

Abstract
Autophagy is a major innate immune defense pathway in both plants and animals. In mammals, this cascade can be elicited by cytokines (IFN-γ) or pattern recognition receptors (TLRs and nucleotide-binding oligomerization domain-like receptors [NLRs]). Many signaling components in TLR- and NLR-induced autophagy are now known; however, those involved in activating autophagy via IFN-γ remain to be elucidated. Here, we engineered macrophages encoding a tandem fluorescently tagged LC3b (tfLC3) autophagosome reporter along with stably integrated shRNAs to demonstrate IFN-γ-induced autophagy required JAK 1/2, PI3K, and p38 MAPK but not STAT1. Moreover, the autophagy-related GTPase Irgm1 proved dispensable in both stable tfLC3-expressing RAW 264.7 and tfLC3-transduced Irgm1-/- primary macrophages, revealing a novel p38 MAPK-dependent, STAT1-independent autophagy pathway that bypasses Irgm1. These unexpected findings have implications for understanding how IFN-γ-induced autophagy is mobilized within macrophages for inflammation and host defense.
INTRODUCTION
Macrophages are involved in many essential host functions including clearance of infection, tissue homeostasis and repair, and resolution of inflammation (1). The type II IFN, IFN-γ, remains the prototypic macrophage-activating factor following its secretion by proliferating type1 T helper cells and NK cells (2). IFN-γ initiates its signal transduction cascade via tyrosine phosphorylation of STAT1 by JAK1 and JAK2 (3-5). Subsequently, STAT1 dimers bind to IFN-stimulated response elements and induce the transcription of several hundred IFN-stimulated genes (3, 4). In addition to the JAK-STAT1 pathway, IFN-γ induces gene expression via STAT1-independent pathways (5-7) and modulates various signaling cascades, including MyD88 (8), p38 MAPK (9), PI3K (10), and protein kinase C (11, 12). In macrophages, the major immune target genes of IFN-γ include MHC class I and II, inflammatory and pyrogenic cytokines (TNF-α and IL-6), chemokines, and antimicrobial proteins such as inducible nitric oxide synthase 2 (NOS2), phagocyte oxidase and immune GTPases (3-5, 13, 14). Notably, IFN-γ is critical for cell-autonomous innate immunity against bacteria, protozoa, viruses, and fungi (14-16).
Autophagy has emerged as a major immune defense pathway in both plant and animal kingdoms (17, 18). In mammals, this cascade can be elicited by host-derived cytokines, such as IFN-γ, or pattern recognition receptors, including TLRs and nucleotide-binding oligomerization domain (NOD)-like receptors (NLRs) (18-24). IFN-γ-induced autophagy is also posited to control intracellular pathogens via several members of the immunity-related guanosine triphosphatase (GTPase) family (IRG proteins, formerly known as p47 GTPases) (16, 19, 20, 25-28) and more recently by members of the 65 kDa guanylate binding protein family (Gbps) (29). The mouse IRG family consists of 17-20 Irg genes, and their expression is driven by IFN-stimulated response sequences and γ-activated sequences (30). One IRG protein in particular, Irgm1, has been touted to exert critical functions in IFN-γ-induced autophagy (20). A human ortholog of Irgm1, IRGM (30), reportedly shares similar biologic duties which may include mitophagy (20, 28). Unlike its mouse counterpart, however, human IRGM is not elicited by IFN-γ. Instead, its expression is driven by the endogenous retrovirus element ERV9 (30, 31). This raises the possibility of an IRGM-independent signaling pathway for IFN-γ-induced autophagy.
In this study, we generated macrophages harboring integrated chromosomal copies of a tandem red fluorescent protein (RFP)-green fluorescent protein (GFP) tagged LC3b (tfLC3) autophagosome reporter (32). This reporter exploits the loss of fluorescence of GFP (pKa, 6.0) but not RFP (pKa, 4.5) in the acidic environment of lysosomes to label autophagic compartments during their maturation to autolysosomes, which form by fusing sequentially or independently with endosomes and lysosomes (18). Moreover, introduction of the tandem fluorescent reporter into primary bone-marrow derived macrophages (BMMs) from Irgm1-deficient mice (Irgm1-/-) also demonstrated that this GTPase is dispensible during the early stages of autophagosome biogenesis. Our results suggest that IFN-γ accelerates not only the formation but also the maturation of autophagosomes and does so via a pathway different to the one implicating Irgm1 described above. This novel pathway uses JAK1/2 and p38 MAPK signaling but does not require STAT1. Our findings open up the possibility of searching for new STAT1- and Irgm1-independent genes during IFN-γ-induced autophagic control of infection.
MATERIALS AND METHODS
Mice
Wild-type (WT), STAT-/-, and Irgm1-/- mice were bred at Yale University School of Medicine, and primary BMMs were derived from the mice as reported previously (25). All animal experiments were undertaken according to Yale institutional guidelines for the handling and care of such mice.
