

Abstract
There is a mutualistic symbiotic relationship between the components of the photoreceptor/retinal pigment epithelium (RPE)/Bruch’s membrane (BrMb)/choriocapillaris (CC) complex that is lost in AMD. Which component in the photoreceptor/RPE/BrMb/CC complex is affected first appears to depend on the type of AMD. In atrophic AMD (~85–90% of cases), it appears that large confluent drusen formation and hyperpigmentation (presumably dysfunction in RPE) are the initial insult and the resorption of these drusen and loss of RPE (hypopigmentation) can be predictive for progression of geographic atrophy (GA). The death and dysfunction of photoreceptors and CC appear to be secondary events to loss in RPE.
In neovascular AMD (~10–15% of cases), the loss of choroidal vasculature may be the initial insult to the complex. Loss of CC with an intact RPE monolayer in wet AMD has been observed. This may be due to reduction in blood supply because of large vessel stenosis. Furthermore, the environment of the CC, basement membrane and intercapillary septa, is a proinflammatory milieu with accumulation of complement components as well as proinflammatory molecules like CRP during AMD. In this toxic milieu, CC die or become dysfunction making adjacent RPE hypoxic. These hypoxic cells then produce angiogenic substances like VEGF that stimulate growth of new vessels from CC, resulting in choroidal neovascularization (CNV). The loss of CC might also be a stimulus for drusen formation since the disposal system for retinal debris and exocytosed material from RPE would be limited. Ultimately, the photoreceptors die of lack of nutrients, leakage of serum components from the neovascularization, and scar formation.
Therefore, the mutualistic symbiotic relationship within the photoreceptor/RPE/BrMb/CC complex is lost in both forms of AMD. Loss of this functionally integrated relationship results in death and dysfunction of all of the components in the complex.
1.0 Basic Normal Anatomy of the Photoreceptor/Retinal pigment epithelium (RPE)/Bruch’s membrane (BrMb)/Choriocapillaris (CC) complex
The components of the photoreceptor/retinal pigment epithelium (RPE)/Bruch’s membrane (BrMb)/Choriocapillaris (CC) complex have a mutualistic symbiotic relationship (Figure 1). Each is dependent on the other components in the complex and each contributes to the well being of the others. We will first discuss each component in the complex in healthy human eyes and then discuss how each is changed in age-related macular degeneration. Finally, we will consider the breakdown of the mutualistic symbiotic relationship in AMD.
Figure 1.
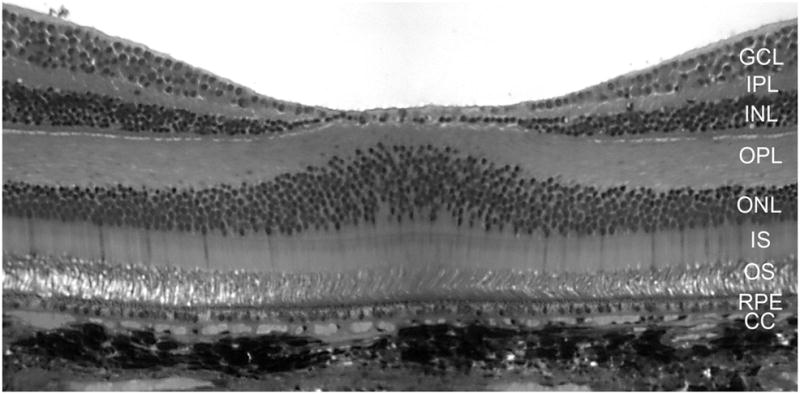
A cross section of the fovea from a Macaque monkey demonstrates the layers of retina and the morphological relationship of photoreceptor/RPE/BrMb/choroid complex. To the left and right of the foveal pit, the center of macula, the layers of the sensory retina are clearly visible. The inner most neuronal nuclei are of ganglion cells (GCL). The inner plexiform layer (IPL) separates the inner nuclear layer of neurons (INL) from the ganglion cell soma. The outer plexiform layer (OPL) represents the synapses between photoreceptors in the outer nuclear layer (ONL) and secondary neurons in the INL. The photoreceptor inner segments (IS) are mitochondria-rich and their outer segments (OS) make close contact with the retinal pigment epithelium (RPE), the outer most layer of retina. Bruch’s membrane (not discernible at this magnification) separates the RPE from the choriocapillaris (CC). The melanocytes of choroid are the extremely dark structures below the CC.
1.1 Photoreceptors
The photoreceptors are a specialized type of neuron in the posterior retina that are capable of phototransduction. Photoreceptors convert light into signals that can stimulate neuronal impulse transmission by triggering a change in the cell membrane potential after absorbing a photon. The two classes of photoreceptor cells are rods and cones and the signal they generate are converted to vision. The rods are narrower than the cones and distributed differently across the retina, but the chemical process in each that supports phototransduction is similar. However, rods are extremely sensitive and can be triggered by a very small number of photons. At very low light levels, visual experience is calculated solely from the rod signal. Cones require significantly brighter light in order to produce a signal. In humans, there are three different types of cone cells (red, green blue), distinguished by their pattern of response to different wavelengths of light. The human anatomical macula is only 6 mm in diameter and it contains a small cone dominated fovea (0.8 mm) (Figure 1) surrounded by a rod-dominated parafovea (Curcio et al., 1996; Curcio, 2001; Curcio et al., 1990). It is estimated by Curcio that only two rods/mm2 of retina die per year. Barron and associates found mitochondrial DNA deletions and cytochrome c oxidase-deficient cones accumulate in the aging retina, particularly in the foveal region. These defects may contribute to the changes in macular function observed in aging and age-related maculopathy (Barron et al., 2001).
The photoreceptor inner segments are rich in mitochondria, which are needed to provide energy for these highly metabolically active cells. The photoreceptors consume more oxygen per gram of tissue weight than any cell in body and have a tissue oxygen level close to zero in the dark (Wangsa-Wirawan and Linsenmeier, 2003).
1.2 Retinal Pigment Epithelium (RPE)
Just posterior to the photoreceptors, the RPE are polarized epithelial cells found at the base of the retina. The RPE consists of a single layer of hexagonal cells that are densely packed with pigment granules (melanosomes). The RPE are firmly attached to their underlying basement membrane, which is the inner or anterior layer of Bruch’s membrane. At the ora serrata, the RPE continues as a membrane passing over the ciliary body and continuing as the posterior surface of the iris. At its apical surface of the RPE faces the photoreceptor outer segments making a complex of close structural interactions. With its basolateral surface, the RPE faces Bruch’s membrane (BrMb), which separates the RPE from fenestrated endothelium of the choriocapillaris (CC). When viewed in cross section, each RPE cell consists of a basal outer non-pigmented part containing a large oval nucleus and an anterior-pigmented portion, which extends as a series of finger-like processes between the photoreceptor outer segments.
The RPE provides the nutrients needed to maintain visual function by light-sensitive outer segments of the photoreceptors. For example, the RPE supplies omega-3 fatty acids for building photoreceptor outer segment membranes and glucose for energy metabolism (Strauss, 2005). The RPE also transports ions, water, and metabolic end products from the subretinal space to the choroid.
The RPE also plays a key role in retinal physiology by forming the outer blood-retinal barrier that prevents nonspecific diffusion and transport of material from the choroid. RPE melanosomes absorb excess incoming light, which protects the retina from light damage. The RPE also serves as the limiting transport system that maintains the outer retinal environment by supplying small molecules such as amino acids, ascorbic acid, and D-glucose while remaining a tight barrier to choroidal blood borne substances. Homeostasis of the ionic environment in and around the RPE is maintained by this delicate, specific transport exchange system.
Another critical role for the RPE involves phagocytosing shed outer segments, scavenging photoreceptor debris, thus, serving as part of the waste disposal system for the retina in general. An RPE cell services 20–30 photoreceptor outer segments. An RPE cell ingests and degrades about 300,000,000 discs through a 70-year life span (Marshall, 1987). In addition to removal of waste from the PR by either recycling it or completely degrading it, the RPE can exocytose the remains for the CC for clearance from the retina.
The RPE is known to produce and to secrete a variety of growth factors: fibroblast growth factors (FGF-1, FGF-2, and FGF-5) and transforming growth factor-β (TGF-β) (Bost et al., 1992; Bost et al., 1994; Caruelle et al., 1989; Connolly et al., 1992; Dunn et al., 1998; Hageman et al., 1991; Ishigooka et al., 1993; Khaliq et al., 1995; Matsumoto et al., 1994; Sternfeld et al., 1989; Tanihara et al., 1993), insulin-like growth factor-I (IGF-I) (Martin and Haywood, 1992; Slomiany and Rosenzweig, 2004), ciliary neurotrophic factor (CNTF) (Cao et al., 1997; Walsh et al., 2001), platelet-derived growth factor (PDGF) (Campochiaro et al., 1994; Campochiaro et al., 1989), VEGF (Adamis et al., 1993), lens epithelium-derived growth factor (LEDGF) (Ahuja et al., 2001), members of the interleukin family (Streilein et al., 2002; Wenkel and Streilein, 2000), and pigment epithelium-derived factor (PEDF) to help build and sustain the choroid and photoreceptors (Streilein et al., 2002; Wenkel and Streilein, 2000). The RPE is also important in wound healing. It is obvious that the RPE performs a variety of complex functions that are essential for proper visual function.
1.3 Bruch’s Membrane (BrMb)
BrMb is a thin (2–4 um) connective tissue strategically located between the metabolically active retinal pigment epithelium (RPE) and its source of nutrition, the choriocapillaris. BrMb is an elastin- and collagen-rich extracellular matrix (ECM) that acts as a molecular sieve. This is penta-laminar structure; the basement membrane of the RPE (0.14–0.15 um thick) most anterior; the inner collagenous layer (ICL; approximately 1.4 um in diameter); a porous elastic layer (EL); the outer collagenous layer (OCL; 0.7 um in diameter); and the basement membrane of the endothelium of the choriocapillaris most posterior (Hogan, 1961). The main components of BrMb are collagens type I, III, IV, V and VI (Chen et al., 2003), fibronectin (Pauleikhoff et al., 1992), laminin (Aisenbrey et al., 2006), heparan sulphate proteoglycans (HSPG) and chondroitin/dermatan sulphate (Hewitt et al., 1989).
BrMb assumes importance in the physiology of the eye by virtue of its strategic location. Bruch’s membrane is a stratified ECM complex that functions as a physical as well as biochemical barrier for normal physiological processes and pathological processes like choroidal neovascularization (CNV). The primary functions of BrMb include: 1) regulating the reciprocal diffusion of bio-molecules, minerals, antioxidant components, trace elements and serum constituents between the choroid and RPE; 2) Providing physical support for RPE cell adhesion (Del Priore et al., 2002; Del Priore and Tezel, 1998) and a surface for migration and perhaps differentiation of RPE (Gong et al., 2008); 3) wound healing (Tezel et al., 2004); and 4) acting as a barrier that restricts retinal and choroidal cellular migration.
Given the acellular nature of BrMb, transport through it is primarily by passive diffusion (Grindle and Marshall, 1978). Diffusion across BrMb depends on its molecular composition, which is influenced by several factors like age and location in the retina. It also depends on hydrostatic pressure on both sides of BrMb and on concentration of specific bio-molecules and organic ions (Grindle and Marshall, 1978). Any alteration in the structure or composition of BrMb could influence its diffusion properties and, ultimately, the function of the RPE and outer retina. Accumulating evidence suggests that the molecular, structural, and functional properties of BrMb are dependent on age, genetics, environmental factors, retinal location, and disease state. As a result, part of the properties of BrMb are unique to each human individual at a given age and, therefore, uniquely affect the progression of ocular disease in the photoreceptor/RPE/BrMb/CC complex.
1.4 Choriocapillaris (CC)
The choriocapillaris (CC), the capillary component of the choroidal vasculature, lies adjacent and posterior to Bruch’s membrane. The CC has an unusual disposition in choroid, arranged in a single layer restricted to the inner portion of the choroid with feeding arterioles and draining venules entering the capillary plexus from below at right angles in the posterior pole. In the equatorial and more peripheral choroid, arterioles and veins are located in the plane of the CC, and the CC is segmental in arrangement. Inner choroidal vessels (CC and medium-sized vessels) are sandwiched between two pigmented cell types, apical RPE of neuroepithelial origin and outer choroidal melanocytes of neural crest origin. The adult CC is unique in that it is fenestrated mostly on the retinal side and its basement membrane comprises the most posterior layer of BrMb. Fenestrated endothelial cells is a characteristic of tissues that are involved in secretion and/or filtration. The sidedness of the CC and its endothelial cells is also true in terms of receptor expression. VEGF receptor-1 and –2 (VEGFR-1 & -R2) are reported to be expressed on the retinal side of the vasculature (Blaauwgeers et al., 1999). The CC is also unique in that it is the only capillary system in which the endothelial cells constitutively express intracellular adhesion molecule-1 (ICAM-1 )(McLeod et al., 1995). ICAM-1 is responsible for firm adherence to endothelial cells by leukocytes with CD11b/CD18 on their surface, like macrophages and neutrophils.
It is assumed that RPE are responsible for transporting nutrients into the photoreceptors (PR) from the CC and removal of waste from the PR by either recycling it, completely degrading it, or degrading it and exocytosing the remains for CC to remove. The juxtaposition of the RPE/BrMb/CC permits the CC to provide all of the metabolic needs from serum for the PR including 90% of the O2 consumed by the PR in darkness (Wangsa-Wirawan and Linsenmeier, 2003). Since photoreceptors are almost anoxic in the dark, any disruption in choroidal blood flow would be detrimental to these cells (Wangsa-Wirawan and Linsenmeier, 2003).
1.4a Physiology and blood flow in choriocapillaris
The choroid lacks metabolic regulation and, therefore, it lacks autoregulation (Friedman and Chandra, 1972). Even exposure of neonatal dogs to 100% oxygen for 4 days does not constrict the choroidal vasculature (McLeod et al., 1996). The PO2 in cat choroid is around 70 mm Hg and it can increase to 250 mm Hg in hyperoxia (Linsenmeier and Yancy, 1989). As a consequence, decreases in choroidal PO2 can be caused by systemic hypoxia or elevated intraocular pressure. Choroidal nonperfusion is extremely detrimental to proper photoreceptor function and viability and retinal detachment from choroid results in photoreceptor death. Wangsa-Wirawan and Linsenmeier have suggested that hyperoxia after retinal detachment might be used therapeutically to rescue photoreceptors (Wangsa-Wirawan and Linsenmeier, 2003).
1.4b Innervations of the choroid
There is an intrinsic network of nitrergic ganglion cells [NADPH-diaphorase and nitric oxide synthase (NOS) positive] in the human choroidal stroma, whose neurons are connected to each other and to the perivascular network (Bron et al., 1997). This intrinsic plexus appears to be present in the choroid of animals with foveas because ganglion cells are found only rarely in non-foveate species.
The ganglion cells in human choroid are concentrated mainly in the temporal and central regions of the human choroid. The majority of the ganglion cells are solitary, polygonal cells, with diameters ranging from 10 to 40 μm. Most are located close to the walls of large arteries, and none are observed near the choriocapillaris (Flügel et al., 1994). Nitric oxide (NO) is a mediator of endothelium-derived vascular relaxation, and the release of NO from perivascular nerves in various organs, including the cat choroid, causes vasodilatation (Bill, 1991; Flügel et al., 1994; Lauber, 1936; Mann et al., 1993; Morris, 1993; Nilsson et al., 1985; Stjernschantz and Bill, 1980). In the choroid, NO is colocalized with vasointestinal polypeptide (VIP), which also has a vasodilator action (Furness et al., 1992; Kummer et al., 1992; Miller et al., 1983; Talmage and Mawe, 1993). Bhutto et al found endothelial nitric oxide synthase (eNOS) associated with blood vessels in choroid while neuronal nitric oxide synthase (nNOS) was localized to perivascular neurons, scattered cells throughout the choroid, and the RPE (Bhutto et al., 2009). The perivascular and ganglionic neural plexuses express nNOS and appear to serve a vasodilator role in the choroid, perhaps adjusting blood flow in response to reduction in arterial blood pressure or protecting the retina from thermal damage associated with light exposure. Flugel et al (Flügel et al., 1994) have suggested that the submacular location of the intrinsic choroidal ganglionic plexus in foveate animals may afford additional protection from light damage at a site where light is focused and the photoreceptors and RPE are most susceptible.
