

Abstract
The unfolded protein response (UPR) is a signaling pathway from the endoplasmic reticulum (ER) to the nucleus that protects cells from the stress caused by misfolded or unfolded proteins [1, 2]. As such, ER stress is an ongoing challenge for all cells given the central biologic importance of secretion as part of normal physiologic functions. This is especially the case for cells that are highly dependent upon secretory function as part of their major duties. Within mucosal tissues, the intestinal epithelium is especially dependent upon an intact UPR for its normal activities [3]. This review will discuss the UPR and the special role that it provides in the functioning of the intestinal epithelium and, when dysfunctional, its implications for understanding mucosal homeostasis and intestinal inflammation, as occurs in inflammatory bowel disease (IBD).
Keywords: Unfolded protein response, endoplasmic reticulum stress, inflammatory bowel disease, intestinal epithelial cells, Paneth cells
The unfolded protein response (UPR)
The UPR is initiated in response to ER stress as a consequence of the accumulation of misfolded proteins in this cellular locale. The UPR is a highly conserved cell biologic process in organisms that range from yeasts to metazoans [1, 2, 4]. In yeasts, ER stress is managed by a single pathway that includes inositol requiring enzyme 1 (IRE1), an ER resident transmembrane protein whose ER luminal amino terminus is involved in sensing the accumulation of unfolded proteins. In the presence of unfolded protein accumulation in the ER, IRE1 displays a novel endoribonuclease activity that leads to the removal of a fragment of the Hac1p transcript within the cytosol resulting in an alternate splice product that encodes a transcriptionally active isoform [5]. Once mobilized to the nucleus, active Hac1p is responsible for directing a transcriptional program which leads to adaptive changes in the cell that aids in resolving the accumulation of the misfolded proteins and its consequences [4, 5]. As such, the IRE1-Hac1p pathway is essential for the maintenance of homeostasis.
In higher metazoans, the UPR is significantly more complex. In addition to a role for an IRE1 homologue that signals through a Hac1p orthologue (X box binding protein 1; XBP1) higher metazoans also use two other signaling pathways from the ER to the nucleus. These include pancreatic ER kinase (PERK) and activation transcription factor 6 (ATF6) [2, 3]. In metazoans, the presence of misfolded proteins in the ER lumen is determined by a complex relationship between glucose regulated protein 78 (grp78 or B cell immunoglobulin protein, BiP) and the three arms of the aforementioned UPR: IRE1, PERK and ATF6. Studies to date support a model wherein grp78 functions both as a chaperone for misfolded proteins in the lumen of the ER as well as a negative regulator of PERK, IRE1 and ATF6 activity [6-8]. In this model, when misfolded proteins exceed the chaperoning capacity of grp78, the misfolded proteins are capable of binding to the three arms of the UPR effector pathway resulting in their activation. In the case of PERK, the sensing of the accumulation of misfolded proteins leads to activation of its kinase function that results in the phosphorylation of elongation initiation factor 2α (eIF2α). eIF2α phosphorylation blocks its ability to function as an initiator of translation leading to a halt in protein synthesis [9]. Some transcripts such as ATF4 evade this inhibition and direct a transcription program as part of the UPR. In the case of ATF4, this includes directing the transcription of CCAAT/enhancer binding homologous protein (CHOP), a factor that promotes programmed cell death [10]. In the case of ATF6, sensing of misfolded proteins in the ER results in its translocation to the Golgi apparatus where it is subject to proteolysis by site one and site two proteases (S1P or S2P) and the proteolytic cleavage of its cytoplasmic tail. The cytoplasmic tail of ATF6 (ATF6-N) is transcriptionally active and directs a UPR-associated, transcriptional program necessary for an appropriate response to ER stress [2, 11].
The final UPR-associated pathway to be discussed is IRE1, which in mammals has two isoforms. These are the ubiquitously expressed IRE1α and IRE1β, which is expressed by the intestinal epithelium and arguably consistent with the special role that the UPR plays in intestinal epithelial homeostasis [6]. Upon activation, IRE1 dimerizes and undergoes autophosphorylation resulting in its gaining competence to function as an unconventional endoribonuclease and kinase for other substrates. The endoribonuclease activity of IRE1α is focused on the removal of a 26 base pair fragment from an intron within the unspliced XBP1 (XBP1u) transcript. This generates a spliced alternative reading frame that encodes a transcriptionally active protein (XBP1s) [12, 13]. XBP1 is the predominant RNA substrate for IRE1α, but other mRNAs may also be cleaved, although with lower efficiency. IRE1β has a broader endoribonuclease activity, which results in the degradation of a large array of transcripts, which as such possesses poorly understood regulatory functions that are relevant to the UPR [14]. IRE1α also exhibits kinase activity upon activation. This kinase activity is directed at apoptosis signal regulating kinase 1 (ASK1) and inhibitor of κ B (IκB) leading to the phosphorylation of Jun related kinase (JNK) and NFκB, respectively. Activation of ASK1 and NFκB through phosphorylation of IκB is dependent upon the ability of IRE1 to recruit TNF-receptor-associated factor 2 (TRAF2) and Iκ kinase (IKK), respectively [15, 16] (Figure 1).
Figure 1. Three arms of the unfolded protein response effector pathway.
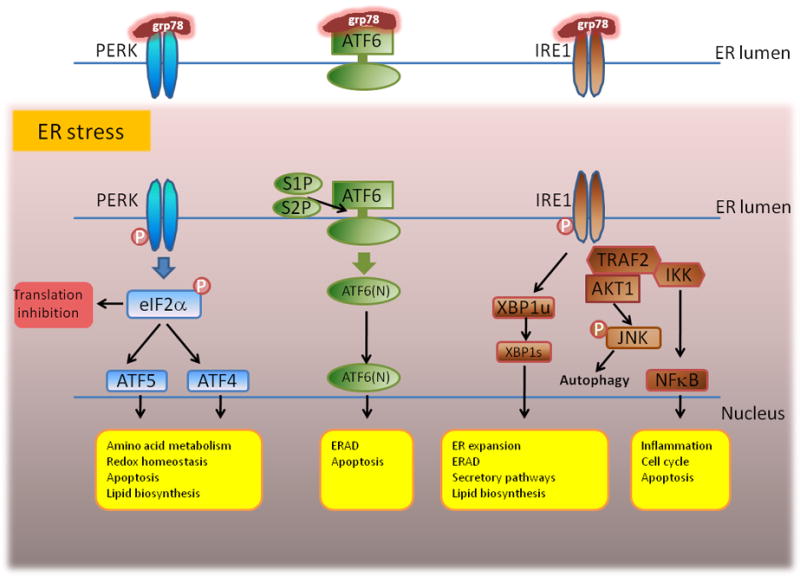
Please refer to text for specific details. Figure was adapted from reference 30.
