neurons. Interestingly, this result is in contrast to what was found for SVs, where CDK-5 was required for polarized SV trafficking in DA but not DB motor neurons (Ou et al., 2010). We focused the rest of our study on DB motor neurons because (1) we can cleanly measure DCV signal in DB axons and dendrites, (2) cdk-5 mutants have a stronger DCV polarity defect in DB neurons, and (3) polarized trafficking has not been studied in detail in DB neurons.
CDK-5 does not affect axon/dendrite morphology or markers
The defect in the polarized distribution of DCVs observed in cdk-5 mutants could be due to defects in axon outgrowth or presynaptic development. To test this possibility, we analyzed the number and morphology of motor neurons in wild-type and cdk-5 mutants. We found no change in the number of motor neurons expressing INS-22::Venus under the control of the Punc-129 promoter (number of cell bodies ± SEM: wild type, 10.2 ± 0.2; cdk-5(gm336), 9.9 ± 0.2; p > 0.05) or in the gross morphology of their axonal and dendritic processes (data not shown). In addition, since the density of INS- 22::Venus puncta in the axons of these motor neurons is unchanged in cdk-5 and cdka-1 mutant animals compared with controls (Fig. 2C), the number of presynaptic sites is likely unaltered. To further test for changes in presynaptic development, we examined the axonal distribution of the presynaptic active zone protein UNC-10/RIM-1. Fluorescently tagged UNC-10 (under the control of the Punc-129 promoter) localizes to synapses in the axons of DB motor neurons and is largely absent from the dendrites (Fig. 3A,D) (Sieburth et al., 2005). We found no significant change in UNC-10::GFP puncta fluorescence intensity (Fig. 3B) or density (Fig. 3C) in DB motor neuron axons of cdk-5(gm336) mutants compared with wild-type controls. We also examined the distribution of the SV marker GFP::RAB-3 in cdk-5 mutants. GFP::RAB-3 localizes to synaptic sites in axons but not dendrites of wild-type DB motor neurons (Fig. 3E,H) (Sieburth et al., 2005), and this polarized distribution of SVs was unaltered in cdk-5 mutants (Fig. 3E,H). We found no change in GFP::RAB-3 puncta density (Fig. 3G) and no decrease in GFP::RAB-3 puncta intensity (Fig. 3F) in cdk-5 mutant DB axons, and no increase in GFP::RAB-3 puncta in cdk-5 mutant DB dendrites (Fig. 3H) compared with controls. These data are consistent with a recent study showing that cdk-5 mutants do not have decreased SVs at presynaptic sites in DB motor neurons (Ou et al., 2010). These results suggest that the defect in the polarized distribution of DCVs observed in cdk-5 mutants is not due to gross defects in axonal development. Thus, CDK-5 is required for the polarized distribution of DCVs, but not SVs, in DB motor neurons.
Figure 3.
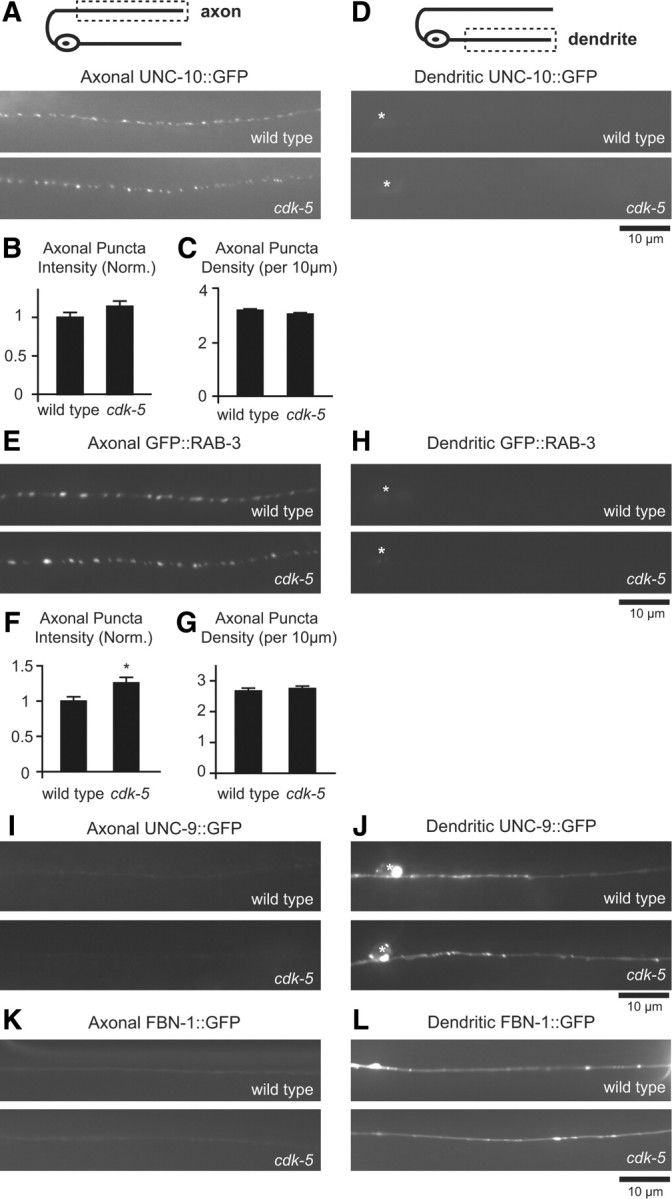
The polarized distribution of axonal and dendritic markers in DB motor neurons is not affected in cdk-5 mutants. A, Schematic diagram of a DB motor neuron (top panel). The boxed region denotes that the axon was imaged for data presented in A–C, E–G, I, and K. Representative images of UNC-10::GFP in DB axons of wild-type and cdk-5 mutant animals (bottom panels). B, C, Quantification of UNC-10::GFP puncta intensity (B) and density (C) in axons of wild-type (n = 21) and cdk-5(gm336) (n = 20) mutant animals. D, Schematic diagram of a DB motor neuron (top panel). The boxed region denotes that the dendrite was imaged for data presented in D, H, J, and L. Representative images of UNC-10::GFP in DB dendrites of wild-type and cdk-5(gm336) mutant animals (bottom panels). E, Representative images of GFP::RAB-3 in DB axons of wild-type and cdk-5(gm336) mutant animals. F, G, Quantification of GFP::RAB-3 puncta intensity (F) and density (G) in axons of wild-type (n = 22) and cdk-5(gm336) (n = 21) mutant animals. H, Representative images of GFP::RAB-3 in DB dendrites of wild-type and cdk-5(gm336) mutant animals. Values that differ significantly from wild-type (Student's t test) are denoted on graphs (*p ≤ 0.01). I, Representative images of UNC-9::GFP in DB axons of wild-type and cdk-5(gm336) mutant animals. J, Representative images of UNC-9::GFP in DB dendrites of wild-type and cdk-5(gm336) mutant animals. K, Representative images of FBN-1::GFP in DB axons of wild-type and cdk-5(gm336) mutant animals. L, Representative images of FBN-1::GFP in DB dendrites of wild-type and cdk-5(gm336) mutant animals.
The increased abundance of DCVs in motor neuron dendrites could be the result of a general defect in dendritic trafficking that could, for example, be caused by a loss of dendrite identity. We tested this possibility by analyzing the distribution of two markers that have been shown to localize to the somatodendritic domain of DB motor neurons, Fibrillin FBN-1::GFP and the invertebrate GAP junction innexin protein UNC-9::GFP (Sieburth et al., 2005; Poon et al., 2008). We found normal somatodendritic localization of both UNC-9::GFP and FBN-1::GFP in cdk-5 mutant animals, suggesting that a general defect in trafficking of dendritic proteins is unlikely (Fig. 3I–L). Together, these data indicate that the DCV localization defect observed in cdk-5 mutants is not likely due to general defects in axon or dendrite development or the establishment and maintenance of axonal–dendritic polarity.