1.4c Blood Flow in Choroid
Whereas retinal vasculature supplies oxygen to the inner retina, the choroidal vasculature supplies oxygen to outer retina. There is little disagreement that the majority of the blood from the ophthalmic artery (approximately 80%) enters the choroidal vasculature. Based on the circulation of radioactive microspheres (Alm and Bill, 1972) and krypton 85 (Friedman et al., 1964), blood flow rate in cat choroid was found to be at least 10 times higher than retinal blood flow. High blood flow was thought necessary to provide enough oxygen to retina. However, two studies demonstrate that, at least in rat, the RBC velocity in choriocapillaris is actually 4 times slower than in inner retinal capillaries (Braun et al., 1997; Wajer et al., 2000). The reported differences in flow may be due to species differences but it is more likely due to techniques employed where the former values are based on flow in all choroidal vessels whereas, the RBC velocities were measured directly in capillaries. Greater retention of microspheres is due to the large blood volume in the entire system. The lobular nature of choriocapillaris (no linear vascular segments) and the unusual disposition of the large blood vessels posterior to and at right angles to the capillaries probably contribute to slow RBC velocities in choriocapillaris. Another contributing factor may be that the lumens of capillaries are broad laterally but flat in an anterior to posterior position.
2.0 Basic definitions of exudative and dry age-related macular degeneration
Age-related macular degeneration (AMD) is the leading cause of blindness in the United States, and 10–18% of individuals between 65 and 75 have lost some central vision as a result of AMD (Friedman, 2008) while 30% of those aged 75 years or older have lost vision (Klein et al., 1992). There are approximately 15 million people affected by AMD in the United States. Age-related macular degeneration (AMD) is a disease associated with aging in which there is a decline in sharp, central vision since the affected area is most often the small central portion of the retina called the macula. The central vision is needed for seeing objects clearly and for common tasks of daily living such as reading and driving. AMD causes no pain. The diagnosis of AMD is based on visual dysfunction and characteristic macular findings. In some cases, AMD advances so slowly that people notice little change in their vision. In others, the disease progresses faster and may lead to a loss of vision in both eyes.
There are many forms of AMD but the two major types are exudative or neovascular or “wet” and non-exudative or “dry” AMD. Approximately 10–15% of the cases of macular degeneration are the exudative type. Most patients with AMD have the non-exudative form of the disease and will not lose central vision but have substantial functional limitations, including fluctuating vision, difficulty reading, limited vision at night or under conditions of reduced illumination. Sunness et al found that total atrophy area enlarged a median of 0.9 disc areas per year in geographic atrophy (GA) (Sunness, 1999). In GA, one form of “dry” AMD, the fovea is often spared from degeneration initially. It tends to progress more slowly than the neovascular type. However, the nonexudative form of AMD can lead to the neovascular form, which is more aggressive and severe. .
The nonexudative form of AMD is characterized by the presence of yellow deposits, called drusen, in the macula and sharply defined focal areas of RPE atrophy, which are associated with varying degrees of loss of the CC. A few small drusen may not cause changes in vision; nearly all people over the age of 50 years have at least one small druse in one or both eyes. However, as drusen grow in size and increase in number, they may lead to a dimming or distortion of vision that people find most noticeable when they read. The early stage of dry AMD is characterized by drusen and pigmentary abnormalities in the macula. Only subjects with large drusen are at risk for late AMD. In more advanced stages of dry AMD, there is loss in RPE (Figure 2) and thinning of the photoreceptors in the macula leading to atrophy, or tissue death. In the atrophic form of dry AMD, patients may have blind spots in the center of their vision. In the advanced stages, patients lose all central vision. There is no known cure for the “dry” type of AMD. Patients may use antioxidants and zinc to slow progression of AMD (AED Study Research Group, 2001; Klein et al., 2008)(also see review by Weikel in this issue).
Figure 2.

Alkaline phosphatase (APase)-incubated choroid from a 88 year-old Caucasian male with GA showing the nonatrophic region (A,D&G), border region (B,E&H) and atrophic region (C,F&I) in flat view prior to embedment (A-C) and in cross sections stained with PAS and hematoxylin (D–I). Blue APase reaction product is present in viable blood vessels only and is most prominent in neovascularization. (A) APase stained capillaries in the nonatrophic region have broad diameter lumens filled with serum APase (arrow in D&G), with endothelial cells and pericytes underlying viable RPE (arrowhead in D&G). At the border region, RPE appear hypertrophic (arrowhead in E&H), capillaries appear constricted (arrow in E&H), and some have completely degenerated (paired arrows H). A thin basal laminar deposit is associated with Bruch’s membrane (open arrow). In the atrophic region, many capillaries have degenerated leaving only remnants of basement membrane material (arrows in F&I). (scale bar = 100 μm in A–C, 30 μm in D–F, 10 μm in G–I) [Figure 10 from McLeod et al, Invest. Ophthalmol. Vis. Sci 50:4982-4991, 2009 (McLeod et al., 2009) with permission]
The “wet” form of AMD is characterized by choroidal neovascularization (CNV), the growth of abnormal blood vessels from the choroid underneath the macula (Figure 3). These new blood vessels leak blood into the retina, causing distortion of vision that makes straight lines look wavy, as well as blind spots and loss of central vision. However, some patients do not notice any such changes, despite the onset of CNV. Clinically, CNV associated with wet AMD may include classic or occult neovascular leakage patterns. Classic CNV is distinct or well demarcated during fluorescein angiography whereas occult CNV is obscured or poorly demarcated on fluorescein angiography. These abnormal blood vessels eventually result in disciform scar, leading to permanent loss of central vision.
Figure 3.

APase-incubated choroid from an 81 year-old Caucasian female with wet AMD. A submacular sea fan-like CNV formation is shown (arrowheads) using epi-illumination to analyze RPE (A) and transillumination (B) to analyze viable blood vessels. Areas of choriocapillaris dropout are located in advance of the CNV (asterisks). Percent RPE and vascular area measurements made in 2 mm intervals from the CNV (C) show that capillary dropout is severe (<20% vascular area) immediately in advance of the sea fan and present well beyond the area with neovascularization. (NH = optic nerve head, scale bar = 2 mm) [Figure 6 from McLeod et al, Invest. Ophthalmol. Vis. Sci 50:4982-4991, 2009 (McLeod et al., 2009) with permission]
2.1 Changes in Photoreceptor/RPE/BrMb/CC complex in AMD
2.1a Changes in and loss of photoreceptors in AMD
AMD is characterized clinically by loss of visual function. Curcio and associates observed more rod than cone loss in AMD (Curcio et al., 1996; Curcio, 2001). They suggested that photoreceptor degeneration and loss occurs before disease in the RPE/Bruch’s membrane complex (Curcio, 2001). Dunaief observed apoptotic photoreceptors, RPE, and inner nuclear layer cells in human AMD (Dunaief et al., 2002; Xu et al., 1996). Furthermore Dunaief observed TUNEL-positive RPE and photoreceptor cells that were at the edges of atrophy, where expansion of atrophic regions is associated with vision loss in patients with GA (Dunaief et al., 2002). Dunaief also suggested the Fas/Fas ligand system may be involved in photoreceptor apoptosis in AMD (Dunaief et al., 2002). Maeda et al observed that the loss of photoreceptors was directly related to loss of RPE after intravitreal ornithine-induced degeneration, suggesting the importance of RPE in photoreceptor viability (Maeda et al., 1998). In geographic atrophy, Kim et al observed that photoreceptors loss was associated with RPE loss (Kim et al., 2002a). In another study, Kim and associates found that photoreceptor loss in wet AMD was associated with scar formation and it was less severe when RPE were present on the scar (Kim et al., 2002b). Green and Enger found that the thickness of the scar was related to the degree of photoreceptor degeneration (Green and Enger, 1993). The thicker the scar, the greater the distance of the photoreceptors from surviving choriocapillaris, their source of nutrients.
2.1b RPE changes in AMD
Many diseases of the retina and choroid affect RPE. Degeneration of RPE cells severely impairs the visual function of retina photoreceptors. Age-related alterations of the RPE include changes in pigmentation and the reduction of melanosomes, cell density of RPE as well as an increase in the number of lipofuscin granules (Delori et al., 2001). The reduction in cell density itself may result from apoptosis, which is caused by accumulation of toxic substances (Hageman et al., 2001; Mullins et al., 2000). Some of this stress is clearly oxidative. Oxidative stress in RPE melanin may be attributed to complexing of melanin with lipofuscin, pigment granules composed of lipid-containing residues of lysosomal digestion, which generate reactive oxygen species upon excitation with blue light, thereby making the aged RPE more susceptible to oxidative damage (Strauss, 2005)(also see reviews in this issue by Boulton, Handa, Sparrow). An increase in oxidative stress due to a reduction in protective mechanisms of RPE or an increase in number and concentration of active photo-oxidative reaction species are believed to contribute to the pathogenesis of AMD. This is enhanced by an age-related reduction in one of the most important antioxidants, α-tocopherol (Friedrichson et al., 1995)(See review by Weikel in this issue).
A change in the ECM environment can affect several aspects of RPE cellular function, including adhesion, proliferation, differentiation, and migration. A significant contribution to the ECM environment for RPE cells comes from BrMb. It has been reported that oxidative stress can be seen as the accumulation of advanced-glycation end products (AGEs) in RPE and BrMb (Handa et al., 1999; Uchiki et al., 2012; Weikel et al., 2011). Drusen also contain AGEs (Crabb et al., 2002). These AGEs may also play an important role in the induction of CNV, a hallmark of wet AMD. Other deposits associated with BrMb include basal laminar deposits that are located between RPE and Bruch’s membrane and basal linear deposits that are located inside Bruch’s membrane. These deposits include metabolic end products such as lipoproteins and other hydrophobic materials. These might be a consequence of incomplete degradation of metabolic end products from both photoreceptors and RPE. Alternatively, they could be related to the inability of aged BrMb to transport material.
Much of the pigmentary change that is visible clinically in retinal disorders takes place in the RPE rather than in the retina or melanosomes of choroid. Dysfunction of the RPE in AMD may lead to photoreceptor loss and blindness. In geographic atrophy (GA), the photoreceptors and RPE degenerate in a horse shoe-shaped pattern surrounding the fovea; the loss of choroidal vasculature appears to be a secondary event (Sarks et al., 1988; Sunness, 1999).
2.1c Bruch’s membrane changes in AMD and drusen formation
Generally Bruch’s membrane becomes thicker with increasing age as a result of an accumulation of as yet unidentified substances in the collagenous areas and intercapillary region (Guymer et al., 1998). The age-associated thickening of the BrMb undergoes diffuse thickening, with thickness being reported to increase by 135% in 10 decades. A prolonged choroidal filling phase during fluorescein angiography signals the presence of diffuse BrMb thickening. The maximum thickness occurs in the ICL, followed by the OCL. However, there is marked heterogeneity in the BrMb thickness among individuals at a given age. Disease risk is associated with age-related thickness of this structure (Spraul et al., 1996; Spraul et al., 1999). In general, BrMb thickening is caused by increased deposition and cross-linking of (less soluble) collagen fibers and increased deposition of biomolecules, the majority being (oxidized) waste products of RPE metabolism. There is an age-related accumulation of PAS-positive (periodic-acid-Schiff) granular, membranous, filamentous and vesicular material eventually resulting in focal deposits and drusen (Guymer et al., 1998)(Figure 4). Obviously, the thickening of BrMb eventually leads to several functional changes, such as changes in elasticity and hydraulic permeability. Marshall hypothesizes that two discrete processes occur in Bruch’s membrane which results in reduced transport with age: membrane remodeling occurs with a programmed decay rate leading to a decrease in hydraulic conductivity by the fifth decade; and lipid content begins to increase by the fourth decade (Starita et al., 1996). The decline in hydraulic conductivity implies a decreased capacity for exchange of fluids between RPE and choroid (Moore et al., 1995). Hussain et al have shown that there is a 44% decline in transport of dextrin through peripheral BrMb in the 9th decade compared to 1st decade (Hussain et al., 2010).
Figure 4.

APase-incubated choroid from the 81 year-old Caucasian female (higher magnification than shown in Figure 3) with wet AMD showing submacular CNV using epi-illumination (A&C) and transillumination (B&D). The front of growing vessels is closely associated with viable RPE (arrows in A-D). Areas of CC dropout are evident in advance of the CNV (asterisks A&B). In PAS and hematoxylin stained sections, the equatorial region (E&H) has broad capillaries (arrows) containing serum APase with both endothelial cells and pericytes. The RPE has a normal morphology (arrowhead) and Bruch’s membrane is free of deposits. In sections taken 1 mm beyond the CNV (F&I), only a few capillaries are viable (arrows) and many degenerative capillaries are seen (asterisks in I). The RPE is hypertrophic (arrowheads) and a basal laminar deposit is present. Sections taken through the edge of the CNV (G&J) show degenerative capillaries (asterisk in J), sub-RPE neovascularization (open arrow) and hypertrophic RPE overlying the leading edge of the CNV. (scale bar=0.5mm A&B, 100 μm C&D, 30 μm E–G, and 10 μm H–J)[Figure 11 from McLeod et al, Invest. Ophthalmol. Vis. Sci 50:4982-4991, 2009 (McLeod et al., 2009) with permission]
Yuan et al recently performed mass spectroscopy on Bruch’s membrane/choriocapillaris complex and found 901 proteins in the complex and 56 were elevated in AMD while 43 were reduced in AMD compared to aged controls (Yuan et al., 2010). About 60% of the elevated proteins are involved in immune response and galectin 3 was the most elevated protein in dry AMD. Galectin 3 is an advanced glycosylation end product (AGE) receptor so its elevation is consistent with a role for AGEs in dry AMD. The alterations progress throughout life, so that in the eyes of many elderly persons, BrMb shows extensive thickening and formation of deposits. The increase in thickness is greater in the posterior pole than in the periphery (Ramrattan et al., 1994). It is not certain whether these changes precede development of senescent changes in the macular retina, nor is it clear whether the changes result from an accumulation of material from the CC or from the RPE, or whether they develop from the collagen and other matrix components in situ. Karwatowski et al found a linear decline in collagen solubility with age: 100% soluble in the first decade of life and 40% soluble in the ninth decade (Karwatowski et al., 1995). AGEs are associated with BM and CC basement membrane and intercapillary septa in aging and AMD (Handa et al., 1999). Levels of AGEs are also environmental, their levels increasing when consuming higher glycemic index diets. Curcio has found greatly increased lipids in AMD Bruch’s membrane, which she hypothesizes to be made by RPE (Curcio et al., 2009a, b; Curcio et al., 2001). All of these changes are associated with the increasing basophilia of the membrane. Such changes are more prominent in the macular and periphery regions, becoming less intense toward the equator (Guymer et al., 1998). It is possible that the serum proteins we have observed in choroidal stroma (Lutty et al, unpublished results) accumulate there because they do not reach the RPE for transport due to deposits in BrMb.
The progressive accumulation of debris, lipid deposition and alteration of the ECM are the three anatomical changes that occur in the BrMb with age. There are three morphological forms of sub-RPE deposits: hard drusen, soft drusen, and basal deposits in and on Bruch’s membrane. Drusen is believed by some to be incompletely digested material from the RPE, which cannot traverse Bruch’s membrane for removal by the CC. Crabb et al identified 129 proteins in drusen by mass spectroscopy (Crabb et al., 2002). The proteome Crabb found in drusen of AMD subjects had oxidative protein modifications and carboxyethyl pyrrole (CEP) protein adducts. These adducts are formed by the oxidation of docosahexaenoate-containing lipids, which are abundant in the photoreceptor outer segments. When Hollyfield and colleagues injected CEP adducts into mice, AMD-like changes were observed in the mice (Hollyfield, 2010; Hollyfield et al., 2008; Hollyfield et al., 2010). Gu et al had previously found circulating antibodies to CEP in AMD subjects suggesting that CEP antibodies might be used as a biomarker for AMD (Gu et al., 2003). One hypothesis on the genesis of drusen states that the deposit of the debris on/in Bruch’s membrane is a result of choriocapillaris insufficiency. Alternatively, debris may accumulate on/in Bruch’s membrane and CC atrophy results since it is not needed or able to perform transport any more (Bird, 1992; Lutty et al., 1999).
Hard drusen are refractile, pinpoint (less than 63 um), sharp edged deposits apparent in the fundus as yellow white deposits. We have found that hard drusen are present almost always over intercapillary septa and not over CC lumens (Bhutto and Lutty, 2004), suggesting that this material accumulates on/in Bruch’s membrane where transport is least likely. These structures may be present in any aged eye in small numbers without any consequence but numerous hard drusen are independent risk factor for vision loss in AMD (Bird et al., 1995; Klein et al., 1991). Soft drusen are larger with indistinct edges. The deposit itself has a fluffy appearance in cross section. There is a tendency for soft drusen to be confluent with each other and large confluent soft drusen are independent risk factors for AMD (Ambati et al., 2003). Interestingly, there can be spontaneous resolution of soft drusen in GA.
