

Abstract
The development of small-molecule inhibitors for perturbing enzyme function requires assays to confirm that the inhibitors interact with their enzymatic targets in vivo. Determining target engagement in vivo can be particularly challenging for poorly characterized enzymes that lack known biomarkers (e.g., endogenous substrates and products) to report on their inhibition. Here, we describe a competitive activity-based protein profiling (ABPP) method for measuring the binding of reversible inhibitors to enzymes in animal models. Key to the success of this approach is the use of activity-based probes that show tempered rates of reactivity with enzymes, such that competition for target engagement with reversible inhibitors can be measured in vivo. We apply the competitive ABPP strategy to evaluate a newly described class of piperazine amide reversible inhibitors for the serine hydrolases LYPAL1 and LYPLA2, two enzymes for which selective, in vivo-active inhibitors are lacking. Competitive ABPP identified individual piperazine amides that selectively inhibit LYPLA1 or LYPLA2 in mice. In summary, competitive ABPP adapted to operate with moderately reactive probes can assess the target engagement of reversible inhibitors in animal models to facilitate the discovery of small-molecule probes for characterizing enzyme function in vivo.
Enzyme inhibitors are valuable chemical probes to perturb biochemical pathways in living systems and, where appropriate, can be developed into new medicines to treat human disease. Most enzymes in the human proteome, however, still lack selective inhibitors1. While advances in high-throughput screening2 combined with chemoproteomic platforms1 have begun to accelerate the development of enzyme inhibitors with excellent potency and selectivity, it is often challenging to determine whether these inhibitors interact with their targets in living systems, especially for poorly characterized enzymes that lack endogenous substrate/product biomarkers.
We, and others have previously shown that activity-based protein profiling (ABPP), a chemoproteomic method that uses active site-directed, covalent probes to evaluate enzyme function in native biological systems,3 can determine target engagement for enzyme inhibitors in cell and animal models.4–7 But, so far, this approach has been limited to assessing irreversible inhibitors for multiple reasons. First, many activity-based probes show poor bioavailablity in vivo, which restricts their utility to the ex vivo analysis of tissue proteomes from inhibitor-treated animals, an assay protocol that involves considerable sample processing and dilution that can compromise the detection of reversible inhibitor-enzyme interactions. Second, even for activity-based probes that show suitable bioavailability, managing their rate of reactivity in vivo is difficult, which, if not properly controlled, can result in the probes “outcompeting” reversible inhibitors and failing to record their target interactions. Here, we sought to address these challenges by creating kinetically tuned activity-based probes that enable direct competitive profiling of reversible inhibitors in vivo. We applied this strategy to assess newly described inhibitors for the serine hydrolases lysophospholipase 1 (LYPLA1) and lysophospholipase 2 (LYPLA2), enzymes that, to date, have lacked selective and in vivo-active inhibitors.
LYPLA1 and LYPLA2 were initially characterized as phospholipases based on in vitro substrate assays.8–9 LYPLA1 has since been found to also act as protein palmitoyl thioesterase (PPT) that removes palmitate modifications from cysteine residues on proteins, such as the Ras GTPases.10 LYPLA2 shares 65% sequence identity with LYPLA1, but its role as a PPT remains unknown. While considerable biochemical and cellular data support a role for LYPLA1 as a PPT,11–13 little is known about the functions of LYPLA1 and LYPLA2 in vivo. Several inhibitors of LYPLA1 have been described,11,14 but none of these agents have been demonstrated to selectively inhibit LYPLA1 over LYPLA2, nor have they been shown to inhibit LYPLA1 in animal models. This latter goal is further challenged by a lack of robust substrate/product biomarkers that can be used to confirm LYPLA1/2 inhibition in vivo.
