

Abstract
AZD6244 is a small molecule inhibitor of the MEK kinase pathway currently in clinical trials. However, the mechanisms mediating intrinsic resistance to MEK inhibition are not fully characterized. To define molecular mechanisms of MEK inhibitor resistance, we analyzed responses of 38 lung cancer cell lines following AZD6244 treatment and their genome-wide gene expression profiles and identified a panel of genes correlated with sensitivity or resistance to AZD6244 treatment. In particular, Ingenuity pathway analysis revealed that activation of the STAT3 pathway was associated with MEK inhibitor resistance. Inhibition of this pathway by JSI-124, a STAT3-specific small molecule inhibitor, or with STAT3-specific siRNA sensitized lung cancer cells to AZD6244 and induced apoptosis. Moreover, combining a STAT3 inhibitor with AZD6244 induced expression of BIM and polyADP-ribose polymerase (PARP) cleavage, whereas activation of the STAT3 pathway inhibited BIM expression and elicited resistance to MEK inhibitors. We found that the STAT3-regulated microRNA miR-17 played a critical role in MEK inhibitor resistance, such that miR-17 inhibition sensitized resistant cells to AZD6244 by inducing BIM and PARP cleavage. Together, these results indicated that STAT3-mediated overexpression of miR-17 blocked BIM expression and caused resistance to AZD6244. Our findings suggest novel approaches to overcome resistance to MEK inhibitors by combining AZD6244 with STAT3 or miR-17 inhibitors.
Keywords: Gene expression profiling, MEK inhibitor resistance, AZD6244, STAT3 pathway, miR-17
Introduction
The MEK/extracellular signal–regulated kinase (ERK) pathway is a critical downstream signal-transduction cascade of most growth factor receptors and is essential for cell growth, survival, differentiation, and transformation. Consequently, MEK inhibitors have been actively investigated in clinical trials for the treatment of various solid tumors (1, 2). Promising results in disease stabilization with the use of tolerable doses of MEK inhibitors have been observed in some groups of patients with lung, pancreatic, or colon cancers or with melanoma (3, 4).
Although MEK-inhibitor treatment has produced clinical responses in some patients, a subset of tumors is resistant to this agent, indicating the presence of intrinsic resistance or sensitivity to MEK inhibition. Some reports have shown that BRAF mutations, especially the BRAF (V600E) mutation, are correlated with sensitivity to MEK inhibitors in melanoma and other cancer cells (5, 6). Other studies have indicated that mutations in MEK1 or activation of the phosphatidylinositol 3-kinase (PI3K) pathway due to mutations in the PI3K p110-α catalytic subunit, epidermal growth factor receptor (EGFR), or phosphatase and tensin homolog (PTEN) predict resistance to the MEK inhibitor in KRAS-mutated cells (7-11). However, the molecular mechanism of MEK inhibitor resistance has not been fully elucidated. Specific genetic mutations are good predictors of sensitivity to MEK inhibitors; however, some tumor growth may not totally depend on signaling caused by a specific genetic mutation but rather on multiple signaling pathways. More and more evidence indicates that activation of functionally redundant pathways are responsible for resistance to targeted therapy and that identifying and cotargeting those pathways may overcome resistance and induce synergistic antitumor effects (12). Recently, we and other groups found that a high level of AKT activity is associated with resistance to the MEK inhibitor AZD6244 in lung cancer and that simultaneous inhibition of the AKT and ERK pathways induced increased antitumor activity by AZD6244 (8, 13). Therefore, identifying the signaling pathway associated with MEK inhibitor resistance will help determine the biomarkers that predict responses to MEK inhibitor treatment and develop potential combination strategies to overcome resistance.
In this study, we used genome-wide gene expression profiling followed by Ingenuity Pathway Analysis (IPA) and found that activation of the signal transducer and activator of transcription 3 (STAT3) pathway was associated with AZD6244 resistance. Thus, inhibition of the STAT3 pathway sensitized resistant cells to AZD6244 treatment, whereas activation of STAT3 induced resistance to AZD6244 in sensitive cells. We further observed that STAT3-mediated MEK inhibitor resistance occurred though microRNA (miRNA) miR-17. Our results suggest that miR-17, which is regulated by the STAT3 pathway, mediated MEK inhibitor resistance by suppressing BIM expression.
Material and Methods
Chemicals and reagents
AZD6244 was synthesized in Dr. William Bornmann’s laboratory at The University of Texas MD Anderson Cancer Center. Antibodies against Janus kinase 1 (JAK1), interleukin-6 signal transducer (IL6ST), phosphorylated STAT3 (p-STAT3), phosphorylated JAK1 (p-JAK1), phosphorylated ERK (p-ERK), phosphorylated AKT (p-AKT), phosphorylated JNK (p-JNK), phosphorylated p38 MAPK (p-p38 MAPK), AKT, ERK, and poly (ADP-ribose) polymerase (PARP) were purchased from Cell Signaling Technology (Danvers, MA). Antibodies against BIM were obtained from Calbiochem (San Diego, CA). Protease inhibitor cocktail, β-actin antibody, and sulforhodamine B (SRB) were obtained from Sigma Chemical Corporation (St. Louis, MO). Plasmid pcDNA3–miR-17 was from Dr. Joshua Mendell (Johns Hopkins University School of Medicine, Baltimore, MD). MiR-17 inhibitor, anti–miR-17, and anti-miRNA negative control were purchased from Ambion (Austin, TX). Reagents for the real-time quantitative polymerase chain reaction (qPCR) assay of miR-17 were purchased from Qiagen (Valencia, CA). Plasmids pcDNA-3.1-STAT3-CA was from Dr. James E. Darnell (The Rockefeller University, New York) (14). STAT3 siRNA was designed on the basis of work by Konnikova et al. and synthesized by Qiagen (15).
Cell lines
All human lung cancer cell lines were from Hamon Center for Therapeutic Oncology Research, The University of Texas Southwestern Medical Center, Dallas. Cell lines were maintained in high-glucose RPMI 1640 supplemented with 5% fetal bovine serum, the cells were cultured at 37°C in a humidified atmosphere containing 5% CO2 and 95% air.
Cell viability assay
The inhibitory effects of AZD6244 on cell growth were determined using the MTS (Promega) or Sulforhodamine B (SRB) colorimetric assay, as described previously (16, 17). Each experiment was performed in octuplicate and repeated at least three times. The relative cell viability (%) was calculated using the equation ODT/ODC×100%, where ODT represents the absorbance of the treatment group and ODC represents the absorbance of the control group. Median inhibitory concentration (IC50) values were determined using in-house software (DIVISA) and plotted in dose-response curves.