A third kind of deposit appears to be within Bruch’s membrane itself or a basal deposit. One form is a basal linear deposit (BLinD), which has been associated specifically with severity and progression of AMD. Green and Enger found that BLinD formed a thin layer of membranous profiles below the RPE (Green and Enger, 1993). Sarks et al suggested that they were caused by entrapment of membranous bodies released from the basal plasma membrane of the RPE, but were unable to enter the ICL owing to the tight meshwork of collagen fibrils beneath them (Sarks et al., 2007). Starita et al found that the inner collagenous zone of Bruch’s Membrane imparts the major resistance to fluid movement between RPE and choroid (Starita et al., 1997). Curcio and Millican demonstrated that BLinD and large drusen with membranous contents are strongly associated with early age-related maculopathy compared with basal laminar deposits (Curcio and Millican, 1999). This deposit stains with PAS stain and has high lipid content. At the ultrastructural level, this deposit is located between the inner collagenous zone of Bruch’s membrane and the basal lamina of the RPE.
Basal laminar deposits (BLamD) are another form of sub-RPE deposit found in the BrMb, consisting of diffuse heterogenous material that lies internal to the RPE basal lamina. It appears as a flocculent PAS positive material at the light microscopic level. Unfortunately, it is only possible to distinguish between BLamD and BLinD by ultrastructural analysis since the former is between the RPE cytoplasmic membrane and the basal lamina of RPE, while the BLinD is found between the basal lamina of RPE and the inner collagenous layer of Bruch’s membrane (Curcio and Millican, 1999). Ultrastructural and histochemical analyses suggest long spaced collagen to be a dominant constituent of these deposits whereas BLinD are composed primarily of membranous debris (Curcio and Millican, 1999; Sarks et al., 2007).
2.1d Choroid and choriocapillaris in AMD
The role of choroid in AMD has been controversial. Using enhanced depth imaging (EDI) spectral domain OCT, Spaide has observed a 16 μm decrease in choroidal thickness per decade of life (Margolis and Spaide, 2009; Spaide et al., 2008). Ding et al observed a more extreme thinning (equivalent to 54 μm reduction in thickness of choroid per decade)(Ding et al., 2011 ). Our impression is that choroidal thinning during AMD is present in almost all of the 20 AMD subjects documented in our histological collection (Imran Bhutto and Gerard Lutty, unpublished data, 2011).
In comparison with age-related thinning of the choroid, Kim et al found no difference in choroidal thickness between wet AMD and control subjects (Kim et al., 2011). Wood et al also observed no choroidal thickening in AMD but found retinal thickness was reduced in AMD subjects compared to controls (Wood et al., 2011), which may be due to loss in photoreceptors. Others have questioned the accuracy of measuring choroidal thickness with spectral domain OCT (Rahman et al., 2011).
Friedman hypothesized that vascular insufficiency resulted in loss of PR and RPE (Friedman, 1997). Furthermore, loss in CC resulted in accumulation of waste at and on Bruch’s membrane, i.e. lack of transport resulted in drusen formation (Friedman et al., 1963). Studies have shown that the density and diameter of the CC and medium-sized choroidal vessels substantially decline with age, resulting in decreased choroidal blood volume and blood flow. Foveolar blood flow declines in with aging and declines further in AMD (Grunwald et al., 1998a; Grunwald et al., 1998b; Grunwald et al., 2005; Metelitsina et al., 2008). Friedman also advanced a hypothesis that AMD is a hemodynamic sequela of atherosclerotic changes affecting the postcapillary resistance of the choroidal vasculature (Friedman et al., 1995). This and other vascular insufficiencies like hypertension could explain changes in the angiographic choroidal filling defects and prolonged filling times in AMD using ICG angiography (Pauleikhoff et al., 1990; Staurenghi et al., 1992)
An image analysis technique was developed to quantify changes in RPE and CC in postmortem human eyes of subjects with AMD (McLeod et al., 2002). The entire choroid was incubated for alkaline phosphatase activity (APase), an indicator of endotherilal cell viability which is found in all viable choroidal blood vessels (McLeod and Lutty, 1994). The choroid is partially bleached so that RPE melanin was beige not black and melanin in choroidal melanocytes is completely bleached. The loss of RPE and CC was quantified by using two methods of illumination to capture images and Adobe Photoshop to determine the number of blue pixels from APase stained choroidal blood vessels (% vascular area or area of vasculature) or the number of tan pixels (% of Bruch’s membrane covered by RPE) (McLeod et al., 2002). This permitted the loss of RPE to be related to loss of CC. The APase-incubated choroid was then flat-embedded and sectioned following image analysis and morphometric analysis to view the areas documented by image analysis in cross section.
The photoreceptors and RPE degenerate in a horse shoe-shaped pattern surrounding the fovea in geographic atrophy (GA), often sparing the fovea; the loss of choroidal vasculature appeared to be a secondary event in earlier studies (Sarks et al., 1988; Sunness, 1999). Using our APase flat-embedding technique, we found a linear relationship between the loss of CC and RPE in GA. A 50% reduction in vascular area was found in regions of complete RPE atrophy in GA but no area was completely devoid of CC (McLeod et al., 2009)(Figure 2). The border of the RPE defect was clearly delineated and coincided closely with the area of decreased choroidal vascular density but there were areas with RPE loss at the border that had normal appearing CC pattern, suggesting that RPE loss occurred in advance of CC death. This agrees with the experimental observations of Korte et al that loss in RPE results in loss of CC (Korte et al., 1984).
Extreme constriction of remaining viable capillaries was apparent in flat mounts and cross sections of areas devoid of RPE (McLeod et al., 2009)(Figure 2). Surviving CC in the area of RPE atrophy had significantly smaller diameters than CC in control subjects and in normal areas of the GA eyes (P<0.0001) (ex: Figure 1C-D). In another study by Bhutto et al, we found a significant reduction in vascular eNOS as well as nNOS in neurons and RPE in AMD choroid (Bhutto et al., 2009). The presumed reduction in nitric oxide in AMD could explain the severe constriction in these surviving capillaries.
Clinically undetected CNV was observed in the periphery and the macula in some of our GA subjects and this was always associated with surviving RPE cells. Sunness has documented the formation of CNV in GA patients (Sunness et al., 1999). She observed that 11% of the GA subjects developed CNV in the study eye by 4 years. If patients had CNV in the fellow eye, 18% developed CNV in the study eye by 2 years and 34% by 4 years. The association of surviving RPE cells with CNV in our GA specimens suggests that RPE cells may furnish a stimulus for new vessel formation or stabilization.
APase analysis of exudative AMD resulted in a very different scenario for viability of CC in wet AMD. We observed loss of CC adjacent to CNV (Figure 3), which we previously observed in diabetic choroidopathy using the APase technique (Cao et al., 1998; McLeod and Lutty, 1994). In wet AMD, large areas that lacked APase+ CC were completely covered with RPE (Figure 4E-F). Areas with significant attenuation of CC around active CNV and disciform scars were areas associated with viable RPE cells. Areas without RPE, had greatly reduced viable CC as we as observed in GA. Areas with active CNV always had RPE associated with it. In the fan-shaped CNV formation shown in Figure 3, the anterior tips of the viable neovascular channels have intense APase activity (transmitted light, Figure 4F) and there was a 50% reduction in viable CC in advance of the CNV (far right, Figure 3H). So the % vascular area was 40% instead of 80% in controls, while the RPE density was 100%. In sections through different areas (Figure 4G), it was apparent that RPE were present over areas with extremely attenuated CC suggesting that exudative AMD may have a vascular etiology.
The final pathologic insult after CNV formation and leakage of serum proteins is formation of disciform scar. The scars often contain active CNV that appears stabilized, i.e. not leaking and not growing. The vessels may be stabilized by the components of scar. We have observed that scars are rich in PEDF and thrombospondin-1, two endogenous inhibitors of angiogenesis.
In summary, some areas in GA had a normal CC pattern yet RPE cells had atrophied. Therefore, in this form of AMD in which the photoreceptors and RPE degenerate in a horse shoe shaped pattern surrounding the fovea, the loss of choroidal vasculature appears to be a secondary event suggesting that GA does not have a vascular etiology. Interestingly, even in areas with complete RPE atrophy, some CC segments remained viable (APase positive) but severely constricted (Figure 2). Surviving CC segments in areas with complete RPE atrophy contradicts the data of Korte and Henkind in animal models that suggests that RPE are essential for survival of CC (Korte et al., 1984). Every example of active CNV we have observed had surviving RPE associated with it as represented by the Figure 5 schematic. In wet AMD, loss of CC occurs in the presence of RPE. We hypothesize that CC loss results in ischemic RPE and then in turn RPE produce hypoxia-inducible angiogenic factors like VEGF, which stimulate growth of CNV from CC, venules, or arterioles and grow through Bruch’s membrane and spread under the RPE (Figure 5).
Figure 5.

Schematic of a normal RPE/BrMb/CC complex (A) and the changes that occur in dry (B) and wet (C) AMD. (A) The CC (purple) lie under BrMb (Black line) and large choroidal blood vessels (blue/red) are below the CC. RPE reside on top of BrMb. In GA (B), RPE are lost and then the CC become attenuated but some CC survive but are constricted. In wet AMD (C), CC is lost while RPE remain. The RPE become hypoxic and produce hypoxia-inducible growth factors like VEGF, which stimulate the formation of CNV (solid purple blood vessel).
2.1f Angiogenic factors and CNV
CNV represents the growth of new blood vessels from the choroid into the subretinal space or sub-RPE. CNV is a common pathological endpoint in a heterogeneous variety of chorioretinal diseases (Green and Wilson, 1986) including diabetic retinopathy (Cao et al., 1998). Nearly any pathologic process that involves the RPE and damages Bruch’s membrane can be complicated by CNV. The exact mechanism underlying the pathogenesis of CNV is not yet well understood. The natural history of CNV can be divided into three stages: 1) the initiation stage, endothelial cells (EC) derived from the choriocapillaris proliferate and migrate towards the retina through the Bruch’s membrane; 2) the active stage when CNV expands; 3) the involution stage when the CNV becomes fibrotic and forms a disciform scar (Gass, 1971).
Angiogenesis, the outgrowth of new capillaries from preexisting vessels, is tightly controlled by a finely tuned balance between factors that either stimulate or inhibit vessel growth. In most normal tissues, inhibitory factors predominate and vessels remain quiescent. In contrast, in a variety of pathologic state such as exudative AMD and tumor growth, neovascularization occurs because of decreased production of inhibitors and/or increased production of angiogenic stimulators (Folkman, 1995). Angiogenesis also requires that matrix metalloproteins and other enzymes break down the surrounding extracellular matrix or basement membrane, permitting endothelial cell migration from the existing blood vessel into the surrounding tissue.
CNV is stimulated by angiogenic factors like, vascular endothelial growth factor (VEGF), but there are many other growth factors like insulin-like growth factors, the fibroblast growth factor family members, interleukins, and angiopoietins that might participate in angiogenesis. Among the angiogenic factors, VEGF is the most important contributor to the angiogenesis in exudative AMD. Endothelial cells (EC), pericytes, glial cells, Muller cells, ganglion cells, photoreceptors, and RPE are potential sources of VEGF production (Ishibashi et al., 1997; Kvanta et al., 1996; Lopez et al., 1996; Otani et al., 2002; Wada et al., 2001). RPE secretes VEGF in a polarized manner, with higher basal secretion towards Bruch’s membrane than apical secretion towards photoreceptors in normal subjects (Blaauwgeers et al., 1999). VEGF acts as both a specific EC mitogen and promoter of vascular permeability. Immunohistochemical expression of VEGF has been shown in surgically excised CNV (Frank et al., 1996; Grossniklaus et al., 1992; Kvanta et al., 1996; Lopez et al., 1996) and the vitreous levels of VEGF were significantly higher in patients with neovascular AMD (Aiello et al., 1994). Anti-VEGF therapies were able to inhibit the laser-induced CNV in animal models (Krzystolik et al., 2002; Saishin et al., 2003) and CNV in human AMD subjects (Jyothi et al., 2010).
Hypoxia inducible factor-1 (HIF-1) is a key signal for overproduction of VEGF with subsequent abnormal growth of the neovascular component of submacular tissue repair. The presence of HIF-1alpha and HIF-2alpha has been recently discovered in active CNV specimens (Sheridan et al., 2009). In addition to hypoxia, hyperglycemia increases the level of HIF and glycemic control can down regulate it (Chiu and Taylor, 2011). In addition to angiogenesis, vasculogenesis, incorporation of progenitors, appears to be implicated in CNV development. VEGF is a chemoattractant for EC precursors, inducing their mobilization and promoting their differentiation (Asahara et al., 1999). The chemokine stromal cell-derived factor 1-alpha (SDF-1 α) and its receptor CXCR4, are HIF inducible and involved in the recruitment of EC precursors into CNV (Guerin et al., 2008; Sengupta et al., 2010). VEGF also represents a potent chemotactic signal for macrophages (Barleon et al., 1996; Clauss et al., 1990).
In the initiation stage of CNV, macrophages and their production of cytokines are important players in the growth of CNV. The second stage, neovascular enlargement, may depend on the presence of infiltrating inflammatory cells responding to cytokines in the area or producing cytokines in an autocrine/paracrine manner. Interleukin-2, -6, and -10 might participate to CNV expansion, but their exact roles have not been investigated thoroughly yet (Apte et al., 2006; Izumi-Nagai et al., 2007).
Vascular endothelium and macrophages produce MMPs, which, in turn, degrade extracellular matrix allowing CNV infiltration through Bruch’s membrane[Grossniklaus HE and Green WR, 2004]. It is unknown whether macrophages actively cause breaks in Bruch’s membrane (via production of collagenase/elastase), or they are introduced into the compromised areas after the CNV breaks through Bruch’s membrane (Grossniklaus and Green, 2004). In an experimental model of CNV, macrophages depletion diminishes both size and severity of the neovascular lesion (Espinosa-Heidmann et al., 2003). Leukocytes are recruited in CNV not only by VEGF but also by vascular adhesion protein-1, an EC adhesion molecule (Noda et al., 2008).
Insulin-like growth factor (IGF)-1 is a growth-promoting polypeptide that can act as an angiogenic agent in the eye; the expression of IGF-1 and its receptor (IGF-1R) mRNA and IGF-1R protein in situ in normal human eye and in eyes with neovascular AMD has been reported (Lambooij et al., 2003). In vivo, both VEGF expression and CNV activity in mice were downregulated by a specific inhibitor of IGF-1R (picropodophyllin) (Economou, 2008).
Nitric oxide is a signaling molecule with pleiotropic effects, and it is a well-known mediator of vascular dilatation and permeability (Moncada et al., 1991). Experimental findings indicate that nitric oxide is an important CNV stimulator, and that its reduction represents a potential therapeutic strategy for CNV (Ando et al., 2002a; Ando et al., 2002b). However, in contrast to the prediction that diminishing NO levels would be salutary, we have observed that reduction of nitric oxide synthases in AMD choroid could be associated with vasoconstriction observed in GA and hemodynamic changes in AMD (Bhutto et al., 2009).
At the involutional stage of CNV, the most important players are TGF-β and TIMP-3, produced by RPE. These markedly influence both the secretion of extracellular matrix and the tissue remodeling. The outcomes of these processes are the maturation of established vessels and formation of scar tissue. The origin of cells and factors contributing to the subretinal fibrosis is not yet completely clear. However it is known that RPE cells themselves, directed by TNF-α, TGF-β, and other growth factors, dedifferentiate and proliferate and, together with choroidal fibroblasts, initiate wound repair or scar formation (Kent and Sheridan, 2003). Stabilization of the blood vessels that remain in the scars may be due to anti-angiogenic factors present in the scar, which is discussed in the next section.
2.1g. Loss in anti-angiogenic factors in the RPE/BrMb/CC complex
The physiological stability of the ocular vascular system is a balance between angiogenic and anti-angiogenic or angiostatic factors. The vasculature is stable or quiescent as long as the angiogenic factors do not overwhelm the endogenous angiostatic factors. Pathologic conditions such as ischemia or inflammation shift the balance towards angiogenic factors that are released by the damaged cells (Folkman and Klagsbrun, 1987; Gao et al., 2001).
Several endogenous angiogenesis inhibitors have been found in the eye: pigment epithelium-derived factor (PEDF), endostatin, and thrombospondin-1 (TSP-1). These anti-angiogenic factors are predominantly extracellular proteins that are part of the matrix or bind to the matrix and quite frequently require proteolytic processing for their activation (Carmeliet and Jain, 2000). Possibly tipping the balance toward vessel formation, we have observed a decrease in pigment epithelium-derived factor (PEDF), endostatin, and thrombospondin-1 (TSP-1) in BrMb/CC complex during AMD (Bhutto et al., 2008)(Figures 6 and 7).