In their totality, the signaling functions of the UPR assist in promoting the adaptive changes in the cell that aid in resolving the ER stress. These are mediated by a variety of cellular changes that include a temporary halt in protein translation, improvements in the secretory machinery such as through enhanced lipogenesis and expansion of the ER, augmentation of the protein folding machinery, and enhancements in the ER associated degradation apparatus (ERAD) [2, 3]. Studies in lower order metazoans such as Caenorhabiditis elegans have shown that the UPR is a carefully choreographed process with differential activation of the PERK, ATF6 and IRE1-associated pathways in different order and for various durations of time in resolving the ER stress [4]. Given the fundamental importance of the UPR in cellular homeostasis, the numbers of physiologic and pathophysiologic functions which are determined by the UPR are extensive and a subject of a large number of reviews [1-3]. The remainder of this review will focus specifically on the role of the UPR in the homeostasis of intestinal tissues and in particular the role played in the intestinal epithelium.
The dependence of highly secretory cells on the UPR
Highly secretory cells are vulnerable to ER stress and thus dependent on a proper UPR for their homeostasis. In metazoans, this was first shown for plasma cells. Loss of XBP1 function, for example, results in an inability of B cells to differentiate into immunoglobulin secreting plasma cells [17]. This dependency has been shown for a wide variety of other secretory cell types including pancreatic acinar cells, pancreatic β cells dedicated to insulin synthesis, salivary gland epithelial cells, hepatocytes, plasmacytoid dendritic cells, macrophages as well as effector CD8+ T cells [3]. In all cases where this has been examined, mild disruptions of the UPR or inability of the cell to elicit an adequate UPR, such as might occur during environmental stresses (e.g. an infection), lead to diminished secretory functions or even entire loss of secretory function as a consequence of cellular apoptosis due to unabated ER stress. For this reason, it is not surprising that homozygotic deletion of XBP1, for example, is embryonic lethal due to abnormalities in hepatocyte function as well as defects in other secretory populations [18]. These observations in a wide variety of cell types confirm and highlight the general importance of a properly functioning UPR in both the physiologic activities of hematopoeitic and parenchymal, and especially epithelial cells. In the case of hematopoietic cells, a properly functioning UPR is necessary for normal innate and adaptive immune functions. For example, in lower metazoans such as C. elegans, the ability to survive a bacterial infection such as Pseudomonas aeruginosa is dependent upon an intact UPR [19]. Similarly, in mice, the ability of a macrophage to survive a bacterial challenge and participate in antimicrobial defense or a dendritic cell to secrete an appropriate quantity of effector cytokines are dependent on the UPR, These latter observations further highlight the integration between innate immune signaling and the UPR given the dependence and regulation of the UPR by toll-like receptor (TLR) signaling. Thus, during an innate immune response, TLR engagement of the UPR helps to promote the ability of innate immune cells to drive and survive an innate immune response [20]. In a similar manner, the ability of the epithelium to effect and survive its own immunologic responsibilities are also dependent upon an intact UPR [3, 21]. In the next section, we will therefore discuss the UPR and its relationship to intestinal epithelial cell homeostasis.
The UPR and the intestinal epithelium
The columnar, polarized epithelium that lines all mucosal surfaces (e.g. gastric, intestinal, biliary, genitourinary, pancreatic, salivary, pulmonary) are amongst the most highly secretory group of cells in metazoans. This is consistent with the aforementioned observation that deletion and disruption of the UPR leads to a common theme that abnormalities in this type of cells are observed in a variety of different tissues [3]. The epithelium has two major functions in all of these sites: role in regionally specific organ functions (e.g. gas exchange in the lung or digestion in intestines) and in a variety of immunologic functions that are important in managing the environmental interface and especially the relationship with both commensal and pathogenic microbes. In line with this, the intestinal and respiratory epithelium have been increasingly appreciated to play a central role in the pathogenesis of organ specific pathologic conditions such as IBD and asthma, respectively. Over the past two decades, there has been an increasing body of literature concerning the innate and adaptive immunologic capacities of both epithelial surfaces [22-24]. With respect to innate functions these properties largely reflect the importance of the epithelium as the earliest cell type involved in both sensing and regulating the composition of the microbial milieu. This occurs by virtue of the epithelial cell’s ability to exhibit polarized biologic functions along the apical surface (apposed to the lumen) and basolateral surface (apposed to the lamina propria surfaces). The epithelium expresses a wide spectrum of pattern recognition receptors that, in response to their ligation, secrete a large variety of antimicrobial peptides in a vectoral manner into the lumen. Similarly, the epithelium interrogates the microbially derived signals and expresses cell surface molecules or secretes a variety of important biologically activated soluble mediators in a vectoral direction into the basolateral space. These mediators regulate the vast array of immune cells that populate the epithelial and subepithelial compartments within the lamina propria.