Microarrays
Five micrograms of RNAs were labeled and hybridized to Affymetrix GeneChips HG-U133A and HG-U133B according to the manufacturer’s protocol (http://www.affymetrix.com). Array data were pre-processed with MAS5 (Affymetrix algorithm for probe summarization) and then quantile-normalized and log-transformed.
Plasmids and anti-miRNA transfection
Lipofectamine 2000 (Invitrogen, Carlsbad, California) was used for the transfection of plasmid DNA or anti-miRNA inhibitor. Forty-eight hours after transfection, cells were treated with AZD6244 for response and apoptosis analysis, or cell lysates and total RNA were prepared for Western blotting and real-time PCR.
Real-time qPCR
Total RNA was isolated from cultured cells using TRIzol reagent (Invitrogen). For analysis of JAK1 and IL6ST and BIM, real-time qPCR was done using the first-strand reverse transcription kit (Invitrogen) followed by the Real Time PCR kit from Bio-Rad (Berkeley, California) in accordance with the manufacturer’s protocol. Glyceraldehyde-3-phosphate dehydrogenase (GAPDH) served as the internal control. For miRNA expression analysis, real-time qPCR was done using the miRNA reverse transcription kit (Qiagen) and miRNA assays kit (Qiagen) following the manufacturer’s protocols. miRNA SNORD44 served as an internal control. The sequences of the primers used were as follows: JAK1, 5′ primer (5′-GAATGACGCCACACTGACTG-3′) and 3′ primer (5′-GATGACAAGATGTCCCTCCG-3′); IL6ST, 5′ primer (5′-GATGACAAGATGTCCCTCCG-3′) and 3′ primer (5′-AAA GGACAGGATGTTGCAGG-3′); and GAPDH, 5′ primer (5′-ATCCCATCACCATCTTCCAG-3′) and 3′ primer (5′-ATGAGTCCTTCCACGATACC-3′).
siRNA knockdown experiments
Transfection was performed using Lipofectamine 2000 (Invitrogen) according to the manufacturer’s instructions. Forty-eight hours after transfection, the cells were treated with AZD6244 for the dose response assay and cell cycle analysis. Cell lysates and RNA were prepared for further analysis by Western blotting and real-time qPCR.
Western blotting analysis
Standard Western blotting was performed using antibodies described above in the reagent section and as described previously (17). The immunoreactive bands were visualized with the Odyssey Imager (Li-COR Biosciences, Lincoln, NE).
Cell cycle and apoptosis assay
Our cell cycle and apoptosis assays have been described previously (13). The cells were treated with AZD6244 alone or combination with JSI-124 for 48 hours and harvested by trypsinization. Cells were analyzed on an EPICS Profile II flow cytometer (Coulter Corp., Hialeah, FL) using the Multicycle AV software (Phoenix Flow Systems, San Diego, CA). Experiments were repeated at least three times.
Pathway analysis
The panel of genes significantly correlated with resistance or sensitivity (P<0.05) to the MEK inhibitor were imported into Ingenuity Pathways Analysis (IPA; Ingenuity® Systems, http://www.ingenuity.com) to analyze network interactions. Networks of these significantly correlated genes were then algorithmically generated on the basis of their connectivity.
Animal study
Female BALB/c nude mice (6- to 8-weeks old) were purchased from Charles River Laboratories (Wilmington, MA). Animal experiments were performed according to the protocol (11-03-09933) approval by the MD Anderson institutional review board and in accordance with the Guidelines for the Care and Use of Laboratory Animals published by the National Institutes of Health. A total of 2×106 H460 cells were inoculated subcutaneously into the right dorsal flanks of the nude mice. Treatments were started when tumors reached an average volume of about 0.1 cm3, which was around 7 days after cells were inoculated. Mice were randomly divided into control and treatment groups (n = 5 animals per group). The treatment groups were administered 20 mg/kg AZD6244, 1 mg/kg JSI-124, or combination of AZD6244 (20 mg/kg) and JSI-124 (1mg/kg), all of which had been solubilized in a medium containing 0.5% hydroxypropyl methylcellulose and 0.1% polysorbate buffer. AZD6244 was administered once daily by oral gavage. JSI-124 was administered intraperitoneally. The control group received the buffer alone. Tumor size was measured by calipers every other day. The tumor volume was calculated, with the formula: length × width2 × 0.52. Mice were euthanized when their tumor volume was larger than 2000 mm3.
Immunohistochemical staining
Formalin-fixed, paraffin-embedded tissue sections of animal tumor tissue specimens were used for immunostaining with antibodies against p-ERK and p-STAT3 with AEC immunohistochemical staining kit (Invitrogen) following the manufacturer’s instructions.
Statistical analyses
Association between gene expression and responses to treatment with MEK inhibitor was determined by calculating log2 ratios of resistant vs sensitive cell lines, and its significance was determined using Student’s t test. Ingenuity software was used to perform signaling pathway analysis. Pathways linked with correlated genes were identified as described in the pathway analysis part. The significance of the in vivo animal study data was determined by using the Mann-Whitney U test.
Results
Gene expression profiling identified that activation of the STAT pathway correlated with AZD6244 resistance in lung cancer cell lines
To determine the molecular mechanism underlying MEK inhibitor resistance, previously we have tested responses to AZD6244 in 38 non–small cell lung cancer cell lines. Susceptibility to AZD6244 differed dramatically between the cell lines, with IC50 values ranging from 0.1 to 250 μM indicating that human lung cancer cells have a range of degrees of intrinsic resistance or sensitivity to MEK-inhibitor treatment (18). Analysis of genetic gene mutations of the cell lines did not find correlation between the gene mutation status of EGFR, KRAS, BRAF or PI3K and the sensitivity to AZD6244 (18). We then analyzed the gene expression profiles and MEK-inhibitor responses of the five cell lines that were the most sensitive (Calu-6, H2087, H596, H2073, and H2887), whose IC50 values ranged from 0.0287 to 0.519 μM, and of the 12 most resistant cell lines (H441, H1650, H1819, H1993, HCC366, H460, HCC1359, H1155, H1299, HCC15, HCC95, and HCC2450) whose IC50 values ranged from 50 to more than 100 μM. Through this analysis, we identified many genes whose expression levels correlated strongly (with normal P<0.05) with their sensitivity or resistance to AZD6244 (Figure 1A and supplementary table 1). Further pathway analysis of those genes showed that activation of the STAT3 pathway correlated most strongly with resistance to AZD6244 (Fig. 1B).