To identify new classes of LYPLA1 and LYPLA2 inhibitors we first established enzyme assays compatible with high-throughput screening. As serine hydrolases, LYPLA1 and LYPLA2 react with fluorophosphonate (FP) activity-based probes and can therefore be assayed by a recently described, high-throughput screening (HTS)-compatible competitive ABPP platform that measures the reactivity of fluorophore-conjugated FP probes with serine hydrolases by fluorescence polarization (fluopol).15 In fluopol-ABPP, small-molecules compete with FP-probes for binding to serine hydrolases and are recorded as hits by their ability to reduce fluopol signal. We expressed and purified human LYPLA1 and LYPLA2 enzymes and confirmed that the wild-type, but not the catalytically dead serine mutants (S119A and S112A, respectively) reacted with a rhodamine-conjugated FP probe (FP-Rh) to generate time-dependent increases in fluopol signal (Figure 1). In collaboration with the Molecular Libraries Production Centers Network (MLCPN), we then screened 315,004 compounds for LYPLA1 or LYPLA2 inhibition. Compounds were classified as potential hits if they produced a >30% reduction of fluopol signal (Figure S1). Following a confirmation screen, 331 and 790 compounds were found to inhibit LYPLA1 and LYPLA2, respectively. We then manually filtered these compounds for those with the highest inhibition and removed non-selective compounds identified as hits in previous fluopol-ABPP screens of other serine hydrolases (PME1 and RBBP9).
Figure 1.
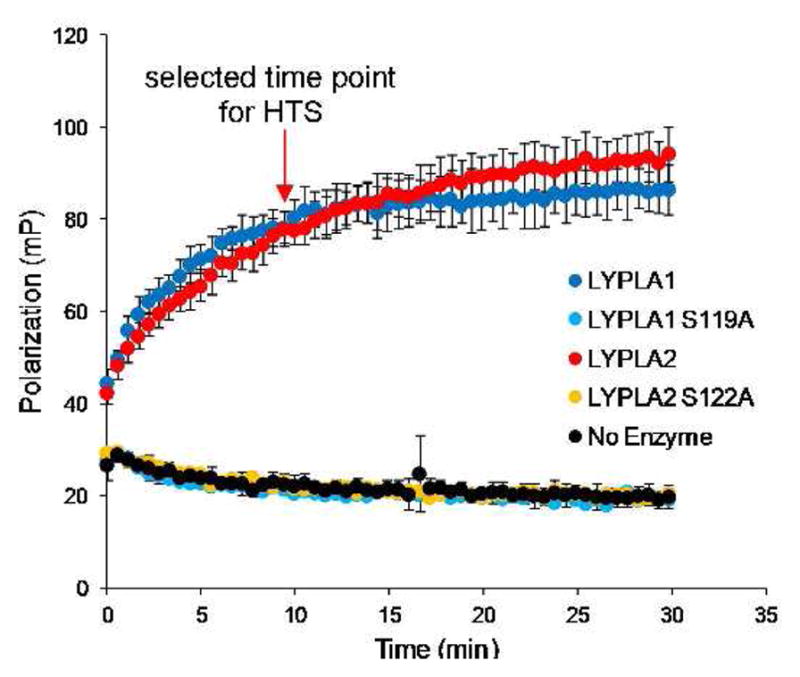
A fluopol-ABPP assay to screen for LYPLA1 and LYPLA2 inhibitors. Time course of FP-Rh labeling for LYPLA1 and LYPLA2 as determined by fluopol-ABPP. No fluopol increase was observed in the absence of enzymes or with the S119A LYPLA1 and S122A LYPLA2 catalytic mutants.
The remaining 91 LYPLA1, 61 LYPLA2, and 95 dual LYPLA1/LYPLA2 inhibitors were analyzed by gel-based ABPP in HEK293T cell proteomes, which express high endogenous levels of LYPLA1 and LYPLA2. We established assay parameters compatible with assessing both reversible and irreversible inhibitors by using a pegylated FP-Rh probe (FP-peg-Rh), which reacts more slowly with LYPLA1 and LYPLA2 compared to the FP-Rh probe to produce only partial enzyme labeling under the reaction conditions employed for inhibitor evaluation (Figure S2). From this analysis, a diverse set of piperazine amides emerged as attractive inhibitors for LYPLA1 and LYPLA2. The p-methoxyphenyl piperazine amide 1, for instance, completely inhibited LYPLA2 when tested at 1 uM, while showing no inhibition of LYPLA1 or other serine hydrolases detected in the HEK293T proteome (Figure 2A–C). A complementary profile was observed for the 2-furoyl piperazine amide 21, which produced near-complete inhibition of LYPLA1 without showing any cross-reactivity with LYPLA2 or other serine hydrolases (Figure 2A–C).