Figure 6.

Serial sections of submacular choroid from normal aged control (left) and retina and submacular choroid from an AMD subject (right) incubated with TSP-1, endostatin, and PEDF antibodies. Right panels are high magnification photos of left panels. (A–D) Hematoxylin and eosin (H&E) staining show morphological features of retina and choroid like migration of RPE cells into retina in AMD (C,D). Pigment in immunostained sections was bleached from RPE and choroidal melanocytes. Immunostaining of CD-34 (E–H) is associated with the retinal and choroidal blood vessels including CC (arrow). In aged control choroid, TSP-1 immunoreactivity (I and J) is intense especially in BrMb (arrowhead). Both endostatin (M and N) and PEDF (Q and R) are prominent in RPE basal lamina, BrMb, and CC basement membrane and show similar pattern and intensity of immunostaining. In contrast, expression of TSP-1 (K and L), endostatin (O and P), and PEDF (S and T) is greatly reduced in AMD choroid compared to the aged control and the reaction product of endostatin and PEDF appears more diffuse in choroidal stroma. (arrowhead, Bruch’s membrane; arrow, choriocapillaris).
Figure 7.
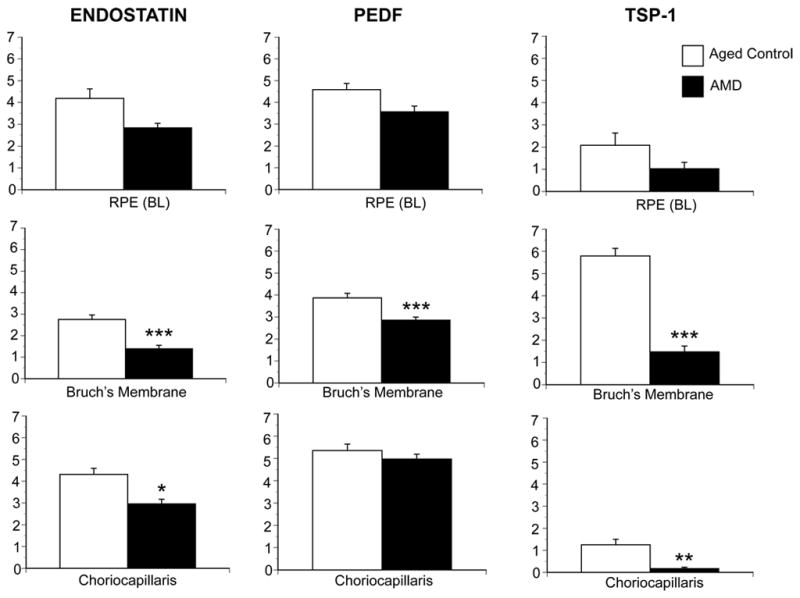
Graphic representation of data from 8 aged control subjects and 12 AMD subjects, represented by the immunohistochemistry in Figure 6. The scores (0–7) on the Y-axis represent the mean scores from three masked observers. Aged control subject data is represented by open bars and AMD subjects by black bars. A significant difference (***, P<0/01) was found for all three inhibitors in BrMb between control and AMD subjects.
PEDF is an RPE-secreted glycoprotein and a member of the serpin family that lacks serine protease inhibitor activity. PEDF is a multifunctional protein with demonstrable neurotrophic, neuroprotective, gliastatic, anti-tumorigenic, anti-angiogenic and antivasopermeability properties (Amaral and Becerra, 2010; Bouck, 2002; Dawson et al., 1999). It also has immunomodulatory features. Different parts of the PEDF molecule mediate these different activities, and peptides corresponding to these regions retain the corresponding activity of native PEDF. The role of endogenous PEDF in the eye is uncertain. Several studies have demonstrated that intraocular injection of viral vectors that express PEDF or injection of recombinant PEDF suppresses retinal or choroidal neovascularization (Gehlbach et al., 2003; Saishin et al., 2005). However, high doses of PEDF may have the opposite effect since continuous infusion of low doses suppressed CNV but high doses were stimulatory. We investigated VEGF and pigment epithelial growth factor (PEDF) in aging and AMD to evaluate the balance between these substances and if there was a shift toward angiogenesis (Gao et al., 2001; Kim et al., 2003). We found that PEDF levels were reduced significantly during AMD in RPE cells, Bruch’s membrane and choroidal stroma (Bhutto et al., 2006). However, VEGF immunoreactivity was not significantly reduced in the RPE/Bruch’s membrane/CC complex. The most intense VEGF immunoreactivity was observed in leukocytes in choroid.
Thrombospondin-1 (TSP-1) is a large (180 kDa), secreted glycoprotein that, like PEDF, has multiple domains and functions, including inhibition of angiogenesis, neuroprotection, and axon guidance. The receptor through which TSP-1 mediates its effects on endothelial cells, CD36, is also a multiligand scavenger receptor. TSP-1 knockout mice show increased vascular density during retinal vascular development. Both PEDF and TSP-1 selectively induce apoptosis in endothelial cells participating in neovascularization, but have no effect on endothelial cells in mature vessels. Cultured RPE produce Tsp-1 (Carron et al., 2000; Miyajima-Uchida et al., 2000) and it is found in vitreous and aqueous humor (Sheibani et al., 2000). Our study of TSP-1 in aging and AMD demonstrated that TSP-1 in Bruch’s membrane declines with age (Uno et al., 2006). We found that it was almost absent in AMD Bruch’s membrane and CC basement membrane. Additionally, we found high levels of TSP-1 as well as PEDF in disciform scars that a few quiescent blood vessels present in them.
Other proteins, such as angiostatin and endostatin, have domains that acquire antiangiogenic activity only after proteolytic cleavage. Endostatin, another endogenous inhibitor of angiogenesis, is generated by the cleavage of a collagen XVIII fragment (O’Reilly et al., 1997). Several proteases, including cathepsin L and elastase, have been demonstrated to be involved in the cleavage of the C-terminal noncollagenous domain (NC1) of collagen type XVIII to produce endostatin. Corroborating its role as an anti angiogenic factor, intravenous administration of adenoviral vectors containing an expression construct for endostatin resulted in prevention of laser-induced CNV in mice (Mori et al., 2001). We have recently found that endostatin is reduced in AMD choroid, compared with aged control choroids, while collagen type XVIII levels were comparable in the two groups (Bhutto et al., 2004).
We combined the analysis of the three angiogenic agents and found all three significantly reduced or absent in AMD BrMb/CC complex (Bhutto et al., 2008)(Figure 6 and 7). These findings suggested that different angiogenic inhibitors were present in the RPE/BrMb/CC complex and may play a significant role in wet AMD. The reduction in all three anti-angiogenic agents in AMD suggests that Bruch’s membrane is susceptible to invasion by CNV. Combinations of two or more angiogenic inhibitors with different molecular mechanisms or targets may achieve synergistic effects in treating CNV.
3.0 Role of inflammation in AMD
3. 1 Proinflammatory RPE/BrMb/CC complex
Despite intensive basic and clinical research, the pathogenesis of AMD remains unclear and likely is multifactorial. A growing body of evidence indicates that inflammation and especially the complement system play key roles in the etiology of AMD. If AMD is caused by inflammation, it is not a classical inflammatory disease per se but a growing body of evidence suggests that chronic, abnormal inflammatory response and immunologic events play an important role in progression of AMD (Jager et al., 2008). Components of the complement system including C3 complement fragments, C5, and the membrane attack complex (MAC) C5b-9 have been clearly shown to be present in ocular drusen and the capillary pillars of the choroid (Crabb et al., 2002; Johnson et al., 2001; Johnson et al., 2000) and in the vitreous of human eyes. Other molecules that mediate local inflammation have also been identified in AMD tissue (Anderson et al., 2002).
The complement system is a biochemical cascade that helps or “complements” the ability of antibodies to destroy pathogens. As part of the innate (or non-adaptable) immune system, the complement system promotes inflammation, eliminates pathogens, and enhances an individual’s immune response. The complement system consists of a group of more than 20 proteins, generally synthesized by the liver, which circulate as inactive precursors, or pro-proteins. When stimulated by one of several triggers, proteases in the complement system can cleave specific proteins, in turn resulting in the release of specific cytokines and consequent initiation of an amplifying cascade of further cleavages. These result in activation of the cell-killing membrane attack complex (MAC), which creates perforations in cell membranes. There are three distinct complement pathways: the classical pathway, the alternative pathway, and the mannose-binding lectin pathway.
The classical complement pathway is triggered by antigen-antibody complexes. The alternative and mannose-binding lectin pathways can be activated without the presence of antibodies (nonspecific immune response). Although each pathway is triggered differently, the common goal of these pathways is to deposit clusters of C3b on target pathogens. C3-convertase cleaves C3, creating C3a and C3b. C3b then binds to the pathogen surface, leading to internalization by macrophages and other phagocytic cells. C5a is an important chemotactic protein, helping recruit inflammatory cells. Both C3a and C5a have anaphylatoxin activity, leading to mast cell degranulation and increased vascular permeability. C5b also starts the membrane attack pathway, which leads to the formation of the end product of the complement cascade: the MAC. The MAC, consisting of C5b, C6, C7, C8, and C9, forms a transmembrane channel within cells leading to osmotic lysis (Dunkelberger and Song, 2010; Walport, 2001). Mullins and associates have found that there is increased attack complex in AMD eyes that have the high risk complement factor H (CFH) genotypes (Mullins et al., 2011a).
The complement system is continuously activated at low levels in the normal eye and intraocular complement regulatory proteins (CD35, CD46, and CFH) tightly regulate this spontaneous complement activation to maintain complement activity at a level that promotes elimination of potential pathogens without damaging healthy tissue. Dysregulation of complement leads to overactive complement activity that can cause immune-mediated ocular damage. One thought on complement activation from Janet Sparrow is that photo-oxidation of A2E, a component of lipofuscin, results in products that activate complement (Zhou et al., 2006)(also see review in this issue by Sparrow). Weismann and associates have demonstrated that CFH binds malondialdehyde epitopes and protects against oxidative stress (Weismann et al., 2011).
Using DNA sequence data from the Human Genome Project, three independent groups demonstrated that a polymorphism (Tyr402His) in the CFH gene increases the risk of developing AMD (Edwards et al., 2005; Haines et al., 2005; Klein et al., 2005b). Polymorphisms in complement factor B (CFB) and factor 3 (C3) genes have been reported as novel risk factors for AMD. Another gene called HTRA1 (encoding a secreted serine protease) has been identified that has implications for the AMD as well (Dewan et al., 2006; Yang et al., 2006).
CFH mainly functions to control the alternative complement activation in plasma, host cells and tissue, and sites of tissue inflammation. CFH is involved in inhibiting the inflammatory response mediated via C3b (the alternative pathway of complement) both by acting as a cofactor for cleavage of C3b to its inactive form, C3bi, and by weakening the active complex that forms between C3b and factor B. C-reactive protein and glycosaminoglycans, the polyanionic surface markers, normally improve the ability of CFH to inhibit complement. The mutation in CFH (Tyr402His) reduces the affinity of CFH for CRP and probably also alters the ability of factor H to recognize specific glycosaminoglycans. This change could result in reduced ability of CFH to regulate the alternative pathway permitting it to run uncontrolled. The failure of CFH-Y402H in the SCR7 domain of CFH to bind to CRP could result in the high levels of unbound CRP in the choroid that we observed (Bhutto et al., 2011), which could be permissive for chronic inflammation (Johnson et al., 2006). Functional analysis indicates that wt but not mutant CFH binds malondialdehyde epitopes and protects against oxidative stress (Weismann et al., 2011).
C-reactive protein (CRP) is an acute-phase protein considered to be a nonspecific serum biomarker for subclinical inflammation, which activates the complement system (Pepys and Hirschfield, 2003). Elevated CRP is considered as a risk for cardiovascular disease (Mainous and Pearson, 2003), adult-onset diabetes (NIDDM) (Dehghan et al., 2007) and, more recently, AMD (Seddon et al., 2004; Seddon et al., 2005). Vine et al (Vine et al., 2005) reported similar findings and concluded that a higher CRP level was a risk factor for AMD, but the mechanism underlying the link between CRP and these diseases is not fully understood. CRP is thought to assist in complement binding to foreign and damaged cells and enhances phagocytosis by macrophages, which express a receptor for CRP. CRP immunoreactivity has also been identified in ocular drusen and other sub-RPE deposits (Anderson et al., 2002; Mullins et al., 2000). However, there have been reports contradicting the association of CRP and AMD (Klein et al., 2005a; McGwin et al., 2005). CRP can be deposited at sites of tissue damage (Parish, 1976) and can possibly form soluble complexes with certain apolipoprotein B (apoB)-containing lipoproteins (Rowe et al., 1984). Recently, we investigated the expression pattern of the CRP and CFH in the submacular RPE/BrMb/CC complex in aged control human eyes and in eyes with early, wet, and dry (geographic atrophy) AMD (Bhutto et al., 2011)(Figure 8). CRP immunoreactivity was prominently localized in and around the CC, ICS and individual cells in choroidal stroma in aged control subjects (Figure 8). In early and wet AMD choroids, CRP was more intense and significantly increased in the RPE/BrMb/CC complex including the ICS. In contrast, immunostaining for CFH was significantly reduced in the BrMb/CC complex including the ICS in eyes with early and wet AMD (Bhutto et al., 2011). Furthermore, there is a significant inverse correlation between the CRP and CFH levels in eyes with advanced AMD (Figure 8). Our results suggest that high levels of CRP and insufficient CFH at the RPE/BrMb/choroid complex may lead to uncontrolled complement activation with associated cell and tissue damage. Another inhibitor of complement, CD46, is also reduced in the RPE during AMD suggesting that the normal regulators of complement are not present in AMD (Vogt et al., 2011).
Figure 8.

Immunolocalization of C-reactive protein (CRP) and complement factor H (CFH) in submacular choroid from aged control, early and late wet AMD eyes. Periodic acid-Schiff’s (PAS) and hematoxylin (Hem) staining shows morphological features of the choroid from aged control (A), drusen (asterisk) in early AMD (B) and CNV (large arrow) anterior to RPE in wet AMD (C). Pigment in immunostained sections was bleached from RPE and choroidal melanocytes. Immunostaining of CD34 is associated with CC (small arrow) and large choroidal vessels appear morphologically normal with broad lumens in aged control (D), whereas CC lumens appear irregular and constricted in early (E) and wet AMD (F). In aged control choroid, CRP (G) and CFH (J) are prominently localized to the CC, intercapillary septa (ICS) and BrM (open arrowhead). CRP immunoreactivity is significantly increased in early (H) and late AMD (I) choroids compared to the aged control and appear more diffuse in choroidal stroma. CFH in early AMD (K) is comparable to aged control, whereas it is significantly decreased in wet AMD (L). Drusen are intensely labeled with CRP and CFH (H and K). Note that in wet AMD the CNV (large arrow), intensely labeled with CD34 antibody (F), has more CRP and less CFH (I and L). Nonimmune rabbit IgG (NIIgG) yields a very weak to negative reaction product except in drusen (M, N, O). [Figure 3 from Bhutto et al British Journal of Ophthalmology 95:1323-1330, 2011 (Bhutto et al., 2011) with permission]
The apolipoproteins LDL and HDL serve a variety of functions including transport of lipoprotein to certain cells or regulating enzymes that package lipids. These proteins can also interact with other systems of the body that have nothing to do with lipid handling. Abnormalities in the amounts or kinds of lipoproteins can cause increased risk of atherosclerosis. Atherosclerosis is a multifactorial disease driven by inflammatory reactions and has been associated with AMD. Lipoprotein(a) or Lp(a) is an atherogenic lipoprotein, similar to low density lipoprotein (LDL), that is attached to apoB-100 by a disulfide bond (Scanu and Fless, 1991; Scanu et al., 1991). Many studies have demonstrated a significant relationship between Lp(a) and CRP (McLaughlin et al., 2002; Rallidis et al., 2002) and other inflammatory factors, such as fibrinogen, interleukin-6 (Park et al., 2002), complement 3 and 4 (Gurbuz et al., 2001) and soluble cellular adhesion molecules (Stenvinkel et al., 2000). We found that Lp(a) was prominently localized to the CC of aged human control choroids (Bhutto and Lutty, unpublished results). Lp(a) was significantly increased in choroidal arteries in eyes with early AMD. The higher levels of Lp(a) in early and wet AMD choroid may represent a possible atherogenic risk factor and may be interpreted as evidence for local inflammation and cellular injury in the RPE/choroid.