Although all lineages of epithelial cells are highly secretory, two cell types stand out in terms of their secretory importance in the intestines. These are the goblet cells which are responsible for the secretion of mucins for generation of an apical barrier that is restrictive to epithelial invasion of commensal and pathogenic microbes as well as factors which both regulate microbial invasion or the activity of the immune system such as resistin like molecule-β (RELM-β) [25]. The second cell type, which is perhaps the most highly secretory cell in the intestinal epithelium that resides deep in the base of the crypt, is the Paneth cell [26]. The Paneth cell is the most long-lived of the stem cell progeny and serves two central functions: delivery of antimicrobial peptides such as the α-defensins (cryptdins) into the lumen [27] and delivery of signals required for proliferation of the stem cell compartment [28]. The central role of these highly secretory cells is revealed by the fact that deletion of goblet cell derived mucins (e.g. Muc2) or abrogation of Paneth cell function leads to a susceptibility to colonic inflammation and an inability to manage both commensalism and pathogenic invasions (e.g. Salmonella sp.), respectively [29-31]. Further, given these properties, it is not surprising that goblet cells and Paneth cells have become woven into the central fabric of our current models regarding the pathogenesis of IBD which states that IBD (and asthma) are dysregulated mucosal immune responses to environmental exposures (e.g. commensal microbiota and allergens, respectively) that occur in a genetically susceptible host which leads to chronic relapsing and remitting cycles of inflammation [3].
These observations underscore the primacy of epithelial cell secretory function in the management of homeostasis and the resolution of inflammatory conditions. Consistent with this, there has been increasing biologic and genetic evidence on the importance of epithelial cell secretory function and its relationship with health and disease [32]. In particular, these studies have also highlighted the important relationships between the UPR, autophagy and innate immune bacterial sensing and IBD[21, 30]. The next section will specifically discuss the UPR and the intestinal epithelium.
The UPR in the maintenance of homeostasis within the intestinal epithelium
Primary (genetic) and secondary (environmental) factors can adversely affect the UPR by either directly inhibiting the function of the UPR or causing the accumulation of unfolded proteins in the ER which exceeds the functional capacity of the UPR within the intestinal epithelium [3]. Moreover, recent studies further suggest that such disruptions may be either a forerunner of intestinal inflammation and/or a perpetuator of intestinal inflammation as is observed in IBD [21, 33, 34].
The identification of biologic pathways linked to abnormalities of the UPR in relation to IBD has come from both a primary interrogation of the genetic basis of human IBD as well as the analysis of a variety of different animal models of intestinal inflammation. Candidate gene and linkage studies suggested association of a region in close proximity to the XBP1 locus to IBD [35-37]. Furthermore, studies with use of deep sequencing have identified rare private variants in XBP1 that are linked to the risk for developing both ulcerative colitis (UC) and Crohn’s disease (CD) [21]. Such rare variants exhibit hypomorphic XBP1 function such that under conditions of ER stress, an ineffective UPR is elicited. Consistent with this, conditional deletion of Xbp1 in intestinal epithelium of mice results in spontaneous enteritis, increased susceptibility to colitis secondary to dextran sodium sulfate (DSS) administration and an inability to manage luminal microbial challenges in association with Paneth cell and to a lesser extent goblet cell dysfunction [21]. In homozygously deleted XBP1-deficient mice, there is near total loss of Paneth cells due to an inability to survive the unabated ER stress and programmed cell death as a consequence of XBP1 deletion as well increased susceptibility of the XBP1-deficient epithelium to inflammatory (e.g. cytokine) and TLR-related signaling which creates a pro-inflammatory milieu [21] (Figure 2). These observations have led to a model where in the context of hypomorphic XBP1 function the epithelium experiences both an inability to manage commensal and pathogenic microbial exposures and at the same time becomes hypersensitive to the consequent microbial signals derived from the dysbiotic commensal community. Thus, it can be envisioned that humans with genetically (or environmentally) determined hypomorphic XBP1 function would be susceptible to Paneth cell and potentially goblet cell dysfunction or loss that may in the proper context initiate and or perpetuate IBD. It is also plausible that hypomorphic XBP1 function will affect other highly secretory cell types, such as dendritic cells and plasma cells, and contribute to the clinical phenotype.
Figure 2. Hypomorphic XBP1 in the intestinal epithelium results in a pro-inflammatory milieu and increased susceptibility to microbial challenges.
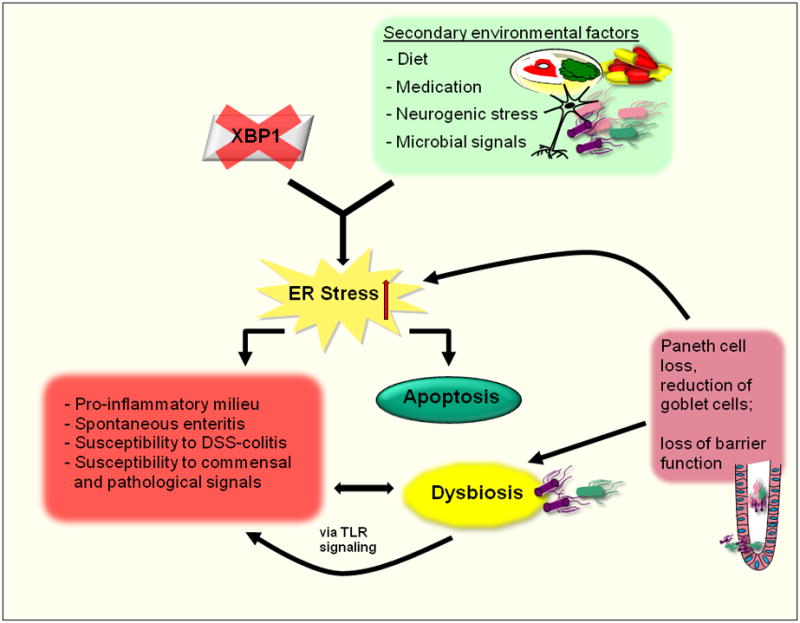
Rare variants of the XBP1 gene result in loss of XBP1 function. In combination with secondary environmental factors, such as diet, medication, neurogenic stress or microbial signals, this generates unabated ER stress, a pro-inflammatory milieu, spontaneous enteritis and increased susceptibility to DSS-induced colitis and commensal and pathogenic pro-inflammatory signals. Subsequent apoptosis of Paneth and goblet cells leads to loss of mucosal barrier function and dysbiosis, further perpetuating inflammation.
