

Abstract
Novel inhibitors of PI3K, Akt and mTOR have been developed recently, some of which have entered clinical trials. Although such compounds inhibit cell proliferation, their effects on cell survival, an important determinant of clinical response, are less clear-cut. Using a broad panel of myeloma cell lines and primary patient samples we show that dual PI3K and mTOR inhibition can induce cell death. The effects are most marked in cells expressing the t(4;14) translocation, whereas t(11;14) cells are largely resistant. Using specific inhibitors of individual pathway components, we show that optimal induction of cell death requires inhibition of both PI3K and mTOR. This is due to a PI3K-independent component of mTOR activation downstream of the MAP Kinase pathway. Novel mTOR kinase inhibitors, which block both TORC1 and TORC2 complexes thereby also reducing Akt activity, are less effective than dual PI3K/mTOR inhibitors because of feedback activation of PI3K signalling. Dual PI3K/mTOR inhibitors sensitise t(4;14) and t(14;16), but not t(11;14), expressing cells to the cytotoxic effects of dexamethasone. We have identified a robust cytogenetic biomarker for response to PI3K/mTOR inhibition - these results will inform the design and prioritisation of clinical studies with novel inhibitors in genetic subgroups of myeloma.
Keywords: PI3Kinase, myeloma, mTOR, translocation
Introduction
Multiple myeloma, a malignancy of plasma cells, shows considerable heterogeneity of pathophysiology, disease tempo and response to therapy. Genetic subtypes which carry prognostic significance can be identified and different classification systems based on myeloma cell biology have been proposed (reviewed in (1)). Abnormal karyotypes are present at a very high frequency and cases of myeloma can be broadly categorised into hyperdiploid and non-hyperdiploid subtypes (1). The latter are enriched for cases with translocations involving the immunoglobulin heavy chain locus on chromosome 14, about 40-50% of all cases, that deregulate partner genes such as c-MAF/MAFB (eg t(14;16)), MMSET/FGFR3 (t(4;14)) and cyclins D1 (t(11;14)) and D3 (t(6;14)) (1). Cytogenetic subtypes are associated with differing outcomes – for example, t(4;14) is associated with an increased incidence of extramedullary disease and a worse outcome with standard therapies (2). Despite the recent advances in treatments for myeloma, cure remains rare, hence new therapeutic approaches are still required.
The PI3-kinase pathway is frequently deregulated in human tumours by a variety of mechanisms (3). Class 1A PI3Ks are the group most clearly implicated in cancer and consist of a regulatory subunit and one of three catalytic subunits, p110α, p110β or p110δ (4). PI3K deregulation in cancer can result from a number of different mechanisms: mutational activation or overexpression of upstream regulators (such as tyrosine kinases and Ras); somatic mutations of the p110α catalytic subunit PIK3CA, the p85 regulatory subunit PIK3R1 or the kinase Akt; and the loss of negative regulators including the lipid phosphatase PTEN (reviewed in (5)). The targets of PI3K signalling include the Akt kinase and related AGC kinases (such as SGK1) and pathway activation can lead to changes in cell growth, survival, metabolism and motility (3). A major downstream target of Akt signalling is TSC2 which controls activity of the mTOR pathway (6). The mTOR serine/threonine kinase is related to the PI3Ks and exists in at least two intracellular multiprotein complexes, mTORC1 and mTORC2 (7). mTORC1, which is inhibited by Rapamycin in complex with FKBP12, is involved in the regulation of protein translation and cell growth via effects on 4EBP-1 and S6-kinase 1. The mTORC2 complex, which is largely Rapamycin-insensitive, is involved in the phosphorylation of several AGC family kinases on a hydrophobic motif which contributes to maximal functional activation. These include Akt (at serine 473), several PKC family members and SGK1 (6).
In the last few years, a large number of novel therapeutics that target PI3K, Akt and mTOR signalling have been developed, in addition to more established compounds such as Rapamycin and its analogues (3, 8). These new agents include inhibitors of individual (p110α, p110β or p110δ) or all class 1 PI3K isoforms, steric or ATP-competitive Akt inhibitors and ATP-competitive inhibitors of mTORC1 and TORC2 signalling. In addition, pan-class 1 PI3K inhibitors with dual mTOR kinase inhibitory activity are available.
The PI3K pathway is frequently activated in myeloma but the mechanisms for this are uncertain as the incidence of PIK3CA mutation and PTEN deletion/mutation is low (9-17). A number of PI3K and mTOR pathway inhibitory compounds have demonstrated activity in pre-clinical studies in myeloma (14, 18-21). In general, mTOR inhibitors such as Rapamycin and its analogues are cytostatic in cell culture assays and have a relatively low response rate as single agents in clinical studies (22). This may be due, in part, to upregulation of PI3K signalling via the insulin-like growth factor pathway due to loss of a negative feedback loop involving S6K1 (7, 23). Disease heterogeneity may also lead to the dilution of therapeutic effects, as molecular subgroups differ in responses to therapy and clinical outcome.(24) Importantly, a key predictor of clinical response is the induction of tumour cell death in pre-clinical assays, as observed by Engelman and others,(25-27) highlighting the need for biomarkers that predict for response to PI3K pathway inhibition. The presence of mutations in the PI3K pathway may indicate a higher likelihood of response, but this is not always the case in other tumour types (28). Increased levels of Akt phosphorylation may also predict for a biological effect of Akt inhibitors (17), however growing evidence suggests that the detection of phosphorylated proteins in primary tumour samples is fraught with technical difficulties (29-32). Hence the identification of more robust and widely applicable biomarkers is needed.
The availability of such a wide range of potential novel therapeutic agents makes it vital to obtain robust pre-clinical information, as it is unlikely that all compounds or combinations could be tested in clinical trials. We have carried out a direct comparison of class 1 PI3K inhibitors, dual PI3K/mTOR inhibitors, novel mTOR inhibitors, rapamycin and analogues, and Akt inhibitors to define optimal induction of myeloma cell death and to assess if cytogenetic subtypes may predict for response. We show that optimal killing requires dual PI3K/mTOR inhibition, is associated with tumours bearing the t(4;14) translocation and with increased Akt phosphorylation, and is negatively associated with the t(11;14) translocation. Our findings have important implications for the design of clinical studies in myeloma.
Materials and Methods
Cells and reagents
The MM1S cell line was donated by Dr S. Rosen (Northwestern University, Chicago, USA), KMS cell lines by Dr Otsuki (Kawasaki Medical School, Okayama, Japan) and LP-1 and JIM-1 cell lines by Prof G. Morgan, Institute of Cancer Research, London, UK. Other human myeloma cell lines (HMCL) were obtained from the American Type Culture Collection (ATCC; LGC Promochem, Teddington, United Kingdom) or from the Deutsche Sammlung von Mikroorganismen und Zellkulturen (DSMZ, Braunschweig, Germany). Primary MM cells isolated from BM aspirates were obtained from MM patients, after informed consent in accordance with the Declaration of Helsinki with ethical approval from the Joint UCL/UCLH institutional review boards. CD138+ plasma cells were isolated using magnetic-activated cell sorting CD138 MicroBeads (Miltenyi Biotech, Surrey, UK). Stromal HS-5 cells were purchased from the ATCC and transduced to express blue fluorescent protein (BFP). Cell lines and primary cells were grown in RPMI-1640/10% (v/v) fetal calf serum, 1% (v/v) penicillin/streptomycin. All cells were cultured at 37°C under 5 % CO2 in a humidified incubator. Inhibitors were obtained as follows and were prepared as 10 mM or 100 mM stock in DMSO and stored at −20°C until use: PI103 and AKTi1/2 (Merck Chemicals, Darmstadt, Germany); PD184352, BEZ235 and PIK90 (Axon Medchem, Groningen, The Netherlands); Rapamycin and Everolimus (LC laboratories, Woburn, MA, USA); PP242, WYE-354 and KU-0063 (Chemdea, Ridgewood, NJ, USA) and Dexamethasone (Sigma-Aldrich, Gillingham, UK).