3.2 Other changes in innate immunity during AMD
Forrester et al suggested that chronic para-inflammation contributes to the initiation of AMD (Xu et al., 2009). Para-inflammation is a tissue adaptive response to noxious stress or cellular dysfunction and it has characteristics of the state between basal inflammation state and true inflammatory state (Medzhitov, 2008). The triggers for para-inflammation are dead cells, oxidative stress, AGEs and oxidized lipoproteins, all of which exist in the photoreceptor/RPE/BrMb/CC complex in early AMD. One characteristic of para-inflammation is activation of microglia, which have been reported in the subretinal space in AMD. Forrester has shown that activated microglia in the subretinal space are associated with break down of the outer blood retinal barrier occurs (Xu et al., 2009). Para-inflammatory changes in choroid may contribute to changes in choroidal thickness, abnormalities in choroidal melanocytes, and fibrosis of choroidal tissue, all of which have received little attention in AMD research.
Ambati’s lab has introduced another unique insight into the role for inflammation in AMD. They have found that RPE in AMD die after exposure to Alu RNA (Kaneko et al., 2011). Normally Alu or noncoding or junk RNA makes up about 11% of the human genome but it is normally cleaved by DICER1 in RPE cells. However, Ambati’s group has found that DICER1 is dramatically reduced in RPE of GA subjects (Kaneko et al., 2011).
4.0 Conclusions
In light of the close association of the photoreceptors, RPE, BrMb, and CC, and their mutualistic symbiotic relationship, it is not surprising that an alteration in a single component of this complex compromises the normal RPE/choroid interaction and ultimately leads to disease and death of photoreceptors. However, the two types of AMD, atrophic and neovascular, may have different etiologies in regard to the death or dysfunction of the photoreceptor/RPE/BrMb/choroid complex.
In neovascular AMD the loss of choroidal vasculature may be the initial insult to the complex. We have observed loss in CC with an intact RPE monolayer in wet AMD (McLeod et al., 2009). This may be due to reduction in blood supply because of large vessel stenosis (Friedman, 1997). In addition, the CC are susceptible to inflammation in that they constitutively express ICAM-1 (McLeod et al., 1995), so firm adhesion of activated neutrophils is always possible. Furthermore, the environment of the CC, basement membrane and intercapillary septa, is a proinflammatory milieu with accumulation of complement components (Anderson et al., 2002) as well as proinflammatory molecules like CRP (Bhutto et al., 2011). Mullins recently reported a relationship between acellular capillaries and drusen (Mullins et al., 2011b). In this toxic milieu, CC die or become dysfunction making adjacent RPE hypoxic. These hypoxic cells would then produce angiogenic substances like VEGF to stimulate regrowth of new vessels, CNV. This loss of CC might also be a stimulus for drusen formation since the disposal system would be limited. Ultimately, the photoreceptors would die of lack of nutrients, leakage of serum, and scar formation.
In atrophic AMD, it appears that large confluent drusen formation and hyperpigmentation (presumably dysfunction in RPE) are the initial insult and the resorption of these drusen and loss of RPE (hypopigmentation) can be predictive for progression of GA (Klein et al., 2008), i.e. RPE and BrMb appear to be dysfunctional first. In our studies, it appeared that the RPE died first in GA and were hypertrophic at the edge of the atrophy (McLeod et al., 2009). Ambati’s work suggests that reduction in DICER1 in RPE during GA and subsequent accumulation of Alu RNA may be the cause of their demise (Kaneko et al., 2011). Another reason for RPE death and dysfunction may be toxic products accumulated by RPE (Chiu and Taylor, 2011; Uchiki et al., 2012; Zhou et al., 2006). These toxic products are said to activate complement, which could be the cause of death for many components in the photoreceptor/RPE/BrMb/CC complex. Loss of CC is secondary to loss of RPE in atrophic AMD.
Encouraging progress has been made in replacing lost RPE cells by RPE transplantation. However, it remains unclear how to restore RPE-retinal interactions or re-establish a blood-retinal barrier. RPE derived from human embryonic stem (hES) cells can be an important source of this tissue for transplantation to prevent further photoreceptor loss after death of RPE. The chances of success may be increased by the survival of some CC segments in GA, which could provide the seed for reformation of the CC after transplantation (McLeod et al., 2009). A hurdle to success may be the inability of the new RPE/progenitor cells to repopulate an aged, thickened BrMb.
The death of photoreceptors by apoptosis, starting with loss of rods, is well documented in AMD (Dunaief et al., 2002) (Curcio et al., 1996; Curcio, 2001). Linsenmeier has shown that photoreceptors live at the edge because their oxygen content drops to near zero in the dark (Wangsa-Wirawan and Linsenmeier, 2003). Any perturbation of that supply would make the photoreceptors dysfunctional. That perturbation may occur because their nutrient transporter and waste removal and recycler, the RPE, become dysfunctional or die. Alternatively the ability of nutrients from the CC or debris from RPE to traverse an aged, cross-linked, lipid-filled BrMb is reduced with aging and AMD, resulting in accumulation of debris in and on BrMb, i.e. drusen form. The fact that photoreceptor loss appears to be related to the size of the scar (Green and Enger, 1993) says that access to nutrients from RPE and CC is critical to photoreceptor survival.
In summary the mutualistic symbiotic relationship within the photoreceptor/RPE/BrMb/CC complex is lost in both forms of AMD. Loss of this functionally integrated relationship results in death and dysfunction of all of the components in the complex. Restoration of the relationship can be accomplished therapeutically by targeting the initial insult. Control of inflammation could prevent loss of CC and increase blood flow in supply blood vessels to the CC. Reduction in oxidative stress could have positive effects on CC and RPE death. This strategy appeared to work in the AREDS study at some level (Age-related Eye Disease Study Research Group, 2001). Restoration of reduced anti-angiogenic agents in the complex by gene therapy might inhibit CNV in wet AMD. Anti-VEGF therapies are already having a profound effect on progression of wet AMD, but better delivery or longer lasting agents are needed to avoid monthly injections of the therapy into the eye (Jyothi et al., 2010). Either strategy might restore the angiogenic/antiangiogenic balance and achieve quiescence in CC. Anti-apoptotic therapies might prevent the death of any of the cells in the complex, preventing the retinal and choroidal degeneration. Finally, controlling glycemic index (avoiding hyperglycemia) and including DHA in diet (Chiu et al., 2009a; Chiu et al., 2009b) as well as avoidance of smoking and maintaining a regular exercise regimen could decrease levels of HIF, reduce oxidative stress, and increase blood flow respectively and, therefore, reduce the risk for AMD and its progression.
Acknowledgments
Studies from the Lutty lab were funded by NIH grants EY016151 (GL), EY09357 (GL), EY01765 (Wilmer); the Altsheler-Durell Foundation; American Health Assistance Foundation; and an unrestricted gift from RPB (Wilmer). Gerard Lutty is an RPB Senior Investigator. The authors acknowledge the D. Scott McLeod for creation of the Figures.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Adamis AP, Shima DT, Yeo KT, Yeo TK, Brown LF, Berse B, D’Amore PA, Folkman J. Synthesis and secretion of vascular permeability factor/vascular endothelial growth factor by human retinal pigment epithelial cells. Biochem Biophys Res Commun. 1993;193 (2):631–638. doi: 10.1006/bbrc.1993.1671. [DOI] [PubMed] [Google Scholar]
- Age-related Eye Disease Study Research Group. A randomized, placebo-controlled, clinical trial of high-dose supplementation with vitamins C and E, beta carotene, and zinc for age-related macular degeneration and vision loss: AREDS report no. 8. Arch Ophthalmol. 2001;119 (10):1417–1436. doi: 10.1001/archopht.119.10.1417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ahuja P, Caffe AR, Holmqvist I, Soderpalm AK, Singh DP, Shinohara T, van Veen T. Lens epithelium-derived growth factor (LEDGF) delays photoreceptor degeneration in explants of rd/rd mouse retina. Neuroreport. 2001;12 (13):2951–2955. doi: 10.1097/00001756-200109170-00039. [DOI] [PubMed] [Google Scholar]
- Aiello LP, Avery RL, Arrigg PG, Keyt BA, Jampel H, Shah ST, Pasquale LR, Thieme H, Iwamoto MA, Park JE, Nguyen HV, Aiello LM, Ferrara N, King G. Vascular endothelial growth factor in ocular fluid of patients with diabetic retinopathy and other retinal disorders. N Engl J Med. 1994;331:1480–1487. doi: 10.1056/NEJM199412013312203. [DOI] [PubMed] [Google Scholar]
- Aisenbrey S, Zhang M, Bacher D, Yee J, Brunken WJ, Hunter DD. Retinal pigment epithelial cells synthesize laminins, including laminin 5, and adhere to them through alpha3- and alpha6-containing integrins. Invest Ophthalmol Vis Sci. 2006;47 (12):5537–5544. doi: 10.1167/iovs.05-1590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alm A, Bill A. The oxygen supply to the retina, II: effects of high intraocular pressure and of increased arterial carbon dioxide tension on uveal and retinal blood flow in cats. Acta Physiol Scand. 1972;84:306–319. doi: 10.1111/j.1748-1716.1972.tb05182.x. [DOI] [PubMed] [Google Scholar]
- Amaral J, Becerra SP. Effects of human recombinant PEDF protein and PEDF-derived peptide 34-mer on choroidal neovascularization. Invest Ophthalmol Vis Sci. 2010;51 (3):1318–1326. doi: 10.1167/iovs.09-4455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ambati J, Ambati B, Yoo S, Ianchulev S, Adamis A. Age-related macular degeneration: etiology, pathogenesis, and therapeutic strategies. Surv Ophthalmol. 2003;48:257–293. doi: 10.1016/s0039-6257(03)00030-4. [DOI] [PubMed] [Google Scholar]
- Anderson D, Mullins R, Hageman G, Johnson L. A role for local inflammation in the formation of drusen in the aging eye. Am J Ophthalmol. 2002;134:411–431. doi: 10.1016/s0002-9394(02)01624-0. [DOI] [PubMed] [Google Scholar]
- Ando A, Yang A, Mori K, Yamada H, Yamada E, Takahashi K, Saikia J, Kim M, Melia M, Fishman M, Huang P, Campochiaro PA. Nitric oxide is proangiogenic in the retina and choroid. J Cell Physiol. 2002a;191 (1):116–124. doi: 10.1002/jcp.10083. [DOI] [PubMed] [Google Scholar]
- Ando A, Yang A, Nambu H, Campochiaro PA. Blockade of nitric-oxide synthase reduces choroidal neovascularization. Mol Pharmacol. 2002b;62 (3):539–544. doi: 10.1124/mol.62.3.539. [DOI] [PubMed] [Google Scholar]
- Apte RS, Richter J, Herndon J, Ferguson TA. Macrophages inhibit neovascularization in a murine model of age-related macular degeneration. PLoS Med. 2006;3 (8):e310. doi: 10.1371/journal.pmed.0030310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Asahara T, Takahashi T, Masuda H, Kalka C, Chen D, Iwaguro H, Inai Y, Silver M, Isner JM. VEGF contributes to postnatal neovascularization by mobilizing bone marrow-derived endothelial progenitor cells. EMBO J. 1999;18 (14):3964–3972. doi: 10.1093/emboj/18.14.3964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barleon B, Sozzani S, Zhou D, Weich HA, Mantovani A, Marme D. Migration of human monocytes in response to vascular endothelial growth factor (VEGF) is mediated via the VEGF receptor flt-1. Blood. 1996;87 (8):3336–3343. [PubMed] [Google Scholar]
- Barron MJ, Johnson MA, Andrews RM, Clarke MP, Griffiths PG, Bristow E, He LP, Durham S, Turnbull DM. Mitochondrial abnormalities in ageing macular photoreceptors. Invest Ophthalmol Vis Sci. 2001;42 (12):3016–3022. [PubMed] [Google Scholar]
- Bhutto IA, Baba T, Merges C, Juriasinghani V, McLeod DS, Lutty GA. C-reactive protein and complement factor H in aged human eyes and eyes with age-related macular degeneration. Br J Ophthalmol. 2011;95 (9):1323–1330. doi: 10.1136/bjo.2010.199216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bhutto IA, Baba T, Merges C, McLeod DS, Lutty GA. Low nitric oxide synthases (NOS) in eyes with age-related macular degeneration (AMD) Exp Eye Res. 2009 doi: 10.1016/j.exer.2009.10.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bhutto IA, Kim SY, McLeod D, Merges SCA, Fukai N, Olsen BR, Lutty GA. Localization of collagen XVIII and the endostatin portion of collagen XVIII in aged human control eyes and eyes with age-related macular degeneration. Invest Ophthalmol Vis Sci. 2004;45:1544–1552. doi: 10.1167/iovs.03-0862. [DOI] [PubMed] [Google Scholar]
- Bhutto IA, Lutty GA. The vasculature of choroid. In: Schepro D, D’Amore PA, editors. Encyclopedia of Microvasculatures. Elsevier; 2004. [Google Scholar]
- Bhutto IA, McLeod DS, Hasegawa T, Kim SY, Merges C, Tong P, Lutty GA. Pigment epithelium-derived factor (PEDF) and vascular endothelial growth factor (VEGF) in aged human choroid and eyes with age-related macular degeneration. Exp Eye Res. 2006;82:99–110. doi: 10.1016/j.exer.2005.05.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bhutto IA, Uno K, Merges C, Zhang L, McLeod DS, Lutty GA. Reduction of endogenous angiogenesis inhibitors in Bruch’s membrane of the submacular region in eyes with age-related macular degeneration. Arch Ophthalmol. 2008;126:670–678. doi: 10.1001/archopht.126.5.670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bill A. The 1990 Endre Balazs Lecture: Effects of some neuropeptides on the uvea. Exp Eye Res. 1991;53:3–11. doi: 10.1016/0014-4835(91)90138-5. [DOI] [PubMed] [Google Scholar]
- Bird AC. Bruch’s membrane changes with age. Br J Ophthalmol. 1992;76:166–168. doi: 10.1136/bjo.76.3.166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bird AC, Bressler NM, Bressler SB, Chisholm IH, Coscas G, Davis MD, de Jong PT, Klaver CC, Klein BE, Klein R, et al. An international classification and grading system for age-related maculopathy and age-related macular degeneration. The International ARM Epidemiological Study Group. Surv Ophthalmol. 1995;39 (5):367–374. doi: 10.1016/s0039-6257(05)80092-x. [DOI] [PubMed] [Google Scholar]