The other primary genetic factors that have been linked to a hypomorphic UPR and intestinal epithelial cell function are anterior gradient gene 2 (AGR2) [38-40] and potentially, orosomucoid gene-like 3 (ORMDL3)[41]. AGR2 is an ER-resident protein that functions as a disulfide isomerase and as such regulates protein folding. A candidate gene study has identified AGR2 variants that result in decreased AGR2 levels and which are associated with both CD and UC [40]. Interestingly, AGR2-deficient mice exhibit ER stress in the intestinal epithelium as shown by increased grp78 and XBP1s together with an absence of detectible goblet cell associated mucus and expansion of the Paneth cell compartment. Studies in inducible Agr2-/- mice revealed that Paneth cell hypertrophy is the first observable defect after Agr2 deletion followed by loss of goblet cells with subsequent inflammatory infiltrates that finally result in decrease of enterocyte proliferation and increased apoptosis. These changes occur prior to the development of spontaneous ileitis and colitis that is observed in constitutive Agr2-/- mice and which is characterized by acute and chronic inflammation with multinucleated giant cells and crypt abnormalities [39]. ORMDL3 is an approximately 20-kDa, two-membrane spanning ER-resident protein. ORMDL3 has been shown to prevent the uptake of calcium from the cytosol into the ER resulting in inadequate calcium levels in the ER. Calcium regulation in the ER is essential in the proper folding of nascent proteins. As such, deletion of multiple ORMDL-like related proteins in yeast or silencing ORMDL3 in human model epithelial cells promotes ER stress [42]. Although not yet identified as an ER stress related pathway in intestinal epithelia in vivo, polymorphisms in the ORMDL3 gene are associated with risk for asthma, primary biliary cirrhosis, insulin dependent diabetes mellitus, CD and UC [41, 43-45]. These observations with XBP1, AGR2 and ORMDL3 are consistent with the importance of primary genetic abnormalities in determining the ability of a cell to manage the UPR, which is important in maintaining homeostasis in intestinal and potentially other epithelial cells such that when abnormal the consequent dysfunction lays the foundation for intestinal inflammation. Consistent with this, although not yet defined as genetic risk factors, loss of IRE1β or S1P through genetic deletion or missense mutation, respectively, in mice leads to increased susceptibility to DSS colitis [6, 46].
Secondary alterations in UPR function and their effects on the intestinal epithelium
In addition to primary genetic abnormalities that are associated with hypofunction of the UPR with its attendant consequences for the maintenance of homeostasis, a variety of secondary factors can lead to protein instability and as a consequence predispose to ER stress and loss of homeostasis and susceptibility to inflammation [3]. These include genetic factors that affect protein structure and consequently their stability biogenesis and those environmental factors that impede protein folding or the capability of the UPR to properly function [3].
Examples of primary abnormalities in the encoded structure of proteins that may lead to ER stress include the biology of the HLA-B27 molecule and mucins [33, 47, 48]. The HLA-B27 protein is unique. Like all major histocompatibility complex (MHC) class 1 molecules – the HLA-B27 protein consists of a human leukocyte antigen (HLA) heavy chain that is noncovalently associated with β2 microglobulin (β2m). The HLA-B27 heavy chain displays an intrinsic tendency to misfold during its assembly in the ER [49]. All MHC class 1 molecules are assembled in the ER in a similar manner. After translocation into the ER during biogenesis, proper folding is assisted by interactions with chaperones such as calnexin, calreticulin and Erp57, followed by association with β2m and acquisition of the cognate peptide that is delivered into the groove of the MHC class 1 molecule through the activities of transporter associated protein 1 and 2 which function in the translocation of peptides derived from proteosomal degradation of polypeptides within the cytosol [50]. The HLA-B27/β2m heterodimer is intrinsically unstable [48]. Interestingly, rats that are transgenic for the human HLA-B27 and human β2m transgene develop a spontaneous colitis and spondylitis that is both dependent upon the microbiota and the hematopoietic system [47]. Recent studies have further shown that susceptibility to inflammation in this animal model correlates directly with the level of HLA-B27 expression and the level of ER stress in the hematopoietic cells [49]. Specifically, HLA-B27/human β2m expression in macrophages of the transgenic rats is associated with an excessive UPR together with increased secretion of interleukin (IL)-23 that is linked to the activation of T helper 17 cells [49]. HLA-B27 is interestingly associated with ankylosing spondylitis (AS) [51] and both AS and IBD are genetically associated with polymorphisms in the IL-23 receptor [52]. Together, these observations highlight the potential importance of a dysregulated UPR in the hematopoietic system within intestinal tissues as a predisposing pathophysiologic pathway in the development of intestinal and joint inflammation [47]. Another excellent example that highlights the importance of primary protein structure in relation to the UPR and intestinal inflammation is that related to recent observations on mucin proteins in mouse models. Using a forward genetics approach, two strains of mice (Eeyore and Winnie) have been developed that contain independent missense mutations in Muc2 [33]. Muc2 is a major constituent of mucin derived from goblet cells in the intestine. In both of these mice there is a spontaneous colitis due to Muc2 protein misfolding together with depletion of goblet cell associated mucin. More directly, the epithelial cells of these mice exhibit evidence of increased ER stress as revealed by elevations of grp78 and XBP1s [33]. Although the chronic inflammation could be due to a loss of MUC2 as Muc2-/- mice develop spontaneous colitis, they strongly point towards a model wherein an inability to properly fold these mucin proteins leads to ER stress which in the environmental milieu associated with the intestine leads not only to intestinal epithelial cell dysfunction but also intestinal inflammation. The direct relationship to human IBD is yet to be defined but it should be noted that polymorphisms in the vicinity of mucin related genes have been identified as risk factors for the development of IBD [53].
The observations with HLA-B27 and mucins highlight the importance of protein instability and the ability to manage this through an appropriate UPR as predisposing factors in the development of intestinal inflammation. These studies taken together with the observations associated with primary genetic alterations of the UPR as discussed in relationship to XBP1 and AGR2, for example, also highlight the uniqueness of the intestinal epithelium and the mucosal environment [21, 40]. They specifically suggest that in the context of a wide variety of inflammatory signals provided by either commensal microbiota and/or other host derived mediators that are normally part of the intestinal milieu are inflammatory in the context of a disordered UPR [30].