Figure 2.

Identification of acyl piperidines 1 and 21 as potent and selective reversible inhibitors of LYPLA2 and LYPLA1, respectively. (A) Gel-based competitive ABPP of HTS hits 1 and 21 and structurally related compounds 2-13 and 22-33 tested in a HEK293T proteome. (B) Structures of profiled compounds. (C) Concentration-dependent blockade of FP-peg-Rh labeling of LYPLA2 and LYPLA1 by 1 and 21, respectively, in a HEK 293T proteome. Fluorescent gels shown in grayscale are representative of at least three independent experiments. (D) Competitive ABPP analysis of gel filtration experiments to determine the reversibility of LYPLA2 and LYPLA1 inhibition by 1 and 21; also shown is the profile for the irreversible inhibitor 1,2,3-triazole urea AA26-9. (E) Kinetic values for inhibition of recombinant human LYPLA2 and LYPLA1 by 1 and 21, respectively, determined using a fluorescent substrate assay. Data represent means ± s.d. (n = 4).
To better understand the structure-activity relationship (SAR) of 1 and 21, we purchased and tested the related compounds 2-13 and 22-33 (Figure 2A and 2B). Compounds 7, 9, and 10, all bearing para-substituted phenyl groups, inhibited LYPLA2 by 70–90%, while ortho-, meta- or unsubstituted aryl piperazines did not exhibit inhibitory activity at 1 μM. Both the 2-furoyl piperazine and 2,5-substituted anilide groups of 21 were required for maximal and selective inhibition of LYPLA1, as revealed by the profiles of disubstituted anilides 28-30, which showed moderate cross-inhibition of LYPLA2 (50–70 % at the concentration of 1 μM), and the structurally related amide 26, which did not inhibit LYPLA1 or LYPLA2. We resynthesized 1 and 21 as described in the Supplementary Methods and confirmed their structural assignment and inhibitory activity by 1H-NMR and competitive ABPP assays, respectively (Figure S3).
Neither 1 nor 21 possess any obvious electrophilic groups for reaction with LYPLA1 or LYPLA2, which led us to conclude that they were likely acting as reversible inhibitors. We confirmed the reversibility of enzyme inhibition for 1 and 21 by gel filtration experiments, which showed complete recovery of FP-peg-Rh labeling of LYPLA1 and LYPLA2 after two consecutive gel filtrations (Figure 2D). In contrast, no recovery of FP-peg-Rh labeling was observed for the 1,2,3-triazole urea irreversible inhibitor AA26-9,7 even after three gel filtration events (Figure 2D).16 1 and 21 showed good potency for LYPLA2 and LYPLA1 with IC50 values of 144 and 210 nM, respectively, derived from gel-based competitive ABPP assays (Figure 2C, Figure S5) and Ki values of 230 and 300 nM, respectively, derived from a fluorogenic substrate assay using purified enzymes. (Figure 2E, Figure S6). We further verified the activity and selectivity of 1 and 21 using the LC-MS platform ABPP-SILAC4,7, which revealed >95% inhibition of LYPLA2 and LYPLA1, respectively, without any changes in the activity of ~25 other serine hydrolases detected in the mouse BW5147 T-cell hybridoma proteome (Figure S7 and Table S1).