The effect of CDK-5 on DCV polarity is not likely a secondary consequence of synaptic transmission defects
CDK-5 can regulate synaptic transmission in other systems (Dhavan and Tsai, 2001; Cheung et al., 2006), and neuronal activity can regulate DCV trafficking (Shakiryanova et al., 2006). Thus, we tested whether the defect in the polarized distribution of DCVs in cdk-5 mutants was a secondary consequence of a role for CDK-5 in synaptic transmission. In C. elegans, we found that cdk-5 mutants have defects in synaptic transmission at the neuromuscular junction based on a behavioral paralysis assay using the acetylcholine esterase inhibitor aldicarb (Nguyen et al., 1995; Miller et al., 1996). Specifically, we found that cdk-5 mutants have reduced sensitivity to aldicarb compared with wild-type animals [animals paralyzed on aldicarb at 180 min (percentage ± SEM): WT, 98 ± 1.6%; cdk-5(gm336), 45 ± 0.74%], suggesting that cdk-5 mutants have reduced synaptic transmission at the neuromuscular junction. If the changes in DCV polarity observed in cdk-5 mutants were a secondary consequence of reduced synaptic transmission, then we would expect other synaptic transmission mutants to have similar defects in DCV polarity. Therefore, we analyzed whether INS-22::Venus-containing DCVs accumulate in motor neuron dendrites of unc-10(e102) mutants. UNC-10/RIM-1 is a RAB-3 interacting protein involved in synaptic vesicle priming, and loss-of-function mutations in unc-10(e102) mutants have stronger defects in synaptic transmission than cdk-5 mutants based on the aldicarb paralysis assay [animals paralyzed on aldicarb at 180 min (percentage ± SEM): WT, 98 ± 1.6%; cdk-5(gm336), 45 ± 0.74%; unc-10(e102), 3.3 ± 3.3%] (Nguyen et al., 1995; Miller et al., 1996; Koushika et al., 2001). We found that, in contrast to our results in cdk-5 mutants, there was no difference in the distribution of INS-22::Venus fluorescence in motor neuron dendrites of unc-10 mutants [dendritic puncta intensity (norm.) ± SEM: WT, 1.0 ± 0.06; unc-10, 0.87 ± 0.07; p > 0.05; dendritic puncta density (per 10 μm) ± SEM: WT, 1.74 ± 0.16; unc-10, 1.85 ± 0.22; p > 0.05]. This result indicates that the defect in the polarized distribution of DCVs observed in cdk-5 mutant animals is not likely an indirect consequence of decreased synaptic transmission.
CDK-5 regulates microtubule polarity in motor neuron dendrites
Because the cytoskeleton plays a fundamental role in neuronal polarity and trafficking, we investigated whether CDK-5 affects microtubule orientation in DB motor neurons. In mammals, microtubules in the axon are oriented plus-end out from the cell body, whereas microtubules in the dendrite are of mixed polarity (Baas et al., 1988; Burton, 1988; Stepanova et al., 2003). In Drosophila motor neurons and C. elegans amphid sensory neurons, while microtubules in the axon are also oriented plus-end out, the majority of microtubules in dendrites are minus-end out (Rolls et al., 2007; Stone et al., 2008; Maniar et al., 2012). We used microtubule plus-end binding protein dynamics to determine the orientation of microtubules in axons and dendrites of DB motor neurons (Mimori-Kiyosue et al., 2000; Stepanova et al., 2003; Stone et al., 2008). We used time-lapse microscopy and kymographs to analyze the movements of GFP-tagged EBP-1 (EBP-1::GFP), a C. elegans homolog of the microtubule plus-end binding protein EB1, in axons and dendrites of wild-type and cdk-5 mutants (see Materials and Methods). EBP-1::GFP puncta migrated at a velocity of 0.21 ± 0.01 μm/s, consistent with reports of EB1 motion in other systems (Mimori-Kiyosue et al., 2000; Morrison et al., 2002). In wild-type DB axons, we found that microtubule orientation was predominantly (96%) plus-end out from the cell body (Fig. 4A,B), which is consistent with previous reports of axonal microtubule orientation in C. elegans, Drosophila, and mammals (Baas et al., 1988; Stepanova et al., 2003; Rolls et al., 2007; Stone et al., 2008; Maniar et al., 2012). In wild-type DB dendrites, we found that the majority of microtubules (91%) were oriented minus-end out from the cell body (Fig. 4C,D), which is similar to the dendritic microtubule orientation found in Drosophila motor neurons and C. elegans amphid sensory neurons (Rolls et al., 2007; Stone et al., 2008; Maniar et al., 2012). In cdk-5 mutant axons, we found no change in the plus-end-out microtubule orientation compared with wild-type axons (Fig. 4A,B). However, in cdk-5 mutant dendrites, we found a significant increase (p < 0.001) in the percentage of plus-end-out microtubules compared with wild-type dendrites (Fig. 4C,D). These results indicate that CDK-5 regulates microtubule polarity in DB motor neuron dendrites.
CDK-5 regulates DCV trafficking in axons and dendrites
Because changes in microtubule polarity could lead to changes in DCV trafficking and ultimately DCV polarity, we next investigated whether CDK-5 regulates the trafficking of DCVs in DB motor neurons. DCVs are highly mobile and move bidirectionally in a saltatory fashion in both axons and dendrites of C. elegans, Drosophila, and mammalian neurons (Gauthier et al., 2004; Zahn et al., 2004; Barkus et al., 2008; de Jong et al., 2008; Kwinter et al., 2009). Similarly, we observed a large fraction of INS-22::Venus puncta moving bidirectionally along motor neuron axons and dendrites (Fig. 5). To gain insight into why the polarized distribution of DCVs is disrupted in cdk-5 mutant motor neurons, we performed time-lapse microscopy and kymograph analysis of mobile INS-22::Venus-containing vesicles in DB axon commissures and dendrites of wild-type and cdk-5 mutant young adult animals (see Materials and Methods). We used these kymographs to calculate the velocity of mobile DCVs and to quantitate the number of DCVs moving away from (anterograde) or toward (retrograde) the cell body. Although we were unable to calculate accurate velocities of DCVs in the axon commissure due to its curved trajectory, we found that, in wild-type dendrites, the average anterograde and retrograde DCV velocities were 1.29 ± 0.07 and 0.97 ± 0.05 μm/s, respectively, which is in agreement with previous reports of DCV velocities in C. elegans and mammals (Zahn et al., 2004; Kwinter et al., 2009).
Figure 5.
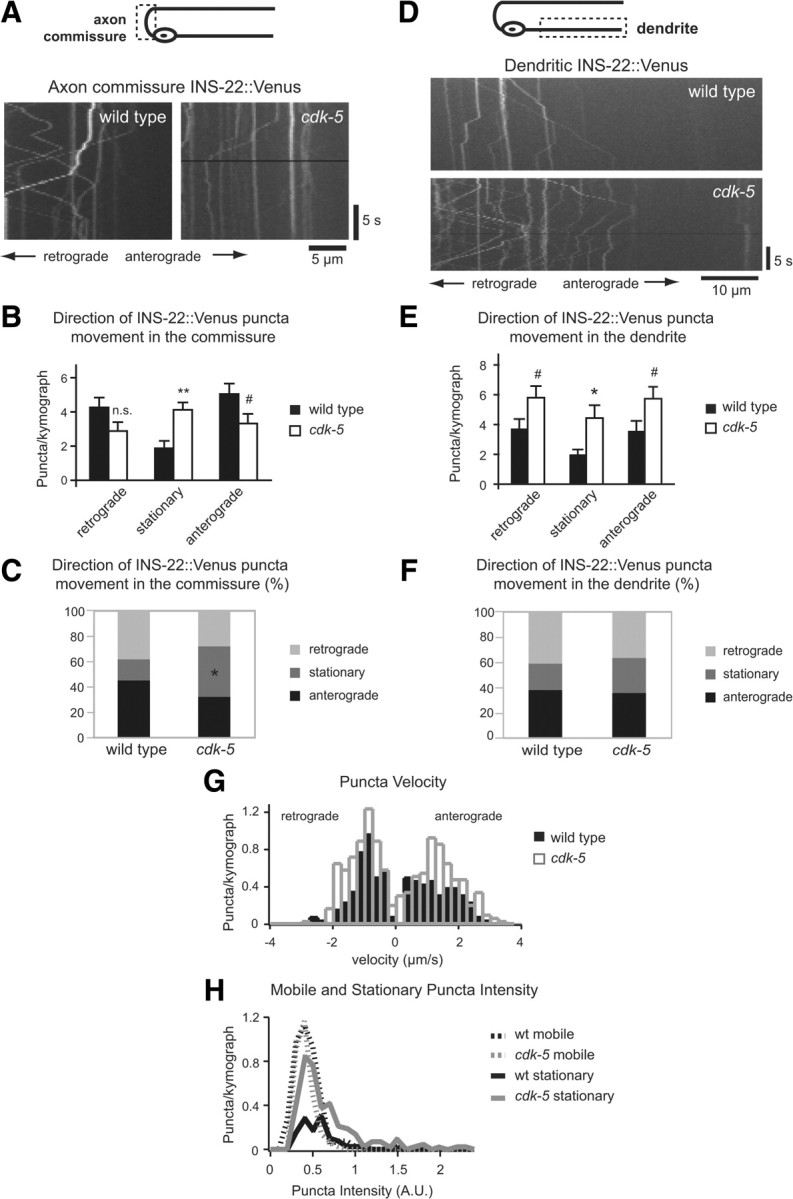
CDK-5 regulates DCV trafficking in DB motor neuron axons and dendrites. A, Schematic diagram of a DB motor neuron (top panel). The boxed region denotes that the axon commissure was imaged for data presented in A–C. Representative kymographs generated from 20 s movies of INS-22::Venus puncta movement in DB axon commissures of wild-type and cdk-5(gm336) mutant animals (bottom panels). B, Quantification of the average number of INS-22::Venus puncta moving anterogradely, retrogradely, or remaining stationary in each kymograph from wild-type (n = 23) and cdk-5 (n = 25) mutant axon commissures. C, Quantification of the direction of INS-22::Venus puncta movement as a percentage of total puncta, in wild-type (n = 257 puncta) and cdk-5 (n = 258) mutant axon commissures. D, Schematic diagram of a DB motor neuron (top panel). The boxed region denotes that the dendrite was imaged for data presented in D–H. Representative kymographs generated from 20 s movies of INS-22::Venus movement in DB dendrites of wild-type and cdk-5(gm336) mutant animals (bottom panels). E, Quantification of the average number of INS-22::Venus puncta moving anterogradely, retrogradely, or remaining stationary in each kymograph from wild-type (n = 26) and cdk-5 (n = 29) mutant dendrites. F, Quantification of the direction of INS-22::Venus puncta movement, as a percentage of total puncta, in wild-type (n = 238 puncta) and cdk-5 (n = 479) mutant dendrites. G, Histogram of INS-22::Venus puncta velocities in dendrites of wild-type and cdk-5 mutant animals. Positive velocities represent anterograde movements, and negative velocities represent retrograde movements. H, Histogram of mobile and stationary INS-22::Venus puncta intensity in dendrites of wild-type and cdk-5 mutant animals. Values that differ significantly [Student's t test (B, E) and χ2 test (C)] from wild type are denoted on graphs (**p < 0.001; *p < 0.01; #p < 0.05).