- Blaauwgeers HG, Holtkamp GM, Rutten H, Witmer AN, Koolwijk P, Partanen TA, Alitalo K, Kroon ME, Kijlstra A, van Hinsbergh VW, Schlingemann RO. Polarized vascular endothelial growth factor secretion by human retinal pigment epithelium and localization of vascular endothelial growth factor receptors on the inner choriocapillaris. Evidence for a trophic paracrine relation. Am J Pathol. 1999;155 (2):421–428. doi: 10.1016/S0002-9440(10)65138-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bost LM, Aotaki-Keen AE, Hjelmeland LM. Coexpression of FGF-5 and bFGF by retinal pigment epithelium in vitro. Exp Eye Res. 1992;55:727–734. doi: 10.1016/0014-4835(92)90177-t. [DOI] [PubMed] [Google Scholar]
- Bost LM, Aotaki-Keen AE, Hjelmeland LM. Cellular adhesion regulates bFGF gene expression in human retinal pigment epithelial cells. Exp Eye Res. 1994;58 (5):545–552. doi: 10.1006/exer.1994.1048. [DOI] [PubMed] [Google Scholar]
- Bouck N. PEDF: anti-angiogenic guardian of ocular function. Trends Mol Med. 2002;8 (7):330–334. doi: 10.1016/s1471-4914(02)02362-6. [DOI] [PubMed] [Google Scholar]
- Braun RD, Dewhirst MW, Hatchell DL. Quantification of erythrocyte flow in the choroid of the albino rat. Am J Physiol. 1997;272(3 Pt 2):H1444–1453. doi: 10.1152/ajpheart.1997.272.3.H1444. [DOI] [PubMed] [Google Scholar]
- Bron AJ, Tripathi RC, Tripathi BJ. Anatomy of the eye and orbit. Chapman and Hall; London: 1997. [Google Scholar]
- Campochiaro PA, Hackett SF, Vinores SA, Freund J, Csaky C, LaRochelle W, Henderer J, Johnson M, Rodriguez IR, Friedman Z, et al. Platelet-derived growth factor is an autocrine growth stimulator in retinal pigmented epithelial cells. J Cell Sci. 1994;107 ( Pt 9):2459–2469. doi: 10.1242/jcs.107.9.2459. [DOI] [PubMed] [Google Scholar]
- Campochiaro PA, Sugg R, Grotendorst G, Hjelmeland LM. Retinal pigment epithelial cells produce PDGF-like proteins and secrete them into their media. Exp Eye Res. 1989;49 (2):217–227. doi: 10.1016/0014-4835(89)90092-4. [DOI] [PubMed] [Google Scholar]
- Cao J, McLeod S, Merges CA, Lutty GA. Choriocapillaris degeneration and related pathologic changes in human diabetic eyes. Arch Ophthalmol. 1998;116 (5):589–597. doi: 10.1001/archopht.116.5.589. [DOI] [PubMed] [Google Scholar]
- Cao W, Wen R, Li F, Lavail MM, Steinberg RH. Mechanical injury increases bFGF and CNTF mRNA expression in the mouse retina. Exp Eye Res. 1997;65 (2):241–248. doi: 10.1006/exer.1997.0328. [DOI] [PubMed] [Google Scholar]
- Carmeliet P, Jain RK. Angiogenesis in cancer and other diseases. Nature. 2000;407:249–257. doi: 10.1038/35025220. [DOI] [PubMed] [Google Scholar]
- Carron JA, Hiscott P, Hagan S, Sheridan CM, Magee R, Gallagher JA. Cultured human retinal pigment epithelial cells differentially express thrombospondin-1, -2, -3, and -4. Int J Biochem Cell Biol. 2000;32 (11–12):1137–1142. doi: 10.1016/s1357-2725(00)00065-0. [DOI] [PubMed] [Google Scholar]
- Caruelle D, Groux-Muscatelli B, Gaudric A, Sestier C, Coscas G, Caruelle JP, Barritault D. Immunological study of acidic fibroblast growth factor (aFGF) distribution in the eye. J Cell Biochem. 1989;39 (2):117–128. doi: 10.1002/jcb.240390204. [DOI] [PubMed] [Google Scholar]
- Chen L, Miyamura N, Ninomiya Y, Handa JT. Distribution of the collagen IV isoforms in human Bruch’s membrane. Br J Ophthalmol. 2003;87 (2):212–215. doi: 10.1136/bjo.87.2.212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chiu CJ, Klein R, Milton RC, Gensler G, Taylor A. Does eating particular diets alter the risk of age-related macular degeneration in users of the Age-Related Eye Disease Study supplements? Br J Ophthalmol. 2009a;93 (9):1241–1246. doi: 10.1136/bjo.2008.143412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chiu CJ, Milton RC, Klein R, Gensler G, Taylor A. Dietary compound score and risk of age-related macular degeneration in the age-related eye disease study. Ophthalmology. 2009b;116 (5):939–946. doi: 10.1016/j.ophtha.2008.12.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chiu CJ, Taylor A. Dietary hyperglycemia, glycemic index and metabolic retinal diseases. Prog Retin Eye Res. 2011;30 (1):18–53. doi: 10.1016/j.preteyeres.2010.09.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clauss M, Gerlach M, Gerlach H, Brett J, Wang F, Familletti PC, Pan YC, Olander JV, Connolly DT, Stern D. Vascular permeability factor: a tumor-derived polypeptide that induces endothelial cell and monocyte procoagulant activity, and promotes monocyte migration. J Exp Med. 1990;172 (6):1535–1545. doi: 10.1084/jem.172.6.1535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Connolly SE, Hjelmeland LM, LaVail MM. Immunohistochemical localization of basic fibroblast growth factor in mature and developing retinas of normal and RCS rats. Curr Eye Res. 1992;11 (10):1005–1017. doi: 10.3109/02713689209033499. [DOI] [PubMed] [Google Scholar]
- Crabb JW, Miyagi M, Gu X, Shadrach K, West KA, Sakaguchi H, Kamei M, Hasan A, Yan L, Rayborn ME, Salomon RG, Hollyfield JG. Drusen proteome analysis: an approach to the etiology of age-related macular degeneration. Proc Natl Acad Sci U S A. 2002;99 (23):14682–14687. doi: 10.1073/pnas.222551899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Curcio C, Medeiros ANE, Millican CL. Photoreceptor loss in age-related macular degeneration. Invest Ophthal Vis Sci. 1996;37:1236–1249. [PubMed] [Google Scholar]
- Curcio C, Millican C. Basal linear deposit and large drusen are specific for early age-related maculopathy. Arch Ophthalmol. 1999;117:329–339. doi: 10.1001/archopht.117.3.329. [DOI] [PubMed] [Google Scholar]
- Curcio CA. Photoreceptor topography in ageing and age-related maculopathy. Eye (Lond) 2001;15 (Pt 3):376–383. doi: 10.1038/eye.2001.140. [DOI] [PubMed] [Google Scholar]
- Curcio CA, Johnson M, Huang JD, Rudolf M. Aging, age-related macular degeneration, and the response-to-retention of apolipoprotein B-containing lipoproteins. Prog Retin Eye Res. 2009a;28 (6):393–422. doi: 10.1016/j.preteyeres.2009.08.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Curcio CA, Johnson M, Huang JD, Rudolf M. Apolipoprotein B-containing lipoproteins in retinal aging and age-related macular degeneration. J Lipid Res. 2009b doi: 10.1194/jlr.R002238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Curcio CA, Millican CL, Bailey T, Kruth HS. Accumulation of cholesterol with age in human Bruch’s membrane. Invest Ophthalmol Vis Sci. 2001;42 (1):265–274. [PubMed] [Google Scholar]
- Curcio CA, Sloan KR, Kalina RE, Hendrickson AE. Human photoreceptor topography. J Comp Neurol. 1990;292 (4):497–523. doi: 10.1002/cne.902920402. [DOI] [PubMed] [Google Scholar]
- Dawson DW, Volpert OV, Gillis P, Crawford SE, Xu H, Benedict W, Bouck NP. Pigment epithelium-derived factor: a potent inhibitor of angiogenesis. Science. 1999;285:245–248. doi: 10.1126/science.285.5425.245. [DOI] [PubMed] [Google Scholar]
- Dehghan A, van Hoek M, Sijbrands EJ, Stijnen T, Hofman A, Witteman JC. Risk of type 2 diabetes attributable to C-reactive protein and other risk factors. Diabetes Care. 2007;30 (10):2695–2699. doi: 10.2337/dc07-0348. [DOI] [PubMed] [Google Scholar]
- Del Priore LV, Geng L, Tezel TH, Kaplan HJ. Extracellular matrix ligands promote RPE attachment to inner Bruch’s membrane. Curr Eye Res. 2002;25 (2):79–89. doi: 10.1076/ceyr.25.2.79.10158. [DOI] [PubMed] [Google Scholar]
- Del Priore LV, Tezel TH. Reattachment rate of human retinal pigment epithelium to layers of human Bruch’s membrane. Arch Ophthalmol. 1998;116 (3):335–341. doi: 10.1001/archopht.116.3.335. [DOI] [PubMed] [Google Scholar]
- Delori FC, Goger DG, Hammond BR, Snodderly DM, Burns SA. Macular pigment density measured by autofluorescence spectrometry: comparison with reflectometry and heterochromatic flicker photometry. J Opt Soc Am A Opt Image Sci Vis. 2001;18 (6):1212–1230. doi: 10.1364/josaa.18.001212. [DOI] [PubMed] [Google Scholar]
- Dewan A, Liu M, Hartman S, Zhang SS, Liu DT, Zhao C, Tam PO, Chan WM, Lam DS, Snyder M, Barnstable C, Pang CP, Hoh J. HTRA1 promoter polymorphism in wet age-related macular degeneration. Science. 2006;314 (5801):989–992. doi: 10.1126/science.1133807. [DOI] [PubMed] [Google Scholar]
- Ding X, Li J, Zeng J, ma W, Liu R, Li T, Yu S, Tang S. Choroidal thickness in Chinese subjects. Invest Ophthalmol Vis Sci. 2011;52 (13):9555–9560. doi: 10.1167/iovs.11-8076. [DOI] [PubMed] [Google Scholar]
- Dunaief JL, Dentchev T, Ying GS, Milam AH. The role of apoptosis in age-related macular degeneration. Arch Ophthalmol. 2002;120 (11):1435–1442. doi: 10.1001/archopht.120.11.1435. [DOI] [PubMed] [Google Scholar]
- Dunkelberger JR, Song WC. Complement and its role in innate and adaptive immune responses. Cell Res. 2010;20 (1):34–50. doi: 10.1038/cr.2009.139. [DOI] [PubMed] [Google Scholar]
- Dunn KC, Marmorstein AD, Bonilha VL, Rodriguez-Boulan E, Giordano F, Hjelmeland LM. Use of the ARPE-19 cell line as a model of RPE polarity: basolateral secretion of FGF5. Invest Ophthalmol Vis Sci. 1998;39 (13):2744–2749. [PubMed] [Google Scholar]
- Economou MA. Uveal melanoma and macular degeneration: molecular biology and potential therapeutic applications. Acta Ophthalmol. 2008;86 (8):930–931. doi: 10.1111/j.1755-3768.2008.01188.x. [DOI] [PubMed] [Google Scholar]
- Edwards AO, Ritter R, 3rd, Abel KJ, Manning A, Panhuysen C, Farrer LA. Complement factor H polymorphism and age-related macular degeneration. Science. 2005;308 (5720):421–424. doi: 10.1126/science.1110189. [DOI] [PubMed] [Google Scholar]
- Espinosa-Heidmann DG, Suner IJ, Hernandez EP, Monroy D, Csaky KG, Cousins SW. Macrophage depletion diminishes lesion size and severity in experimental choroidal neovascularization. Invest Ophthalmol Vis Sci. 2003;44 (8):3586–3592. doi: 10.1167/iovs.03-0038. [DOI] [PubMed] [Google Scholar]
- Flügel C, Tamm ER, Mayer B, Lütjen-Drecoll E. Species differences in choroidal vasodilative innervation: Evidence for specific intrinsic nitrergic and VIP-positive neurons in the human eye. Invest Ophthalmol Vis Sci. 1994;35:592–599. [PubMed] [Google Scholar]
- Folkman J. Clinical applications of research on angiogenesis. N Engl J Med. 1995;333:1757–1763. doi: 10.1056/NEJM199512283332608. [DOI] [PubMed] [Google Scholar]
- Folkman J, Klagsbrun M. Angiogenic factors. Science. 1987;235:442–447. doi: 10.1126/science.2432664. [DOI] [PubMed] [Google Scholar]
- Frank RN, Amin RH, Eliott D, Puklin JE, Abrams GW. Basic Fibroblast growth factor and vascular endothelial growth factor are present in epiretinal and choroidal neovascular membranes. Am J Ophthalmol. 1996;122:393–403. doi: 10.1016/s0002-9394(14)72066-5. [DOI] [PubMed] [Google Scholar]
- Friedman E. A hemodynamic model of the pathogenesis of age-related macular degeneration. Am J Ophthalmol. 1997;124 (5):677–682. doi: 10.1016/s0002-9394(14)70906-7. [DOI] [PubMed] [Google Scholar]
- Friedman E. The pathogenesis of age-related macular degeneration. Am J Ophthalmol. 2008;146 (3):348–349. doi: 10.1016/j.ajo.2008.05.017. [DOI] [PubMed] [Google Scholar]
- Friedman E, Chandra SR. Choroidal blood flow III: effects of oxygen and carbon dioxide. Arch Ophthalmol. 1972;87:70–71. doi: 10.1001/archopht.1972.01000020072015. [DOI] [PubMed] [Google Scholar]
- Friedman E, Kopald HH, Smith TR. Retinal and choroidal blood flow determined with krypton-85 anesthetized animals. Invest Opthalmol. 1964;3:539–547. [PubMed] [Google Scholar]
- Friedman E, Krupsky S, Lane AM, Oak SS, Friedman ES, Egan K, Gragoudis ES. Ocular blood flow velocity in age-related macular degeneration. Ophtahlmology. 1995;102:640–646. doi: 10.1016/s0161-6420(95)30974-8. [DOI] [PubMed] [Google Scholar]
- Friedman E, Smith TR, Kuwabara T. Senile choroidal vascular patterns and drusen. Arch Ophthalmol. 1963;69:220–230. doi: 10.1001/archopht.1963.00960040226014. [DOI] [PubMed] [Google Scholar]
- Friedrichson T, Kalbach HL, Buck P, van Kuijk FJ. Vitamin E in macular and peripheral tissues of the human eye. Curr Eye Res. 1995;14 (8):693–701. doi: 10.3109/02713689508998497. [DOI] [PubMed] [Google Scholar]
- Furness JB, Pompolo S, Shuttleworth CW, Burleigh DE. Light- and electron-microscopic immunochemical analysis of nerve fiber types innervating the taenia of the guinea pig cecum. Cell Tissue Res. 1992;270:125–137. doi: 10.1007/BF00381887. [DOI] [PubMed] [Google Scholar]
- Gao G, Li Y, Zhang D, Gee S, Crosson C, Ma J. Unbalanced expression of VEGF and PEDF in ischemia-induced retinal neovascularization. FEBS Lett. 2001;489:270–276. doi: 10.1016/s0014-5793(01)02110-x. [DOI] [PubMed] [Google Scholar]
- Gass JD. Photocoagulation of macular lesions. Trans Am Acad Ophthalmol Otolaryngol. 1971;75 (3):580–608. [PubMed] [Google Scholar]
- Gehlbach P, Demetriades AM, Yamamoto S, Deering T, Duh EJ, Yang HS, Cingolani C, Lai H, Wei L, Campochiaro PA. Periocular injection of an adenoviral vector encoding pigment epithelium-derived factor inhibits choroidal neovascularization. Gene Ther. 2003;10:637–646. doi: 10.1038/sj.gt.3301931. [DOI] [PubMed] [Google Scholar]
- Gong J, Sagiv O, Cai H, Tsang SH, Del Priore LV. Effects of extracellular matrix and neighboring cells on induction of human embryonic stem cells into retinal or retinal pigment epithelial progenitors. Exp Eye Res. 2008;86 (6):957–965. doi: 10.1016/j.exer.2008.03.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Green WR, Enger C. Age-related macular degeneration histopathologic studies. Ophthalmology. 1993;100:1519–1535. doi: 10.1016/s0161-6420(93)31466-1. [DOI] [PubMed] [Google Scholar]
- Green WR, Key SN. Senile macular degeneration: a histopathological study. Trans Am Ophthalmol Soc. 1977;75:180–254. [PMC free article] [PubMed] [Google Scholar]
- Green WR, Wilson DJ. Choroidal neovascularization. Ophthalmology. 1986;93:1169–1176. doi: 10.1016/s0161-6420(86)33609-1. [DOI] [PubMed] [Google Scholar]
- Grindle C, Marshall J. Ageing changes in bruch’s membrane and their functional implications. Trans Ophthalmol Soc UK. 1978;98:14–16. [PubMed] [Google Scholar]
- Grossniklaus HE, Green WR. Choroidal neovascularization. Am J Ophthalmol. 2004;137 (3):496–503. doi: 10.1016/j.ajo.2003.09.042. [DOI] [PubMed] [Google Scholar]
- Grossniklaus HE, Martinez JA, Brown VB, Lambert HM, Sternberg P, Jr, Capone A, Jr, Aaberg TM, Lopez PF. Immunohistochemical and histochemical properties of surgically excised subretinal neovascular membranes in age-related macular degeneration. Am J Ophthalmol. 1992;114 (4):464–472. doi: 10.1016/s0002-9394(14)71859-8. [DOI] [PubMed] [Google Scholar]