This latter concept highlights the potential role that environmental factors likely play in these pathways. Specifically, environmental challenges to the UPR may convert a genetic susceptibility into pathologic inflammation [30]. A wide variety of secondary environmental factors have been described as being deleterious to the UPR. These include factors derived from microbes, dietary sources, medications, neurogenic stress as well as inflammation [3]. Specifically, microbes have been shown to express metabolic products (e.g. trierixin production by Streptomyces species) [54] or toxins (e.g. Shigatoxigenic Escherichia coli that produce AB5 cytotoxins) that are capable of directly inhibiting XBP1 or cleaving grp78, respectively [55]. Similarly, inflammatory mediators such as oxygen free radicals or TNF promote ER stress at least in part through challenging the ability of the ER to maintain a milieu that is conducive to proper protein folding [56]. Interestingly, cytokines such as IL-10 have been observed to protect the cell from the deleterious effects of ER stress [34]. It is therefore interesting that in humans who experience ischemia reperfusion (I/R) injury, the Paneth cell is the cell type most susceptible to the hypoxia associated with I/R injury. In this context, the Paneth cell is highly predisposed to apoptosis leading to increased bacterial translocation and potentially sepsis [57]. Together, these observations highlight the important role of environmental factors in either causing or worsening ER stress. They further suggest that an individual’s genetic ability to manage these environmental assaults is critical to determining whether homeostasis or inflammation will prevail.
Summary
In summary, highly secretory cells are extremely dependent upon an unfolded protein response in pursuit of their normal physiologic functions. In the intestinal epithelium, this is highly related to the ability of this cell type to mediate its important innate immunologic functions at the mucosal interface with the microbial milieu. Recent studies have shown that primary and secondary genetic and environmental factors which either effect the ability the UPR to function or the tendency of proteins to misfold, respectively, can lead to a loss of homeostasis and when unabated, to inflammation in the unique environmental context of intestinal tissues.
Acknowledgments
Support is received from NIH DK44319, DK51362, DK53056 and DK088 and the Harvard Digestive Diseases Center (RSB), T32 (MT) and Innsbruck Medical University (MFI 2007-407, AK), the Austrian Science Fund and Ministry of Science P21530 and START Y446 (AK), the European Research Council under the European Community’s Seventh Framework Programme (FP7/2007-2013) / ERC Grant agreement n° 260961 (AK) and the National Institute for Health Research Cambridge Biomedical Research Centre (AK).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Schroder M, Kaufman RJ. The mammalian unfolded protein response. Annu Rev Biochem. 2005;74:739–89. doi: 10.1146/annurev.biochem.73.011303.074134. [DOI] [PubMed] [Google Scholar]
- 2.Todd DJ, Lee AH, Glimcher LH. The endoplasmic reticulum stress response in immunity and autoimmunity. Nat Rev Immunol. 2008;8:663–74. doi: 10.1038/nri2359. [DOI] [PubMed] [Google Scholar]
- 3.Kaser A, Blumberg RS. Endoplasmic reticulum stress and intestinal inflammation. Mucosal Immunol. 2010;3:11–6. doi: 10.1038/mi.2009.122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Mori K. Signalling pathways in the unfolded protein response: development from yeast to mammals. J Biochem. 2009;146:743–50. doi: 10.1093/jb/mvp166. [DOI] [PubMed] [Google Scholar]
- 5.Cox JS, Walter P. A novel mechanism for regulating activity of a transcription factor that controls the unfolded protein response. Cell. 1996;87:391–404. doi: 10.1016/s0092-8674(00)81360-4. [DOI] [PubMed] [Google Scholar]
- 6.Bertolotti A, Wang X, Novoa I, Jungreis R, Schlessinger K, Cho JH, West AB, Ron D. Increased sensitivity to dextran sodium sulfate colitis in IRE1beta-deficient mice. J Clin Invest. 2001;107:585–93. doi: 10.1172/JCI11476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Kimata Y, Kimata YI, Shimizu Y, Abe H, Farcasanu IC, Takeuchi M, Rose MD, Kohno K. Genetic evidence for a role of BiP/Kar2 that regulates Ire1 in response to accumulation of unfolded proteins. Mol Biol Cell. 2003;14:2559–69. doi: 10.1091/mbc.E02-11-0708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Shen J, Chen X, Hendershot L, Prywes R. ER stress regulation of ATF6 localization by dissociation of BiP/GRP78 binding and unmasking of Golgi localization signals. Dev Cell. 2002;3:99–111. doi: 10.1016/s1534-5807(02)00203-4. [DOI] [PubMed] [Google Scholar]
- 9.Harding HP, Novoa I, Zhang Y, Zeng H, Wek R, Schapira M, Ron D. Regulated translation initiation controls stress-induced gene expression in mammalian cells. Mol Cell. 2000;6:1099–108. doi: 10.1016/s1097-2765(00)00108-8. [DOI] [PubMed] [Google Scholar]