Kymograph analysis of INS-22::Venus-containing DCVs in DB axon commissures revealed a decrease in the number of DCVs moving in the anterograde direction (p < 0.05) and an increase in the number of stationary DCVs (p < 0.001) in cdk-5 mutants compared with controls (Fig. 5A,B). This trafficking defect correlates with a significant change (p < 0.001) in the relative proportions of anterograde, retrograde, and stationary DCVs in cdk-5 mutants compared with controls (Fig. 5C), with no change in the total number of DCVs in the axon commissure [average number of total DCVs/kymograph ± SEM: WT (n = 23), 9.65 ± 0.9; cdk-5(gm336) (n = 25), 9.40 ± 0.9; p > 0.05]. These data, together with the decrease in INS-22::Venus fluorescence at presynaptic sites in cdk-5 mutant axons (Fig. 2A,B), are consistent with the idea that CDK-5 promotes anterograde DCV trafficking in DB axon commissures.
We also observed changes in DCV trafficking in DB dendrites of cdk-5 mutants compared with wild-type controls (Fig. 5D–F). We discovered an increase in the total number of DCVs in dendrites of cdk-5 mutants [average number of total DCVs/kymograph ± SEM: WT (n = 34), 7.35 ± 1.00; cdk-5 (n = 36), 12.47 ± 1.23; p < 0.01]. Consistent with this, we found an increase in the number of stationary DCVs (p ≤ 0.01), and an increase in the number of DCVs moving in both anterograde (p < 0.05) and retrograde (p < 0.05) directions in cdk-5 mutant dendrites (Fig. 5E). However, there was no significant change (p > 0.05) in the relative proportions of anterograde, retrograde, and stationary DCVs in cdk-5 mutant dendrites (Fig. 5F). Histogram analysis of the distribution of DCV velocities revealed an increase in the number of anterograde DCVs moving at a velocity of ∼1–1.5 μm/s (Fig. 5G), although there was no change in the average anterograde and retrograde DCV velocities in cdk-5 mutants [average anterograde DCV velocity ± SEM (μm/s): WT, 1.29 ± 0.07; cdk-5, 1.42 ± 0.06; p > 0.05, KS test; average retrograde DCV velocity ± SEM (μm/s): WT, 0.97 ± 0.05; cdk-5, 1.08 ± 0.04; p > 0.05, KS test]. These results imply that, in both wild-type and cdk-5 mutant dendrites, DCVs are transported on a motor that travels at 1–1.5 μm/s, but in cdk-5 mutants there are more DCV trafficking events on this motor. Together, these data suggest that cdk-5 mutants have increased DCVs in DB dendrites due to an increase in trafficking of DCVs into dendrites (i.e., anterograde trafficking), an increase in DCV accumulation (i.e., stationary DCVs), and a compensatory increase in retrograde DCV trafficking. Our time-lapse data, together with the increase in INS-22::Venus fluorescence in cdk-5 mutant dendrites (Fig. 2D,F), are consistent with the idea that CDK-5 prevents trafficking of DCVs into DB dendrites.
We also used kymographs of INS-22::Venus movement to test whether cdk-5 mutants have defects in the packaging of neuropeptides into DCVs in addition to their defects in DCV trafficking. Our steady-state imaging data show that INS-22::Venus puncta fluorescence intensity changes in cdk-5 mutants (Fig. 2 and numbers reported in the text). Because each DCV contains many molecules of INS-22::Venus, changes in fluorescence intensity could represent either changes in the number of DCVs in each cluster (i.e., puncta) or changes in the amount of INS-22::Venus neuropeptides contained in each DCV. To distinguish between these possibilities, we measured the fluorescence intensity of mobile versus stationary INS-22::Venus puncta. Assuming that mobile puncta likely represent single DCVs, this analysis allows us to estimate the amount of INS-22::Venus neuropeptide contained in each DCV in wild-type and cdk-5 mutants. Histogram analysis of INS-22::Venus puncta fluorescence of mobile versus stationary puncta in wild-type and cdk-5 mutants supports the idea that cdk-5 mutants have changes in the number of DCVs in each cluster (Fig. 5H). First, the average INS-22::Venus fluorescence intensity of mobile puncta was not altered in cdk-5 mutants compared with wild-type controls [average puncta intensity (norm.) ± SEM: wild type, 1.0 ± 0.02; cdk-5, 0.95 ± 0.02; p > 0.05] (Fig. 5H). Second, INS-22::Venus fluorescence intensities of stationary puncta in cdk-5 mutants shifted to higher values, revealing multiple peaks with increased fluorescence intensities compared with the intensities of mobile puncta (Fig. 5H). Assuming that mobile puncta largely represent single DCVs (see Materials and Methods), these data are consistent with the idea that changes in INS-22::Venus puncta fluorescence observed in cdk-5 mutant axons and dendrites are not due to changes in INS-22::Venus packaging, but likely represent changes in the number of DCVs per cluster. Thus, our data suggest that CDK-5 regulates polarized trafficking of DCVs in motor neurons. Together, our time-lapse analysis of DCVs in axons and dendrites are consistent with a model in which CDK-5 regulates the polarized distribution of DCVs by both promoting DCV trafficking in axons and inhibiting DCV trafficking into dendrites.
Role of kinesin UNC-104/KIF1A in DCV trafficking
The increase in anterograde DCV trafficking and plus-end-out microtubules in cdk-5 mutant dendrites raises the question of whether a plus-end-directed axonal motor, such as UNC-104/KIF1A (Hall and Hedgecock, 1991), is responsible for trafficking DCVs into dendrites. The kinesin UNC-104/KIF1A is required for the transport of DCVs to axons in C. elegans and Drosophila and has been reported to localize to dendrites as well as axons (Zhou et al., 2001; Jacob and Kaplan, 2003; Shin et al., 2003; Zahn et al., 2004; Sieburth et al., 2005; Pack-Chung et al., 2007; Barkus et al., 2008). In addition, a recent study showed that, in PVD sensory neurons in C. elegans, UNC-104 is responsible for mistrafficking axonal cargo into dendrites of unc-33/CRMP mutant animals (Maniar et al., 2012). We tested whether the increase in dendritic DCVs observed in cdk-5 mutants was dependent on UNC-104 by analyzing cdk-5;unc-104 double-mutant animals. Consistent with its role in axonal DCV transport, we found little to no INS-22::Venus fluorescence in motor neuron axons (Fig. 6A) and a corresponding increase in the amount of INS-22::Venus in the cell bodies and dendrites of unc-104(e1265) loss-of-function mutants (Fig. 6B–F) (Sieburth et al., 2005). If UNC-104-dependent trafficking is responsible for the increased accumulation of DCVs in cdk-5 mutant dendrites, then this increase should be blocked by unc-104 mutations. In contrast, we found a significant increase in INS-22::Venus fluorescence intensity in cdk-5;unc-104 double-mutant dendrites compared with either single mutant or wild-type controls (Fig. 6E). However, we did not observe a corresponding additive increase in INS-22::Venus puncta density in the double mutants (Fig. 6F). These data, together with our histogram analysis of INS-22::Venus content in DCVs (Fig. 5H), suggests that the number of DCV accumulation sites in cdk-5;unc-104 double-mutant dendrites may have reached its upper limit and that the further increase in intensity represents the addition of more DCVs to existing clusters. Thus, the increased abundance of INS-22::Venus fluorescence in cdk-5;unc-104 double-mutant dendrites suggests that kinesin UNC-104 is not required for the DCV accumulation in cdk-5 mutant dendrites. Interestingly, the accumulation of DCVs in the cell bodies of unc-104 mutants decreased in unc-104;cdk-5 double mutants concomitantly with the increase in the dendrites (Fig. 6B,C). These results suggest that CDK-5 may function at the cell body to inhibit trafficking of DCVs into dendrites.