Immunoblotting
Proteins were extracted from cells lysed [lysis buffer; 50 mM Hepes pH 7.5, 100 mM NaCl, 1% Triton X-100, 0·5% sodium deoxycholate, 20 mM NaF, 2 mM EDTA, 1mM Na orthovanadate, complete protease inhibitors (P2714, Sigma-Aldrich)] on ice for 30 min. Proteins were separated by SDS-polyacrylamide gel electrophoresis (SDS-PAGE) and blotted onto a nitrocellulose membrane (GE Healthcare, Buckingham, UK). Anti-phospho-AKT S473 (D9E), phospho-AKT T308, phospho-p70 S6 kinase T389, phospho-S6 S235/236, PTEN, phospho-p44/42 MAPK (137F5), p4EBP1 T37/46 and GAPDH (14C10) antibodies were purchased from Cell Signaling (New England Biolabs, Hertfordshire, UK). All antibodies were used at 1:1000. After a second incubation with peroxidase-conjugated anti-rabbit IgG (GE Healthcare) the blots were developed with the ECL system (GE Healthcare).
Immunofluorescence assays
HMCLs were fixed with 2% paraformaldehyde and permeabilised with 90% methanol before staining with anti-phospho-AKT (Cell Signaling, S473) followed by APC-conjugated goat anti-rabbit IgG antibody (Cell Signaling). Samples were analysed by flow cytometry using a Cyan ADP (Beckman Coulter, Bucks, UK) and data expressed as median cell fluorescence.
Proliferation and Apoptosis Analysis
A 200 μL aliquot of cells was added to 200 μL Annexin V binding buffer (10 mM HEPES pH 7.4, 150 mM NaCl, 10 mM CaCl2) and stained with 5 μL of propidium iodide (Sigma-Aldrich) at 50 g/L and 0.25 μL of FITC conjugated Annexin V (Roche Diagnostics, Burgess Hill, UK). A fixed number of Flowcheck Fluorospheres (Beckman Coulter) were added to each sample in order to quantify viable cell number. The fraction of live cells (Annexin and PI negative) and the absolute number of live cells was quantified by flow cytometry (Cyan ADP, Beckman Coulter) and treated samples compared with control cells to give information on apoptosis rates and cell proliferation.
2D direct contact HS-5 stromal co-culture
HS-5 cells modified to express BFP were cultured as a monolayer until 80% confluence. Medium was discarded and replaced by HMCLs in suspension. After treatment, suspension and trypsinised cells were pooled and centrifuged. The supernatant was carefully removed to ensure that the ratio of beads to cells was not variable between samples. Cells were resuspended in Annexin-V buffer and analysed for proliferation and apoptosis as described above. BFP-positive HS-5 cells were gated and excluded from analysis.
JC-1 assay
To monitor the mitochondrial membrane potential, JC-1 assays were carried out with the BD MitoScreen kit from BD Biosciences (San Diego, CA, USA) following the manufacturer’s recommendations.
Caspase assay
Cells were sonicated for 5 min and incubated 1 h at 37°C in Caspase 3 Assay buffer (20mM HEPES, 2mM EDTA, 0.1% CHAPS, 5mM DTT, pH7.4) containing 8.25μM Caspase 3 substrate Ac-DEVD-Amc (Sigma). Caspase 3 activity was then monitored by spectroscopy with an excitation wavelength of 340nm and an emission wavelength of 435nm.
PIP2 and PIP3 assays
Lipids were extracted from HMCLs and analysed by PIP2 and PIP3 Elisa assays (Echelon Biosciences Inc, Salt Lake City, USA) according to the manufacturer’s recommendations. PIP3 data were normalised to PIP2 levels to control for variability in lipid extraction procedures.
Statistical analysis
The statistical analytic functions of the Prism software package (Graphpad, La Jolla, CA, USA) were used as indicated in the results.
Results
Activation of the PI3K/Akt pathway is associated with t(4;14) in HMCL
Initial investigation of the activation of PI3K signalling, as judged by Akt phosphorylation on S473 by immunoblotting, suggested that HMCL with t(4;14) translocation showed higher levels of PI3K activity (Figure 1A) and that this activity was low in t(11;14) bearing cells. The t(4;14) cell line OPM2 had loss of PTEN as previously described (10) and there was minimal/absent protein expression in H929 cells, but all other cell lines were PTEN replete (supplementary information, Figure S1A). There was no clear correlation between Akt phosphorylation and activation of mTOR signalling as judged by phosphorylation of the ribosomal S6 protein.
Figure 1. Activation of the PI3K pathway in a panel of HMCL representing the major non-hyperdiploid IgH translocation subtypes.
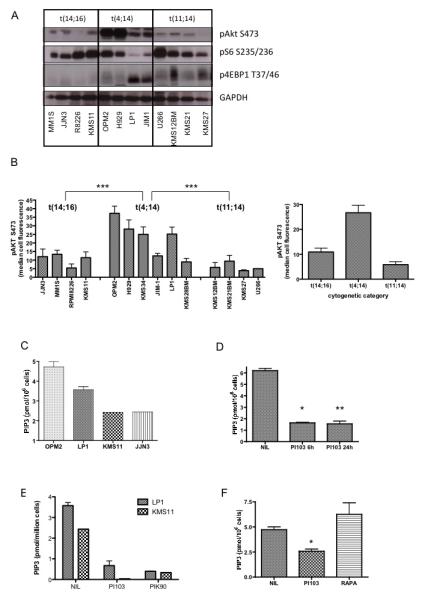
A. Lysates from a selection of HMCL were immunoblotted with the indicated antibodies. The translocation subtypes are labelled above the top panel. The KMS11 cell line is positive for both t(14;16) and t(4;14).
B. Akt phosphorylation at serine 473 was quantified by flow cytometry in the individual indicated cell lines (mean ± SEM of replicates of at least 3 separate experiments for each cell line). Akt phosphorylation was significantly higher in t(4;14) cells compared with either t(14;16) (p=0.0003 by Mann-Whitney test) or t(11;14) cells (p-0.0002). The difference between t(14;16) and t(11;14) cells was of borderline statistical significance (p=0.06). Pooled data for all the cell lines in each cytogenetic category is shown in the right hand panel.
C – F. Cell extracts were analysed for PIP3 levels by ELISA.
C. Steady state PIP3 levels in two t(4;14) cell lines (OPM2 and LP1) and two t(14:16) cell lines (KMS11, JJN3) were measured.
D. OPM2 cells were incubated with 1uM PI103 for the indicated times and PIP3 measured.
E. KMS11 and LP1 cells were incubated with either 1uM PI103 or 1uM PIK90 for 6 hours and residual PIP3 levels measured.
F. OPM2 cells were incubated with 1uM PI103 or 20nM Rapamycin for 6 hours and residual PIP3 levels measured.
In order to use a more quantitative assay of PI3K pathway activation, we evaluated relative levels of pAkt by flow cytometry in a larger panel of cell lines (Figure 1B and Figure S1B). This confirmed the significantly higher levels of pAkt in t(4;14) cells, in comparison with the low levels in t(11;14) cells. Levels of pAkt in t(14;16) cell lines were slightly higher than in t(11;14) cells and this was of borderline statistical significance (p=.06)
In order to assess if increased Akt phosphorylation was caused by elevated PI3K activity, direct measurement of cellular phosphatidylinositol (3,4,5)-trisphosphate (PIP3) levels was carried out in a subset of cell lines. This showed higher levels of PIP3 in t(4;14) cells compared with t(14;16) cells, consistent with the pAkt results (Figure 1C). PIP3 levels in t(11;14) cells were below the limit of detection of the assay. Incubation of cells with the PI3K/mTOR inhibitor PI103 (33) or the PI3K inhibitor PIK90 (Table 1), but not the mTOR inhibitor Rapamycin, led to a marked and sustained reduction in PIP3 levels in t(4;14) and t(14;16) bearing cells (Figure 1D-F).