- 10.Vattem KM, Wek RC. Reinitiation involving upstream ORFs regulates ATF4 mRNA translation in mammalian cells. Proc Natl Acad Sci U S A. 2004;101:11269–74. doi: 10.1073/pnas.0400541101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Haze K, Yoshida H, Yanagi H, Yura T, Mori K. Mammalian transcription factor ATF6 is synthesized as a transmembrane protein and activated by proteolysis in response to endoplasmic reticulum stress. Mol Biol Cell. 1999;10:3787–99. doi: 10.1091/mbc.10.11.3787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Calfon M, Zeng H, Urano F, Till JH, Hubbard SR, Harding HP, Clark SG, Ron D. IRE1 couples endoplasmic reticulum load to secretory capacity by processing the XBP-1 mRNA. Nature. 2002;415:92–6. doi: 10.1038/415092a. [DOI] [PubMed] [Google Scholar]
- 13.Yoshida H, Matsui T, Yamamoto A, Okada T, Mori K. XBP1 mRNA is induced by ATF6 and spliced by IRE1 in response to ER stress to produce a highly active transcription factor. Cell. 2001;107:881–91. doi: 10.1016/s0092-8674(01)00611-0. [DOI] [PubMed] [Google Scholar]
- 14.Nakamura D, Tsuru A, Ikegami K, Imagawa Y, Fujimoto N, Kohno K. Mammalian ER stress sensor IRE1beta specifically down-regulates the synthesis of secretory pathway proteins. FEBS Lett. 2011;585:133–8. doi: 10.1016/j.febslet.2010.12.002. [DOI] [PubMed] [Google Scholar]
- 15.Nishitoh H, Matsuzawa A, Tobiume K, Saegusa K, Takeda K, Inoue K, Hori S, Kakizuka A, Ichijo H. ASK1 is essential for endoplasmic reticulum stress-induced neuronal cell death triggered by expanded polyglutamine repeats. Genes Dev. 2002;16:1345–55. doi: 10.1101/gad.992302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Urano F, Wang X, Bertolotti A, Zhang Y, Chung P, Harding HP, Ron D. Coupling of stress in the ER to activation of JNK protein kinases by transmembrane protein kinase IRE1. Science. 2000;287:664–6. doi: 10.1126/science.287.5453.664. [DOI] [PubMed] [Google Scholar]
- 17.Iwakoshi NN, Lee AH, Vallabhajosyula P, Otipoby KL, Rajewsky K, Glimcher LH. Plasma cell differentiation and the unfolded protein response intersect at the transcription factor XBP-1. Nat Immunol. 2003;4:321–9. doi: 10.1038/ni907. [DOI] [PubMed] [Google Scholar]
- 18.Lee AH, Chu GC, Iwakoshi NN, Glimcher LH. XBP-1 is required for biogenesis of cellular secretory machinery of exocrine glands. Embo J. 2005;24:4368–80. doi: 10.1038/sj.emboj.7600903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Richardson CE, Kooistra T, Kim DH. An essential role for XBP-1 in host protection against immune activation in C. elegans. Nature. 2010;463:1092–5. doi: 10.1038/nature08762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Martinon F, Chen X, Lee AH, Glimcher LH. TLR activation of the transcription factor XBP1 regulates innate immune responses in macrophages. Nat Immunol. 2010;11:411–8. doi: 10.1038/ni.1857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kaser A, Lee AH, Franke A, Glickman JN, Zeissig S, Tilg H, Nieuwenhuis EE, Higgins DE, Schreiber S, Glimcher LH, Blumberg RS. XBP1 links ER stress to intestinal inflammation and confers genetic risk for human inflammatory bowel disease. Cell. 2008;134:743–56. doi: 10.1016/j.cell.2008.07.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Henderson P, van Limbergen JE, Schwarze J, Wilson DC. Function of the intestinal epithelium and its dysregulation in inflammatory bowel disease. Inflamm Bowel Dis. 2011;17:382–95. doi: 10.1002/ibd.21379. [DOI] [PubMed] [Google Scholar]
- 23.Santaolalla R, Fukata M, Abreu MT. Innate immunity in the small intestine. Curr Opin Gastroenterol. 2011;27:125–31. doi: 10.1097/MOG.0b013e3283438dea. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Tam A, Wadsworth S, Dorscheid D, Man SP, Sin DD. The airway epithelium: more than just a structural barrier. Ther Adv Respir Dis. 2011 doi: 10.1177/1753465810396539. [DOI] [PubMed] [Google Scholar]
- 25.Artis D, Wang ML, Keilbaugh SA, He W, Brenes M, Swain GP, Knight PA, Donaldson DD, Lazar MA, Miller HR, Schad GA, Scott P, Wu GD. RELMbeta/FIZZ2 is a goblet cell-specific immune-effector molecule in the gastrointestinal tract. Proc Natl Acad Sci U S A. 2004;101:13596–600. doi: 10.1073/pnas.0404034101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Selsted ME, Ouellette AJ. Mammalian defensins in the antimicrobial immune response. Nat Immunol. 2005;6:551–7. doi: 10.1038/ni1206. [DOI] [PubMed] [Google Scholar]
- 27.Ayabe T, Satchell DP, Wilson CL, Parks WC, Selsted ME, Ouellette AJ. Secretion of microbicidal alpha-defensins by intestinal Paneth cells in response to bacteria. Nat Immunol. 2000;1:113–8. doi: 10.1038/77783. [DOI] [PubMed] [Google Scholar]