Plasmids and tfLC3 macrophage cell lines
mRFP-GFP-LC3b plasmid (a generous gift from Dr. Tamotsu Yoshimori, Osaka University) was transfected into RAW 264.7 murine macrophages or primary BMMs using Lipofectamine LTX (Invitrogen, Carlsbad, CA) or Nucleofector system (Amaxa Biosystems, Cologne, Germany) to stably express tfLC3. To generate pLKD.neo, a lentiviral vector to express short hairpin RNA (shRNA), a fragment of the human U6 promoter was PCR amplified with the primers 5′-CGGAATTCTTCCAATGCATTGGCTGCAGACCGGTGTTTCGTCCTTTCCAC-3′ and 5′-TGAATTCTCGACCTCGAGAC-3′, and pLKO.1-puro/shGqα (Sigma, St Louis, MO) as the template. The amplicon was digested with NruI and EcoRI (Takara Bio Inc., Tokyo, Japan) before cloning into pLKO.1-puro/shGqα to generate the pLKD.pur vector. The neomycin resistance gene was PCR amplified with the primers 5′-CGGGATCCCGCATGATTGAACAAGATGG-3′ and 5′-GGGGTACCTCAGAAGAACTCGTCAAGAAG-3′ and pEGFP-N1 (Clontech, Palo Alto, CA) as the template. The amplicon was digested with BamHI and KpnI (Takara Bio Inc.) before cloning into pLKD.pur to obtain the pLKD.neo vector. Subsequently, pLKD.pur or pLKD.neo lentiviruses were constructed using the Addgene pLKO.1 protocol (www.addgene.org). The RNAi sequences were as follows: Atg7, 5’-TTCTGTCACGGTTCGATAATG-3’; IFN-γR1, 5’-GCCAGAGTTAAAGCTAAGGTT-3’ (shIFN-γR1-1) and 5’-CCACATAGAATATCAGACTTA-3’ (shIFN-γR1-2); STAT1, 5’-CCGAAGAACTTCACTCTCTTA-3’ (shSTAT1-1) and 5’-TTGCAAGAGCTGAACTATAAC-3’ (shSTAT1-2); Irgm1, 5’-CCAGAAATCAAGAGATACAAA-3’ (shIrgm1-1) and 5’-CGACTGATATTTGGTGTAGAT-3’ (shIrgm1-2). The ViraPower Lentiviral Expression system (Invitrogen) was used to co-transfect the viral vector into 293FT cells (Invitrogen) to produce lentiviruses. The resulting viral supernatant was used to infect RAW 264.7 cells, and then stable knockdown cells were selected with Puromycin or G418 (Clontech).
Reagents
Recombinant mouse IFN-γ was purchased from R&D Systems (Minneapolis, MN). p38 MAPK inhibitor, E64d, and JAK inhibitor I were bought from Calbiochem (Darmstadt, Germany). LY294002, a PI3K inhibitor, was from Cell Signaling (Danvers, MA). Pepstatin A was obtained from Sigma.
Fluorescent staining
RAW 264.7 cells or primary BMMs were fixed with 3% paraformaldehyde in PBS for 10 min, washed 3 times with PBS, and then stained with DAPI (Invitrogen) to detect nuclei. The cells were observed using Nikon ECLIPSE Ti2000 a fluorescence microscope (Nikon Instech Co, Kawasaki, Japan).
Western blotting
After IFN-γ treatment, macrophages were washed with PBS, lysed in buffer (50 mM Tris, pH 7.5, 1% Triton X-100, 150 mM NaCl) plus protease inhibitor cocktail (Roche, Mannheim, Germany) and the lysates subjected to SDS-PAGE before transferring to polyvinylidene fluoride membranes. Western blotting was performed with the following antibodies: rabbit anti-Atg7 polyclonal Ab (pAb) (GeneTex, Irvine, CA), mouse anti-LC3b mAb (2G6; Nano Tools, Hamburg, Germany), rabbit anti-NOS2 rabbit pAb (ENZO Life Sciences, Plymouth Meeting, PA), mouse anti-GAPDH mAb (6C5; Santa Cruz Biotechnology), rabbit anti-STAT1 pAb (GenScript, Piscataway, NJ), rabbit anti-phospho STAT1 pAb (GenScript), anti-IFN-γR pAb (GeneTex), goat anti-Irgm1 pAb (A-19; Santa Cruz Biotechnology), anti-actin pAb (Sigma), rabbit anti-p38, phospho-p38, Erk1/2, phospho-Erk1/2, SAPK/JNK, and phospho-SAPK/JNK mAbs (Cell Signaling), HRP-conjugated anti-mouse IgG Ab (Jackson ImmunoResearch Laboratories, West Grove, PA), HRP-conjugated anti-rabbit IgG Ab (Jackson Immuno Research Laboratories), and HRP-conjugated anti-goat IgG Ab (Santa Cruz Biotechnology). The expression of GAPDH and actin was used as the internal control. The intensity of bands was quantified using ImageJ software.
RESULTS
IFN-γ enhances autophagosome biogenesis in tfLC3 reporter macrophages
Using clonally derived RAW 264.7 macrophage cells expressing tfLC3 (RAW-tfLC3), we found both the number and size of autophagosomes increased in a dose-dependent manner following treatment with IFN-γ (0.1-300 U/ml) for 24 h (Fig. 1, A, B, and F). Furthermore, time-course analysis revealed that autophagosome formation began within ~60 minutes of IFN-γ stimulation (200 U/ml) and peaked between 2-6 h after cytokine treatment (Fig. 1H). Similar analyses revealed that IFN-γ promoted the maturation of autophagosomes to autolysosomes (detected only as RFP vacuoles), a process that began 2 h after IFN-γ stimulation and peaked slightly later, between 4-8 h after stimulation (Fig. 1, B, G, and I). Both processes could be blocked by provision of shRNA against the core autophagic adaptor protein, Atg7, confirming fidelity of the bipartite reporter (Fig. 1B-E). These results suggest that IFN-γ promotes the formation of autophagosomes as well as their maturation to autolysosomes via canonical Atg pathways. Western blotting with an anti-LC3b mAb confirmed that the activation of LC3b-I to LC3b-II in IFN-γ-treated RAW 264.7 cells on treatment with the lysosomal protease inhibitors E64d and pepstatin A to prevent LC3b degradation within autolysosomes (Fig. 1J). This also peaked between 2-4 h after IFN-γ treatment. Hence we measured the IFN-γ-induced formation and maturation of autophagosomes at this time in genetic and pharmacologic loss-of-function experiments described below.