The increase in DCVs observed in cdk-5 mutant dendrites requires cytoplasmic dynein
Because mutations in unc-104 did not block the DCV polarity defect in cdk-5 mutants and the majority (67%) of microtubules in cdk-5 mutant dendrites are still oriented minus-end out from the cell body, we tested whether the major minus-end-directed motor, cytoplasmic dynein, was responsible for trafficking DCVs into cdk-5 mutant dendrites. Cytoplasmic dynein has been shown to traffic DCVs carrying BDNF and has recently been shown to traffic presynaptic and postsynaptic cargoes into dendrites (Gauthier et al., 2004; Colin et al., 2008; Kapitein et al., 2010; Ou et al., 2010). We found that loss-of-function mutations in the heavy chain of dynein, dhc-1(js319) (Koushika et al., 2004), or light-intermediate chain of dynein, dli-1(ku266) (Yoder and Han, 2001), completely block the increase in DCVs in cdk-5 mutant dendrites (Fig. 7D,F) (data not shown). This result indicates that cytoplasmic dynein is required for the increase in DCVs observed in the dendrites of cdk-5 mutants.
Figure 7.

Cytoplasmic dynein is required for the increase in dendritic DCVs in cdk-5 mutant DB dendrites. A, Schematic diagram of a DB motor neuron (top panel). The boxed region denotes that the axon was imaged for data presented in A–C and G. Representative images of INS-22::Venus DB axons of wild-type, cdk-5(gm336), dhc-1(js319), and cdk-5;dhc-1 double-mutant animals (bottom panels). B, C, Quantification of INS-22::Venus puncta intensity (B) and density (C) in axons of wild-type (n = 30), cdk-5 (n = 35), dhc-1 (n = 22), cdk-5;dhc-1 (n = 20) animals. D, Schematic diagram of a DB motor neuron (top panel). The boxed region denotes that the dendrite was imaged for data presented in D–F and H. Representative images of INS-22::Venus in DB dendrites of wild-type, cdk-5(gm336), dhc-1(js319), and cdk-5;dhc-1 double-mutant animals (bottom panels). E, F, Quantification of INS-22::Venus puncta intensity (E) and density (F) in dendrites of wild-type (n = 27), cdk-5 (n = 19), dhc-1 (n = 12), and cdk-5;dhc-1 (n = 19) animals. G, Quantification of percentage plus-end-out and minus-end-out microtubules in DB axons of wild-type (n = 76 puncta), cdk-5(gm336) (n = 93), dhc-1(js319) (n = 36), and cdk-5;dhc-1(n = 61) double mutants based on analysis of EBP-1::GFP movement in axons. H, Quantification of percentage plus-end-out and minus-end-out microtubules in DB dendrites of wild-type (n = 107 puncta), cdk-5(gm336) (n = 195), dhc-1(js319) (n = 60), and cdk-5;dhc-1 (n = 218) double-mutant animals based on analysis of EBP-1::GFP movement in dendrites. Values that differ significantly [Tukey–Kramer test (B, C, E, F) and χ2 test (G, H)] from wild type (marked by asterisks above each bar) or from other genotypes (comparisons marked by brackets) are denoted on the graphs (**p < 0.001; *p < 0.01; #p < 0.05; n.s., p > 0.05).
Our results show that CDK-5 regulates two aspects of neuronal polarity: polarized DCV trafficking and microtubule orientation. Because dynein motor function has been reported to be required for the uniform plus-end-out microtubule orientation in Drosophila axons (Zheng et al., 2008), and microtubule organization in non-neuronal cells (Smith et al., 2000), we investigated whether the effects of CDK-5 on neuronal polarity were related. Specifically, we tested whether dynein mutations suppress the change in microtubule orientation observed in cdk-5 mutant dendrites by analyzing kymographs of EBP-1::GFP dynamics in DB axons and dendrites of dhc-1 single and cdk-5;dhc-1 double mutants. We found no significant changes in the plus-end-out microtubule orientation in axons of dhc-1 single or dhc-1;cdk-5 double mutants (Fig. 7G). Similarly, we found no differences in the largely minus-end-out microtubule orientation in dendrites of dhc-1 mutants compared with wild-type controls (Fig. 7H). Finally, there was no significant difference (p > 0.05) between the increased proportion of plus-end-out microtubules observed in cdk-5 single- and cdk-5;dhc- 1 double-mutant dendrites (Fig. 7H). These results suggest that the dhc-1(js319) dynein mutation does not affect microtubule orientation in DB axons and dendrites. Thus, the ability of the dhc-1(js319) dynein mutation to suppress the accumulation of DCVs in cdk-5 mutant dendrites cannot be attributed to potential effects of dynein on microtubule polarity, and our findings are more consistent with a model in which CDK-5 inhibits dynein-dependent trafficking of DCVs into dendrites.
We next analyzed the role of dynein in DB axons of wild-type and cdk-5 mutant animals. We found a significant increase in INS-22::Venus puncta fluorescence intensity in dhc-1 single-mutant axons (Fig. 7A,B), consistent with a role for dynein in retrograde DCV trafficking in the axon. As described above, cdk-5 single mutants have decreased abundance of DCVs in DB axons (Figs. 2A,B, 7A,B). If the decrease in axonal DCVs observed in cdk-5 mutants was mediated by dynein, then this decrease should be blocked by mutations in dynein. However, we found that dhc-1 mutation only partially suppressed the decrease in DCV accumulation observed in cdk-5 mutant axons (Fig. 7A,B). This result suggests that in DB motor neuron axons CDK-5 and dynein may function in separate pathways to regulate DCV accumulation. This result is consistent with our time-lapse analysis showing that CDK-5 does not inhibit retrograde DCV trafficking in axons but rather promotes anterograde DCV trafficking (Fig. 5B). However, because the dhc-1(js319) allele is not a null mutation, we cannot exclude the possibility that the partial effect of dhc-1 mutation on DCV trafficking in cdk-5 mutant axons may be due to an incomplete loss of dynein function.
Dynein is required for increased trafficking of DCVs in cdk-5 mutant dendrites
Our time-lapse analysis of DCV movement shows that cdk-5 mutants have increased numbers of mobile (anterogradely and retrogradely moving DCVs) and stationary DCVs in dendrites (Fig. 5E). To investigate whether dynein regulates DCV trafficking in dendrites, we performed time-lapse analysis of mobile INS-22::Venus puncta in dhc-1 single and cdk-5;dhc-1 double mutants. We found a significant decrease in the number of DCVs moving in the anterograde and retrograde directions in dhc-1 single-mutant dendrites (Fig. 8A). These data indicate that dynein is required for DCV trafficking in the dendrites of wild-type animals. Furthermore, dhc-1 mutation blocked the increase in anterograde and retrograde DCV trafficking (Fig. 8A) and total number of DCVs observed in cdk-5 mutant dendrites (number of DCVs/kymograph: WT, 7.35 ± 1.00; cdk-5, 12.47 ± 1.23; dhc-1, 3.43 ± 0.52; cdk-5;dhc-1, 5.96 ± 1.2; cdk-5;dhc-1 vs cdk-5, p < 0.001; cdk-5;dhc-1 vs wt and dhc-1, p > 0.05). These results are consistent with our steady-state imaging data in cdk-5;dhc-1 double mutants (Fig. 7D,F) and suggest that dynein is required for anterograde DCV trafficking in dendrites. Although dynein may also participate in retrograde DCV trafficking, the decrease in the total number of DCVs in dendrites of cdk-5;dhc-1 double mutants (Fig. 7D,F) suggest that changes in retrograde trafficking may be a secondary consequence of decreased anterograde DCV trafficking. Thus, our data suggest a model in which CDK-5 inhibits anterograde trafficking of dynein in general, or more specifically, inhibits trafficking of dynein–DCV complexes into dendrites.
Figure 8.