Figure 1. Pathway analysis identified that the STAT pathway was associated with resistance to the MEK inhibitor AZD6244.
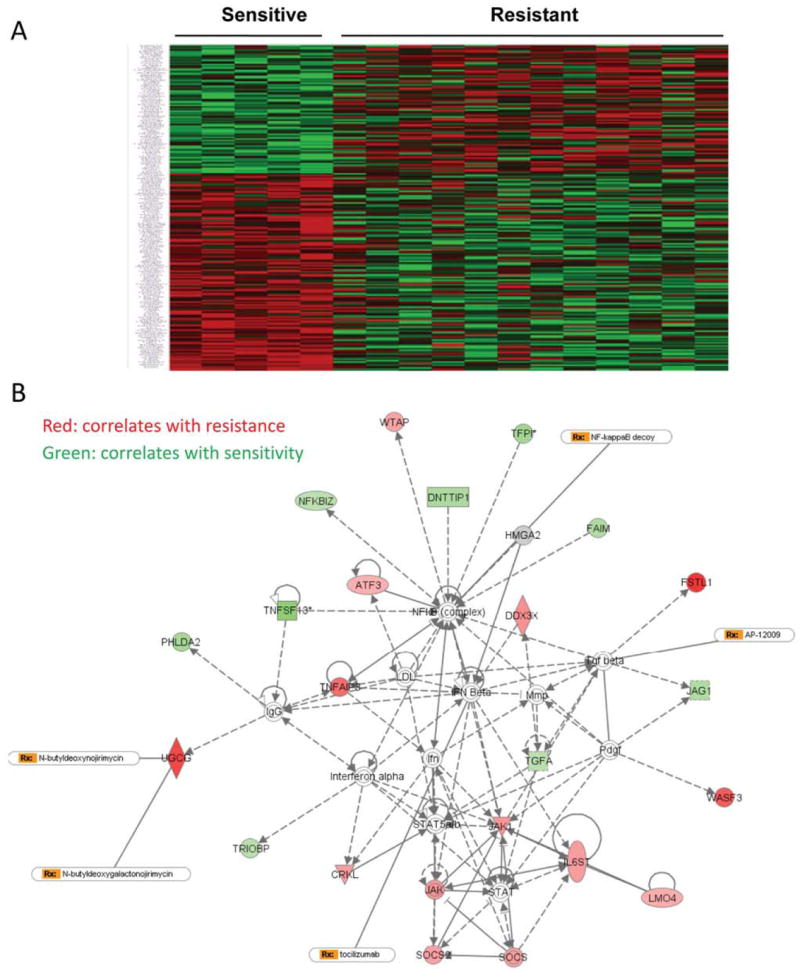
A, Heat map shows the correlation of gene expression with sensitivity to AZD6244. B, The signaling pathway associated with sensitivity or resistance to AZD6244 obtained by using Ingenuity software analysis. STAT pathway is showed in the map to be associated with MEK inhibitor resistance.
Validate the correlation of gene expression with sensitivity to AZD6244
To confirm that activation of the STAT3 pathway was correlated with MEK inhibitor resistance, real-time PCR was performed to determine the expression of JAK1 and IL6ST, which are related to STAT pathways and were higher in resistant cell lines than that in sensitive cell lines, as determined by our gene expression mRNA array. We checked the expression of these molecules in randomly selected sensitive and resistant cell lines (4 each) using real-time qPCR and Western blotting. Real-time qPCR results showed that expression of both JAK1 and IL6ST was much higher in resistant cell lines than in sensitive cell lines (Fig. 2A and 2B). Western blotting also confirmed that levels of p-STAT3, and IL6ST were higher in resistant cell lines than in sensitive cell lines, indicating that the STAT3 pathway was constitutively activated in resistant cell lines (Fig. 2C). Based on the database of Cancer Cell line Project (http://www.sanger.ac.uk/genetics/CGP/CellLines/), we did not find the correlation of EGFR, BRAF, and KRAS Mutation with sensitivity to AZD6244 (data not show).
Figure 2. Validation of STAT3 pathway associated with resistance to AZD6244.

A, B, Real-time PCR was performed to check the expression of JAK1 and IL6ST; C, Western blotting confirmed the activation of STAT3 pathways in resistant cell lines. Cell lysates were collected from randomly chosen sensitive and resistant cell lines (4 each), and Western blotting was performed to check the levels of JAK1, IL6ST, and pSTAT3 with specific antibodies. EGFR, BRAF, and KRAS mutation status of the cell lines are labeled at the bottom of Fig. 2C. (Grey square indicates mutation)
STAT3 pathway regulates sensitivity of lung cancer cells to MEK inhibitor treatment
To further test if STAT3 pathway mediated MEK inhibitor resistance, STAT3 was knocked down in resistant cell lines H460 and H226 (Fig. 3A), and the constitutively active form of STAT3 was overexpressed in two AZD6244-sensitive cell lines Calu6 and H1437 (Fig. 3B). However we did not observe changes in JAK1 and IL6ST expression in the cells with overexpression of the constitutively active form of STAT3. Both p-STAT3 and total STAT3 were up-regulated in H460 cells after treatment with AZD6244 at time points up to 72 hours (Fig. 3C). SRB assay were performed to assess the responses to AZD6244 in cells with STAT3 knockdown or overexpression. The results showed that knockdown of STAT3 in H460 and H226 cells significantly sensitized the cells to AZD6244 (Fig.3D). Knockdown of STAT3 decreased IC50 values from >50 μM to <1 μM in both the H460 and H226 cell lines. Activation of the STAT3 pathway in two sensitive cell lines, Calu6 and H1437, with the constitutively active form of STAT3 induced resistance to AZD6244 (Fig. 3E). These results suggested that exogenous activation of the STAT3 pathway caused MEK inhibitor in lung cancer cells.
Figure 3. STAT3 pathway regulates sensitivity of lung cancer cells to MEK inhibitor treatment.

A, Western blotting confirmed that STAT3 was inhibited with STAT3-specific siRNA. B, Western blotting confirmed that overexpression of constitutive STAT3 in Calu6 and H1437 cells. C, Time course experiment was performed to determine the change of STAT3 expression in H460 cells treated with AZD6244. D, Knockdown of STAT3 sensitized H460 and H226 lung cancer cells to AZD6244. Cells were transfected with 50 nM STAT3-specific siRNA or negative control with Lipofectamine 2000. Then, 24 h after transfection, cells were treated with AZD6244 with different doses as indicated for an additional 72 h. Cell viability was determined by SRB assay. E, Forty-eight hours after transfection with constitutive active STAT3, cells were treated with AZD6244 for another 72 hours, and SRB was performed to determine cell viability.