Fig. 1.
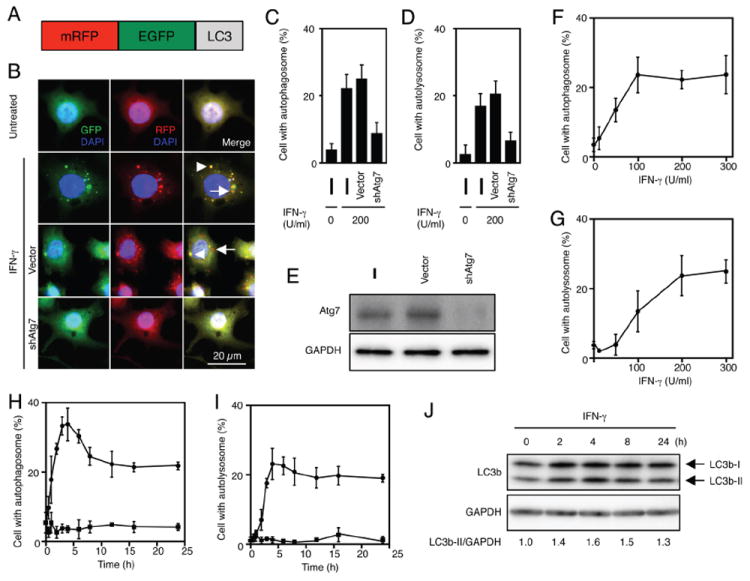
IFN-γ enhances autophagosome biogenesis in tfLC3 reporter macrophages. A, Diagram of tfLC3 reporter. B-D, RAW-tfLC3 cells and stable Atg7 knockdown cells that expressed tfLC3 were incubated with 200 U/ml IFN-γ for 24 h. The cells were fixed and stained with DAPI to detect nuclei. Representative fluorescent images are shown, and arrowheads or arrows indicate autophagosomes or autolysosomes, respectively (B). The proportions of RAW 264.7 cells that formed autophagosomes (C) and autolysosomes (D) are shown. The proportions were calculated using at least 300 cells, and the values are expressed as the mean ± SD from three independent experiments. E, The cells were lysed in a cell lysis buffer with a protease inhibitor cocktail, and then the lysates were subjected to SDS-PAGE analysis and western blotting. F-I, RAW-tfLC3 were treated with 200 U/ml or the indicated concentration of IFN-γ (circles) or vehicle alone (squares) for 24 h or the indicated times, and were then fixed. The proportions of RAW 264.7 cells that formed autophagosomes (F and H) and autolysosomes (G and I) are shown. The proportions were calculated using at least 300 cells, and the values are expressed as the mean ± SD from three independent experiments. J, RAW 264.7 cells were treated with 200 U/ml IFN-γ in the presence of E64d plus pepstatin A (10 μg/ml each) for the indicated times, and then lysed in cell lysis buffer with a protease inhibitor cocktail. Subsequently, the cell lysates were subjected to SDS-PAGE analysis and western blotting. Densitometric LC3b-II/GAPDH ratios are shown underneath the blot.
JAK1/2 is required for autophagy early after IFN-γ stimulation
IFN-γ engagement of its receptor results in transactivation of JAK1 and JAK2, phosphorylation and translocation of STAT1 molecules to the nucleus where they regulate gene transcription (5). To test whether JAKs are required for IFN-γ-induced autophagy, we treated IFN-γ-activated RAW-tfLC3 cells with the JAK inhibitor I, 2-(1,1-dimethylethyl)-9-fluoro-3,6-dihydro-7H-benz[h]-imidaz[4,5-f]isoquinolin-7-one. This inhibitor is highly and selectively potent for JAK1-3 (IC50, approximately 1-15nM) as well as for the tyrosine kinase, Tyk2, that operates downstream of the type I IFN receptor, IFN-αR. It completely abolished autophagosome formation and maturation together with expression of the antimicrobial enzyme, inducible NOS2, a known JAK1/2 target gene that served as a positive control (Fig. 2). Thus, JAK1/2 appear obligate for the early signaling cascade downstream of IFN-γ that promotes autophagic induction.
Fig. 2.