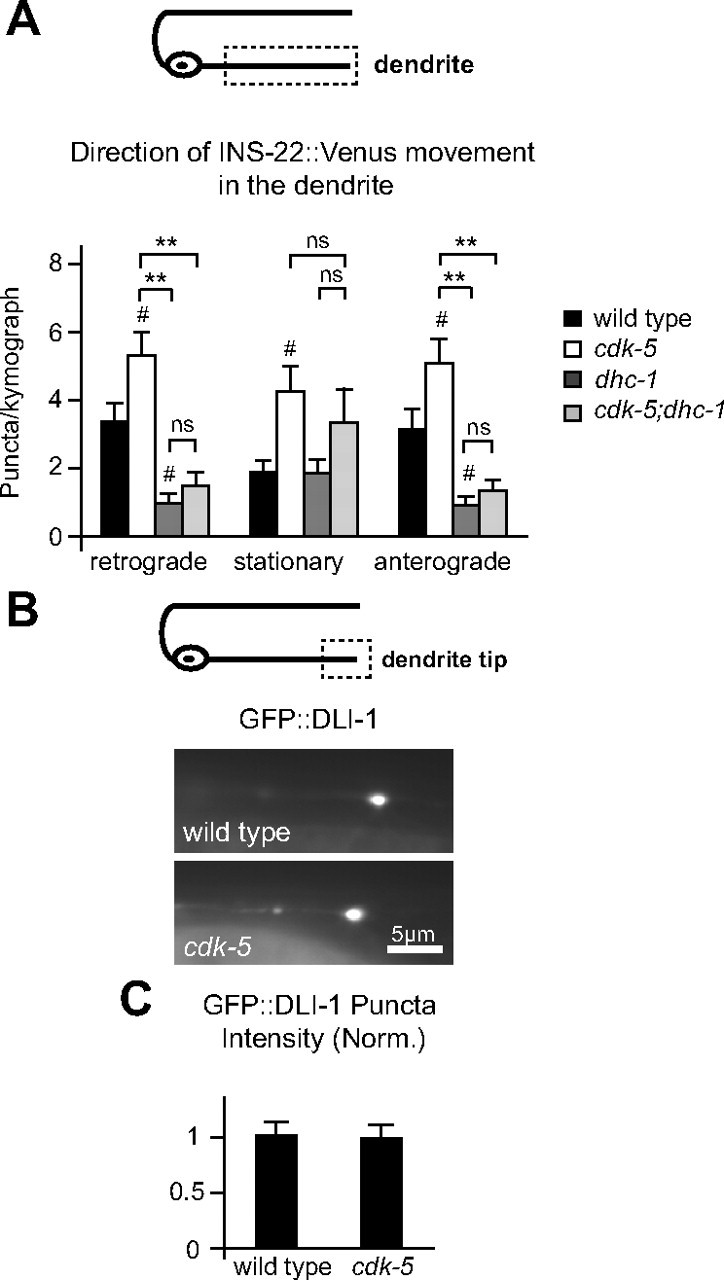
Cytoplasmic dynein is required for DCV trafficking in DB motor neuron dendrites in wild-type and cdk-5 mutant animals. A, Schematic diagram of a DB motor neuron (top panel). The boxed region denotes that the dendrite was imaged for data presented in A. Quantification of the direction of INS-22::Venus puncta movement in DB dendrites of wild-type (n = 34), cdk-5(gm336) (n = 36), dhc-1(js319) (n = 21), and cdk-5;dhc-1 (n = 29) double-mutant dendrites (bottom panel). B, Schematic diagram of a DB motor neuron (top panel). The boxed region denotes that the distal dendrite tip was imaged for data presented in B and C. Representative images of GFP::DLI-1 accumulations at the distal tip of DB7 dendrites in wild-type and cdk-5(gm336) mutant animals (bottom panels). C, Quantification of GFP::DLI-1 puncta intensity at the tip of DB7 dendrites in wild-type (n = 29) and cdk-5 (n = 23) mutant animals. Values that differ significantly (Tukey–Kramer test) from wild type (marked by symbols above each bar) or from other genotypes (comparisons marked by brackets) are denoted on the graphs (**p < 0.001; #p < 0.05; ns, p > 0.05).
To test whether CDK-5 inhibits general trafficking of dynein into dendrites, we analyzed the distribution of a dynein-associated protein, GFP-tagged dynein light-intermediate chain (DLI-1::GFP) (Arimoto et al., 2011), in DB dendrites of wild-type and cdk-5 mutants. We found that the distribution of DLI-1::GFP was diffuse throughout most of the dendrite, but accumulated at the distal tips of DB dendrites, consistent with their predominantly minus-end-out microtubule orientation and DLI-1 association with dynein. We found no change in the amount of DLI-1::GFP fluorescence at the distal dendrite tips in cdk-5 mutants compared with wild-type controls (Fig. 8B,C). Thus, CDK-5 may not regulate overall trafficking of dynein into dendrites, but may have a more specific effect on the trafficking of DCVs by dynein. Together, these results suggest that cytoplasmic dynein is required for the increased trafficking and accumulation of DCVs in cdk-5 mutant dendrites, and are consistent with a model in which CDK-5 inhibits loading of DCVs onto dynein or trafficking of dynein-DCV complexes into dendrites.
Discussion
CDK-5 regulates polarized trafficking of DCVs
We investigated the role of CDK-5 in neuronal polarity and found that CDK-5 and its activator CDKA-1/p35 are required for the polarized distribution of neuropeptide-containing DCVs in DA and DB cholinergic motor neurons (Fig. 2). Time-lapse microscopy of mobile DCVs in DB neurons revealed that cdk-5 mutation results in decreased trafficking of DCVs into axons and increased trafficking and accumulation of DCVs in dendrites (Fig. 5). In DB neurons, this polarity defect appears to be specific for DCVs because cdk-5 mutants had no obvious defect in the polarized distribution of axonal markers, such as SVs, or dendritic markers (Fig. 3). There are several potential models to explain the mechanism by which CDK-5 regulates polarized DCV trafficking as follows.
First, the axonal DCV motor UNC-104 mistraffics into dendrites in cdk-5 mutants. In unc-104 mutants, DCVs do not traffic properly to axons and accumulate in motor neuron cell bodies and dendrites (Fig. 6) (Sieburth et al., 2005). If this model were correct, then genetically removing unc-104 in cdk-5 mutants should not increase the DCV trafficking defect of cdk-5 single mutants. Instead, we found that cdk-5;unc-104 double mutants had an increased abundance of DCVs in dendrites compared with either single mutant (Fig. 6D–F). Thus, we think this model is unlikely.
Second, DCVs are trafficked to both axons and dendrites and CDK-5 promotes retrograde trafficking of DCVs from dendrites back to the cell bodies. If this model were correct, then cdk-5 mutants should have decreased numbers of retrogradely moving DCVs in dendrites. Our time-lapse analysis of mobile DCVs showed that cdk-5 mutants do not have decreased retrograde DCV trafficking but instead have increased amounts of DCV trafficking in dendrites (Fig. 5E), suggesting that this model is also unlikely.
Third, CDK-5 positively regulates axonal trafficking of DCVs on kinesin UNC-104. For example, CDK-5 might directly promote the loading of DCVs onto UNC-104 or promote anterograde trafficking of UNC-104 into axons. If this model were correct, then cdk-5 mutants should have decreased anterograde DCV trafficking in the axon and cdk-5;unc-104 double mutants should have a nonadditive effect on DCVs in the dendrites. Consistent with this model, we found that cdk-5 mutants have decreased anterograde DCV trafficking in axon commissures (Fig. 5B); however, we also found that cdk-5;unc-104 double mutants have additive increases in DCVs in dendrites (Fig. 6D–F). Thus, CDK-5 appears to promote UNC-104-dependent anterograde trafficking of DCVs in axons; however, our genetic data suggest that this cannot be the only mechanism by which CDK-5 regulates DCV polarity.
Fourth, CDK-5 inhibits the transport of DCVs into dendrites. Our data are also consistent with this model. First, cdk-5 mutants have increased amounts of DCVs trafficking into and accumulating in dendrites (Figs. 2, 5). Second, the cell body accumulation of DCVs observed in unc-104 single mutants is reduced to wild-type levels in cdk-5;unc-104 double mutants along with a concomitant increase of DCVs in dendrites (Fig. 6). Third, mutations in the minus-end-directed motor cytoplasmic dynein completely block the increased trafficking of DCVs in cdk-5 mutant dendrites (Figs. 7, 8).
In summary, our data support a model in which CDK-5 regulates DCV polarity by both promoting axonal DCV trafficking and inhibiting dendritic DCV trafficking.
CDK-5 regulates microtubule polarity
This study also identifies CDK-5 as a novel regulator of microtubule polarity. We used the microtubule plus-end binding protein EBP-1 to describe the orientation of microtubules in cholinergic motor neurons and found that, in wild-type animals, DB axons have predominantly plus-end-out microtubule polarity, whereas DB dendrites have a slightly less uniform orientation with the vast majority of microtubules being oriented with their minus-ends distal to the cell body (Fig. 4). These results are consistent with the microtubule polarity inferred from analysis of tailless motors in DA motor neurons (Ou et al., 2010), and of EB protein dynamics in amphid sensory neurons in C. elegans (Maniar et al., 2012) and Drosophila (Rolls et al., 2007; Stone et al., 2008; Zheng et al., 2008). Although cdk-5 mutants do not affect microtubule orientation in DB motor neuron axons (Fig. 4A,B), we found, surprisingly, that cdk-5 mutants have an increase in plus-end-out microtubules in DB dendrites (Fig. 4C,D). While the precise molecular mechanism involved is not known, it will be interesting to test in future studies whether the effect of CDK-5 on microtubule polarity is mediated by its ability to phosphorylate microtubule-associated proteins (Wada et al., 1998; Patrick et al., 1999; Dhavan and Tsai, 2001; Tanaka et al., 2004; Hou et al., 2007; Kaminosono et al., 2008).