STAT3 inhibitor JSI-124 sensitized lung cancer cell to AZD6244 in vitro and in vivo
To determine whether combining AZD6244 with a small-molecule STAT3 inhibitor could overcome MEK inhibitor resistance, we tested inhibition of p-STAT3 in H460 cells with different doses of JSI-124, a STAT3 inhibitor (19), and results showed that 50 nM JSI-124 inhibited p-STAT3 levels by approximately 80% (Fig. 4A). Furthermore, inhibition of the STAT3 pathway with 20 and 40 nM JSI-124 significantly sensitized the resistant cell lines H460 and H226 to AZD6244 (P<0.05) (Fig. 4B, and 4C). We also tested several other resistant cell lines, including H2882, HCC827, HCC193, and HCC515, and in all cell lines tested, treatment with a very low concentration of JSI-124 (40 nM) sensitized cells to AZD6244 (data not shown). To confirm these results in vivo, a study was performed in a mouse human xenograft lung tumor model derived from the H460 cell lines. Combination treatment with AZD6244 and JSI-124 significantly inhibited tumor growth compared to treatment with each drug alone (Fig. 4D, and 4E). We also observed that JSI-124 alone has some effect on the tumor growth inhibition, which was consistent with a previous report (19). Immunohistochemical staining further confirmed that p-ERK and p-STAT3 were significantly inhibited in animal tumor tissue specimens treated with AZD6244, JSI-124 or both compared with that treated with vehicle, indicating that both targets of AZD6244 and JSI-124 can be effectively inhibited with the combination treatment in vivo (Fig. 4F).
Figure 4. STAT3 inhibitor JSI-124 sensitized lung cancer cell to AZD6244 in vitro and in vivo.
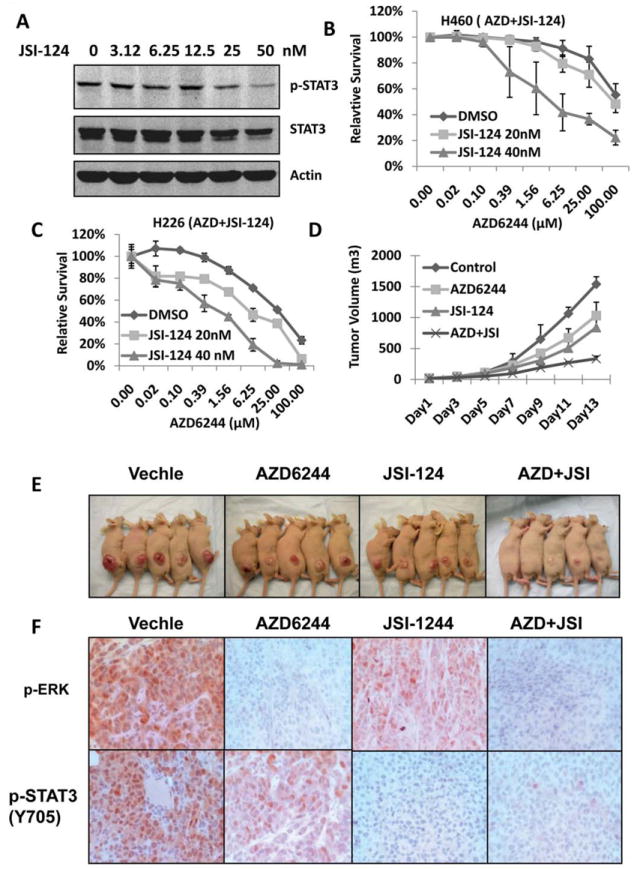
A, H460 cells were treated with JSI-124 for 24 hours; cell lysates were collected for Western blotting assay to determine p-STAT3 (Y705) and STAT3 expression. B, and C, Inhibition of the STAT3 pathway with JSI-124 sensitized cells to AZD6244 treatment. H460 and H226 cell lines were treated with 20 or 40 nM JSI-124 and AZD6244 at indicated doses for 72 hours. Cell viability was determined by SRB assay. D, Combination with AZD6244 and JSI-124 enhanced antitumor activity in vivo. Treatments were started when tumors reached an average volume of about 0.1 cm3. The treatment groups were administered 20 mg/kg AZD6244, 1 mg/kg JSI-124, or combination of AZD6244 (20 mg/kg) and JSI-124 (1mg/kg). E, Xenograft tumors in the four treatment groups with 2 weeks treatment. F, Immunohistochemical staining for p-ERK and p-STAT3 (Y705) with animal tumor tissue specimens.
Combination of JSI-124 and AZD6244 induced cell apoptosis through BIM
To further analyze how JSI-124 sensitized the resistance cells to AZD6244 treatment, cell cycle analysis were performed. The results showed that a substantial amount of cell apoptosis was induced by combination treatment with AZD6244 and JSI-124 and not by treatment with AZD6244 alone (Fig.5A). JSI-124 (50nM) had no effect on the levels of pAKT, pJNK, or pp38MAPK (Fig.5B). Previous studies indicated that induction of BIM expression is a critical step in MEK inhibitor–induced cell apoptosis (20-22). To determine whether apoptosis induction by combination treatment with JSI-124 and AZD6244 is also due to BIM induction, Western blotting was performed to assess BIM expression in cells treated with AZD6244 alone or combination with JSI-124. The results showed that neither AZD6244 nor JSI-124 alone induced BIM expression in H460 cells, whereas combination treatment with both dramatically induced BIM expression, including BIM-EL, BIM-L, and BIM-SL, in the H460 resistant cell line (Fig. 5C). BIM-EL in particular showed the greatest up-regulation in cells with combination treatment Results also showed that combination treatment with JSI-124 and AZD6244 induced cleavage of PARP, the protein marker for cell apoptosis. However, there was no induction of PARP cleavage in cells treated with AZD6244 alone and only minimal induction of PARP cleavage in cells treated with JSI-124 alone (Fig. 5C,). In addition, activation of the STAT3 pathway inhibited the induction of BIM and PARP cleavage by AZD6244 in sensitive Calu6 cells (Fig. 5D). These results indicated that activation of the STAT3 pathway induced resistance to AZD6244 through inhibiting BIM induction, whereas blocking the STAT3 pathway overcame resistance to AZD6244 and induced cell apoptosis through inducing BIM expression. BIM is regulated by FoxO3A at the transcriptional level (23). ERK also directly phosphorylates BIM at S69 and promotes its degradation (24). However, inhibition of ERK alone in MEK inhibitor resistant cells could only induce minor BIM expression in AZD6244 resistant cells, although p-FoxO3A was inhibited (Fig.5E). Inhibition of STAT3 alone had no effect on the phosphorylation of FoxO3A and BIM (Fig.5E) or any effect on nuclear translocation of FoxO3A (s-Fig.1) suggesting that regulation of BIM by the STAT3 pathway may be not through FoxO3A or phosphorylation of BIM.