We next asked whether 1 and 21 could also inhibit LYPLA2 and LYPLA1 in living cells. As mentioned previously, it is not straightforward to answer this question for reversible inhibitors using standard competitive ABPP protocols, which involve incubation of living cells with an inhibitor followed by ex vivo treatment of cell lysates with an activity-based probe. While, in principle, we could perform competitive ABPP in situ using alkynylated FP probes,17 we were concerned that the high rates of reactivity displayed by FP probes with most serine hydrolases would complicate the analysis of reversible inhibitors. Consistent with this premise, we found that LYPLA1, LYPLA2, and several other serine hydrolases were rapidly inactivated (within 5 min) by an FP-alkyne probe 34 (Figure 3A, Figure S8). We hypothesized that implementing an alternative probe with more tempered reactivity could facilitate competitive ABPP in situ under kinetically controlled conditions. We tested probe 35, which is based on a recently discovered triazole urea scaffold for serine hydrolase inhibitors,7 and found that it reacted much more slowly than FP-alkyne with LYPLA1, LYPLA2, and most serine hydrolases (Figure 3A, Figure S8). We next incubated HEK293T and mouse T cells with 1 and 21 (5 μM) or DMSO for 3 hours followed by a 1 hr treatment with probe 35 (50 μM). Cells were harvested, lysed, and probe-labeled enzymes visualized by click chemistry18,19 conjugation with a Rh-azide reporter tag.20 Gel-based ABPP revealed the selective and near-complete (>95%) inhibition of LYPLA2 and LYPLA1 in cells treated with 1 and 21, respectively (Figure 3B, Figure S9). The in situ activity of both inhibitors was further confirmed by ABPP-SILAC, which revealed selective inhibition of LYPLA2 and LYPLA1 by 1 and 21, respectively, across the 15+ serine hydrolases detected in this analysis (Figure 3C, Table S1, and Figure S10).
Figure 3.

Competitive ABPP of 1 and 21 in living cells. (A) Time-dependent competition of FP-Rh labeling by the click-able probes FP-alkyne 34 and triazole urea 35 in a mouse brain membrane proteome. (B) Gel-based competitive ABPP of HEK293T cells treated with 1 and 21 (5 μM, 3 h) following by probe 35 (50 μM, 1 h) to measure in situ target engagement. (C) ABPP-SILAC analysis of serine hydrolase activities from inhibitor-treated HEK 293T cells. Bars represent means ± s.d. of light/heavy ratios for the multiple peptides observed for each enzyme; data are derived from two independent biological replicates.
We finally asked whether competitive ABPP with probe 35 could also be used to determine the inhibition of LYPLA1 and LYPLA2 in vivo. Mice were administered 1 and 21 (each at 50 mg/kg in PEG300, i.p.) or vehicle and, after 3 hr, treated with probe 35 (100 mg/kg in PEG300, i.p.) for 1 hr. Mice were then sacrificed and their tissues removed, processed, reacted with Rh-azide, and analyzed by gel-based ABPP. The gel profiles confirmed that 1 and 21 selectively and near-completely (>90%) inhibited LYPLA2 and LYPLA1, respectively, in lung, heart, and kidney (Figure 4). In contrast, only partial blockade (~50%) was observed for LYPLA1 and LYPLA2 in brain, suggesting limited CNS penetration for the inhibitors. No detectable inhibition of LYPLA1 or LYPLA2 was observed in liver, perhaps reflecting rapid metabolism of 1 and 21 in this organ. These data indicate that competitive ABPP can quantitatively assess target engagement in vivo across numerous organs in parallel to reveal cases of tissue-restricted inhibition.
Figure 4.