Because CDK-5 affects the polarized trafficking of DCVs and microtubule orientation, we tested whether these two polarity effects were related. Our data suggest that the effect of CDK-5 on microtubule polarity cannot explain the increased trafficking of DCVs into dendrites. Dynein mutations completely block the increased trafficking and accumulation of DCVs in cdk-5 mutant dendrites (Figs. 7, 8) without affecting the cdk-5-dependent change in microtubule polarity (Fig. 7H). Interestingly, the normal polarized distribution of several axonal and dendritic markers (Fig. 3) and the normal accumulation of the dynein complex protein DLI-1::GFP at the distal tips of dendrites in cdk-5 mutants (Fig. 8B,C), suggest that these mutants do not have gross defects in polarized transport. Thus, robust and redundant cellular mechanisms may exist to establish and maintain neuronal polarity. Alternatively, the magnitude of the effect of CDK-5 on microtubule polarity may not be large enough to result in obvious polarity defects, or the effect of cdk-5 on microtubule orientation may result in defects in the polarized trafficking of other cargo or aspects of neuronal polarity that we did not examine.
Comparison of SV and DCV trafficking by CDK-5
SVs are also trafficked in a polarized manner to presynaptic sites in axons and many genes and mechanisms involved in this trafficking have been described in cultured hippocampal neurons (West et al., 1997) and C. elegans motor neurons (Byrd et al., 2001; Crump et al., 2001; Sakamoto et al., 2005; Tanizawa et al., 2006; Hung et al., 2007; Sakaguchi-Nakashima et al., 2007; Poon et al., 2008; Ou et al., 2010; Ou and Shen, 2011). A recent study showed that CDK-5 regulates the polarized trafficking of SV precursors to axons by inhibiting cytoplasmic dynein (Ou et al., 2010). Similarly, we found that polarized DCV trafficking is also dependent on CDK-5 and dynein. Thus, despite the differences between SVs and DCVs, CDK-5 can promote the polarized trafficking of both vesicle types. There are several interesting differences between CDK-5 regulation of SV and DCV polarity. First, Ou et al. (2010) found that CDK-5 was required for polarized trafficking of SVs in DA class motor neurons but, surprisingly, not in DB class motor neurons (Ou et al., 2010). In contrast, we found that the polarized distribution of DCVs requires cdk-5 in both DA and DB motor neurons. The precise reason for this difference is not clear. However, these studies suggest that SV polarity is regulated by multiple and redundant mechanisms that differ between DA and DB neurons, whereas DCV polarity requires CDK-5 activity in both cell types. Second, Ou et al. (2010) used time-lapse analysis of SVs in DA axons to show that CDK-5 inhibits trafficking of SVs back to the cell body in a dynein-dependent manner (Ou et al., 2010). In contrast, we found that CDK-5 functions to promote DCV trafficking in DB axons, suggesting that CDK-5 may promote loading of DCVs onto UNC-104 or trafficking of UNC-104-DCV complexes into axons. While positive regulation of UNC-104 by CDK-5 does not appear to be relevant for SV polarity in DA axons, CDK-5 can promote UNC-104-dependent trafficking in axons of GABAergic DD motor neurons (Park et al., 2011). In addition to promoting axonal DCV trafficking, we show that CDK-5 also prevents trafficking of DCVs into DB dendrites in a dynein-dependent manner. These data are consistent with the idea that CDK-5 may prevent the trafficking of DCVs into dendrites either by inhibiting the loading of DCVs onto dynein or by inhibiting trafficking of dynein-DCV complexes into dendrites. One simple model that may explain these two effects of CDK-5 on DCV trafficking in DB axons and dendrites would be as follows: CDK-5 phosphorylates a protein (e.g., a specific DCV adaptor), which then promotes association of DCVs with UNC-104 in the cell body and also inhibits association with dynein. In this manner, CDK-5 phosphorylation of this DCV-specific protein would act as a switch to both promote trafficking of DCVs into axons and inhibit trafficking of DCVs into dendrites. By analogy, CDK-5 might phosphorylate a SV-specific protein to differentially regulate its polarized trafficking. Further experiments will be necessary to identify the relevant SV- and DCV-specific proteins to test this model.
In closing, while CDK-5 can regulate the polarized trafficking of SV precursors and DCVs destined for axons, the dependence on CDK-5 and the precise trafficking mechanisms involved appear to differ between neuronal cell types and vesicular cargos. Because DCVs are diverse and can transport cargo destined for secretion in dendrites, much remains to be learned about the molecular mechanisms involved in trafficking of different DCV populations to specific destinations.
Footnotes
This work was supported by National Institutes of Health (NIH) Grant NS059953 (P.J.), a Basil O'Connor March of Dimes Scholar Award (P.J.), and the Tufts Center for Neuroscience Research (NIH Grant P30 NS047243). P.R.G. was supported by a Dean's Fellowship and the Synapse Neurobiology Training Program (NIH Grant T32 NS061764). We thank Josh Kaplan, Lars Dreier, QueeLim Ch'ng, Derek Sieburth, and Jeremy Dittman for advice, reagents, and comments on this manuscript. We thank Michele Jacob, Victor Hatini, Jennifer Kowalski, and members of the Juo Laboratory for helpful discussions and comments on this manuscript, and Jeremy Dittman and Dan Cox for data analysis software. We also thank Kang Shen for generously sharing unpublished data, and Gian Garriga, Ken Miller, Naoki Hisamoto, the Caenorhabditis Genetics Center, and Shohei Mitani for plasmids and strains.
References
- Arimoto M, Koushika SP, Choudhary BC, Li C, Matsumoto K, Hisamoto N. The Caenorhabditis elegans JIP3 protein UNC-16 functions as an adaptor to link kinesin-1 with cytoplasmic dynein. J Neurosci. 2011;31:2216–2224. doi: 10.1523/JNEUROSCI.2653-10.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baas PW, Deitch JS, Black MM, Banker GA. Polarity orientation of microtubules in hippocampal neurons: uniformity in the axon and nonuniformity in the dendrite. Proc Natl Acad Sci U S A. 1988;85:8335–8339. doi: 10.1073/pnas.85.21.8335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barkus RV, Klyachko O, Horiuchi D, Dickson BJ, Saxton WM. Identification of an axonal kinesin-3 motor for fast anterograde vesicle transport that facilitates retrograde transport of neuropeptides. Mol Biol Cell. 2008;19:274–283. doi: 10.1091/mbc.E07-03-0261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Borgonovo B, Ouwendijk J, Solimena M. Biogenesis of secretory granules. Curr Opin Cell Biol. 2006;18:365–370. doi: 10.1016/j.ceb.2006.06.010. [DOI] [PubMed] [Google Scholar]
- Brenner S. The genetics of Caenorhabditis elegans. Genetics. 1974;77:71–94. doi: 10.1093/genetics/77.1.71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burbea M, Dreier L, Dittman JS, Grunwald ME, Kaplan JM. Ubiquitin and AP180 regulate the abundance of GLR-1 glutamate receptors at postsynaptic elements in C. elegans. Neuron. 2002;35:107–120. doi: 10.1016/s0896-6273(02)00749-3. [DOI] [PubMed] [Google Scholar]
- Burton PR. Dendrites of mitral cell neurons contain microtubules of opposite polarity. Brain Res. 1988;473:107–115. doi: 10.1016/0006-8993(88)90321-6. [DOI] [PubMed] [Google Scholar]
- Byrd DT, Kawasaki M, Walcoff M, Hisamoto N, Matsumoto K, Jin Y. UNC-16, a JNK-signaling scaffold protein, regulates vesicle transport in C. elegans. Neuron. 2001;32:787–800. doi: 10.1016/s0896-6273(01)00532-3. [DOI] [PubMed] [Google Scholar]
- Cai T, Fukushige T, Notkins AL, Krause M. Insulinoma-associated protein IA-2, a vesicle transmembrane protein, genetically interacts with UNC-31/CAPS and affects neurosecretion in Caenorhabditis elegans. J Neurosci. 2004;24:3115–3124. doi: 10.1523/JNEUROSCI.0101-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chalfie M, Sulston JE, White JG, Southgate E, Thomson JN, Brenner S. The neural circuit for touch sensitivity in Caenorhabditis elegans. J Neurosci. 1985;5:956–964. doi: 10.1523/JNEUROSCI.05-04-00956.1985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheung ZH, Fu AK, Ip NY. Synaptic roles of Cdk5: implications in higher cognitive functions and neurodegenerative diseases. Neuron. 2006;50:13–18. doi: 10.1016/j.neuron.2006.02.024. [DOI] [PubMed] [Google Scholar]