Table 1.
| COMPOUND | TARGET |
|---|---|
| PI103, BEZ-235 | PI3K, mTORC1, mTORC2 |
| PIK90 | PI3K |
| WYE654, KU0063, PP242 | mTORC1, mTORC2 |
| RAPAMYCIN | mTORC1 |
| AKTi1/2 | AKT |
| PD184352 | MAPKK |
Induction of cell death in t(4;14) HMCL by dual inhibition of PI3K and mTOR
HMCL were incubated with different concentrations of PI103, a dual inhibitor of PI3K and mTOR, in order to assess the effect on cell survival (Annexin V/PI staining by flow cytometry). Variable responses were seen in an initial panel of cell lines, with some cells showing marked sensitivity and others resistance (Figure 2A) and an indication that t(4;14) cells may be more sensitive. The complete HMCL panel was subsequently exposed to an optimal PI103 concentration for 72 hours. Results showed that t(4;14) cells were most sensitive to PI103, while t(11;14) cells were largely resistant (Figure 2B). t(14;16) cells showed intermediate sensitivity. (Figure 2B) Cell death was significantly higher in t(4;14) cells compared with either t(14;16) (p=0.0001 by Mann-Whitney test) or t(11;14) cells (p=0.0001). The difference between t(14;16) and t(11;14) cells was small but significant (p=0.02) (Figure 2C)
Figure 2. Effects of the dual PI3K/mTOR inhibitor PI103 on myeloma cell survival.
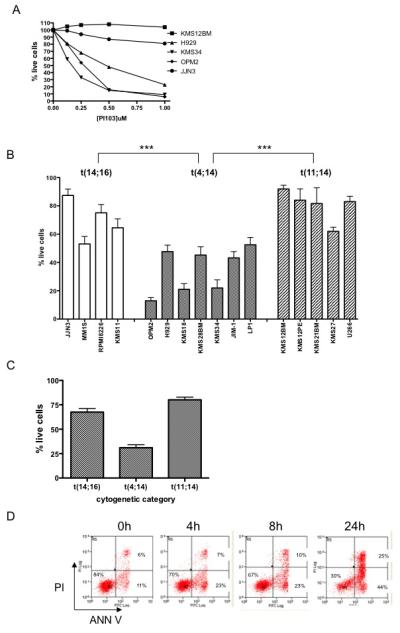
A. Myeloma cell lines were incubated with varying concentrations of PI103 for 72 hours and then analysed by flow cytometry (Annexin V/PI staining) to quantify cell survival (normalised to the respective untreated control sample). KMS12BM express t(11;14); JJN3 t(14;16) and KMS34, OPM2 and H929 express t(4;14).
B. Myeloma cell lines were incubated with 1μM PI103 for 72 hours and cell survival was quantified by Annexin V/PI flow cytometry in the individual indicated cell lines. PI103 induced significantly higher cell death in t(4;14) cells compared with either t(14;16) (p=0.0001 by Mann-Whitney test) or t(11;14) cells (p-0.0001). The difference between t(14;16) and t(11;14) cells was also statistically significant (p=0.02).
C. Pooled survival data after incubation with 1μM PI103 for 72 hours for all cell lines from each cytogenetic category.
D. OPM2 cells incubated with 1μM PI103 were analysed by Annexin V/PI staining at the indicated time-points showing the appearance of an Annexin-positive PI-negative apoptotic population.
E. HMCL were incubated with the indicated compounds and mitochondrial membrane potential (left panel) and Caspase activity (right panel, JIM1 cell line) analysed. PI103 at 1μM, PIK90 at 1μM, Rapamycin at 20nM.
F. Data for cell death induced by 1μM PI103 after 72 hours in individual cell lines plotted against steady-state level of phosphorylated Akt S473 (as measured by flow cytometry). Cell death induced by PI103 is significantly correlated with Akt phosphorylation (r=0.718, p=0.0017 by Spearman correlation test).
Cell death was apoptotic as shown by the early appearance of Annexin V positive/PI negative cells, loss of mitochondrial membrane potential and Caspase-3 activation (Figure 2D, E). In order to assess if the degree of PI3K pathway activation could predict for the induction of cell death by PI103, we compared the basal level of Akt phosphorylation with the proportion of cells killed after 72 hours incubation. We found a significant correlation between constitutive Akt phosphorylation and subsequent PI103-induced cell death (Figure 2F).
Optimal induction of cell death requires inhibition of both PI3K and mTOR pathways
A wide range of compounds that target PI3K and mTOR are in pre-clinical and clinical evaluation and are likely to have differing toxicity profiles. Having identified a cytotoxic effect of dual PI3K/mTOR inhibition in the t(4;14) MMSET subset of myeloma cells, we wished to investigate which components of this pathway are required for optimal induction of cell death. Therefore, we utilised highly selective inhibitors of PI3K, Akt and mTOR to assess the comparative effects of blocking individual components of these pathways (see Table 1). Dose-finding assays were first carried out to confirm target inhibition and the lowest concentration that caused target inhibition was used in order to minimise the risk of off-target effects. (data not shown and supplementary data Figure S2)
Results depicted in Figure 3A show that individual blockade of PI3K with optimal concentrations of PIK90 (34), Akt with the allosteric inhibitor AKTi1/2 (35) or mTOR with Rapamycin was significantly less effective than dual blockade of PI3K and mTOR with PI103 in t(4;14) cells. Higher concentrations of PIK90 did not result in enhanced cell killing (supplementary data Figure S3). A molecularly distinct dual PI3K/mTOR inhibitor, BEZ-235 (36), produced similar results to PI103 on t(4;14) cells (Figure 3A and supplementary data Figure S4). The effect of BEZ-235 on t(11;14) and t(14;16) expressing cells was also tested and was shown to be similar to PI103 in being more effective at inducing cell death compared with PIK90 or rapamycin, although effects in these sub-groups were smaller than in t(4;14) cells (Figure 3B). BEZ-235 was more active than PI103 in inducing cell death in the t(11;14) group – this compound has additional activity against class II and III PI3Ks and this may be responsible for the slight difference compared to PI103.
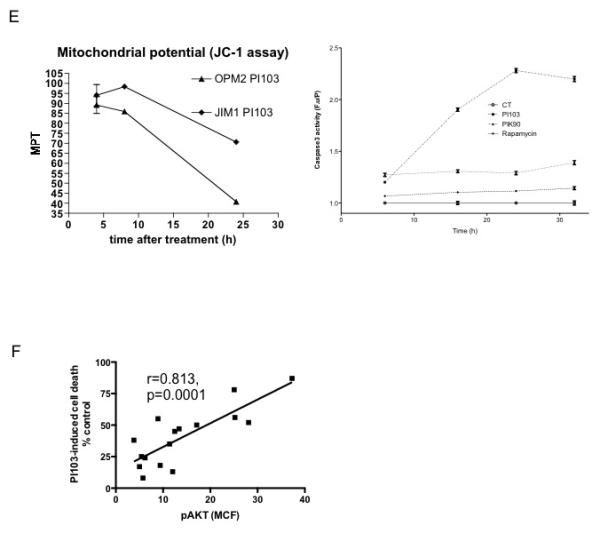
Figure 3. Optimal induction of cell death in t(4;14) myeloma cell lines requires blockade of both PI3K and mTOR.
A. t(4;14) expressing myeloma cell lines were incubated with 1μM PI103 (dual PI3K/mTOR inhibitor), 1μM PIK90 (PI3K inhibitor), 5μM Akti (Akt inhibitor), 20nM Rapamycin (mTOR inhibitor) or 1μM BEZ235 (dual PI3K/mTOR inhibitor) for 72 hours and cell survival quantified by Annexin V/PI flow cytometry. The cell death induced by PI103 is significantly greater than that seen with either PIK90 (p<0.0001 by Mann-Whitney test) or Rapamycin (p<0.0001) but not different to BEZ235 (p=0.975). The data are pooled from all t(4;14) cell lines.
B. t(11;14) or t(14;16) expressing myeloma cell lines were incubated with 1μM PI103, 1μM BEZ235, 1μM PIK90 or 20nM Rapamycin for 72 hours and cell survival quantified by Annexin V/PI flow cytometry.
C. The t(4;14) expressing cell line JIM-1 was incubated with 1μM PI103, 1μM PIK90, 1μM BEZ235, 20nM rapamycin or the combination of PIK90 and Rapamycin for 4 or 8 hours and cell lysates immunoblotted with the indicated antibodies.