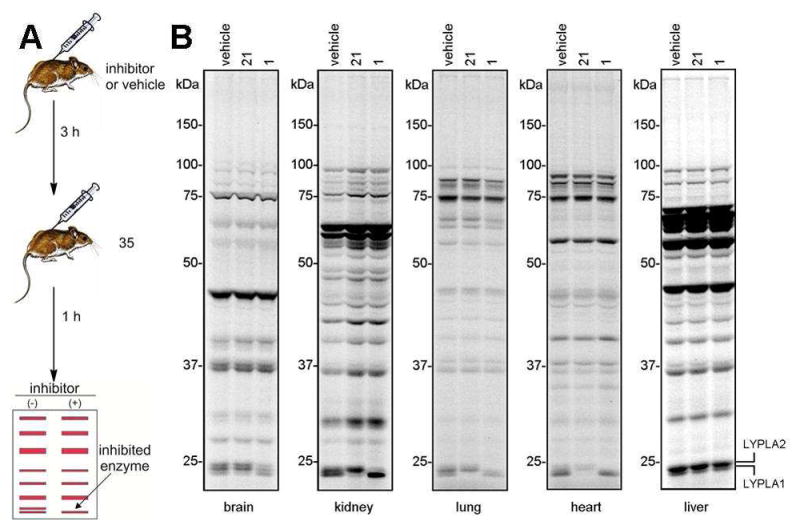
Evaluating target engagement for 1 and 21 in vivo. (A) General layout of a competitive ABPP experiment to measure target engagement for reversible inhibitors in mice. (B) Competitive ABPP gels of tissue proteomes from mice treated with 1 and 21 (50 mg/kg, 3 hr) followed by probe 35 (100 mg/kg, 1 hr) ; n = 3 mice/group.
In summary, we have described herein an advanced strategy for competitive ABPP that uses kinetically tuned probes to determine target engagement for reversible inhibitors in living cells and mice. We applied this approach to identify and evaluate acyl piperidines 1 and 21 as, to our knowledge, the first selective and in vivo-active inhibitors for serine hydrolases LYPLA2 and LYPLA1, respectively. We anticipate that future application of 1 and 21 in conjunction with global methods to assess protein palmitoylation 17,21 and lipid metabolites,22 respectively, should help to define the endogenous functions of LYPLA1/2. The information on target engagement provided by competitive ABPP should help to guide these studies by distinguishing tissues where maximal enzyme inhibition was achieved by 1 and 21 (e.g., heart, lung, kidney) from those where enzyme blockade was incomplete (brain) or absent (liver). Projecting forward, we believe that the strategy described herein should also be applicable to characterize reversible inhibitors for additional serine hydrolases and other classes of enzymes for which activity-based probes have been developed.3 One limitation that we should mention, however, is that kinetically tuned activity-based probes may sacrifice some of the breadth of proteomic coverage provided by more broadly reactive probes, which is especially pertinent to large enzyme classes like the serine hydrolases.23 It is also not clear, in many instances, why certain probes show greater or lesser rates of reactivity with individual enzymes, which means that identifying a matched probe is still largely an empirical process. Nonetheless, we anticipate that these issues can be addressed by synthesizing and screening a suite of probes to identify reagents that are tailored to profile complementary subsets of serine hydrolases such that most of these enzymes can be evaluated for reversible inhibition across a wide range of biological systems.
Supplementary Material
Acknowledgments
We thank P. Baillargeon, L. Deluca and D. Davda for MLSMR compound management, and D. Milliken and K. Masuda for technical assistance. This work was supported by NIH grants CA087660 (B.F.C.), MH084512 (H.R.), CA151460 (B.R.M.); the German Academic Exchange Service (postdoctoral fellowship to A.A.); California Institute for Regenerative Medicine (J.W.C.), Hewitt Foundation for Medical Research (K.L.H.); and the National Science Foundation (D.A.B.).