- Ch'ng Q, Sieburth D, Kaplan JM. Profiling synaptic proteins identifies regulators of insulin secretion and lifespan. PLoS Genet. 2008;4:e1000283. doi: 10.1371/journal.pgen.1000283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Colin E, Zala D, Liot G, Rangone H, Borrell-Pagès M, Li XJ, Saudou F, Humbert S. Huntingtin phosphorylation acts as a molecular switch for anterograde/retrograde transport in neurons. EMBO J. 2008;27:2124–2134. doi: 10.1038/emboj.2008.133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crump JG, Zhen M, Jin Y, Bargmann CI. The SAD-1 kinase regulates presynaptic vesicle clustering and axon termination. Neuron. 2001;29:115–129. doi: 10.1016/s0896-6273(01)00184-2. [DOI] [PubMed] [Google Scholar]
- de Jong EK, Vinet J, Stanulovic VS, Meijer M, Wesseling E, Sjollema K, Boddeke HW, Biber K. Expression, transport, and axonal sorting of neuronal CCL21 in large dense-core vesicles. FASEB J. 2008;22:4136–4145. doi: 10.1096/fj.07-101907. [DOI] [PubMed] [Google Scholar]
- Dhavan R, Tsai LH. A decade of CDK5. Nat Rev Mol Cell Biol. 2001;2:749–759. doi: 10.1038/35096019. [DOI] [PubMed] [Google Scholar]
- Dikeakos JD, Reudelhuber TL. Sending proteins to dense core secretory granules: still a lot to sort out. J Cell Biol. 2007;177:191–196. doi: 10.1083/jcb.200701024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edwards SL, Charlie NK, Richmond JE, Hegermann J, Eimer S, Miller KG. Impaired dense core vesicle maturation in Caenorhabditis elegans mutants lacking Rab2. J Cell Biol. 2009;186:881–895. doi: 10.1083/jcb.200902095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fisher JM, Sossin W, Newcomb R, Scheller RH. Multiple neuropeptides derived from a common precursor are differentially packaged and transported. Cell. 1988;54:813–822. doi: 10.1016/s0092-8674(88)91131-2. [DOI] [PubMed] [Google Scholar]
- Gauthier LR, Charrin BC, Borrell-Pagès M, Dompierre JP, Rangone H, Cordelières FP, De Mey J, MacDonald ME, Lessmann V, Humbert S, Saudou F. Huntingtin controls neurotrophic support and survival of neurons by enhancing BDNF vesicular transport along microtubules. Cell. 2004;118:127–138. doi: 10.1016/j.cell.2004.06.018. [DOI] [PubMed] [Google Scholar]
- Hall DH, Hedgecock EM. Kinesin-related gene unc-104 is required for axonal transport of synaptic vesicles in C. elegans. Cell. 1991;65:837–847. doi: 10.1016/0092-8674(91)90391-b. [DOI] [PubMed] [Google Scholar]
- Hammarlund M, Watanabe S, Schuske K, Jorgensen EM. CAPS and syntaxin dock dense core vesicles to the plasma membrane in neurons. J Cell Biol. 2008;180:483–491. doi: 10.1083/jcb.200708018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haspel G, O'Donovan MJ, Hart AC. Motoneurons dedicated to either forward or backward locomotion in the nematode Caenorhabditis elegans. J Neurosci. 2010;30:11151–11156. doi: 10.1523/JNEUROSCI.2244-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Horton AC, Ehlers MD. Neuronal polarity and trafficking. Neuron. 2003;40:277–295. doi: 10.1016/s0896-6273(03)00629-9. [DOI] [PubMed] [Google Scholar]
- Hou Z, Li Q, He L, Lim HY, Fu X, Cheung NS, Qi DX, Qi RZ. Microtubule association of the neuronal p35 activator of Cdk5. J Biol Chem. 2007;282:18666–18670. doi: 10.1074/jbc.C700052200. [DOI] [PubMed] [Google Scholar]
- Hung W, Hwang C, Po MD, Zhen M. Neuronal polarity is regulated by a direct interaction between a scaffolding protein, Neurabin, and a presynaptic SAD-1 kinase in Caenorhabditis elegans. Development. 2007;134:237–249. doi: 10.1242/dev.02725. [DOI] [PubMed] [Google Scholar]
- Jacob TC, Kaplan JM. The EGL-21 carboxypeptidase E facilitates acetylcholine release at Caenorhabditis elegans neuromuscular junctions. J Neurosci. 2003;23:2122–2130. doi: 10.1523/JNEUROSCI.23-06-02122.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Juo P, Harbaugh T, Garriga G, Kaplan JM. CDK-5 regulates the abundance of GLR-1 glutamate receptors in the ventral cord of Caenorhabditis elegans. Mol Biol Cell. 2007;18:3883–3893. doi: 10.1091/mbc.E06-09-0818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaminosono S, Saito T, Oyama F, Ohshima T, Asada A, Nagai Y, Nukina N, Hisanaga S. Suppression of mutant Huntingtin aggregate formation by Cdk5/p35 through the effect on microtubule stability. J Neurosci. 2008;28:8747–8755. doi: 10.1523/JNEUROSCI.0973-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kapitein LC, Schlager MA, Kuijpers M, Wulf PS, van Spronsen M, MacKintosh FC, Hoogenraad CC. Mixed microtubules steer dynein-driven cargo transport into dendrites. Curr Biol. 2010;20:290–299. doi: 10.1016/j.cub.2009.12.052. [DOI] [PubMed] [Google Scholar]
- Koushika SP, Richmond JE, Hadwiger G, Weimer RM, Jorgensen EM, Nonet ML. A post-docking role for active zone protein Rim. Nat Neurosci. 2001;4:997–1005. doi: 10.1038/nn732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koushika SP, Schaefer AM, Vincent R, Willis JH, Bowerman B, Nonet ML. Mutations in Caenorhabditis elegans cytoplasmic dynein components reveal specificity of neuronal retrograde cargo. J Neurosci. 2004;24:3907–3916. doi: 10.1523/JNEUROSCI.5039-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kwinter DM, Lo K, Mafi P, Silverman MA. Dynactin regulates bidirectional transport of dense-core vesicles in the axon and dendrites of cultured hippocampal neurons. Neuroscience. 2009;162:1001–1010. doi: 10.1016/j.neuroscience.2009.05.038. [DOI] [PubMed] [Google Scholar]
- Landry M, Vila-Porcile E, Hokfelt T, Calas A. Differential routing of coexisting neuropeptides in vasopressin neurons. Eur J Neurosci. 2003;17:579–589. [PubMed] [Google Scholar]
- Lessmann V, Brigadski T. Mechanisms, locations, and kinetics of synaptic BDNF secretion: an update. Neurosci Res. 2009;65:11–22. doi: 10.1016/j.neures.2009.06.004. [DOI] [PubMed] [Google Scholar]
- Li C, Kim K. Neuropeptides. WormBook. 2008;2008:1–36. doi: 10.1895/wormbook.1.142.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maniar TA, Kaplan M, Wang GJ, Shen K, Wei L, Shaw JE, Koushika SP, Bargmann CI. UNC-33 (CRMP) and ankyrin organize microtubules and localize kinesin to polarize axon-dendrite sorting. Nat Neurosci. 2012;15:48–56. doi: 10.1038/nn.2970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller KG, Alfonso A, Nguyen M, Crowell JA, Johnson CD, Rand JB. A genetic selection for Caenorhabditis elegans synaptic transmission mutants. Proc Natl Acad Sci U S A. 1996;93:12593–12598. doi: 10.1073/pnas.93.22.12593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mimori-Kiyosue Y, Shiina N, Tsukita S. The dynamic behavior of the APC-binding protein EB1 on the distal ends of microtubules. Curr Biol. 2000;10:865–868. doi: 10.1016/s0960-9822(00)00600-x. [DOI] [PubMed] [Google Scholar]
- Morfini G, Szebenyi G, Brown H, Pant HC, Pigino G, DeBoer S, Beffert U, Brady ST. A novel CDK5-dependent pathway for regulating GSK3 activity and kinesin-driven motility in neurons. EMBO J. 2004;23:2235–2245. doi: 10.1038/sj.emboj.7600237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morrison EE, Moncur PM, Askham JM. EB1 identifies sites of microtubule polymerisation during neurite development. Brain Res Mol Brain Res. 2002;98:145–152. doi: 10.1016/s0169-328x(01)00290-x. [DOI] [PubMed] [Google Scholar]
- Nguyen M, Alfonso A, Johnson CD, Rand JB. Caenorhabditis elegans mutants resistant to inhibitors of acetylcholinesterase. Genetics. 1995;140:527–535. doi: 10.1093/genetics/140.2.527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ou CY, Shen K. Neuronal polarity in C. elegans. Dev Neurobiol. 2011;71:554–566. doi: 10.1002/dneu.20858. [DOI] [PubMed] [Google Scholar]