- Grunwald J, Hariprasad S, DuPont J. Effect of aging on foveolar choroidal circulation. Arch Ophthalmol. 1998a;116:150–154. doi: 10.1001/archopht.116.2.150. [DOI] [PubMed] [Google Scholar]
- Grunwald J, Hariprasad S, DuPont J, Maguire M, Fine S, Brucker A, Maguire A, Ho A. Foveolar choroidal blood flow in age-related macular degeneration. Invest Ophthalmol Vis Sci. 1998b;39:385–390. [PubMed] [Google Scholar]
- Grunwald JE, Metelitsina TI, Dupont JC, Ying GS, Maguire MG. Reduced foveolar choroidal blood flow in eyes with increasing AMD severity. Invest Ophthalmol Vis Sci. 2005;46 (3):1033–1038. doi: 10.1167/iovs.04-1050. [DOI] [PubMed] [Google Scholar]
- Gu X, El-Remessy A, Brooks S, Al-Shabrawey M, Tsai N, Caldwell R. Hyperoxia induces retinal vascular endothelial cell apoptosis through formation of peroxynitrite. Am J Physiol Cell Physiol. 2003;285:C546–554. doi: 10.1152/ajpcell.00424.2002. [DOI] [PubMed] [Google Scholar]
- Guerin E, Sheridan C, Assheton D, Kent D, Wong D, Grant M, Hiscott P. SDF1-alpha is associated with VEGFR-2 in human choroidal neovascularisation. Microvasc Res. 2008;75 (3):302–307. doi: 10.1016/j.mvr.2007.12.001. [DOI] [PubMed] [Google Scholar]
- Gurbuz O, Ozdemir Y, Cosar CB, Kural G. Lipoprotein (a) in Behcet’s disease as an indicator of disease activity and in thrombotic complications. Eur J Ophthalmol. 2001;11 (1):62–65. doi: 10.1177/112067210101100112. [DOI] [PubMed] [Google Scholar]
- Guymer R, Luthert P, Bird A. Changes in Bruch’s membrane and related structures with age. Prog Ret Eye Res. 1998;18:59–90. doi: 10.1016/s1350-9462(98)00012-3. [DOI] [PubMed] [Google Scholar]
- Hageman G, Luthert P, Chong N, Johnson L, Anderson D, Mullins R. An integrated hypothesis that considers drusen as biomarkers of immune-mediated processes at the RPE-Bruch’s membrane interface in aging and age-related macular degeneration. Prog Ret Eye Res. 2001;20:705–732. doi: 10.1016/s1350-9462(01)00010-6. [DOI] [PubMed] [Google Scholar]
- Hageman GS, Kirchoff-Rempe MA, Lewis GP, Fisher SK, Anderson DH. Sequestration of basic fibroblast growth factor in the primate retinal interphotoreceptor matrix. Proc Natl Acad Sci U S A. 1991;88 (15):6706–6710. doi: 10.1073/pnas.88.15.6706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haines JL, Hauser MA, Schmidt S, Scott WK, Olson LM, Gallins P, Spencer KL, Kwan SY, Noureddine M, Gilbert JR, Schnetz-Boutaud N, Agarwal A, Postel EA, Pericak-Vance MA. Complement factor H variant increases the risk of age-related macular degeneration. Science. 2005;308 (5720):419–421. doi: 10.1126/science.1110359. [DOI] [PubMed] [Google Scholar]
- Handa JT, Verzijl N, Matsunaga H, Aotaki-Keen A, Lutty GA, te Koppele JM, Miyata T, Hjelmeland LM. Increase in the advanced glycation end product pentosidine in Bruch’s membrane with age. Invest Ophthalmol Vis Sci. 1999;40 (3):775–779. [PubMed] [Google Scholar]
- Hewitt AT, Nakazawa K, Newsome DA. Analysis of newly synthesized Bruch’s membrane proteoglycans. Invest Ophthalmol Vis Sci. 1989;30 (3):478–486. [PubMed] [Google Scholar]
- Hogan M. Ultrastructure of the choroid. Its role in the pathogenesis of chorioretinal diseases. Trans Pacific Coast Oto-ophthalmol Soc. 1961;42:61–87. [PubMed] [Google Scholar]
- Hollyfield JG. Age-related macular degeneration: the molecular link between oxidative damage, tissue-specific inflammation and outer retinal disease: the Proctor lecture. Invest Ophthalmol Vis Sci. 2010;51 (3):1275–1281. doi: 10.1167/iovs.09-4478. [DOI] [PubMed] [Google Scholar]
- Hollyfield JG, Bonilha VL, Rayborn ME, Yang X, Shadrach KG, Lu L, Ufret RL, Salomon RG, Perez VL. Oxidative damage-induced inflammation initiates age-related macular degeneration. Nat Med. 2008;14 (2):194–198. doi: 10.1038/nm1709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hollyfield JG, Perez VL, Salomon RG. A hapten generated from an oxidation fragment of docosahexaenoic acid is sufficient to initiate age-related macular degeneration. Mol Neurobiol. 2010;41 (2–3):290–298. doi: 10.1007/s12035-010-8110-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hussain AA, Starita C, Hodgetts A, Marshall J. Macromolecular diffusion characteristics of ageing human Bruch’s membrane: implications for age-related macular degeneration (AMD) Exp Eye Res. 2010;90 (6):703–710. doi: 10.1016/j.exer.2010.02.013. [DOI] [PubMed] [Google Scholar]
- Ishibashi T, Hata Y, Yoshikawa H, Nakagawa K, Sueishi K, Inomata H. Expression of vascular endothelial growth factor in experimental choroidal neovascularization. Graefes Arch Clin Exp Ophthalmol. 1997;235:159–167. doi: 10.1007/BF00941723. [DOI] [PubMed] [Google Scholar]
- Ishigooka H, Kitaoka T, Boutilier SB, Bost LM, Aotaki-Keen AE, Tablin F, Hjelmeland LM. Developmental expression of bFGF in the bovine retina. Invest Ophthalmol Vis Sci. 1993;34 (9):2813–2823. [PubMed] [Google Scholar]
- Izumi-Nagai K, Nagai N, Ozawa Y, Mihara M, Ohsugi Y, Kurihara T, Koto T, Satofuka S, Inoue M, Tsubota K, Okano H, Oike Y, Ishida S. Interleukin-6 receptor-mediated activation of signal transducer and activator of transcription-3 (STAT3) promotes choroidal neovascularization. Am J Pathol. 2007;170 (6):2149–2158. doi: 10.2353/ajpath.2007.061018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jager RD, Mieler WF, Miller JW. Age-related macular degeneration. N Engl J Med. 2008;358 (24):2606–2617. doi: 10.1056/NEJMra0801537. [DOI] [PubMed] [Google Scholar]
- Johnson LV, Leitner WP, Staples MK, Anderson DH. Complement activation and inflammatory processes in Drusen formation and age related macular degeneration. Exp Eye Res. 2001;73 (6):887–896. doi: 10.1006/exer.2001.1094. [DOI] [PubMed] [Google Scholar]
- Johnson LV, Ozaki S, Staples MK, Erickson PA, Anderson DH. A potential role for immune complex pathogenesis in drusen formation. Exp Eye Res. 2000;70 (4):441–449. doi: 10.1006/exer.1999.0798. [DOI] [PubMed] [Google Scholar]
- Johnson PT, Betts KE, Radeke MJ, Hageman GS, Anderson DH, Johnson LV. Individuals homozygous for the age-related macular degeneration risk-conferring variant of complement factor H have elevated levels of CRP in the choroid. Proc Natl Acad Sci U S A. 2006;103 (46):17456–17461. doi: 10.1073/pnas.0606234103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jyothi S, Chowdhury H, Elagouz M, Sivaprasad S. Intravitreal bevacizumab (Avastin) for age-related macular degeneration: a critical analysis of literature. Eye. 2010;24:816–824. doi: 10.1038/eye.2009.219. [DOI] [PubMed] [Google Scholar]
- Kaneko H, Dridi S, Tarallo V, Gelfand BD, Fowler BJ, Cho WG, Kleinman ME, Ponicsan SL, Hauswirth WW, Chiodo VA, Kariko K, Yoo JW, Lee DK, Hadziahmetovic M, Song Y, Misra S, Chaudhuri G, Buaas FW, Braun RE, Hinton DR, Zhang Q, Grossniklaus HE, Provis JM, Madigan MC, Milam AH, Justice NL, Albuquerque RJ, Blandford AD, Bogdanovich S, Hirano Y, Witta J, Fuchs E, Littman DR, Ambati BK, Rudin CM, Chong MM, Provost P, Kugel JF, Goodrich JA, Dunaief JL, Baffi JZ, Ambati J. DICER1 deficit induces Alu RNA toxicity in age-related macular degeneration. Nature. 2011;471 (7338):325–330. doi: 10.1038/nature09830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karwatowski WS, Jeffries TE, Duance VC, Albon J, Bailey AJ, Easty DL. Preparation of Bruch’s membrane and analysis of the age-related changes in the structural collagens. Br J Ophthalmol. 1995;79 (10):944–952. doi: 10.1136/bjo.79.10.944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kent D, Sheridan C. Choroidal neovascularization: a wound healing perspective. Molecular Vision. 2003;9:747–755. [PubMed] [Google Scholar]
- Khaliq A, Patel B, Jarvis-Evans J, Moriarty P, McLeod D, Boulton M. Oxygen modulates production of bFGF and TGF-beta by retinal cells in vitro. Exp Eye Res. 1995;60 (4):415–423. doi: 10.1016/s0014-4835(05)80098-3. [DOI] [PubMed] [Google Scholar]
- Kim SY, Mocanu C, McLeod DS, Bhutto IA, Merges C, Eid M, Tong P, Lutty GA. Expression of pigment epithelium-derived factor (PEDF) and vascular endothelial growth factor (VEGF) in sickle cell retina and choroid. Exp Eye Res. 2003;77:433–445. doi: 10.1016/s0014-4835(03)00174-x. [DOI] [PubMed] [Google Scholar]
- Kim SY, Sadda S, Humayun MS, de Juan E, Jr, Melia BM, Green WR. Morphometric analysis of the macula in eyes with geographic atrophy due to age-related macular degeneration. Retina. 2002a;22 (4):464–470. doi: 10.1097/00006982-200208000-00011. [DOI] [PubMed] [Google Scholar]
- Kim SY, Sadda S, Pearlman J, Humayun MS, de Juan E, Jr, Melia BM, Green WR. Morphometric analysis of the macula in eyes with disciform age-related macular degeneration. Retina. 2002b;22 (4):471–477. doi: 10.1097/00006982-200208000-00012. [DOI] [PubMed] [Google Scholar]
- Kim YG, Baek SH, Moon SW, Lee HK, Kim US. Analysis of spectral domain optical coherence tomography findings in occult macular dystrophy. Acta Ophthalmol. 2011;89 (1):e52–56. doi: 10.1111/j.1755-3768.2010.01958.x. [DOI] [PubMed] [Google Scholar]
- Klein ML, Ferris FL, 3rd, Armstrong J, Hwang TS, Chew EY, Bressler SB, Chandra SR. Retinal precursors and the development of geographic atrophy in age-related macular degeneration. Ophthalmology. 2008;115 (6):1026–1031. doi: 10.1016/j.ophtha.2007.08.030. [DOI] [PubMed] [Google Scholar]
- Klein R, Davis MD, Magli YL, Segal P, Klein BE, Hubbard L. The Wisconsin age-related maculopathy grading system. Ophthalmology. 1991;98 (7):1128–1134. doi: 10.1016/s0161-6420(91)32186-9. [DOI] [PubMed] [Google Scholar]
- Klein R, Klein BE, Knudtson MD, Wong TY, Shankar A, Tsai MY. Systemic markers of inflammation, endothelial dysfunction, and age-related maculopathy. Am J Ophthalmol. 2005a;140 (1):35–44. doi: 10.1016/j.ajo.2005.01.051. [DOI] [PubMed] [Google Scholar]
- Klein R, Klein BE, Linton KL. Prevalence of age-related maculopathy. The Beaver Dam Eye Study. Ophthalmology. 1992;99 (6):933–943. doi: 10.1016/s0161-6420(92)31871-8. [DOI] [PubMed] [Google Scholar]
- Klein RJ, Zeiss C, Chew EY, Tsai JY, Sackler RS, Haynes C, Henning AK, SanGiovanni JP, Mane SM, Mayne ST, Bracken MB, Ferris FL, Ott J, Barnstable C, Hoh J. Complement factor H polymorphism in age-related macular degeneration. Science. 2005b;308 (5720):385–389. doi: 10.1126/science.1109557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Korte GE, Repucci V, Henkind P. RPE destruction causes choriocapillary atrophy. Invest Ophthalmol Vis Sci. 1984;25:1135–1145. [PubMed] [Google Scholar]
- Krzystolik MG, Afshari MA, Adamis AP, Gaudreault J, Gragoudas ES, Michaud NA, Li W, Connolly E, O’Neill CA, Miller JW. Prevention of experimental choroidal neovascularization with intravitreal anti-vascular endothelial growth factor antibody fragment. Arch Ophthalmol. 2002;120:338–346. doi: 10.1001/archopht.120.3.338. [DOI] [PubMed] [Google Scholar]
- Kummer W, Fischer A, Mundel P, et al. Nitric-oxide synthase in VIP-containing vasodilator nerve fibers in the guinea pig. Neuroreport. 1992;3:653–655. doi: 10.1097/00001756-199207000-00028. [DOI] [PubMed] [Google Scholar]
- Kvanta A, Algvere PV, Berglin L, Seregard S. Subfoveal fibrovascular membranes in age-related macular degeneration express vascular endothelial growth factor. Invest Ophthalmol Vis Sci. 1996;37:1929–1934. [PubMed] [Google Scholar]
- Lambooij AC, van Wely KH, Lindenbergh-Kortleve DJ, Kuijpers RW, Kliffen M, Mooy CM. Insulin-like growth factor-I and its receptor in neovascular age-related macular degeneration. Invest Ophthalmol Vis Sci. 2003;44 (5):2192–2198. doi: 10.1167/iovs.02-0410. [DOI] [PubMed] [Google Scholar]
- Lauber H. Die Aderhaut (choroidea), in handbuch der mikroskopischen anatomie. Springer; Berlin: 1936. [Google Scholar]
- Linsenmeier RA, Yancy CM. Effects of hyperoxia on the oxygen distribution in the intact cat retina. Invest Ophthalmol Vis Sci. 1989;30:612–618. [PubMed] [Google Scholar]
- Lopez PF, Sippy BD, Lambert HM, Thach AB, Hinton DR. Transdifferentiated retinal pigment epithelial cells are immunoreactive for vascular endothelial growth factor in surgically excised age-related macular degeneration-related choroidal neovascular membranes. Invest Ophthalmol Vis Sci. 1996;37:855–868. [PubMed] [Google Scholar]
- Lutty G, Grunwald J, Majji AB, Uyama M, Yoneya S. Changes in choriocapillaris and retinal pigment epithelium in age-related macular degeneration. Mol Vis. 1999;5:35. [PubMed] [Google Scholar]
- Maeda H, Ogata N, Yi X, Takeuchi M, Ohkuma H, Uyama M. Apoptosis of photoreceptor cells in ornithine-induced retinopathy. Graefes Arch Clin Exp Ophthalmol. 1998;236 (3):207–212. doi: 10.1007/s004170050066. [DOI] [PubMed] [Google Scholar]
- Mainous AG, 3rd, Pearson WS. Aspirin and ibuprofen: potential mediators of the cardiovascular risk due to smoking? Fam Med. 2003;35 (2):112–118. [PubMed] [Google Scholar]
- Mann RM, Riva CE, Cranstoun SD, et al. Nitric oxide and choroidal blood flow (chBF) regulation. Invest Ophthalmol Vis Sci. 1993;34 (ARVO Suppl):1394. [PubMed] [Google Scholar]
- Margolis R, Spaide RF. A pilot study of enhanced depth imaging optical coherence tomography of the choroid in normal eyes. Am J Ophthalmol. 2009;147 (5):811–815. doi: 10.1016/j.ajo.2008.12.008. [DOI] [PubMed] [Google Scholar]
- Marshall J. The ageing retina: physiology or pathology. Eye (Lond) 1987;1 ( Pt 2):282–295. doi: 10.1038/eye.1987.47. [DOI] [PubMed] [Google Scholar]
- Martin DS, Haywood JR. Sympathetic nervous system activation by glutamate injections into the paraventricular nucleus. Brain Res. 1992;577 (2):261–267. doi: 10.1016/0006-8993(92)90282-e. [DOI] [PubMed] [Google Scholar]
- Matsumoto M, Yoshimura N, Honda Y. Increased production of transforming growth factor-beta 2 from cultured human retinal pigment epithelial cells by photocoagulation. Invest Ophthalmol Vis Sci. 1994;35 (13):4245–4252. [PubMed] [Google Scholar]
- McGwin G, Hall TA, Xie A, Owsley C. The relation between C reactive protein and age related macular degeneration in the Cardiovascular Health Study. Br J Ophthalmol. 2005;89 (9):1166–1170. doi: 10.1136/bjo.2005.067397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McLaughlin T, Abbasi F, Lamendola C, Liang L, Reaven G, Schaaf P, Reaven P. Differentiation between obesity and insulin resistance in the association with C-reactive protein. Circulation. 2002;106 (23):2908–2912. doi: 10.1161/01.cir.0000041046.32962.86. [DOI] [PubMed] [Google Scholar]