D. t(4;14) expressing myeloma cell lines were incubated with 1μM PI103 (dual PI3K/mTOR inhibitor), 1μM PIK90 (PI3K inhibitor), 20nM Rapamycin (mTOR inhibitor) or the combination of PIK90 and Rapamycin for 72 hours and cell survival quantified by Annexin V/PI flow cytometry. The data are pooled from all t(4;14) cell lines.
E. LP-1 cells were incubated with 1μM PI103, 1μM PIK90, 10μM PD184352 (MAPKK/MEK inhibitor) or the combination of PIK90 and PD184352 and cell lysates immunoblotted with the indicated antibodies.
In many cell types, mTOR signalling is predominantly regulated by PI3K and its target Akt, acting on the TSC complex - inhibition of either PI3K or Akt would be predicted to eliminate mTOR activation. In order to investigate why inhibition of PI3K was significantly less effective at inducing myeloma cell death compared with dual PI3K/mTOR inhibition, we looked at downstream signalling pathways in cells treated with various inhibitors. Incubation of cells with dual inhibitors PI103 or BEZ-235 inhibited PI3K signalling, as shown by abrogated Akt phosphorylation, and mTOR signalling as shown by reduced S6 and 4EBP1 phosphorylation (Figure 3C). The PI3K inhibitor PIK90 was also effective in reducing Akt phosphorylation but despite this, mTOR pathways were only partially inhibited. Similar results were seen in several cell lines (data not shown). This indicates that in myeloma cells, there is significant PI3K-independent activation of mTOR signalling. Rapamycin did not reduce Akt phosphorylation but inhibited S6 phosphorylation. The combination of PIK90 and Rapamycin reduced both Akt and S6 phosphorylation to levels seen with PI103 or BEZ-235 but had varying effects on phosphorylation of 4EBP1, in keeping with recent data on incomplete inhibition of mTORC1 signalling by Rapamycin (37). We next investigated the effect of combined PI3K (with PIK90) and mTORC1 (with Rapamycin) inhibition on cell survival in comparison with PI103 in 6 different t(4;14) cell lines – the results showed that the combination was as effective as the dual inhibitor PI103 (Figure 3D). Similar results were seen with the combination of Akti1/2 plus rapamycin (Supplementary data Figure S5).
To identify which signalling pathway is responsible for PI3K-independent mTOR signalling in myeloma cells, we incubated them with the MAPKK inhibitor PD184352 with or without the PI3K inhibitor PIK90. (Figure 3E) The results show PI3K-independent mTOR signalling in myeloma cells is likely to be due to activation of Ras/MAPK signalling as evidenced by inhibition of S6 phosphorylation by PD184352 and near elimination of S6 phosphorylation by dual inhibition of PI3K and MAPKK with PIK90 and PD184352 respectively (Figure 3E) (38). Blockade of PI3K led to enhanced MAPK phosphorylation – Akt is known to phosphorylate RAF at an inhibitory site, serine 259,(39) and PI3K/Akt inhibition could therefore result in enhanced RAF activity and increased MAPK phosphorylation.
Novel mTOR kinase inhibitors are less effective than dual PI3K/mTOR blockade in inducing cell death
Novel mTOR kinase inhibitors, which inhibit both mTORC1 and mTORC2, have been recently developed (37, 40-42). These have been shown to be more effective TORC1 inhibitors than Rapamycin and in addition reduce Akt activity by inhibiting TORC2 phosphorylation of AktS473, a modification required for maximal kinase activity. In some (40, 41), but not all (42), reported studies, TOR kinase inhibitors can also reduce the phosphorylation of Akt at T308, a site phosphorylated by PDK1, by an unknown mechanism. We utilised three structurally unrelated TORC kinase inhibitors, PP242, WYE-354 and KU-0063794, to assess their effects on myeloma cell growth and survival. Assessment of downstream signalling indicated that mTOR kinase inhibitors blocked the phosphorylation of the mTORC1 targets S6 and 4EBP1 (Figure 4A).
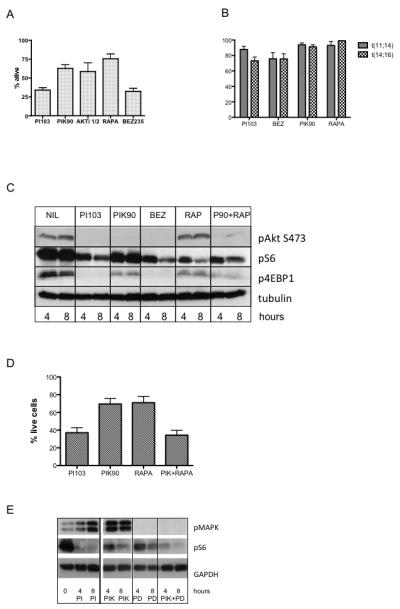
Figure 4. The effects of novel mTOR kinase inhibitors on myeloma cell survival and signalling.
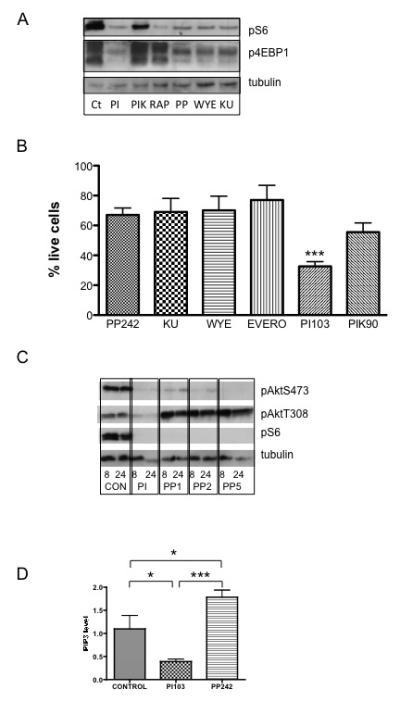
A. OPM2 cells were incubated with 1μM PI103 (dual PI3K/mTOR inhibitor), 1μM PIK90 (PI3K inhibitor), 20nM Rapamycin (mTOR inhibitor), 1μM PP242, 1μM WYE-354, 1μM KU-0063 (all mTOR kinase inhibitors) for 6 hours and cell lysates immunoblotted with the indicated antibodies.
B. The panel of t(4;14) expressing cell lines were incubated with 1uM PP242, KU-0063, WYE-354, 1μM Everolimus, 1μM PI103 or 1μM PIK90 for 72 hours and cell survival quantified by Annexin V/PI flow cytometry. Results are normalised to untreated controls and are mean ± SEM of at least 3 separate experiments. The difference between mTOR kinase inhibitors and PI103 was statistically significant – for example, for PP242 versus PI103, p=0.001.
C. OPM2 cells were incubated with 1μM PI103, or varying concentrations of PP242 (1, 2 or 5 μM) for 8 or 24 hours and cell lysates immunoblotted with the indicated antibodies.
D. OPM2 cells were incubated with 1μM PI103 or 1μM PP242 for 24 hours and cell extracts analysed for PIP3 levels by ELISA. Mean ± SEM of triplicate samples.
Preliminary experiments showed that TORC kinase inhibitors could reduce cell proliferation in a variety of myeloma cells (data not shown). We went on to compare the effect of TORC kinase inhibitors with dual PI3K/mTOR inhibition on cell death. In all t(4;14) myeloma cell lines tested, although mTOR kinase inhibitors could induce cell death, they were significantly less effective than PI103 (Figure 4B and supplementary data Figure S6). Therefore, we went on to analyse the effects of mTOR kinase inhibitors on Akt phosphorylation. PP242, at concentrations ranging from 1 to 5uM, inhibited Akt phosphorylation at S473, as expected for an mTORC2-mediated event (Figure 4C). However, incubation with mTOR kinase inhibitors led to a marked increase in phosphorylation of Akt at T308, the key phosphorylation site for regulating activity (Figure 4C). To investigate this further, we measured PIP3 levels in cells incubated with PP242 and found them to be significantly elevated (Figure 4D) – this is likely to account for the increased AktT308 phosphorylation. Such an increase in PIP3 levels and the increased Akt phosphorylation at T308 is likely to counteract the cytotoxic effects of TORC kinase inhibitors in myeloma and may be due to the known negative feedback effects of TORC1 on insulin and IGF1 signalling (7).