Figure 5. Combination of JSI-124 and AZD6244 induced cell apoptosis through BIM.
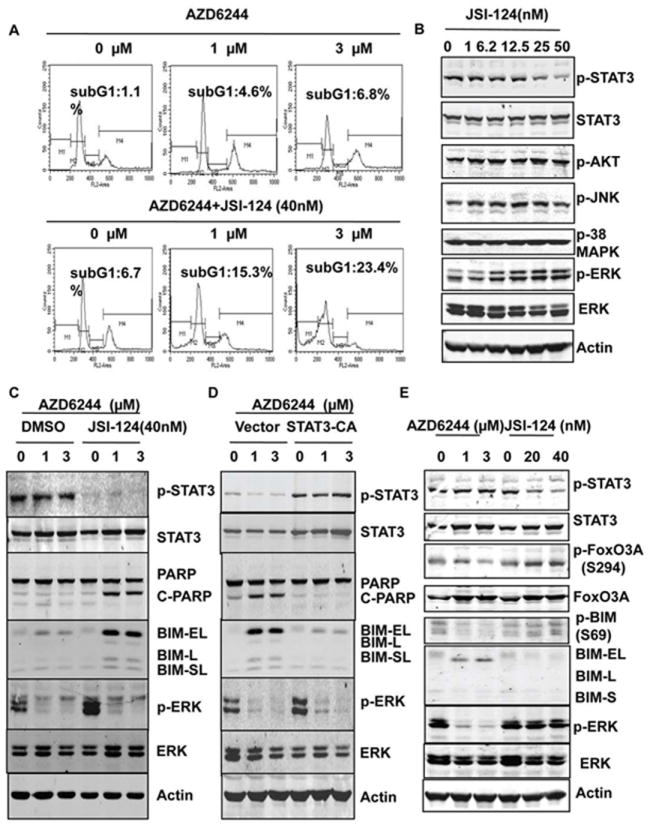
A, Analysis of the apoptotic sub-G1 cell population. H460 cells were treated with AZD6244 only or the combination of AZD6244 and JSI-124 for 48 hours, and cells were collected and stained with propidium iodide for cell cycle analysis. B, Effects of JSI-124 on PI3K/AKT, JNK, and p38MAPK pathways. H460 cells were treated with different doses of JSI-124 for 24 hours, and a Western blot was performed with the antibodies indicated. C, Combination treatment with AZD6244 and JSI-124 induced BIM expression in MEK inhibitor–resistant cells. H460 cells were treated with AZD6244 only or with AZD6244 and JSI-124 for 48 hours, and Western blotting was performed to determine the expression of proteins as indicated. D, Activation of the STAT3 pathway inhibited BIM expression induced by AZD6244. Calu6 cells were transfected with constitutively active STAT3 or control vector. Then, 48 hours after transfection, cells were treated with AZD6244 with different dosages as indicated for another 48 hours. Western blotting was performed to check the protein expression with specific antibodies as indicated. E, Effects of MAPK and STAT3 pathways on FoxO3A and BIM. H460 cells were treated with AZD6244 or JSI-124 alone for 48 hours, and a Western blot was performed with the antibodies indicated.
STAT3 regulates BIM through miR-17
The results above showed that blocking the STAT3 pathway sensitized resistant cells to AZD6244 by inducing cell apoptosis through BIM. However, how STAT3 cooperated with the ERK-regulating BIM gene is unclear. One recent study found that STAT3 regulated the expression of the miRNA cluster miR-17-92 on the transcriptional level (25). Moreover, studies with transgenic animal models indicated that miR-17-92 promotes cell proliferation and induces tumorigenicity through targeting BIM expression. Thus, we hypothesized that STAT3-mediated MEK inhibitor resistance might occur through the up-regulation of miR-17-92, which suppressed BIM by targeting its 3’-untranslated region.
To test this hypothesis, real-time qPCR was performed to determine miR-17 expression in Calu6 and H1437 cells that had overexpression of the constitutively active form STAT3 and its expression in H460 and H226 cells with STAT3 knockdown. The results of real-time PCR showed that overexpression of constitutively active STAT3 up-regulated miR-17 in Calu6 and H1437 cells, whereas knockdown of STAT3 expression in H460 and H226 cells down-regulated the expression of miR-17 (Fig. 6A). Consistent with the levels of miR-17 in cells with STAT3 activation, BIM was down-regulated at the mRNA level (Fig. 6B), whereas inhibition of miR-17 with anti–miR-17 up-regulated BIM RNA in resistant cells (Fig. 6B). To further test whether miR-17 up-regulated by STAT3 plays a role in MEK inhibitor resistance, Calu6 and H1437 cells were transfected with miR-17 expression vector, then treated with AZD6244 and assessed by SRB assay. The result showed that overexpression of miR-17 in Calu6 and H1437 cells induced resistance to AZD6244 (Fig. 6C). As it was expected, inhibition of miR-17 with anti-miR-17 sensitized H460 and H226 cells to treatment with AZD6244 (Fig. 6D). The result of Western blotting further confirmed that overexpression of miR-17 inhibited the BIM expression induced by AZD6244 and inhibited the expression of PARP cleavage in MEK inhibitor–sensitive Calu6 cells (Fig.6E); whereas inhibition of miR-17 with anti-miR-17 combined with AZD6244 induced expression of BIM and PARP cleavage in MEK inhibitor–resistant H460 cells (Fig. 6F). These results indicated that miR-17 regulated by the STAT3 pathway plays an important role in the response of lung cancer to MEK inhibitor treatment.
Figure 6. STAT3 regulates BIM through miR-17.