- 28.Sato T, van Es JH, Snippert HJ, Stange DE, Vries RG, van den Born M, Barker N, Shroyer NF, van de Wetering M, Clevers H. Paneth cells constitute the niche for Lgr5 stem cells in intestinal crypts. Nature. 2011;469:415–8. doi: 10.1038/nature09637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Bergstrom KS, Kissoon-Singh V, Gibson DL, Ma C, Montero M, Sham HP, Ryz N, Huang T, Velcich A, Finlay BB, Chadee K, Vallance BA. Muc2 protects against lethal infectious colitis by disassociating pathogenic and commensal bacteria from the colonic mucosa. PLoS Pathog. 2010;6:e1000902. doi: 10.1371/journal.ppat.1000902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kaser A, Blumberg RS. Endoplasmic reticulum stress in the intestinal epithelium and inflammatory bowel disease. Semin Immunol. 2009;21:156–63. doi: 10.1016/j.smim.2009.01.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Van der Sluis M, De Koning BA, De Bruijn AC, Velcich A, Meijerink JP, Van Goudoever JB, Buller HA, Dekker J, Van Seuningen I, Renes IB, Einerhand AW. Muc2-deficient mice spontaneously develop colitis, indicating that MUC2 is critical for colonic protection. Gastroenterology. 2006;131:117–29. doi: 10.1053/j.gastro.2006.04.020. [DOI] [PubMed] [Google Scholar]
- 32.Round JL, Mazmanian SK. The gut microbiota shapes intestinal immune responses during health and disease. Nat Rev Immunol. 2009;9:313–23. doi: 10.1038/nri2515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Heazlewood CK, Cook MC, Eri R, Price GR, Tauro SB, Taupin D, Thornton DJ, Png CW, Crockford TL, Cornall RJ, Adams R, Kato M, Nelms KA, Hong NA, Florin TH, Goodnow CC, McGuckin MA. Aberrant mucin assembly in mice causes endoplasmic reticulum stress and spontaneous inflammation resembling ulcerative colitis. PLoS Med. 2008;5:e54. doi: 10.1371/journal.pmed.0050054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Shkoda A, Ruiz PA, Daniel H, Kim SC, Rogler G, Sartor RB, Haller D. Interleukin-10 blocked endoplasmic reticulum stress in intestinal epithelial cells: impact on chronic inflammation. Gastroenterology. 2007;132:190–207. doi: 10.1053/j.gastro.2006.10.030. [DOI] [PubMed] [Google Scholar]
- 35.Barmada MM, Brant SR, Nicolae DL, Achkar JP, Panhuysen CI, Bayless TM, Cho JH, Duerr RH. A genome scan in 260 inflammatory bowel disease-affected relative pairs. Inflamm Bowel Dis. 2004;10:513–20. doi: 10.1097/00054725-200409000-00004. [DOI] [PubMed] [Google Scholar]
- 36.Hampe J, Franke A, Rosenstiel P, Till A, Teuber M, Huse K, Albrecht M, Mayr G, De La Vega FM, Briggs J, Gunther S, Prescott NJ, Onnie CM, Hasler R, Sipos B, Folsch UR, Lengauer T, Platzer M, Mathew CG, Krawczak M, Schreiber S. A genome-wide association scan of nonsynonymous SNPs identifies a susceptibility variant for Crohn disease in ATG16L1. Nat Genet. 2007;39:207–11. doi: 10.1038/ng1954. [DOI] [PubMed] [Google Scholar]
- 37.Vermeire S, Rutgeerts P, Van Steen K, Joossens S, Claessens G, Pierik M, Peeters M, Vlietinck R. Genome wide scan in a Flemish inflammatory bowel disease population: support for the IBD4 locus, population heterogeneity, and epistasis. Gut. 2004;53:980–6. doi: 10.1136/gut.2003.034033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Park SW, Zhen G, Verhaeghe C, Nakagami Y, Nguyenvu LT, Barczak AJ, Killeen N, Erle DJ. The protein disulfide isomerase AGR2 is essential for production of intestinal mucus. Proc Natl Acad Sci U S A. 2009;106:6950–5. doi: 10.1073/pnas.0808722106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Zhao F, Edwards R, Dizon D, Afrasiabi K, Mastroianni JR, Geyfman M, Ouellette AJ, Andersen B, Lipkin SM. Disruption of Paneth and goblet cell homeostasis and increased endoplasmic reticulum stress in Agr2-/- mice. Dev Biol. 2010;338:270–9. doi: 10.1016/j.ydbio.2009.12.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Zheng W, Rosenstiel P, Huse K, Sina C, Valentonyte R, Mah N, Zeitlmann L, Grosse J, Ruf N, Nurnberg P, Costello CM, Onnie C, Mathew C, Platzer M, Schreiber S, Hampe J. Evaluation of AGR2 and AGR3 as candidate genes for inflammatory bowel disease. Genes Immun. 2006;7:11–8. doi: 10.1038/sj.gene.6364263. [DOI] [PubMed] [Google Scholar]
- 41.McGovern DP, Gardet A, Torkvist L, Goyette P, Essers J, Taylor KD, Neale BM, Ong RT, Lagace C, Li C, Green T, Stevens CR, Beauchamp C, Fleshner PR, Carlson M, D’Amato M, Halfvarson J, Hibberd ML, Lordal M, Padyukov L, Andriulli A, Colombo E, Latiano A, Palmieri O, Bernard EJ, Deslandres C, Hommes DW, de Jong DJ, Stokkers PC, Weersma RK, Sharma Y, Silverberg MS, Cho JH, Wu J, Roeder K, Brant SR, Schumm LP, Duerr RH, Dubinsky MC, Glazer NL, Haritunians T, Ippoliti A, Melmed GY, Siscovick DS, Vasiliauskas EA, Targan SR, Annese V, Wijmenga C, Pettersson S, Rotter JI, Xavier RJ, Daly MJ, Rioux JD, Seielstad M. Genome-wide association identifies multiple ulcerative colitis susceptibility loci. Nat Genet. 2010;42:332–7. doi: 10.1038/ng.549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Cantero-Recasens G, Fandos C, Rubio-Moscardo F, Valverde MA, Vicente R. The asthma-associated ORMDL3 gene product regulates endoplasmic reticulum-mediated calcium signaling and cellular stress. Hum Mol Genet. 2010;19:111–21. doi: 10.1093/hmg/ddp471. [DOI] [PubMed] [Google Scholar]
- 43.Moffatt MF, Kabesch M, Liang L, Dixon AL, Strachan D, Heath S, Depner M, von Berg A, Bufe A, Rietschel E, Heinzmann A, Simma B, Frischer T, Willis-Owen SA, Wong KC, Illig T, Vogelberg C, Weiland SK, von Mutius E, Abecasis GR, Farrall M, Gut IG, Lathrop GM, Cookson WO. Genetic variants regulating ORMDL3 expression contribute to the risk of childhood asthma. Nature. 2007;448:470–3. doi: 10.1038/nature06014. [DOI] [PubMed] [Google Scholar]