Dual inhibitors of PI3K and mTOR enhance the cytotoxic effect of glucocorticoids in t(4;14) and t(14;16) HMCL, but not in t(11;14) lines
Glucocorticoids are amongst the most active agents in myeloma but resistance may be present, or develop, in particular in relapsed disease. Previous studies have suggested that there is a link between PI3K/mTOR signalling and glucocorticoid-induced apoptosis.(43, 44) Therefore, we examined the effect on cell death induction of the combination of the dual PI3K/mTOR inhibitor PI103 and Dexamethasone in a variety of HMCL. Initial dose-finding experiments showed marked co-operativity in several cell lines (supplementary data Figure S7 ). Further investigation using the broad panel of cell lines showed that co-operativity was restricted to cells expressing t(4;14) and t(14;16), with no significant effect of the combination seen in t(11;14) cell lines (Figure 5A).
Figure 5. The effect of combining PI3K and mTOR inhibitors with Dexamethasone on myeloma cell survival.
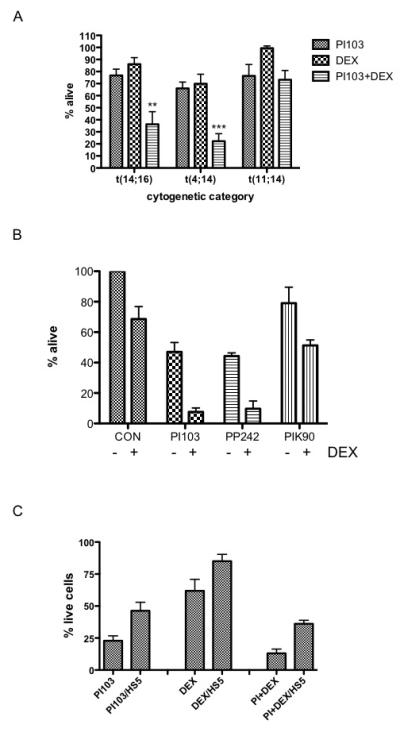
A. Cell lines from each of the three major IgH translocation subtypes were incubated with PI103, Dexamethasone or the combination for 72 hours and cell survival quantified by Annexin V/PI flow cytometry. 1μM PI103 and 1μM Dexamethasone were used to treat t(14;16) and t(11;14) cells. The concentration of PI103 was reduced to 0.25μM for t(4;14) cells to evaluate the combined effects with Dexamethasone as higher concentrations caused significant cell death as a single agent.
B. OPM2 cells were incubated with 1μM Dexamethasone and either 0.25μM PI103, 0.25μM PP242 or 1μM PIK90 for 72 hours and cell survival quantified by Annexin V/PI flow cytometry.
C. OPM2 cells were incubated with 1μM PI103, 1μM Dexamethasone or the combination in standard suspension cultures or in co-cultures with the stromal cell line HS-5. Viable OPM2 cells were quantified by flow cytometry and normalised to untreated controls.
In order to assess the key component of dual PI3K/mTOR inhibition which enhances glucocorticoid responses, comparison was made between PI103, PP242 (mTOR inhibitor) and PIK90 (PI3K inhibitor). Results showed that mTOR kinase inhibition with PP242 was as effective as PI103 in combination with dexamethasone (Figure 5B) and more effective than PIK90. These results suggest that effective inhibition of mTOR signalling is crucial to sensitising myeloma cells to glucocorticoids.
As stromal cells may modify the response to cytotoxic agents in myeloma and other malignancies, we repeated these studies in the presence of the bone marrow stromal cell line HS-5. Although the effects of PI103 as a single agent were attenuated by stroma, significant levels of cell death were still detected (Figure 5C). PI103 also enhanced the effects of dexamethasone in the presence of stromal support.
Effects of PI3K and mTOR inhibitors on primary myeloma cells of different cytogenetic subgroups
To investigate if the results obtained in HMCL were representative of those seen in primary tumour samples, CD138 selected cells from patients from different cytogenetic subgroups were incubated with PI103, BEZ-235, PIK90, Rapamycin or the combination of PIK90 and Rapamycin. Annexin V/PI staining showed that, as with the cell lines, t(4;14) positive cells were the most sensitive to PI3K/mTOR inhibition (Figure 6A). The effects of PI103 were dose-dependent with an IC50 value around 200nM (Figure 6A). Optimal cell killing required the blockade of both PI3K and mTOR either with dual inhibitors (PI103, BEZ-235) or with the combination of PIK90 and Rapamycin (Figure 6B). Immunoblotting showed that primary myeloma cells also demonstrate PI3K-independent activation of S6 phosphorylation which is sensitive to Rapamycin (Figure 6C).
Figure 6. The effect of PI3K and mTOR inhibition on primary myeloma cell survival.
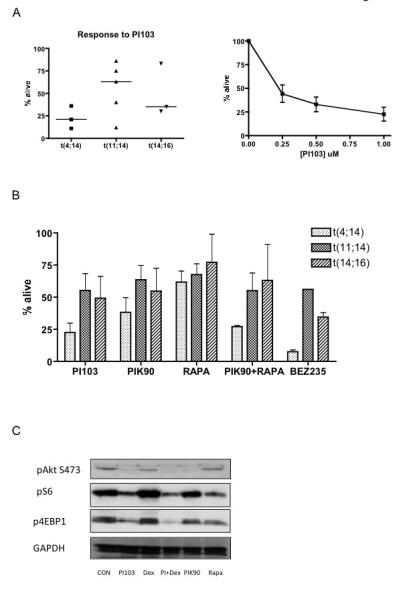
A. CD138 positive primary myeloma cells were selected from bone marrow samples from 3 patients with t(4;14), 5 with t(11;14) and 3 with t(14;16) and incubated with the dual PI3K/mTOR inhibitor PI103 (1μM) for 48-72 hours and cell survival quantified by Annexin V/PI flow cytometry (left panel). A PI103 dose-response curve for the three t(4;14) samples is shown in the right hand panel.
B. Primary myeloma cells from patients in the different cytogenetic subgroups were incubated with either 1μM PI103 (dual PI3K/mTOR inhibitor), 1μM PIK90 (PI3K inhibitor, 20nM Rapamycin (mTOR inhibitor), a combination of PIK90 and Rapamycin or 1μM BEZ235 (dual PI3K/mTOR inhibitor) for 48-72 hours and cell survival quantified by Annexin V/PI flow cytometry.
C. Primary myeloma cells were incubated with either 1μM PI103, 1μM Dexamethasone, a combination of PI103 and Dexamethasone, 1μM PIK90 or Rapamycin for 4 hours and cell lysates immunoblotted with the indicated antibodies.
Discussion
The last few years have seen the rapid development of small molecule inhibitors of various components of the PI3K signalling pathway (3, 8, 28). A number of these are already in early phase clinical trials. These compounds include those that are targeted to individual catalytic isoforms of class 1 PI3K or to all class 1 PI3Ks or indeed to selected combinations. Novel selective kinase inhibitors of mTOR are also available (7) and some molecules can inhibit both PI3K and mTOR. The availability of such a broad range of compounds makes it imperative to identify, in pre-clinical studies, those that have greatest anti-tumour activity, the genetic context in which this occurs, and robust biomarkers for the selection of relevant patient subgroups (28, 45). We have utilised highly selective inhibitors of PI3K and mTOR signalling to carry out a direct comparison in myeloma.