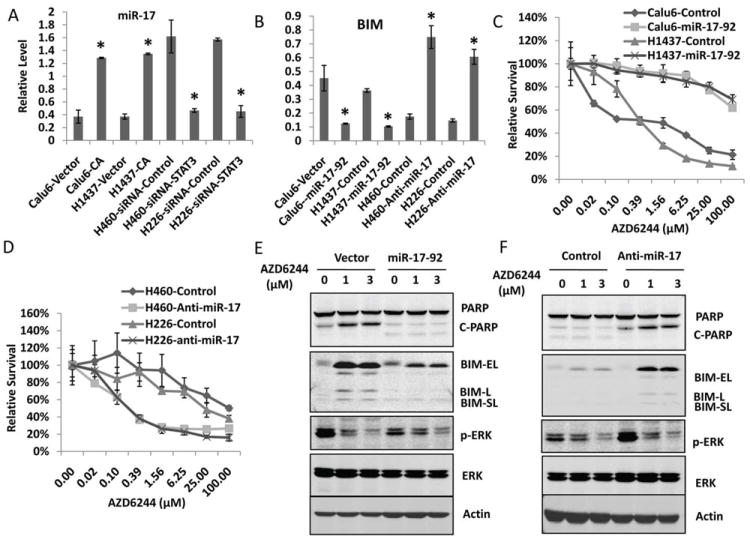
A, STAT3 regulates mir-17 expression in lung cancer cells. A, Real-time qPCR analysis of mature miR-17 expression. B, Real-time qPCR analysis of BIM expression. *, P < 0.05, indicates a significant difference. C, Overexpression of the miR-17-92 cluster induced resistance to AZD6244. Calu6 and H1437 cells were transfected with miR-17-92 expression vector. Then, 24 hours after transfection, cells were treated with AZD6244 at different doses for another 72 hours, and the SRB assay was performed to analyze cell viability. D. Inhibition of miR-17 sensitized cells to AZD6244. H460 and H226 cells were transfected with anti–miR-17 or negative control. Then, 24 hours after transfection, cells were treated with AZD6244 with different doses for another 72 hours and the SRB assay performed to analyze cell viability. E, Overexpression of the miR-17-92 cluster blocked BIM induction by AZD6244. Calu6 cells were transfected with miR-17-92 expression vector or control vector. Then, 48 hours after transfection, cells were treated with AZD6244 at different doses as indicated for another 48 hours. Western blotting was performed to analyze the expression of BIM and PARP. F, Inhibition of miR-17 induced BIM expression by AZD6244. H460 cells were transfected with anti–miR-17 or negative control. Then, 48 hours after transfection, cells were treated with AZD6244 for 48 hours, and Western blotting was performed with specific antibodies as indicated.
Discussion
In this study we tested the MEK inhibitor AZD6244 on a panel of 38 non–small cell lung cancer (NSCLC) cell lines that have been characterized with respect to gene copy number, gene expression, mutation, and protein expression profiles. In our analysis of gene expression profiles, we found one group of genes correlated with MEK inhibitor resistance and another group of genes correlated with MEK inhibitor sensitivity. Analyzing the genes that were significantly correlated with sensitivity or resistance to MEK inhibitors using IPA software, we identified that activation of the STAT3 pathway was associated with MEK inhibitor resistance.
Recently, using a similar gene expression profiling approach, Dry et al. also identified that higher levels of IL6 correlated with resistance to MEK inhibition, indicating that the STAT3 pathway may mediate AZD6244 resistance (26). Although we did not find IL6 to be one of the genes correlated with MEK inhibitor resistance in our study, JAK1, IL6ST, and LIMO4, which are related to JAK-STAT3 pathways, were correlated with MEK inhibitor resistance. JAK1 and IL6ST are molecules directly upstream of STAT3. Overexpression of JAK1 and IL6ST can directly activate STAT3 (27, 28). LIMO4 can bind to and activate IL6ST, thus activating the STAT3 pathway (29). Higher levels of JAK1, IL6ST, and LIMO4 might at least partly contribute to the STAT3 activation and thus induce MEK inhibitor resistance. Another recent study by Yoon et al. showed that feedback activation of STAT3 by MEK inhibitor in the KRAS mutated lung cancer cells results in MEK inhibitor resistance, also indicating agreement with our study (30). We further confirmed that inhibition of the STAT3 pathway with STAT3-specific siRNA, or with JSI-124, a STAT3-specific inhibitor sensitized lung cancer cells to MEK inhibitor treatment in vitro and in vivo.
The STAT3 pathway has been shown to be activated in many types of cancer and is associated with cancer transformation, angiogenesis, invasion, and metastasis and with immune system suppression (31, 32). In this study, we found that the combination of AZD6244 and JSI-124 induced cell apoptosis through inducing dramatic BIM expression and PARP cleavage, whereas activation of the STAT3 pathway by overexpression of constitutively active STAT3 in the sensitive cell lines blocked BIM expression and apoptosis induction. Induction of BIM by simultaneous inhibition of the ERK and STAT3 pathways is consistent with previous reports that induction of BIM expression is required for tumor suppression mediated by MEK inhibitors (20, 33). BIM is regulated by both the AKT and MAPK pathways on the transcriptional level through FoxO3A (23, 34-36). ERK also can directly phosphorylate BIM and thereby promote its degradation (37, 38). However our results showed that JSI-124, a STAT3 inhibitor has no effect on p-FoxO3A or p-BIM, suggesting that STAT3 regulates BIM by other mechanisms.
Recent studies have shown that BIM was regulated not only at the transcriptional and protein levels but also the posttranscriptional level. Several studies have indicated that miR-17 promotes tumorigenicity by inhibiting cell apoptosis through targeting BIM and PTEN (39-41). Importantly, miR-17 has been reported to be highly expressed in lung cancer and to promote the proliferation of cancer cells (42) and to be regulated by STAT3 (25). Recent studies have indicated that miRNAs are also involved in resistance to chemotherapeutic agents and possibly to target therapeutic agents as well (43, 44). In this study we found that up-regulation of miR-17-92 by activation of the STAT3 pathway induced MEK inhibitor resistance, whereas simultaneous inhibition of the MEK and STAT3 pathways or miR-17 significantly sensitized resistant cells to AZD6244 treatment by up-regulating BIM. Our results not only provide insight into the molecular mechanism of MEK inhibitor resistance but also indicate novel alternative approaches for overcoming the MEK inhibitor resistance by combining AZD6244 with miRNA inhibitors. Given that STAT3 inhibitors have serious adverse effects, small-molecule RNA-based miRNA inhibitors have the advantage of less toxicity and will be promising in future cancer treatment either as single agents or in combination with other therapeutic drugs.
In summary, using biochemical and biological methods we identified that the activation of STAT3 pathways mediates MEK inhibitor resistance. We further found that STAT3-mediated MEK inhibitor resistance occurs through the inhibition of BIM by miRNA-17. Our results suggest that the combination of a small molecule–based inhibitor with a STAT3 inhibitor or a miR-17 inhibitor may be productive approaches for lung cancer therapy.
Supplementary Material
Acknowledgments
We thank Virginia M. Mohlere and Deborah L. Waits for scientific editing.