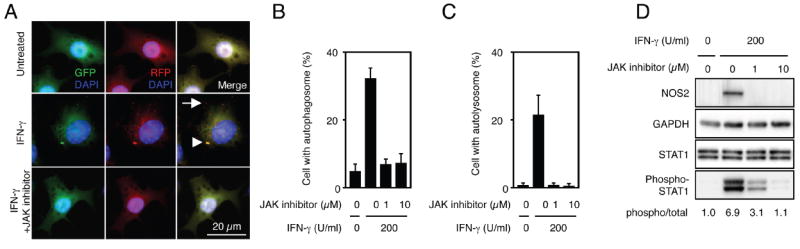
JAK1/2 is required for autophagy early after IFN-γ stimulation. A-C, RAW-tfLC3 was retreated with JAK inhibitor I or left untreated for 2 h, and then the cells were stimulated with 200 U/ml IFN-γ for 4 h. The cells were fixed and stained with DAPI to detect nuclei. Representative fluorescent images are shown, and arrowhead or arrow indicates autophagosome or autolysosome, respectively (A). The proportions of RAW 264.7 cells that formed autophagosomes (B) and autolysosomes (C) are shown. The proportions were calculated using at least 300 cells, and the values are expressed as the mean ± SD from three independent experiments. D, The cells were lysed in a cell lysis buffer with a protease inhibitor cocktail, and then the lysates were subjected to SDS-PAGE analysis and western blotting. Densitometric phosphorylated STAT1/STAT1 ratios are shown underneath the blot.
STAT1 and Irgm1 are dispensible for autophagy activation by IFN-γ stimulation
In the classical IFN-γ signal cascade, JAK1/2 activation leads to STAT1 phsophorylation and translocation to the nucleus to activate target genes like Irgm1 (25), the latter of which has been implicated as a major component in stimulating the autophagic response (19, 20). We therefore took a loss-of-function approach by introducing lentiviral pLKD shRNAs into the RAW-tfLC3 reporter cell line to derive stable macrophages that not only express tfLC3 but are also silenced for either STAT1 or Irgm1. In addition, we generated a third set of stable RAW-tfLC3 macrophage clones lacking IFN-γR1 as a robust negative control.
This assay system allowed us to determine whether STAT1 and Irgm1 are functionally involved in eliciting autophagy under conditions where the host cell is not transiently activated via TLR or NLR sensing of transfected siRNA pools (29). IFN-γ-stimulated formation and maturation of autophagosomes was inhibited only in IFN-γR1-silenced cells; neither STAT1 nor Irgm1 silencing had any effect on autophagosomal induction (Fig. 3, A-E). In contrast, expression of endogenous Irgm1 was completely blocked in IFN-γR1, STAT1 and Irgm1 shRNA-expressing RAW-tfLC3 macrophage cell lines (Fig. 3F). Identical results were obtained with two independent shRNA-expressing RAW-tfLC3 clones for each of the three shRNA-targeted genes. Similar outcomes were also observed using primary BMMs from C57BL/6 wild-type, STAT1-/-, and Irgm1-/- mice that had been nucleoporated with the tfLC3 plasmid (Fig. 3, G-I). Here both autophagosome formation and maturation within the first 4-6 hours was identical between genotypes, as was conversion of endogenous LC3b-I to LC3b-II (Fig. 3J and Fig. 4E). Thus, our genetic loss-of-function results indicate that IFN-γ can activate macrophage autophagy in an STAT1- and Irgm1-independent manner.
Fig. 3.
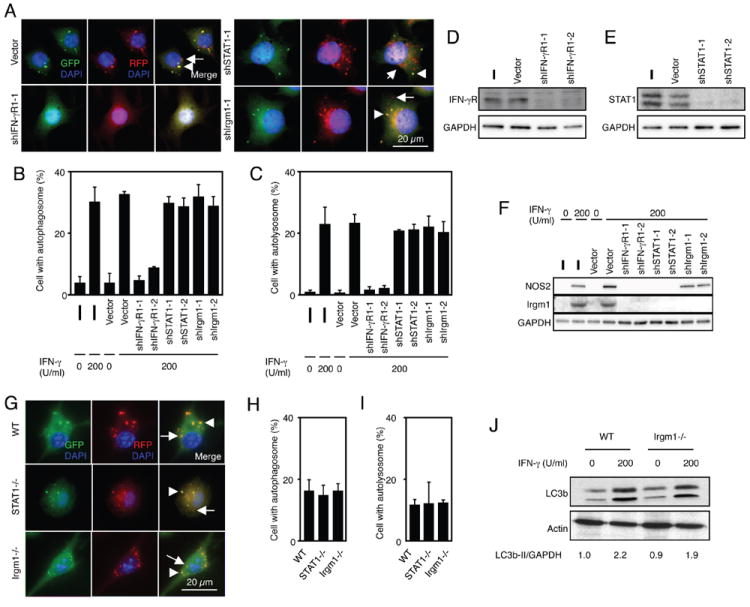
STAT1 and Irgm1 are dispensible for autophagy activation by IFN-γ stimulation. A-C and G-I, RAW 264.7 cells, stable knockdown cells, and BMMs that expressed tfLC3 were stimulated with 200 U/ml IFN-γ for 4 h. The cells were fixed and stained with DAPI to detect nuclei. Representative fluorescent images are shown, and arrowheads or arrows indicate autophagosomes or autolysosomes, respectively (A and G). The proportions of RAW 264.7 cells that formed autophagosomes (B and H) and autolysosomes (C and I) are shown. The proportions were calculated using at least 300 cells, and the values are expressed as the mean ± SD from three independent experiments. D-F and J, The cells were lysed in a cell lysis buffer with a protease inhibitor cocktail, and then the lysates were subjected to SDS-PAGE analysis and western blotting. Densitometric LC3b-II/GAPDH ratios are shown underneath the blot.
Fig. 4.