There is increasing evidence that induction of cell death is important for predicting a beneficial clinical response to a therapeutic compound - most established anti-tumour agents induce cell killing and this is also likely to be necessary for more targeted therapies (5, 8, 26, 27, 45). We found that myeloma cell lines bearing the t(4;14) translocation were significantly more likely to undergo cell death in response to dual inhibition of PI3K and mTOR signalling when compared to the other common IgH translocation subtypes, t(11;14) and t(14;16). This was associated with higher levels of PI3K pathway activity in t(4;14) cells as measured by Akt phosphorylation or by direct quantification of PIP3 levels. The mechanism for increased pathway activation is unclear although we identified higher levels of DEPTOR protein in these cells compared with t(11;14) lines (data not shown). It has previously been shown that myeloma cells, in particular those with deregulated MAF, have higher levels of DEPTOR and that this can enhance PI3K signalling by relieving the negative regulation of IGF1/IRS/PI3K by mTOR/S6K1 (7, 46). t(4;14) cells have also recently been shown to have distinctive methylation profiles, presumed to be a consequence of deregulated MMSET which possesses histone methyltransferase activity.(47) Deregulated methylation of regulators of PI3K activity may contribute to the increased signalling seen in t(4;14) bearing cells - for example, IRS2 which positively mediates PI3K signalling, has been shown to be hypomethylated and overexpressed in this subtype.(47)
In order to define more stringently the component requirements for PI3K pathway blockade induced cell death in myeloma cells, we made use of highly selective PI3K inhibitors that do not affect mTOR signalling directly. Selective inhibitors of p110β and p110δ (48) failed to inhibit proliferation or to trigger significant levels of cell death (data not shown). Pan class 1 PI3K blockade, although effective at reducing proliferation and blocking Akt activation, was significantly less effective at inducing cell death when compared with compounds with dual anti-PI3K and mTOR activity. Regulation of the mTOR module is considered to be one of the main outputs of PI3K and Akt signalling – if mTOR is primarily regulated by the PI3K/Akt pathway, we would predict that blockade of PI3K should be functionally equivalent to dual inhibition of PI3K and mTOR. The data shown here indicate that in myeloma cells, a significant proportion of mTOR activity is not controlled by PI3K (see Figure 3) but emanates from the RAS/MAPK pathway, which can also regulate mTOR signalling via effects on TSC proteins (38). Therefore, PI3K inhibitory compounds that lack significant direct anti-mTOR activity that are already in early clinical trials may be less effective in this tumour type. Of interest, combining a PI3K inhibitor with Rapamycin was as effective as using a dual PI3K/mTOR kinase inhibitor - Rapamycin only affects mTOR in the TORC1 complex and its effects on this complex result in incomplete inhibition of downstream outputs (7). This suggests that the important component of mTOR inhibition by dual inhibitors in this context is the effect on TORC1 and not TORC2. These results are summarised in Figure 7.
Figure 7. Pathway diagram of PI3K, Akt and mTOR signalling and effects of specific inhibitors in this study.
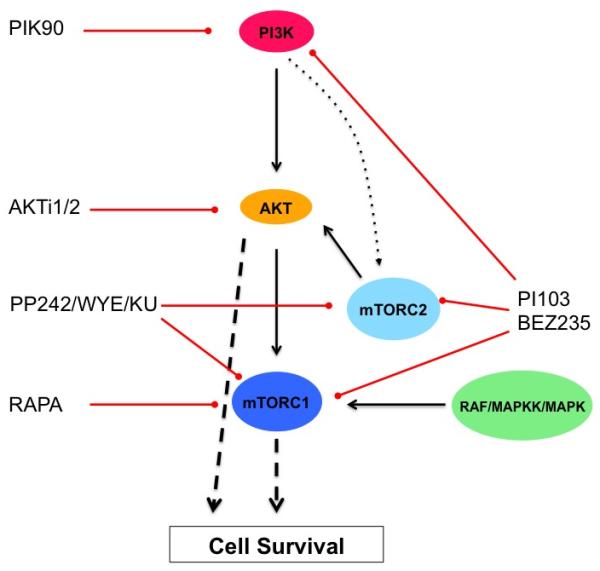
PI3K pathway components showing the level at which the inhibitory compounds used in this study have their activity. Optimal induction of cell death requires inhibition of both PI3K/Akt and of mTORC1 signalling. This can be achieved by dual inhibitors PI103 and BEZ235 or by combining PIK90 or AKTi1/2 with rapamycin.
The development of potent selective mTOR active site kinase inhibitors has also allowed us to address the question of whether complete and effective blockade of this pathway can induce cell death in myeloma cells. A recent publication has shown activity of one of these agents, PP242, in myeloma(49). Our results with three distinct compounds, including PP242, show that although TOR kinase inhibitors have potent anti-proliferative effects (data not shown), these compounds do not induce the levels of cell death seen with dual PI3K/mTOR inhibition. A potential reason for this, as we have demonstrated, is feedback activation of the PI3K pathway resulting from mTOR blockade leading to elevated PIP3 levels and marked enhancement of Akt phosphorylation at threonine 308, a key regulatory site for kinase activity. Such effects may also account for the limited clinical activity of Rapamycin and its analogues (7). Recent papers have shown that mTOR kinase inhibitors, as expected, reduce phosphorylation of Akt at serine 473, an mTORC2 target site. Some, but not all, workers have also shown reduced phosphorylation at Akt threonine 308 by an unknown mechanism, raising the possibility that these compounds may have more potent pro-apoptotic effects than Rapamycin (40-42). However, we have shown that in myeloma cells, mTOR kinase inhibitors do not reduce, but rather increase, Akt T308 phosphorylation, and this is associated with elevated PIP3 levels. The reasons for these differences are not clear but may relate to long-term versus short-term effects of the inhibitors, or to cell type-specific effects.
We have also shown that dual PI3K/mTOR inhibition can potently sensitise myeloma cells to the effects of Dexamethasone. Rapamycin has previously been shown to have similar activity, (44) but direct comparison with PI103 shows the latter to be more effective (data not shown). In this instance, the effects of PP242, an mTOR kinase inhibitor, approach those seen with PI103 suggesting that the main component responsible for the interaction with glucocorticoids is likely to be effective inhibition of mTOR. This is in keeping with previous findings showing that 4EBP1 plays an important part in promoting Dexamethasone-induced apoptosis, (44) as 4EBP1 is more effectively dephosphorylated by mTOR kinase inhibitors compared with Rapamycin. The impact of PI3K/mTOR inhibition on Dexamethasone-induced cell death was seen in t(4;14) and t(14;16), but not in t(11;14) cells. We were unable to detect consistent changes in candidate BCL2 family members including MCL1 and BIM in response to individual compounds and combinations (data not shown). Further studies will be required to understand the mechanisms behind these findings.
Our findings in myeloma cell lines were confirmed in primary CD138 selected tumour cells, which were found to behave in a similar manner - t(4;14) bearing cells are most sensitive and t(11;14) least sensitive to dual PI3K/mTOR inhibition and maximal cell killing is achieved when both PI3K and mTOR kinase activities are blocked. The identification of robust predictive biomarkers is important for the clinical development of targeted therapies. Although previous studies have indicated that histologically detected Akt activation predicts sensitivity to Akt inhibition,(17) the detection of phosphorylated proteins by immunohistochemistry or other techniques from primary tissue is fraught with technical challenges – in particular, there may be rapid de-phosphorylation of key residues during sample preparation and fixation.(29-32) In contrast, translocation subtypes in myeloma are readily identified using FISH probes, thus providing a robust and stable biomarker.
In conclusion, we have shown that cell death can be induced in myeloma cells by dual PI3K and mTOR inhibition. In direct experimental comparisons, dual inhibitors were shown to be more effective than selective inhibitors of PI3K that lack anti-mTOR activity or novel mTOR kinase inhibitors. We have identified the t(4;14) translocation as a biomarker for sensitivity to PI3K/mTOR inhibition and also shown that the t(11;14) subgroup is resistant. Our findings provide a rational platform for the design and evaluation of clinical studies of PI3K and mTOR targeted therapies in myeloma.
Supplementary Material
Acknowledgements
This work was supported by grants from Cancer Research UK and Leukaemia & Lymphoma Research UK. The work was undertaken at University College London/University College London Hospitals, who received a proportion of funding from the Department of Health’s NIHR Comprehensive Biomedical Research Centres funding scheme and is a CRUK Cancer Centre and a Leukaemia and Lymphoma Research Centre of Excellence.
Footnotes
Author contributions AK and KY designed the study; JQ and KY provided vital reagents; CS, CWC and AK carried out experiments; AK wrote the manuscript, which was reviewed by all authors.
Supplementary information is available at Leukemia’s website
The authors declare no conflicts of interest.