Funding: This work was supported in part by the National Institutes of Health through MD Anderson’s Cancer Center Core Grant CA-016672 - Lung Program, Research Animal Support Facility, Specialized Program of Research Excellence (SPORE) Grant CA-070907 (to J. Minna and J. Roth), and R01 grant CA-092487 (to B. Fang).
References
- 1.Adjei AA, Cohen RB, Franklin W, et al. Phase I pharmacokinetic and pharmacodynamic study of the oral, small-molecule mitogen-activated protein kinase kinase 1/2 inhibitor AZD6244 (ARRY-142886) in patients with advanced cancers. J Clin Oncol. 2008;26:2139–46. doi: 10.1200/JCO.2007.14.4956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Rinehart J, Adjei AA, Lorusso PM, et al. Multicenter phase II study of the oral MEK inhibitor, CI-1040, in patients with advanced non-small-cell lung, breast, colon, and pancreatic cancer. J Clin Oncol. 2004;22:4456–62. doi: 10.1200/JCO.2004.01.185. [DOI] [PubMed] [Google Scholar]
- 3.Friday BB, Adjei AA. Advances in targeting the Ras/Raf/MEK/Erk mitogen-activated protein kinase cascade with MEK inhibitors for cancer therapy. Clin Cancer Res. 2008;14:342–6. doi: 10.1158/1078-0432.CCR-07-4790. [DOI] [PubMed] [Google Scholar]
- 4.Wang D, Boerner SA, Winkler JD, LoRusso PM. Clinical experience of MEK inhibitors in cancer therapy. Biochim Biophys Acta. 2007;1773:1248–55. doi: 10.1016/j.bbamcr.2006.11.009. [DOI] [PubMed] [Google Scholar]
- 5.Pratilas CA, Taylor BS, Ye Q, et al. (V600E)BRAF is associated with disabled feedback inhibition of RAF-MEK signaling and elevated transcriptional output of the pathway. Proc Natl Acad Sci U S A. 2009;106:4519–24. doi: 10.1073/pnas.0900780106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Solit DB, Garraway LA, Pratilas CA, et al. BRAF mutation predicts sensitivity to MEK inhibition. Nature. 2006;439:358–62. doi: 10.1038/nature04304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Engelman JA, Chen L, Tan X, et al. Effective use of PI3K and MEK inhibitors to treat mutant Kras G12D and PIK3CA H1047R murine lung cancers. Nat Med. 2008;14:1351–6. doi: 10.1038/nm.1890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Pratilas CA, Hanrahan AJ, Halilovic E, et al. Genetic predictors of MEK dependence in non-small cell lung cancer. Cancer Res. 2008;68:9375–83. doi: 10.1158/0008-5472.CAN-08-2223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Sos ML, Fischer S, Ullrich R, et al. Identifying genotype-dependent efficacy of single and combined PI3K- and MAPK-pathway inhibition in cancer. Proc Natl Acad Sci U S A. 2009;106:18351–6. doi: 10.1073/pnas.0907325106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Emery CM, Vijayendran KG, Zipser MC, et al. MEK1 mutations confer resistance to MEK and B-RAF inhibition. Proc Natl Acad Sci U S A. 2009;106:20411–6. doi: 10.1073/pnas.0905833106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Wee S, Jagani Z, Xiang KX, et al. PI3K Pathway Activation Mediates Resistance to MEK Inhibitors in KRAS Mutant Cancers. Cancer Research. 2009;69:4286–93. doi: 10.1158/0008-5472.CAN-08-4765. [DOI] [PubMed] [Google Scholar]
- 12.Wheeler DL, Dunn EF, Harari PM. Understanding resistance to EGFR inhibitors-impact on future treatment strategies. Nat Rev Clin Oncol Nature Reviews Clinical Oncology. 2010;7:493–507. doi: 10.1038/nrclinonc.2010.97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Meng J, Peng H, Dai B, et al. High level of AKT activity is associated with resistance to MEK inhibitor AZD6244 (ARRY-142886) Cancer Biol Ther. 2009;8:2073–80. doi: 10.4161/cbt.8.21.9844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Ginsberg M, Czeko E, Muller P, Ren Z, Chen X, Darnell JE., Jr Amino acid residues required for physical and cooperative transcriptional interaction of STAT3 and AP-1 proteins c-Jun and c-Fos. Mol Cell Biol. 2007;27:6300–8. doi: 10.1128/MCB.00613-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Konnikova L, Kotecki M, Kruger MM, Cochran BH. Knockdown of STAT3 expression by RNAi induces apoptosis in astrocytoma cells. BMC Cancer. 2003;3:23. doi: 10.1186/1471-2407-3-23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Cai D, Shames DS, Raso MG, et al. Steroid Receptor Coactivator-3 Expression in Lung Cancer and Its Role in the Regulation of Cancer Cell Survival and Proliferation. Cancer Research. 70:6477–85. doi: 10.1158/0008-5472.CAN-10-0005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Guo W, Wu S, Liu J, Fang B. Identification of a small molecule with synthetic lethality for K-ras and protein kinase C iota. Cancer Res. 2008;68:7403–8. doi: 10.1158/0008-5472.CAN-08-1449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Meng J, Dai B, Fang B, et al. Combination treatment with MEK and AKT inhibitors is more effective than each drug alone in human non-small cell lung cancer in vitro and in vivo. PLoS One. 5:e14124. doi: 10.1371/journal.pone.0014124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Blaskovich MA, Sun J, Cantor A, Turkson J, Jove R, Sebti SM. Discovery of JSI-124 (cucurbitacin I), a selective Janus kinase/signal transducer and activator of transcription 3 signaling pathway inhibitor with potent antitumor activity against human and murine cancer cells in mice. Cancer Res. 2003;63:1270–9. [PubMed] [Google Scholar]