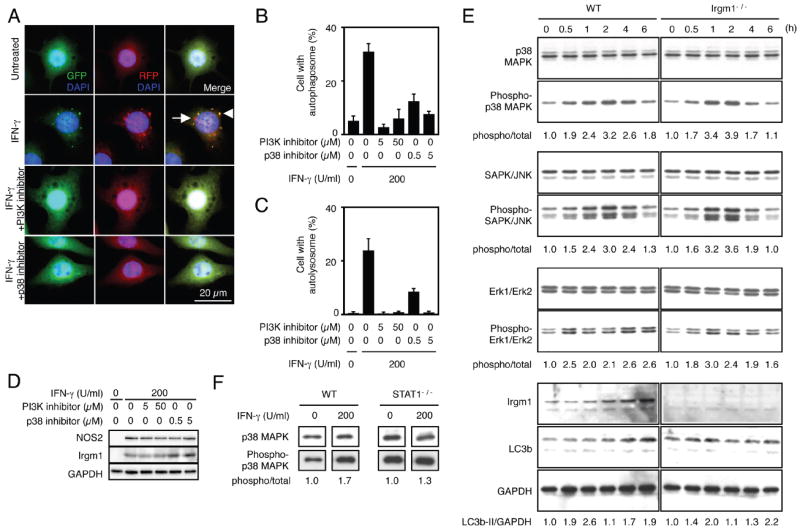
PI3K and p38 MAPK are required for autophagy activation by IFN-γ stimulayion. A-D, RAW-tfLC3 cells were pretreated with the indicated concentrations of a PI3K inhibitor or p38 MAPK inhibitor for 2 h, and then the cells were stimulated with 200 U/ml IFN-γ for 4 h. The cells were fixed and stained with DAPI to detect nuclei. Representative fluorescent images are shown, and arrowhead or arrow indicates autophagosome or autolysosome, respectively (A). The proportions of RAW 264.7 cells that formed autophagosomes (B) and autolysosomes (C) are shown. The proportions were calculated using at least 300 cells, and the values are expressed as the mean ± SD from three independent experiments. The cells were lysed in a cell lysis buffer with a protease inhibitor cocktail, and then the cell lysates were subjected to SDS-PAGE analysis and western blotting (D). E and F, BMMs from WT, Irgm1-/-, or STAT1-/- mice were treated with 200 U/ml IFN-γ for 6 h or the indicated times. The cells were lysed in a cell lysis buffer with a protease inhibitor cocktail, and then the cell lysates were subjected to SDS-PAGE analysis and western blotting. Densitometric LC3b-II/GAPDH and phosphorylation ratios are shown underneath the blot.
PI3K and p38 MAPK are required for autophagy activation by IFN-γ stimulation
The above findings suggested an unconventional pathway being responsible for IFN-γ-induced autophagy. Besides the classic JAK1/2-STAT1 signaling cascade, IFN-γ activates several other downstream pathways. These include p38 MAPK (9) and PI3K signaling cascades (6, 10). The latter cascade, in particular, encompasses some key regulators of autophagy, notably the serine/threonine kinase mammalian target of rapamycin (mTOR) and class I and III PI3Ks (33). Class I PI3Ks phosphorylate phosphatidylinositol-4,5-bisphosphate (PtdIns(4,5)P2) at the plasma membrane to generate phosphatidylinositol-3,4,5-trisphsphate (PtdIns(3,4,5)P3), which activates mTOR (33-35). VPS34, a class III PI3K, also contributes to mTOR activation when amino acids are plentiful. However, its role differs under starved or cytokine-activated conditions where it phosphorylates PtdIns in endomembranes to produce phosphatidylinositol-3-phosphate (PtdIns3P), which is required for recruiting Beclin-1/Atg6 to stimulate autophagy (33, 34). Two PI3K inhibitors, 3-methyladenine and LY294002, have been shown to inhibit autophagy (33), and LY294002 completely blocked IFN-γ-induced autophagy in our RAW-tfLC3 macrophages as well (Fig. 4, A-C). Interestingly, however, it did not alter IFN-γ-induced NOS2 or Irgm1 expression (Fig. 4D), suggesting functional bifurcation of these pathways.
This bifurcation was further reinforced upon examination of p38 MAPK activity. An specific inhibitor of p38 MAPK, 4-[4-(4-fluorophenyl)-2-(4-methylsulfinylphenyl)-1H-imidazol-5-yl]pyridine, completely blocked autophagy but had no effect on NOS2 and Irgm1 expression following IFN-γ activation (Fig. 4, A-D). Such divergence was again underscored in wild-type, STAT1-/-, and Irgm1-/- BMMs (Fig. 4E and F). Early IFN-γ-induced phosphorylation of p38 as well as other MAPK members, Erk1/2 and stress-activated protein kinase (SAPK)/JNK, was still intact in Irgm1-/- BMMs which coincided with normal autophagosome and autolysosome formation (Fig. 3, G-J and 4E). These results reveal both PI3K and p38 MAPK are required for the activation of autophagy in an Irgm1-independent manner.
DISCUSSION
IFN-γ-induced autophagosome formation was previously reported to enlist Irgm1 based on short-term GFP-Irgm1 overexpression or transient siRNA silencing in RAW 264.7 cells (19, 20). By using dual color tfLC3 reporter stably integrated into the chromosome of RAW 264.7 macrophages, combined with genetic or pharmacologic loss-of-function approaches, we found IFN-γ accelerates not only the formation but also the maturation of autophagosomes via JAK1/2 and p38 MAPK signaling pathway.