References
- 1.Fonseca R, Bergsagel PL, Drach J, Shaughnessy J, Gutierrez N, Stewart AK, et al. International Myeloma Working Group molecular classification of multiple myeloma: spotlight review. Leukemia. 2009 Dec;23(12):2210–2221. doi: 10.1038/leu.2009.174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Avet-Loiseau H, Leleu X, Roussel M, Moreau P, Guerin-Charbonnel C, Caillot D, et al. Bortezomib plus dexamethasone induction improves outcome of patients with t(4;14) myeloma but not outcome of patients with del(17p) J Clin Oncol. 2010 Oct 20;28(30):4630–4634. doi: 10.1200/JCO.2010.28.3945. [DOI] [PubMed] [Google Scholar]
- 3.Liu P, Cheng H, Roberts TM, Zhao JJ. Targeting the phosphoinositide 3-kinase pathway in cancer. Nat Rev Drug Discov. 2009 Aug;8(8):627–644. doi: 10.1038/nrd2926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Vanhaesebroeck B, Guillermet-Guibert J, Graupera M, Bilanges B. The emerging mechanisms of isoform-specific PI3K signalling. Nat Rev Mol Cell Biol. 2010 May;11(5):329–341. doi: 10.1038/nrm2882. [DOI] [PubMed] [Google Scholar]
- 5.Wong KK, Engelman JA, Cantley LC. Targeting the PI3K signaling pathway in cancer. Curr Opin Genet Dev. 2010 Feb;20(1):87–90. doi: 10.1016/j.gde.2009.11.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Foster KG, Fingar DC. Mammalian target of rapamycin (mTOR): conducting the cellular signaling symphony. J Biol Chem. 2010 May 7;285(19):14071–14077. doi: 10.1074/jbc.R109.094003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Efeyan A, Sabatini DM. mTOR and cancer: many loops in one pathway. Curr Opin Cell Biol. 2010 Apr;22(2):169–176. doi: 10.1016/j.ceb.2009.10.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Brachmann S, Fritsch C, Maira SM, Garcia-Echeverria C. PI3K and mTOR inhibitors: a new generation of targeted anticancer agents. Curr Opin Cell Biol. 2009 Apr;21(2):194–198. doi: 10.1016/j.ceb.2008.12.011. [DOI] [PubMed] [Google Scholar]
- 9.Ismail SI, Mahmoud IS, Msallam MM, Sughayer MA. Hotspot mutations of PIK3CA and AKT1 genes are absent in multiple myeloma. Leuk Res. 2010 Jun;34(6):824–826. doi: 10.1016/j.leukres.2009.11.018. [DOI] [PubMed] [Google Scholar]
- 10.Ge NL, Rudikoff S. Expression of PTEN in PTEN-deficient multiple myeloma cells abolishes tumor growth in vivo. Oncogene. 2000 Aug 24;19(36):4091–4095. doi: 10.1038/sj.onc.1203801. [DOI] [PubMed] [Google Scholar]
- 11.Hyun T, Yam A, Pece S, Xie X, Zhang J, Miki T, et al. Loss of PTEN expression leading to high Akt activation in human multiple myelomas. Blood. 2000 Nov 15;96(10):3560–3568. [PubMed] [Google Scholar]
- 12.Hsu J, Shi Y, Krajewski S, Renner S, Fisher M, Reed JC, et al. The AKT kinase is activated in multiple myeloma tumor cells. Blood. 2001 Nov 1;98(9):2853–2855. doi: 10.1182/blood.v98.9.2853. [DOI] [PubMed] [Google Scholar]
- 13.Choi Y, Zhang J, Murga C, Yu H, Koller E, Monia BP, et al. PTEN, but not SHIP and SHIP2, suppresses the PI3K/Akt pathway and induces growth inhibition and apoptosis of myeloma cells. Oncogene. 2002 Aug 8;21(34):5289–5300. doi: 10.1038/sj.onc.1205650. [DOI] [PubMed] [Google Scholar]
- 14.Pene F, Claessens YE, Muller O, Viguie F, Mayeux P, Dreyfus F, et al. Role of the phosphatidylinositol 3-kinase/Akt and mTOR/P70S6-kinase pathways in the proliferation and apoptosis in multiple myeloma. Oncogene. 2002 Sep 26;21(43):6587–6597. doi: 10.1038/sj.onc.1205923. [DOI] [PubMed] [Google Scholar]
- 15.Chang H, Qi XY, Claudio J, Zhuang L, Patterson B, Stewart AK. Analysis of PTEN deletions and mutations in multiple myeloma. Leuk Res. 2006 Mar;30(3):262–265. doi: 10.1016/j.leukres.2005.07.008. [DOI] [PubMed] [Google Scholar]
- 16.Younes H, Leleu X, Hatjiharissi E, Moreau AS, Hideshima T, Richardson P, et al. Targeting the phosphatidylinositol 3-kinase pathway in multiple myeloma. Clin Cancer Res. 2007 Jul 1;13(13):3771–3775. doi: 10.1158/1078-0432.CCR-06-2921. [DOI] [PubMed] [Google Scholar]
- 17.Zollinger A, Stuhmer T, Chatterjee M, Gattenlohner S, Haralambieva E, Muller-Hermelink HK, et al. Combined functional and molecular analysis of tumor cell signaling defines 2 distinct myeloma subgroups: Akt-dependent and Akt-independent multiple myeloma. Blood. 2008 Oct 15;112(8):3403–3411. doi: 10.1182/blood-2007-11-119362. [DOI] [PubMed] [Google Scholar]
- 18.Frost P, Moatamed F, Hoang B, Shi Y, Gera J, Yan H, et al. In vivo antitumor effects of the mTOR inhibitor CCI-779 against human multiple myeloma cells in a xenograft model. Blood. 2004 Dec 15;104(13):4181–4187. doi: 10.1182/blood-2004-03-1153. [DOI] [PubMed] [Google Scholar]
- 19.McMillin DW, Ooi M, Delmore J, Negri J, Hayden P, Mitsiades N, et al. Antimyeloma activity of the orally bioavailable dual phosphatidylinositol 3-kinase/mammalian target of rapamycin inhibitor NVP-BEZ235. Cancer Res. 2009 Jul 15;69(14):5835–5842. doi: 10.1158/0008-5472.CAN-08-4285. [DOI] [PubMed] [Google Scholar]
- 20.Shi Y, Gera J, Hu L, Hsu JH, Bookstein R, Li W, et al. Enhanced sensitivity of multiple myeloma cells containing PTEN mutations to CCI-779. Cancer Res. 2002 Sep 1;62(17):5027–5034. [PubMed] [Google Scholar]
- 21.Baumann P, Mandl-Weber S, Oduncu F, Schmidmaier R. The novel orally bioavailable inhibitor of phosphoinositol-3-kinase and mammalian target of rapamycin, NVP-BEZ235, inhibits growth and proliferation in multiple myeloma. Exp Cell Res. 2009 Feb 1;315(3):485–497. doi: 10.1016/j.yexcr.2008.11.007. [DOI] [PubMed] [Google Scholar]
- 22.Coiffier B, Ribrag V. Exploring mammalian target of rapamycin (mTOR) inhibition for treatment of mantle cell lymphoma and other hematologic malignancies. Leuk Lymphoma. 2009 Dec;50(12):1916–1930. doi: 10.3109/10428190903207548. [DOI] [PubMed] [Google Scholar]
- 23.Shi Y, Yan H, Frost P, Gera J, Lichtenstein A. Mammalian target of rapamycin inhibitors activate the AKT kinase in multiple myeloma cells by up-regulating the insulin-like growth factor receptor/insulin receptor substrate-1/phosphatidylinositol 3-kinase cascade. Mol Cancer Ther. 2005 Oct;4(10):1533–1540. doi: 10.1158/1535-7163.MCT-05-0068. [DOI] [PubMed] [Google Scholar]
- 24.Bergsagel PL, Kuehl WM. Molecular pathogenesis and a consequent classification of multiple myeloma. J Clin Oncol. 2005 Sep 10;23(26):6333–6338. doi: 10.1200/JCO.2005.05.021. [DOI] [PubMed] [Google Scholar]