- Ou CY, Poon VY, Maeder CI, Watanabe S, Lehrman EK, Fu AK, Park M, Fu WY, Jorgensen EM, Ip NY, Shen K. Two cyclin-dependent kinase pathways are essential for polarized trafficking of presynaptic components. Cell. 2010;141:846–858. doi: 10.1016/j.cell.2010.04.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pack-Chung E, Kurshan PT, Dickman DK, Schwarz TL. A Drosophila kinesin required for synaptic bouton formation and synaptic vesicle transport. Nat Neurosci. 2007;10:980–989. doi: 10.1038/nn1936. [DOI] [PubMed] [Google Scholar]
- Pandey JP, Smith DS. A cdk5-dependent switch regulates lis1/ndel1/dynein-driven organelle transport in adult axons. J Neurosci. 2011;31:17207–17219. doi: 10.1523/JNEUROSCI.4108-11.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park M, Watanabe S, Poon VY, Ou CY, Jorgensen EM, Shen K. CYY-1/cyclin Y and CDK-5 differentially regulate synapse elimination and formation for rewiring neural circuits. Neuron. 2011;70:742–757. doi: 10.1016/j.neuron.2011.04.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patrick GN, Zukerberg L, Nikolic M, de la Monte S, Dikkes P, Tsai LH. Conversion of p35 to p25 deregulates Cdk5 activity and promotes neurodegeneration. Nature. 1999;402:615–622. doi: 10.1038/45159. [DOI] [PubMed] [Google Scholar]
- Pierce SB, Costa M, Wisotzkey R, Devadhar S, Homburger SA, Buchman AR, Ferguson KC, Heller J, Platt DM, Pasquinelli AA, Liu LX, Doberstein SK, Ruvkun G. Regulation of DAF-2 receptor signaling by human insulin and ins-1, a member of the unusually large and diverse C. elegans insulin gene family. Genes Dev. 2001;15:672–686. doi: 10.1101/gad.867301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Poon VY, Klassen MP, Shen K. UNC-6/netrin and its receptor UNC-5 locally exclude presynaptic components from dendrites. Nature. 2008;455:669–673. doi: 10.1038/nature07291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ratner N, Bloom GS, Brady ST. A role for cyclin-dependent kinase(s) in the modulation of fast anterograde axonal transport: effects defined by olomoucine and the APC tumor suppressor protein. J Neurosci. 1998;18:7717–7726. doi: 10.1523/JNEUROSCI.18-19-07717.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rolls MM, Satoh D, Clyne PJ, Henner AL, Uemura T, Doe CQ. Polarity and intracellular compartmentalization of Drosophila neurons. Neural Dev. 2007;2:7. doi: 10.1186/1749-8104-2-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sakaguchi-Nakashima A, Meir JY, Jin Y, Matsumoto K, Hisamoto N. LRK-1, a C. elegans PARK8-related kinase, regulates axonal-dendritic polarity of SV proteins. Curr Biol. 2007;17:592–598. doi: 10.1016/j.cub.2007.01.074. [DOI] [PubMed] [Google Scholar]
- Sakamoto R, Byrd DT, Brown HM, Hisamoto N, Matsumoto K, Jin Y. The Caenorhabditis elegans UNC-14 RUN domain protein binds to the kinesin-1 and UNC-16 complex and regulates synaptic vesicle localization. Mol Biol Cell. 2005;16:483–496. doi: 10.1091/mbc.E04-07-0553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shakiryanova D, Tully A, Levitan ES. Activity-dependent synaptic capture of transiting peptidergic vesicles. Nat Neurosci. 2006;9:896–900. doi: 10.1038/nn1719. [DOI] [PubMed] [Google Scholar]
- Shin H, Wyszynski M, Huh KH, Valtschanoff JG, Lee JR, Ko J, Streuli M, Weinberg RJ, Sheng M, Kim E. Association of the kinesin motor KIF1A with the multimodular protein liprin-alpha. J Biol Chem. 2003;278:11393–11401. doi: 10.1074/jbc.M211874200. [DOI] [PubMed] [Google Scholar]
- Sieburth D, Ch'ng Q, Dybbs M, Tavazoie M, Kennedy S, Wang D, Dupuy D, Rual JF, Hill DE, Vidal M, Ruvkun G, Kaplan JM. Systematic analysis of genes required for synapse structure and function. Nature. 2005;436:510–517. doi: 10.1038/nature03809. [DOI] [PubMed] [Google Scholar]
- Sieburth D, Madison JM, Kaplan JM. PKC-1 regulates secretion of neuropeptides. Nat Neurosci. 2007;10:49–57. doi: 10.1038/nn1810. [DOI] [PubMed] [Google Scholar]
- Smith DS, Niethammer M, Ayala R, Zhou Y, Gambello MJ, Wynshaw-Boris A, Tsai LH. Regulation of cytoplasmic dynein behaviour and microtubule organization by mammalian Lis1. Nat Cell Biol. 2000;2:767–775. doi: 10.1038/35041000. [DOI] [PubMed] [Google Scholar]
- Solimena M, Dirkx R, Jr, Hermel JM, Pleasic-Williams S, Shapiro JA, Caron L, Rabin DU. ICA 512, an autoantigen of type I diabetes, is an intrinsic membrane protein of neurosecretory granules. EMBO J. 1996;15:2102–2114. [PMC free article] [PubMed] [Google Scholar]
- Stepanova T, Slemmer J, Hoogenraad CC, Lansbergen G, Dortland B, De Zeeuw CI, Grosveld F, van Cappellen G, Akhmanova A, Galjart N. Visualization of microtubule growth in cultured neurons via the use of EB3-GFP (end-binding protein 3-green fluorescent protein) J Neurosci. 2003;23:2655–2664. doi: 10.1523/JNEUROSCI.23-07-02655.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stone MC, Roegiers F, Rolls MM. Microtubules have opposite orientation in axons and dendrites of Drosophila neurons. Mol Biol Cell. 2008;19:4122–4129. doi: 10.1091/mbc.E07-10-1079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sumakovic M, Hegermann J, Luo L, Husson SJ, Schwarze K, Olendrowitz C, Schoofs L, Richmond J, Eimer S. UNC-108/RAB-2 and its effector RIC-19 are involved in dense core vesicle maturation in Caenorhabditis elegans. J Cell Biol. 2009;186:897–914. doi: 10.1083/jcb.200902096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tanaka T, Serneo FF, Tseng HC, Kulkarni AB, Tsai LH, Gleeson JG. Cdk5 phosphorylation of doublecortin ser297 regulates its effect on neuronal migration. Neuron. 2004;41:215–227. doi: 10.1016/s0896-6273(03)00852-3. [DOI] [PubMed] [Google Scholar]
- Tanizawa Y, Kuhara A, Inada H, Kodama E, Mizuno T, Mori I. Inositol monophosphatase regulates localization of synaptic components and behavior in the mature nervous system of C. elegans. Genes Dev. 2006;20:3296–3310. doi: 10.1101/gad.1497806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wada Y, Ishiguro K, Itoh TJ, Uchida T, Hotani H, Saito T, Kishimoto T, Hisanaga S. Microtubule-stimulated phosphorylation of tau at Ser202 and Thr205 by cdk5 decreases its microtubule nucleation activity. J Biochem. 1998;124:738–746. doi: 10.1093/oxfordjournals.jbchem.a022174. [DOI] [PubMed] [Google Scholar]
- West AE, Neve RL, Buckley KM. Targeting of the synaptic vesicle protein synaptobrevin in the axon of cultured hippocampal neurons: evidence for two distinct sorting steps. J Cell Biol. 1997;139:917–927. doi: 10.1083/jcb.139.4.917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- White JG, Southgate E, Thomson JN, Brenner S. The structure of the nervous system of Caenorhabditis elegans. Philos Trans R Soc Lond B Biol Sci. 1986;314:1–340. doi: 10.1098/rstb.1986.0056. [DOI] [PubMed] [Google Scholar]
- Yoder JH, Han M. Cytoplasmic dynein light intermediate chain is required for discrete aspects of mitosis in Caenorhabditis elegans. Mol Biol Cell. 2001;12:2921–2933. doi: 10.1091/mbc.12.10.2921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zahn TR, Angleson JK, MacMorris MA, Domke E, Hutton JF, Schwartz C, Hutton JC. Dense core vesicle dynamics in Caenorhabditis elegans neurons and the role of kinesin UNC-104. Traffic. 2004;5:544–559. doi: 10.1111/j.1600-0854.2004.00195.x. [DOI] [PubMed] [Google Scholar]
- Zheng Y, Wildonger J, Ye B, Zhang Y, Kita A, Younger SH, Zimmerman S, Jan LY, Jan YN. Dynein is required for polarized dendritic transport and uniform microtubule orientation in axons. Nat Cell Biol. 2008;10:1172–1180. doi: 10.1038/ncb1777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou HM, Brust-Mascher I, Scholey JM. Direct visualization of the movement of the monomeric axonal transport motor UNC-104 along neuronal processes in living Caenorhabditis elegans. J Neurosci. 2001;21:3749–3755. doi: 10.1523/JNEUROSCI.21-11-03749.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou KM, Dong YM, Ge Q, Zhu D, Zhou W, Lin XG, Liang T, Wu ZX, Xu T. PKA activation bypasses the requirement for UNC-31 in the docking of dense core vesicles from C. elegans neurons. Neuron. 2007;56:657–669. doi: 10.1016/j.neuron.2007.09.015. [DOI] [PubMed] [Google Scholar]