In the present study, we found permanent loss-of-function approaches using primary BMMs from Irgm1-/- mice or RAW 264.7 macrophages stably expressing a dual tfLC3 autophagosome-autolysosome reporter together with Irgm1 shRNAs, suggest this IRG protein is dispensable during the early stages of autophagy. Our findings raise the question of whether Irgm1 is a bona fide autophagy gene or whether it has additional lysosomal trafficking roles against pathogens such as Mycobacterium tuberculosis and Crohn’s disease-associated Escherichia coli as suggested from several studies (25, 28, 35). Recent evidence in Irgm1-deficient mice crossed with GFP-LC3b transgenic animals found induction of LC3b puncta in myeloid stem cell precursors was actually elevated rather than impaired compared with wild-type controls (36). Thus the precise roles of Irgm1 as part of the IFN-γ-induced autophagic cascade needs further clarification. In this respect it mirrors another member of the IRG family, Irga6, which targets and disrupts the Toxoplasma gondii parasitophorous vacuole with the help of autophagy protein Atg5 in an autophagosome-independent fashion (37). As such the IRGs may fulfill multiple trafficking and signaling functions besides autophagy during infection and inflammatory settings.
It has been reported that members of the MAPK family can be activated by IFN stimulation (5, 9, 38, 39). Moreover, the activation of Erk2, and possibly other MAPKs, by IFN-γ is mediated by JAK2 and Proline-rich tyrosine kinase 2 (Pyk2) (40). Our analysis of IFN-γ-induced autophagy also indicated JAK1/2 and p38 MAPK dependence but STAT1 independence. Autophagy is used to degrade microorganisms that invade intracellularly (18, 41). Taken together, these results suggesting that IFN-γ-induced autophagy activation through JAK2/Pyk2/MAPK-signaling pathway contributes to the host defence. Here as many as 100-150 genes can be expressed downstream of IFN-γ in STAT1-/- fibroblasts. Among these are caspase 7-like Lice-2, IL-1β, Daax and the serine protease PIM-1 that co-ordinate apoptotic/pyroptotic responses possibly in response to autophagic stimuli (7). Likewise, other gene targets may proceed via STAT1 independent, PI3K-dependent signaling, as seen in human peripheral blood monocytes (6). The requirement for p38 MAPK is in accordance with recent observations that this pathway regulates binding of p38IP to Atg9 for trafficking to nascent autopahgosomes (42). Hence our findings may encourage efforts to focus on a subset of IFN-γ-induced target genes that can be transcribed in STAT1-independent fashion while engaging p38 MAPK en route to stimulating autophagosome formation. These efforts should have widespread implications for understanding autophagic and cytokine-based immunity to infection.
Acknowledgments
We thank Tamotsu Yoshimori (Osaka University, Osaka, Japan) for the generous gift of the mRFP-GFP-LC3 plasmid.
Footnotes
This work was supported by a Grant-in-Aid for Challenging Exploratory Research 22659088 (to T.M.), National Institutes of Health NIAID RO1 AI068041-01A1, BWF Investigator in Pathogenesis of Infectious Disease Award and W.W. Winchester Foundation (to J.D.M.).
References
- 1.Nathan CF. Secretory products of macrophages. J Clin Invest. 1987;79:319–326. doi: 10.1172/JCI112815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.MacMicking JD. Recognizing macrophage activation and host defense. Cell Host Microbe. 2009;5:405–407. doi: 10.1016/j.chom.2009.05.006. [DOI] [PubMed] [Google Scholar]
- 3.Stark GR, Kerr IM, Williams BR, Silverman RH, Schreiber RD. How cells respond to interferons. Annu Rev Biochem. 1998;67:227–264. doi: 10.1146/annurev.biochem.67.1.227. [DOI] [PubMed] [Google Scholar]
- 4.Darnell JE., Jr STATs and gene regulation. Science. 1997;277:1630–1635. doi: 10.1126/science.277.5332.1630. [DOI] [PubMed] [Google Scholar]
- 5.Platanias LC. Mechanisms of type-I- and type-II-interferon-mediated signalling. Nat Rev Immunol. 2005;5:375–386. doi: 10.1038/nri1604. [DOI] [PubMed] [Google Scholar]
- 6.Navarro A, Anand-Apte B, Tanabe Y, Feldman G, Larner AC. A PI-3 kinase-dependent, Stat1-independent signaling pathway regulates interferon-stimulated monocyte adhesion. J Leukoc Biol. 2003;73:540–545. doi: 10.1189/jlb.1002508. [DOI] [PubMed] [Google Scholar]
- 7.Ramana CV, Gil MP, Han Y, Ransohoff RM, Schreiber RD, Stark GR. Stat1-independent regulation of gene expression in response to IFN-γ. Proc Natl Acad Sci U S A. 2001;98:6674–6679. doi: 10.1073/pnas.111164198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Sun D, Ding A. MyD88-mediated stabilization of interferon-γ-induced cytokine and chemokine mRNA. Nat Immunol. 2006;7:375–381. doi: 10.1038/ni1308. [DOI] [PubMed] [Google Scholar]