Footnotes
ASSOCIATED CONTENT
Supporting Information. Additional ABPP profiles, substrate data, compound characterization, and experimental protocols. This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.Moellering RE, Cravatt BF. Chem Biol. 2012;19:11. doi: 10.1016/j.chembiol.2012.01.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Inglese J, Johnson RL, Simeonov A, Xia M, Zheng W, Austin CP, Auld DS. Nat Chem Biol. 2007;3:466. doi: 10.1038/nchembio.2007.17. [DOI] [PubMed] [Google Scholar]
- 3.Cravatt BF, Wright AT, Kozarich JW. Annu Rev Biochem. 2008;77:383. doi: 10.1146/annurev.biochem.75.101304.124125. [DOI] [PubMed] [Google Scholar]
- 4.Deu E, Verdoes M, Bogyo M. Nat Struct Mol Biol. 2012;19:9. doi: 10.1038/nsmb.2203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Long JZ, Nomura DK, Cravatt BF. Chem Biol. 2009;16:744. doi: 10.1016/j.chembiol.2009.05.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Chang JW, Nomura DK, Cravatt BF. Chem Biol. 2011;18:476. doi: 10.1016/j.chembiol.2011.02.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Adibekian A, Martin BR, Wang C, Hsu KL, Bachovchin DA, Niessen S, Hoover H, Cravatt BF. Nat Chem Biol. 2011;7:469. doi: 10.1038/nchembio.579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Sugimoto H, Hayashi H, Yamashita S. Journal of Biological Chemistry. 1996;271:7705. doi: 10.1074/jbc.271.13.7705. [DOI] [PubMed] [Google Scholar]
- 9.Toyoda T, Sugimoto H, Yamashita S. Biochimica et Biophysica Acta (BBA) - Molecular and Cell Biology of Lipids. 1999;1437:182. doi: 10.1016/s1388-1981(99)00007-4. [DOI] [PubMed] [Google Scholar]
- 10.Duncan JA, Gilman AG. J Biol Chem. 1998;273:15830. doi: 10.1074/jbc.273.25.15830. [DOI] [PubMed] [Google Scholar]
- 11.Dekker FJ, Rocks O, Vartak N, Menninger S, Hedberg C, Balamurugan R, Wetzel S, Renner S, Gerauer M, Scholermann B, Rusch M, Kramer JW, Rauh D, Coates GW, Brunsveld L, Bastiaens PI, Waldmann H. Nat Chem Biol. 2010;6:449. doi: 10.1038/nchembio.362. [DOI] [PubMed] [Google Scholar]
- 12.Duncan JA, Gilman AG. J Biol Chem. 2002;277:31740. doi: 10.1074/jbc.M202505200. [DOI] [PubMed] [Google Scholar]
- 13.Yeh DC, Duncan JA, Yamashita S, Michel T. Journal of Biological Chemistry. 1999;274:33148. doi: 10.1074/jbc.274.46.33148. [DOI] [PubMed] [Google Scholar]
- 14.Biel M, Deck P, Giannis A, Waldmann H. Chemistry – A European Journal. 2006;12:4121. doi: 10.1002/chem.200501128. [DOI] [PubMed] [Google Scholar]
- 15.Bachovchin DA, Brown SJ, Rosen H, Cravatt BF. Nat Biotechnol. 2009;27:387. doi: 10.1038/nbt.1531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Consistent with a reversible mechanism of inhibition, both 1 and 21 showed greatly attenuated apparent inhibitory activity in competitive ABPP reactions performed with the FP-Rh probe, which reacts much more rapidly with LYPLA1 and LYPLA2 compared to the FP-peg-Rh probe. In contrast, the irreversible inhibitor AA26-9 showed equivalent activity when tested with either FP-Rh or FP-peg-Rh probes (Figure S4).
- 17.Martin BR, Wang C, Adibekian A, Tully SE, Cravatt BF. Nat Methods. 2012;9:84. doi: 10.1038/nmeth.1769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Rostovtsev VV, Green LG, Fokin VV, Sharpless KB. Angew Chem Int Ed Engl. 2002;41:2002. doi: 10.1002/1521-3773(20020715)41:14<2596::AID-ANIE2596>3.0.CO;2-4. [DOI] [PubMed] [Google Scholar]
- 19.Tornoe CW, Christensen C, Meldal M. J Org Chem. 2002;67:3057. doi: 10.1021/jo011148j. [DOI] [PubMed] [Google Scholar]
- 20.Speers AE, Cravatt BF. Chem Biol. 2004;11:535. doi: 10.1016/j.chembiol.2004.03.012. [DOI] [PubMed] [Google Scholar]
- 21.Martin BR, Cravatt BF. Nat Methods. 2009;6:135. doi: 10.1038/nmeth.1293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Gross RW, Han X. Chem Biol. 2011;18:284. doi: 10.1016/j.chembiol.2011.01.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Long JZ, Cravatt BF. Chem Rev. 2011;111:6022. doi: 10.1021/cr200075y. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.