- 44.Liu X, Invernizzi P, Lu Y, Kosoy R, Lu Y, Bianchi I, Podda M, Xu C, Xie G, Macciardi F, Selmi C, Lupoli S, Shigeta R, Ransom M, Lleo A, Lee AT, Mason AL, Myers RP, Peltekian KM, Ghent CN, Bernuzzi F, Zuin M, Rosina F, Borghesio E, Floreani A, Lazzari R, Niro G, Andriulli A, Muratori L, Muratori P, Almasio PL, Andreone P, Margotti M, Brunetto M, Coco B, Alvaro D, Bragazzi MC, Marra F, Pisano A, Rigamonti C, Colombo M, Marzioni M, Benedetti A, Fabris L, Strazzabosco M, Portincasa P, Palmieri VO, Tiribelli C, Croce L, Bruno S, Rossi S, Vinci M, Prisco C, Mattalia A, Toniutto P, Picciotto A, Galli A, Ferrari C, Colombo S, Casella G, Morini L, Caporaso N, Colli A, Spinzi G, Montanari R, Gregersen PK, Heathcote EJ, Hirschfield GM, Siminovitch KA, Amos CI, Gershwin ME, Seldin MF. Genome-wide meta-analyses identify three loci associated with primary biliary cirrhosis. Nat Genet. 2010;42:658–60. doi: 10.1038/ng.627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Barrett JC, Clayton DG, Concannon P, Akolkar B, Cooper JD, Erlich HA, Julier C, Morahan G, Nerup J, Nierras C, Plagnol V, Pociot F, Schuilenburg H, Smyth DJ, Stevens H, Todd JA, Walker NM, Rich SS. Genome-wide association study and meta-analysis find that over 40 loci affect risk of type 1 diabetes. Nat Genet. 2009;41:703–7. doi: 10.1038/ng.381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Brandl K, Rutschmann S, Li X, Du X, Xiao N, Schnabl B, Brenner DA, Beutler B. Enhanced sensitivity to DSS colitis caused by a hypomorphic Mbtps1 mutation disrupting the ATF6-driven unfolded protein response. Proc Natl Acad Sci U S A. 2009;106:3300–5. doi: 10.1073/pnas.0813036106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Hammer RE, Maika SD, Richardson JA, Tang JP, Taurog JD. Spontaneous inflammatory disease in transgenic rats expressing HLA-B27 and human beta 2m: an animal model of HLA-B27-associated human disorders. Cell. 1990;63:1099–112. doi: 10.1016/0092-8674(90)90512-d. [DOI] [PubMed] [Google Scholar]
- 48.Turner MJ, Sowders DP, DeLay ML, Mohapatra R, Bai S, Smith JA, Brandewie JR, Taurog JD, Colbert RA. HLA-B27 misfolding in transgenic rats is associated with activation of the unfolded protein response. J Immunol. 2005;175:2438–48. doi: 10.4049/jimmunol.175.4.2438. [DOI] [PubMed] [Google Scholar]
- 49.DeLay ML, Turner MJ, Klenk EI, Smith JA, Sowders DP, Colbert RA. HLA-B27 misfolding and the unfolded protein response augment interleukin-23 production and are associated with Th17 activation in transgenic rats. Arthritis Rheum. 2009;60:2633–43. doi: 10.1002/art.24763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Paulsson K, Wang P. Chaperones and folding of MHC class I molecules in the endoplasmic reticulum. Biochim Biophys Acta. 2003;1641:1–12. doi: 10.1016/s0167-4889(03)00048-x. [DOI] [PubMed] [Google Scholar]
- 51.Taurog JD, Maika SD, Satumtira N, Dorris ML, McLean IL, Yanagisawa H, Sayad A, Stagg AJ, Fox GM, Le O’Brien A, Rehman M, Zhou M, Weiner AL, Splawski JB, Richardson JA, Hammer RE. Inflammatory disease in HLA-B27 transgenic rats. Immunol Rev. 1999;169:209–23. doi: 10.1111/j.1600-065x.1999.tb01317.x. [DOI] [PubMed] [Google Scholar]
- 52.Rahman P, Inman RD, Gladman DD, Reeve JP, Peddle L, Maksymowych WP. Association of interleukin-23 receptor variants with ankylosing spondylitis. Arthritis Rheum. 2008;58:1020–5. doi: 10.1002/art.23389. [DOI] [PubMed] [Google Scholar]
- 53.Moehle C, Ackermann N, Langmann T, Aslanidis C, Kel A, Kel-Margoulis O, Schmitz-Madry A, Zahn A, Stremmel W, Schmitz G. Aberrant intestinal expression and allelic variants of mucin genes associated with inflammatory bowel disease. J Mol Med. 2006;84:1055–66. doi: 10.1007/s00109-006-0100-2. [DOI] [PubMed] [Google Scholar]
- 54.Tashiro E, Hironiwa N, Kitagawa M, Futamura Y, Suzuki S, Nishio M, Imoto M. Trierixin, a novel Inhibitor of ER stress-induced XBP1 activation from Streptomyces sp. 1. Taxonomy, fermentation, isolation and biological activities. J Antibiot (Tokyo) 2007;60:547–53. doi: 10.1038/ja.2007.69. [DOI] [PubMed] [Google Scholar]
- 55.Paton AW, Beddoe T, Thorpe CM, Whisstock JC, Wilce MC, Rossjohn J, Talbot UM, Paton JC. AB5 subtilase cytotoxin inactivates the endoplasmic reticulum chaperone BiP. Nature. 2006;443:548–52. doi: 10.1038/nature05124. [DOI] [PubMed] [Google Scholar]
- 56.Xue X, Piao JH, Nakajima A, Sakon-Komazawa S, Kojima Y, Mori K, Yagita H, Okumura K, Harding H, Nakano H. Tumor necrosis factor alpha (TNFalpha) induces the unfolded protein response (UPR) in a reactive oxygen species (ROS)-dependent fashion, and the UPR counteracts ROS accumulation by TNFalpha. J Biol Chem. 2005;280:33917–25. doi: 10.1074/jbc.M505818200. [DOI] [PubMed] [Google Scholar]
- 57.Grootjans J, Hodin CM, de Haan JJ, Derikx JP, Rouschop KM, Verheyen FK, van Dam RM, Dejong CH, Buurman WA, Lenaerts K. Level of activation of the unfolded protein response correlates with Paneth cell apoptosis in human small intestine exposed to ischemia/reperfusion. Gastroenterology. 2011;140:529–539. e3. doi: 10.1053/j.gastro.2010.10.040. [DOI] [PubMed] [Google Scholar]