- 9.Valledor AF, Sanchez-Tillo E, Arpa L, Park JM, Caelles C, Lloberas J, Celada A. Selective roles of MAPKs during the macrophage response to IFN-γ. J Immunol. 2008;180:4523–4529. doi: 10.4049/jimmunol.180.7.4523. [DOI] [PubMed] [Google Scholar]
- 10.Choudhury GG. A linear signal transduction pathway involving phosphatidylinositol 3-kinase, protein kinase Cε, and MAPK in mesangial cells regulates interferon-γ-induced STAT1α transcriptional activation. J Biol Chem. 2004;279:27399–27409. doi: 10.1074/jbc.M403530200. [DOI] [PubMed] [Google Scholar]
- 11.Ivaska J, Bosca L, Parker PJ. PKCε is a permissive link in integrin-dependent IFN-γ signalling that facilitates JAK phosphorylation of STAT1. Nat Cell Biol. 2003;5:363–369. doi: 10.1038/ncb957. [DOI] [PubMed] [Google Scholar]
- 12.Hardy PO, Diallo TO, Matte C, Descoteaux A. Roles of phosphatidylinositol 3-kinase and p38 mitogen-activated protein kinase in the regulation of protein kinase C-α activation in interferon-γ-stimulated macrophages. Immunology. 2009;128:e652–660. doi: 10.1111/j.1365-2567.2009.03055.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Boehm U, Klamp T, Groot M, Howard JC. Cellular responses to interferon-γ. Annu Rev Immunol. 1997;15:749–795. doi: 10.1146/annurev.immunol.15.1.749. [DOI] [PubMed] [Google Scholar]
- 14.MacMicking JD, Nathan C, Hom G, Chartrain N, Fletcher DS, Trumbauer M, Stevens K, Xie QW, Sokol K, Hutchinson N, et al. Altered responses to bacterial infection and endotoxic shock in mice lacking inducible nitric oxide synthase. Cell. 1995;81:641–650. doi: 10.1016/0092-8674(95)90085-3. [DOI] [PubMed] [Google Scholar]
- 15.MacMicking JD. IFN-inducible GTPases and immunity to intracellular pathogens. Trends Immunol. 2004;25:601–609. doi: 10.1016/j.it.2004.08.010. [DOI] [PubMed] [Google Scholar]
- 16.Shenoy AR, Kim BH, Choi HP, Matsuzawa T, Tiwari S, MacMicking JD. Emerging themes in IFN-γ-induced macrophage immunity by the p47 and p65 GTPase families. Immunobiology. 2007;212:771–784. doi: 10.1016/j.imbio.2007.09.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Liu Y, Schiff M, Czymmek K, Talloczy Z, Levine B, Dinesh-Kumar SP. Autophagy regulates programmed cell death during the plant innate immune response. Cell. 2005;121:567–577. doi: 10.1016/j.cell.2005.03.007. [DOI] [PubMed] [Google Scholar]
- 18.Levine B, Mizushima N, Virgin HW. Autophagy in immunity and inflammation. Nature. 2011;469:323–335. doi: 10.1038/nature09782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Gutierrez MG, Master SS, Singh SB, Taylor GA, Colombo MI, Deretic V. Autophagy is a defense mechanism inhibiting BCG and Mycobacterium tuberculosis survival in infected macrophages. Cell. 2004;119:753–766. doi: 10.1016/j.cell.2004.11.038. [DOI] [PubMed] [Google Scholar]
- 20.Singh SB, Davis AS, Taylor GA, Deretic V. Human IRGM induces autophagy to eliminate intracellular mycobacteria. Science. 2006;313:1438–1441. doi: 10.1126/science.1129577. [DOI] [PubMed] [Google Scholar]
- 21.Xu Y, Jagannath C, Liu XD, Sharafkhaneh A, Kolodziejska KE, Eissa NT. Toll-like receptor 4 is a sensor for autophagy associated with innate immunity. Immunity. 2007;27:135–144. doi: 10.1016/j.immuni.2007.05.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Sanjuan MA, Dillon CP, Tait SW, Moshiach S, Dorsey F, Connell S, Komatsu M, Tanaka K, Cleveland JL, Withoff S, Green DR. Toll-like receptor signalling in macrophages links the autophagy pathway to phagocytosis. Nature. 2007;450:1253–1257. doi: 10.1038/nature06421. [DOI] [PubMed] [Google Scholar]
- 23.Cooney R, Baker J, Brain O, Danis B, Pichulik T, Allan P, Ferguson DJ, Campbell BJ, Jewell D, Simmons A. NOD2 stimulation induces autophagy in dendritic cells influencing bacterial handling and antigen presentation. Nat Med. 2010;16:90–97. doi: 10.1038/nm.2069. [DOI] [PubMed] [Google Scholar]
- 24.Travassos LH, Carneiro LA, Ramjeet M, Hussey S, Kim YG, Magalhaes JG, Yuan L, Soares F, Chea E, Le Bourhis L, Boneca IG, Allaoui A, Jones NL, Nunez G, Girardin SE, Philpott DJ. Nod1 and Nod2 direct autophagy by recruiting ATG16L1 to the plasma membrane at the site of bacterial entry. Nat Immunol. 2010;11:55–62. doi: 10.1038/ni.1823. [DOI] [PubMed] [Google Scholar]
- 25.MacMicking JD, Taylor GA, McKinney JD. Immune control of tuberculosis by IFN-γ-inducible LRG-47. Science. 2003;302:654–659. doi: 10.1126/science.1088063. [DOI] [PubMed] [Google Scholar]