- 20.Cragg MS, Jansen ES, Cook M, Harris C, Strasser A, Scott CL. Treatment of B-RAF mutant human tumor cells with a MEK inhibitor requires Bim and is enhanced by a BH3 mimetic. J Clin Invest. 2008;118:3651–9. doi: 10.1172/JCI35437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Yang J-Y, Chang C-J, Xia W, et al. Activation of FOXO3a Is Sufficient to Reverse Mitogen-Activated Protein/Extracellular Signal-Regulated Kinase Kinase Inhibitor Chemoresistance in Human Cancer. Cancer Research. 70:4709–18. doi: 10.1158/0008-5472.CAN-09-4524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Meng J, Fang B, Liao Y, Chresta CM, Smith PD, Roth JA. Apoptosis induction by MEK inhibition in human lung cancer cells is mediated by Bim. PLoS One. 5:e13026. doi: 10.1371/journal.pone.0013026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Sunters A, Fernandez de Mattos S, Stahl M, et al. FoxO3a transcriptional regulation of Bim controls apoptosis in paclitaxel-treated breast cancer cell lines. J Biol Chem. 2003;278:49795–805. doi: 10.1074/jbc.M309523200. [DOI] [PubMed] [Google Scholar]
- 24.Sheridan C, Brumatti G, Martin SJ. Oncogenic B-RafV600E inhibits apoptosis and promotes ERK-dependent inactivation of Bad and Bim. J Biol Chem. 2008;283:22128–35. doi: 10.1074/jbc.M800271200. [DOI] [PubMed] [Google Scholar]
- 25.Brock M, Trenkmann M, Gay RE, et al. Interleukin-6 modulates the expression of the bone morphogenic protein receptor type II through a novel STAT3-microRNA cluster 17/92 pathway. Circ Res. 2009;104:1184–91. doi: 10.1161/CIRCRESAHA.109.197491. [DOI] [PubMed] [Google Scholar]
- 26.Dry JR, Pavey S, Pratilas CA, et al. Transcriptional pathway signatures predict MEK addiction and response to selumetinib (AZD6244) Cancer Res. 70:2264–73. doi: 10.1158/0008-5472.CAN-09-1577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Zhang Y, Turkson J, Carter-Su C, et al. Activation of Stat3 in v-Src-transformed fibroblasts requires cooperation of Jak1 kinase activity. J Biol Chem. 2000;275:24935–44. doi: 10.1074/jbc.M002383200. [DOI] [PubMed] [Google Scholar]
- 28.Giese B, Au-Yeung CK, Herrmann A, et al. Long term association of the cytokine receptor gp130 and the Janus kinase Jak1 revealed by FRAP analysis. J Biol Chem. 2003;278:39205–13. doi: 10.1074/jbc.M303347200. [DOI] [PubMed] [Google Scholar]
- 29.Novotny-Diermayr V, Lin B, Gu L, Cao X. Modulation of the interleukin-6 receptor subunit glycoprotein 130 complex and its signaling by LMO4 interaction. J Biol Chem. 2005;280:12747–57. doi: 10.1074/jbc.M500175200. [DOI] [PubMed] [Google Scholar]
- 30.Yoon YK, Kim HP, Han SW, et al. KRAS mutant lung cancer cells are differentially responsive to MEK inhibitor due to AKT or STAT3 activation: Implication for combinatorial approach. Molecular Carcinogenesis. 2010;49:353–62. doi: 10.1002/mc.20607. [DOI] [PubMed] [Google Scholar]
- 31.Darnell JE. Validating Stat3 in cancer therapy. Nat Med. 2005;11:595–6. doi: 10.1038/nm0605-595. [DOI] [PubMed] [Google Scholar]
- 32.Yu H, Pardoll D, Jove R. STATs in cancer inflammation and immunity: a leading role for STAT3. Nat Rev Cancer. 2009;9:798–809. doi: 10.1038/nrc2734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Wang YF, Jiang CC, Kiejda KA, Gillespie S, Zhang XD, Hersey P. Apoptosis induction in human melanoma cells by inhibition of MEK is caspase-independent and mediated by the Bcl-2 family members PUMA, Bim, and Mcl-1. Clin Cancer Res. 2007;13:4934–42. doi: 10.1158/1078-0432.CCR-07-0665. [DOI] [PubMed] [Google Scholar]
- 34.Brunet A, Bonni A, Zigmond MJ, et al. Akt promotes cell survival by phosphorylating and inhibiting a Forkhead transcription factor. Cell. 1999;96:857–68. doi: 10.1016/s0092-8674(00)80595-4. [DOI] [PubMed] [Google Scholar]
- 35.Obsil T, Obsilova V. Structure/function relationships underlying regulation of FOXO transcription factors. Oncogene. 2008;27:2263–75. doi: 10.1038/onc.2008.20. [DOI] [PubMed] [Google Scholar]
- 36.Yang JY, Zong CS, Xia W, et al. ERK promotes tumorigenesis by inhibiting FOXO3a via MDM2-mediated degradation. Nat Cell Biol. 2008;10:138–48. doi: 10.1038/ncb1676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.O’Reilly LA, Kruse EA, Puthalakath H, et al. MEK/ERK-mediated phosphorylation of Bim is required to ensure survival of T and B lymphocytes during mitogenic stimulation. J Immunol. 2009;183:261–9. doi: 10.4049/jimmunol.0803853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Ewings KE, Wiggins CM, Cook SJ. Bim and the pro-survival Bcl-2 proteins: opposites attract, ERK repels. Cell Cycle. 2007;6:2236–40. doi: 10.4161/cc.6.18.4728. [DOI] [PubMed] [Google Scholar]
- 39.Mu P, Han YC, Betel D, et al. Genetic dissection of the miR-17~92 cluster of microRNAs in Myc-induced B-cell lymphomas. Genes Dev. 2009;23:2806–11. doi: 10.1101/gad.1872909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Ventura A, Young AG, Winslow MM, et al. Targeted deletion reveals essential and overlapping functions of the miR-17 through 92 family of miRNA clusters. Cell. 2008;132:875–86. doi: 10.1016/j.cell.2008.02.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Xiao C, Srinivasan L, Calado DP, et al. Lymphoproliferative disease and autoimmunity in mice with increased miR-17-92 expression in lymphocytes. Nat Immunol. 2008;9:405–14. doi: 10.1038/ni1575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Hayashita Y, Osada H, Tatematsu Y, et al. A polycistronic microRNA cluster, miR-17-92, is overexpressed in human lung cancers and enhances cell proliferation. Cancer Res. 2005;65:9628–32. doi: 10.1158/0008-5472.CAN-05-2352. [DOI] [PubMed] [Google Scholar]
- 43.Fornari F, Milazzo M, Chieco P, et al. MiR-199a-3p regulates mTOR and c-Met to influence the doxorubicin sensitivity of human hepatocarcinoma cells. Cancer Res. 70:5184–93. doi: 10.1158/0008-5472.CAN-10-0145. [DOI] [PubMed] [Google Scholar]
- 44.Zhou M, Liu Z, Zhao Y, et al. MicroRNA-125b confers the resistance of breast cancer cells to paclitaxel through suppression of pro-apoptotic Bcl-2 antagonist killer 1 (Bak1) expression. J Biol Chem. 285:21496–507. doi: 10.1074/jbc.M109.083337. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.