- 25.Brachmann SM, Hofmann I, Schnell C, Fritsch C, Wee S, Lane H, et al. Specific apoptosis induction by the dual PI3K/mTor inhibitor NVP-BEZ235 in HER2 amplified and PIK3CA mutant breast cancer cells. Proc Natl Acad Sci U S A. 2009 Dec 29;106(52):22299–22304. doi: 10.1073/pnas.0905152106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Faber AC, Li D, Song Y, Liang MC, Yeap BY, Bronson RT, et al. Differential induction of apoptosis in HER2 and EGFR addicted cancers following PI3K inhibition. Proc Natl Acad Sci U S A. 2009 Nov 17;106(46):19503–19508. doi: 10.1073/pnas.0905056106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Turke AB, Engelman JA. PIKing the right patient. Clin Cancer Res. 2010 Jul 15;16(14):3523–3525. doi: 10.1158/1078-0432.CCR-10-1201. [DOI] [PubMed] [Google Scholar]
- 28.Vanhaesebroeck B, Vogt PK, Rommel C. PI3K: From the Bench to the Clinic and Back. Curr Top Microbiol Immunol. 2010;347:1–19. doi: 10.1007/82_2010_65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Baker AF, Dragovich T, Ihle NT, Williams R, Fenoglio-Preiser C, Powis G. Stability of phosphoprotein as a biological marker of tumor signaling. Clin Cancer Res. 2005 Jun 15;11(12):4338–4340. doi: 10.1158/1078-0432.CCR-05-0422. [DOI] [PubMed] [Google Scholar]
- 30.Burns JA, Li Y, Cheney CA, Ou Y, Franlin-Pfeifer LL, Kuklin N, et al. Choice of fixative is crucial to successful immunohistochemical detection of phosphoproteins in paraffin-embedded tumor tissues. J Histochem Cytochem. 2009 Mar;57(3):257–264. doi: 10.1369/jhc.2008.952911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Pinhel IF, Macneill FA, Hills MJ, Salter J, Detre S, A’Hern R, et al. Extreme loss of immunoreactive p-Akt and p-Erk1/2 during routine fixation of primary breast cancer. Breast Cancer Res. 2010 Sep 28;12(5):R76. doi: 10.1186/bcr2719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Marchio C, Dowsett M, Reis-Filho JS. Revisiting the technical validation of tumour biomarker assays: how to open a Pandora’s box. BMC Med. 2011;9:41. doi: 10.1186/1741-7015-9-41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Raynaud FI, Eccles S, Clarke PA, Hayes A, Nutley B, Alix S, et al. Pharmacologic characterization of a potent inhibitor of class I phosphatidylinositide 3-kinases. Cancer Res. 2007 Jun 15;67(12):5840–5850. doi: 10.1158/0008-5472.CAN-06-4615. [DOI] [PubMed] [Google Scholar]
- 34.Knight ZA, Gonzalez B, Feldman ME, Zunder ER, Goldenberg DD, Williams O, et al. A pharmacological map of the PI3-K family defines a role for p110alpha in insulin signaling. Cell. 2006 May 19;125(4):733–747. doi: 10.1016/j.cell.2006.03.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Lindsley CW, Zhao Z, Leister WH, Robinson RG, Barnett SF, Defeo-Jones D, et al. Allosteric Akt (PKB) inhibitors: discovery and SAR of isozyme selective inhibitors. Bioorg Med Chem Lett. 2005 Feb 1;15(3):761–764. doi: 10.1016/j.bmcl.2004.11.011. [DOI] [PubMed] [Google Scholar]
- 36.Maira SM, Stauffer F, Brueggen J, Furet P, Schnell C, Fritsch C, et al. Identification and characterization of NVP-BEZ235, a new orally available dual phosphatidylinositol 3-kinase/mammalian target of rapamycin inhibitor with potent in vivo antitumor activity. Mol Cancer Ther. 2008 Jul;7(7):1851–1863. doi: 10.1158/1535-7163.MCT-08-0017. [DOI] [PubMed] [Google Scholar]
- 37.Thoreen CC, Kang SA, Chang JW, Liu Q, Zhang J, Gao Y, et al. An ATP-competitive mammalian target of rapamycin inhibitor reveals rapamycin-resistant functions of mTORC1. J Biol Chem. 2009 Mar 20;284(12):8023–8032. doi: 10.1074/jbc.M900301200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Roux PP, Ballif BA, Anjum R, Gygi SP, Blenis J. Tumor-promoting phorbol esters and activated Ras inactivate the tuberous sclerosis tumor suppressor complex via p90 ribosomal S6 kinase. Proc Natl Acad Sci U S A. 2004 Sep 14;101(37):13489–13494. doi: 10.1073/pnas.0405659101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Zimmermann S, Moelling K. Phosphorylation and regulation of Raf by Akt (protein kinase B) Science. 1999 Nov 26;286(5445):1741–1744. doi: 10.1126/science.286.5445.1741. [DOI] [PubMed] [Google Scholar]
- 40.Feldman ME, Apsel B, Uotila A, Loewith R, Knight ZA, Ruggero D, et al. Active-site inhibitors of mTOR target rapamycin-resistant outputs of mTORC1 and mTORC2. PLoS Biol. 2009 Feb 10;7(2):e38. doi: 10.1371/journal.pbio.1000038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Garcia-Martinez JM, Moran J, Clarke RG, Gray A, Cosulich SC, Chresta CM, et al. Ku-0063794 is a specific inhibitor of the mammalian target of rapamycin (mTOR) Biochem J. 2009 Jul 1;421(1):29–42. doi: 10.1042/BJ20090489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Yu K, Toral-Barza L, Shi C, Zhang WG, Lucas J, Shor B, et al. Biochemical, cellular, and in vivo activity of novel ATP-competitive and selective inhibitors of the mammalian target of rapamycin. Cancer Res. 2009 Aug 1;69(15):6232–6240. doi: 10.1158/0008-5472.CAN-09-0299. [DOI] [PubMed] [Google Scholar]
- 43.Wei G, Twomey D, Lamb J, Schlis K, Agarwal J, Stam RW, et al. Gene expression-based chemical genomics identifies rapamycin as a modulator of MCL1 and glucocorticoid resistance. Cancer Cell. 2006 Oct;10(4):331–342. doi: 10.1016/j.ccr.2006.09.006. [DOI] [PubMed] [Google Scholar]
- 44.Yan H, Frost P, Shi Y, Hoang B, Sharma S, Fisher M, et al. Mechanism by which mammalian target of rapamycin inhibitors sensitize multiple myeloma cells to dexamethasone-induced apoptosis. Cancer Res. 2006 Feb 15;66(4):2305–2313. doi: 10.1158/0008-5472.CAN-05-2447. [DOI] [PubMed] [Google Scholar]
- 45.Engelman JA. Targeting PI3K signalling in cancer: opportunities, challenges and limitations. Nat Rev Cancer. 2009 Aug;9(8):550–562. doi: 10.1038/nrc2664. [DOI] [PubMed] [Google Scholar]
- 46.Peterson TR, Laplante M, Thoreen CC, Sancak Y, Kang SA, Kuehl WM, et al. DEPTOR is an mTOR inhibitor frequently overexpressed in multiple myeloma cells and required for their survival. Cell. 2009 May 29;137(5):873–886. doi: 10.1016/j.cell.2009.03.046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Walker BA, Wardell CP, Chiecchio L, Smith EM, Boyd KD, Neri A, et al. Aberrant global methylation patterns affect the molecular pathogenesis and prognosis of multiple myeloma. Blood. 2010 Oct 13; doi: 10.1182/blood-2010-04-279539. [DOI] [PubMed] [Google Scholar]
- 48.Billottet C, Banerjee L, Vanhaesebroeck B, Khwaja A. Inhibition of class I phosphoinositide 3-kinase activity impairs proliferation and triggers apoptosis in acute promyelocytic leukemia without affecting atra-induced differentiation. Cancer Res. 2009 Feb 1;69(3):1027–1036. doi: 10.1158/0008-5472.CAN-08-2608. [DOI] [PubMed] [Google Scholar]
- 49.Hoang B, Frost P, Shi Y, Belanger E, Benavides A, Pezeshkpour G, et al. Targeting TORC2 in multiple myeloma with a new mTOR kinase inhibitor. Blood. 2010 Nov 25;116(22):4560–4568. doi: 10.1182/blood-2010-05-285726. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.