- 26.Ling YM, Shaw MH, Ayala C, Coppens I, Taylor GA, Ferguson DJ, Yap GS. Vacuolar and plasma membrane stripping and autophagic elimination of Toxoplasma gondii in primed effector macrophages. J Exp Med. 2006;203:2063–2071. doi: 10.1084/jem.20061318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Tiwari S, Choi HP, Matsuzawa T, Pypaert M, MacMicking JD. Targeting of the GTPase Irgm1 to the phagosomal membrane via PtdIns(3,4)P(2) and PtdIns(3,4,5)P(3) promotes immunity to mycobacteria. Nat Immunol. 2009;10:907–917. doi: 10.1038/ni.1759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Singh SB, Ornatowski W, Vergne I, Naylor J, Delgado M, Roberts E, Ponpuak M, Master S, Pilli M, White E, Komatsu M, Deretic V. Human IRGM regulates autophagy and cell-autonomous immunity functions through mitochondria. Nat Cell Biol. 2010;12:1154–1165. doi: 10.1038/ncb2119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kim BH, Shenoy AR, Kumar P, Das R, Tiwari S, MacMicking JDg. A family of IFN-γ-inducible 65-kD GTPases protects against bacterial infection. Science. 2011;332:717–721. doi: 10.1126/science.1201711. [DOI] [PubMed] [Google Scholar]
- 30.Bekpen C, Hunn JP, Rohde C, Parvanova I, Guethlein L, Dunn DM, Glowalla E, Leptin M, Howard JC. The interferon-inducible p47 (IRG) GTPases in vertebrates: loss of the cell autonomous resistance mechanism in the human lineage. Genome Biol. 2005;6:R92. doi: 10.1186/gb-2005-6-11-r92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Bekpen C, Marques-Bonet T, Alkan C, Antonacci F, Leogrande MB, Ventura M, Kidd JM, Siswara P, Howard JC, Eichler EE. Death and resurrection of the human IRGM gene. PLoS Genet. 2009;5:e1000403. doi: 10.1371/journal.pgen.1000403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Kimura S, Noda T, Yoshimori T. Dissection of the autophagosome maturation process by a novel reporter protein, tandem fluorescent-tagged LC3. Autophagy. 2007;3:452–460. doi: 10.4161/auto.4451. [DOI] [PubMed] [Google Scholar]
- 33.Nobukuni T, Joaquin M, Roccio M, Dann SG, Kim SY, Gulati P, Byfield MP, Backer JM, Natt F, Bos JL, Zwartkruis FJ, Thomas G. Amino acids mediate mTOR/raptor signaling through activation of class 3 phosphatidylinositol 3OH-kinase. Proc Natl Acad Sci U S A. 2005;102:14238–14243. doi: 10.1073/pnas.0506925102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Byfield MP, Murray JT, Backer JM. hVps34 is a nutrient-regulated lipid kinase required for activation of p70 S6 kinase. J Biol Chem. 2005;280:33076–33082. doi: 10.1074/jbc.M507201200. [DOI] [PubMed] [Google Scholar]
- 35.Lapaquette P, Glasser AL, Huett A, Xavier RJ, Darfeuille-Michaud A. Crohn’s disease-associated adherent-invasive E. coli are selectively favoured by impaired autophagy to replicate intracellularly. Cell Microbiol. 2010;12:99–113. doi: 10.1111/j.1462-5822.2009.01381.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.King KY, Baldridge MT, Weksberg DC, Chambers SM, Lukov GL, Wu S, Boles NC, Jung SY, Qin J, Liu D, Songyang Z, Eissa NT, Taylor GA, Goodell MA. Irgm1 protects hematopoietic stem cells by negative regulation of interferon signaling. Blood. 2011 doi: 10.1182/blood-2011-01-328682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Zhao Z, Fux B, Goodwin M, Dunay IR, Strong D, Miller BC, Cadwell K, Delgado MA, Ponpuak M, Green KG, Schmidt RE, Mizushima N, Deretic V, Sibley LD, Virgin HW. Autophagosome-independent essential function for the autophagy protein Atg5 in cellular immunity to intracellular pathogens. Cell Host Microbe. 2008;4:458–469. doi: 10.1016/j.chom.2008.10.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Roy SK, Hu J, Meng Q, Xia Y, Shapiro PS, Reddy SP, Platanias LC, Lindner DJ, Johnson PF, Pritchard C, Pages G, Pouyssegur J, Kalvakolanu DV. MEKK1 plays a critical role in activating the transcription factor C/EBP-beta-dependent gene expression in response to IFN-γ. Proc Natl Acad Sci U S A. 2002;99:7945–7950. doi: 10.1073/pnas.122075799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Katsoulidis E, Li Y, Mears H, Platanias LC. The p38 mitogen-activated protein kinase pathway in interferon signal transduction. J Interferon Cytokine Res. 2005;25:749–756. doi: 10.1089/jir.2005.25.749. [DOI] [PubMed] [Google Scholar]
- 40.Takaoka A, Tanaka N, Mitani Y, Miyazaki T, Fujii H, Sato M, Kovarik P, Decker T, Schlessinger J, Taniguchi T. Protein tyrosine kinase Pyk2 mediates the Jak-dependent activation of MAPK and Stat1 in IFN-γ, but not IFN-α, signaling. EMBO J. 1999;18:2480–2488. doi: 10.1093/emboj/18.9.2480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Virgin HW, Levine B. Autophagy genes in immunity. Nat Immunol. 2009;10:461–470. doi: 10.1038/ni.1726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Webber JL, Tooze SA. Coordinated regulation of autophagy by p38α MAPK through mAtg9 and p38IP. EMBO J. 2010;29:27–40. doi: 10.1038/emboj.2009.321. [DOI] [PMC free article] [PubMed] [Google Scholar]