- McLeod DS, Brownstein R, Lutty GA. Vaso-obliteration in the canine model of oxygen-induced retinopathy. Invest Ophthalmol Vis Sci. 1996;37:300–311. [PubMed] [Google Scholar]
- McLeod DS, Grebe R, Bhutto I, Merges C, Baba T, Lutty GA. Relationship between RPE and choriocapillaris in age-related macular degeneration. Invest Ophthalmol Vis Sci. 2009;50 (10):4982–4991. doi: 10.1167/iovs.09-3639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McLeod DS, Lefer DJ, Merges C, Lutty GA. Enhanced expression of intracellular adhesion molecule-1 and P-selectin in the diabetic human retina and choroid. Am J Pathol. 1995;147:642–653. [PMC free article] [PubMed] [Google Scholar]
- McLeod DS, Lutty GA. High resolution histologic analysis of the human choroidal vasculature. Invest Ophthalmol Vis Sci. 1994;35:3799–3811. [PubMed] [Google Scholar]
- McLeod DS, Taomoto M, Otsuji T, Green WR, Sunness JS, Lutty GA. Quantifying changes in RPE and choriocapillaris in eyes with age-related macular degeneration. Invest Ophthalmol Vis Sci. 2002;43:1986–1993. [PubMed] [Google Scholar]
- Medzhitov R. Origin and physiological roles of inflammation. Nature. 2008;454 (7203):428–435. doi: 10.1038/nature07201. [DOI] [PubMed] [Google Scholar]
- Metelitsina TI, Grunwald JE, DuPont JC, Ying GS, Brucker AJ, Dunaief JL. Foveolar choroidal circulation and choroidal neovascularization in age-related macular degeneration. Invest Ophthalmol Vis Sci. 2008;49 (1):358–363. doi: 10.1167/iovs.07-0526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller AS, Coster DJ, Costa M, Furness JB. Vasoactive intestinal peptide immunoreactive nerve fibers in the human eye. Aust J Ophthalmol. 1983;11:185–193. doi: 10.1111/j.1442-9071.1983.tb01077.x. [DOI] [PubMed] [Google Scholar]
- Miyajima-Uchida H, Hayashi H, Beppu R, Kuroki M, Fukami M, Arakawa F, Tomita Y, Oshima K. Production and accumulation of thrombospondin-1 in human retinal pigment epithelial cells. Invest Ophthalmol Vis Sci. 2000;41:561–567. [PubMed] [Google Scholar]
- Moncada S, Palmer RM, Higgs EA. Nitric oxide: physiology, pathophysiology, and pharmacology. Pharmacol Rev. 1991;43 (2):109–142. [PubMed] [Google Scholar]
- Moore DJ, Hussain AA, Marshall J. Age-related variation in the hydraulic conductivity of Bruch’s membrane. Invest Ophthalmol Vis Sci. 1995;36 (7):1290–1297. [PubMed] [Google Scholar]
- Mori K, Ando A, Gelbach P, Nesbitt D, Takahashi K, Goldsteen D, Penn M, Chen CT, Melia M, Phipps S, Moffat D, Brazzel KL, Dixon GKH, Campochiaro P. Inhibition of choroidal neovascularization by intravenous injection of adenoviral vectors expressing secretable endostatin. Am J Pathol. 2001;159:313–320. doi: 10.1016/S0002-9440(10)61697-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morris JL. Co-transmission from autonomic vasodilator neurons supplying the guinea pig uterine artery. J Auton Nerv Syst. 1993;42:11–21. doi: 10.1016/0165-1838(93)90337-t. [DOI] [PubMed] [Google Scholar]
- Mullins RF, Dewald AD, Streb LM, Wang K, Kuehn MH, Stone EM. Elevated membrane attack complex in human choroid with high risk complement factor H genotypes. Exp Eye Res. 2011a;93 (4):565–567. doi: 10.1016/j.exer.2011.06.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mullins RF, Johnson MN, Faidley EA, Skeie JM, Huang J. Choriocapillaris vascular dropout related to density of drusen in human eyes with early age-related macular degeneration. Invest Ophthalmol Vis Sci. 2011b;52 (3):1606–1612. doi: 10.1167/iovs.10-6476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mullins RF, Russell SR, Anderson DH, Hageman GS. Drusen associated with aging and age-related macular degeneration contain proteins common to extracellular deposits associated with atherosclerosis, elastosis, amyloidosis, and dense deposit disease. FASEB J. 2000;14 (7):835–846. [PubMed] [Google Scholar]
- Nilsson SF, Linder J, Bill A. Characteristics of uveal vasodilation produced by facial nerve stimulation in monkeys, cats and rabbits. Exp Eye Res. 1985;40:841–852. doi: 10.1016/0014-4835(85)90129-0. [DOI] [PubMed] [Google Scholar]
- Noda K, She H, Nakazawa T, Hisatomi T, Nakao S, Almulki L, Zandi S, Miyahara S, Ito Y, Thomas KL, Garland RC, Miller JW, Gragoudas ES, Mashima Y, Hafezi-Moghadam A. Vascular adhesion protein-1 blockade suppresses choroidal neovascularization. FASEB J. 2008;22 (8):2928–2935. doi: 10.1096/fj.07-105346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O’Reilly MS, Boehm T, Shing Y, Fukai N, Vasios G, Lane wS, Flynn E, Birkhead JR, Olsen BR, Folkman J. Endostatin: an endogenous inhibitor of angiogenesis and tumor growth. Cell. 1997;88:277–285. doi: 10.1016/s0092-8674(00)81848-6. [DOI] [PubMed] [Google Scholar]
- Otani A, Takagi H, Oh H, Koyama S, Ogura Y, Matumura M, Honda Y. Vascular endothelial growth factor family and receptor expression in human choroidal neovascular membranes. Microvasc Res. 2002;64 (1):162–169. doi: 10.1006/mvre.2002.2407. [DOI] [PubMed] [Google Scholar]
- Parish WE. Studies on vasculitis. VII. C-reactive protein as a substance perpetuating chronic vasculitis. Occurrence in lesions and concentrations in sera. Clin Allergy. 1976;6 (6):543–550. doi: 10.1111/j.1365-2222.1976.tb01939.x. [DOI] [PubMed] [Google Scholar]
- Park CW, Shin YS, Kim CM, Lee SY, Yu SE, Kim SY, Choi EJ, Chang YS, Bang BK. Increased C-reactive protein following hemodialysis predicts cardiac hypertrophy in chronic hemodialysis patients. Am J Kidney Dis. 2002;40 (6):1230–1239. doi: 10.1053/ajkd.2002.36891. [DOI] [PubMed] [Google Scholar]
- Pauleikhoff D, Chen J, Chisholm I, Bird A. Choroidal perfusion abnormality with age-related bruch’s membrane change. Am J Ophthalmol. 1990;109:211–217. doi: 10.1016/s0002-9394(14)75989-6. [DOI] [PubMed] [Google Scholar]
- Pauleikhoff D, Zuels S, Sheraidah GS, Marshall J, Wessing A, Bird AC. Correlation between biochemical composition and fluorescein binding of deposits in Bruch’s membrane. Ophthalmology. 1992;99 (10):1548–1553. doi: 10.1016/s0161-6420(92)31768-3. [DOI] [PubMed] [Google Scholar]
- Pepys MB, Hirschfield GM. C-reactive protein: a critical update. J Clin Invest. 2003;111 (12):1805–1812. doi: 10.1172/JCI18921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rahman W, Chen FK, Yeoh J, Patel P, Tufail A, Da Cruz L. Repeatability of manual subfoveal choroidal thickness measurements in healthy subjects using the technique of enhanced depth imaging optical coherence tomography. Invest Ophthalmol Vis Sci. 2011;52 (5):2267–2271. doi: 10.1167/iovs.10-6024. [DOI] [PubMed] [Google Scholar]
- Rallidis LS, Zolindaki MG, Manioudaki HS, Laoutaris NP, Velissaridou AH, Papasteriadis EG. Prognostic value of C-reactive protein, fibrinogen, interleukin-6, and macrophage colony stimulating factor in severe unstable angina. Clin Cardiol. 2002;25 (11):505–510. doi: 10.1002/clc.4960251106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramrattan RS, van der Schaft TL, Mooy CM, de Bruijn WC, Mulder PG, de Jong PT. Morphometric analysis of Bruch’s membrane, the choriocapillaris, and the choroid in aging. Invest Ophthalmol Vis Sci. 1994;35:2857–2864. [PubMed] [Google Scholar]
- Rowe IF, Soutar AK, Trayner IM, Thompson GR, Pepys MB. Circulating human C-reactive protein binds very low density lipoproteins. Clin Exp Immunol. 1984;58 (1):237–244. [PMC free article] [PubMed] [Google Scholar]
- Saishin Y, Silva RL, Kachi S, Aslam S, Gong YY, Lai H, Carrion M, Harris B, Hamilton M, Wei L, Campochiaro PA. Periocular gene transfer of pigment epithelium-derived factor inhibits choroidal neovascularization in a human-sized eye. Hum Gene Ther. 2005;16 (4):473–478. doi: 10.1089/hum.2005.16.473. [DOI] [PubMed] [Google Scholar]
- Saishin Y, Takahashi K, Lima e Silva R, Hylton D, Rudge JS, Wiegand SJ, Campochiaro PA. VEGF-TRAP(R1R2) suppresses choroidal neovascularization and VEGF-induced breakdown of the blood-retinal barrier. J Cell Physiol. 2003;195 (2):241–248. doi: 10.1002/jcp.10246. [DOI] [PubMed] [Google Scholar]
- Sarks JP, Sarks SH, Killingsworth MC. Evolution of geographic atrophy of the retinal pigment epithelium. Eye. 1988;2:552–577. doi: 10.1038/eye.1988.106. [DOI] [PubMed] [Google Scholar]
- Sarks S, Cherepanoff S, Killingsworth M, Sarks J. Relationship of Basal laminar deposit and membranous debris to the clinical presentation of early age-related macular degeneration. Invest Ophthalmol Vis Sci. 2007;48 (3):968–977. doi: 10.1167/iovs.06-0443. [DOI] [PubMed] [Google Scholar]
- Scanu AM, Fless G. The apoB 100-apo(a) complex: relation to triglyceride-rich particles. Adv Exp Med Biol. 1991;285:295–298. doi: 10.1007/978-1-4684-5904-3_35. [DOI] [PubMed] [Google Scholar]
- Scanu AM, Lawn RM, Berg K. Lipoprotein(a) and atherosclerosis. Ann Intern Med. 1991;115 (3):209–218. doi: 10.7326/0003-4819-115-3-209. [DOI] [PubMed] [Google Scholar]
- Seddon JM, Gensler G, Milton RC, Klein ML, Rifai N. Association between C-reactive protein and age-related macular degeneration. JAMA. 2004;291 (6):704–710. doi: 10.1001/jama.291.6.704. [DOI] [PubMed] [Google Scholar]
- Seddon JM, George S, Rosner B, Rifai N. Progression of age-related macular degeneration: prospective assessment of C-reactive protein, interleukin 6, and other cardiovascular biomarkers. Arch Ophthalmol. 2005;123 (6):774–782. doi: 10.1001/archopht.123.6.774. [DOI] [PubMed] [Google Scholar]
- Sengupta N, Afzal A, Caballero S, Chang KH, Shaw LC, Pang JJ, Bond VC, Bhutto I, Baba T, Lutty GA, Grant MB. Paracrine modulation of CXCR4 by IGF-1 and VEGF: implications for choroidal neovascularization. Invest Ophthalmol Vis Sci. 2010;51 (5):2697–2704. doi: 10.1167/iovs.09-4137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sheibani N, Sorenson CM, Cornelius LA, Frazier WA. Thrombospondin-1, a natural inhibitor of angiogenesis, is present in vitreous and aqueous humor and is modulated by hyperglycemia. Biochem Biophys Res Commun. 2000;267:257–261. doi: 10.1006/bbrc.1999.1903. [DOI] [PubMed] [Google Scholar]
- Sheridan CM, Pate S, Hiscott P, Wong D, Pattwell DM, Kent D. Expression of hypoxia-inducible factor-1alpha and -2alpha in human choroidal neovascular membranes. Graefes Arch Clin Exp Ophthalmol. 2009;247 (10):1361–1367. doi: 10.1007/s00417-009-1133-3. [DOI] [PubMed] [Google Scholar]
- Slomiany MG, Rosenzweig SA. Autocrine effects of IGF-I-induced VEGF and IGFBP-3 secretion in retinal pigment epithelial cell line ARPE-19. Am J Physiol Cell Physiol. 2004;287 (3):C746–753. doi: 10.1152/ajpcell.00568.2003. [DOI] [PubMed] [Google Scholar]
- Spaide RF, Koizumi H, Pozzoni MC. Enhanced depth imaging spectral-domain optical coherence tomography. Am J Ophthalmol. 2008;146 (4):496–500. doi: 10.1016/j.ajo.2008.05.032. [DOI] [PubMed] [Google Scholar]
- Spraul CW, Lang GE, Grossniklaus HE. Morphometric analysis of the choroid, Bruch’s membrane, and retinal pigment epithelium in eyes with age-related macular degeneration. Invest Ophthalmol Vis Sci. 1996;37:2724–2735. [PubMed] [Google Scholar]
- Spraul CW, Lang GE, Grossniklaus HE, Lang GK. Histologic and morphometric analysis of the choroid, Bruch’s membrane, and retinal pigment epithelium in postmortem eyes with age-related macular degeneration and histologic examination of surgically excised choroidal neovascular membranes. Surv Ophthalmol. 1999;44(Suppl 1):S10–32. doi: 10.1016/s0039-6257(99)00086-7. [DOI] [PubMed] [Google Scholar]
- Starita C, Hussain AA, Pagliarini S, Marshall J. Hydrodynamics of ageing Bruch’s membrane: implications for macular disease. Exp Eye Res. 1996;62 (5):565–572. doi: 10.1006/exer.1996.0066. [DOI] [PubMed] [Google Scholar]
- Starita C, Hussain AA, Patmore A, Marshall J. Localization of the site of major resistance to fluid transport in Bruch’s membrane. Invest Ophthalmol Vis Sci. 1997;38 (3):762–767. [PubMed] [Google Scholar]
- Staurenghi G, Bottoni F, Lonati C, Autelitano A, Orzalesi N. Drusen and ‘choroidal filling defects’: A cross-sectional survey. Ophthalmologica. 1992;205:178–186. doi: 10.1159/000310337. [DOI] [PubMed] [Google Scholar]
- Stenvinkel P, Lindholm B, Heimburger M, Heimburger O. Elevated serum levels of soluble adhesion molecules predict death in pre-dialysis patients: association with malnutrition, inflammation, and cardiovascular disease. Nephrol Dial Transplant. 2000;15 (10):1624–1630. doi: 10.1093/ndt/15.10.1624. [DOI] [PubMed] [Google Scholar]
- Sternfeld MD, Robertson JE, Shipley GD, Tsai J, Rosenbaum JT. Cultured human retinal pigment epithelial cells express basic fibroblast growth factor and its receptor. Curr Eye Res. 1989;8 (10):1029–1037. doi: 10.3109/02713688908997395. [DOI] [PubMed] [Google Scholar]
- Stjernschantz J, Bill A. Vasomotor effects in facial nerve stimulation: non-cholinergic vasodilation in the eye. Acta Physiol Scand. 1980;109:45–50. doi: 10.1111/j.1748-1716.1980.tb06562.x. [DOI] [PubMed] [Google Scholar]
- Strauss O. The retinal pigment epithelium in visual function. Physiol Rev. 2005;85 (3):845–881. doi: 10.1152/physrev.00021.2004. [DOI] [PubMed] [Google Scholar]
- Streilein JW, Ma N, Wenkel H, Ng TF, Zamiri P. Immunobiology and privilege of neuronal retina and pigment epithelium transplants. Vision Res. 2002;42 (4):487–495. doi: 10.1016/s0042-6989(01)00185-7. [DOI] [PubMed] [Google Scholar]
- Sunness JS. The natural history of geographic atrophy, the advanced atrophic form of age-related macular degeneration. Mol Vis. 1999;5:25. [PubMed] [Google Scholar]
- Sunness JS, Gonzalez-Baron J, Bressler NM, Tian Y, Hawkins B, Applegate CA. The development of choroidal neovascularization in eyes with geographic atrophy form of are-related macular degeneration. Ophthalmology. 1999;106:910–919. doi: 10.1016/S0161-6420(99)00509-6. [DOI] [PubMed] [Google Scholar]
- Talmage EK, Mawe GM. NADPH-diaphorase and VIP are colocalized in neurons of gallbladder ganglia. J Auton Nerv Syst. 1993;43:83–89. doi: 10.1016/0165-1838(93)90324-n. [DOI] [PubMed] [Google Scholar]
- Tanihara H, Yoshida M, Matsumoto M, Yoshimura N. Identification of transforming growth factor-beta expressed in cultured human retinal pigment epithelial cells. Invest Ophthalmol Vis Sci. 1993;34 (2):413–419. [PubMed] [Google Scholar]
- Tezel TH, Del Priore LV, Kaplan HJ. Reengineering of aged Bruch’s membrane to enhance retinal pigment epithelium repopulation. Invest Ophthalmol Vis Sci. 2004;45 (9):3337–3348. doi: 10.1167/iovs.04-0193. [DOI] [PubMed] [Google Scholar]
- Uchiki T, Weikel KA, Jiao W, Shang F, Caceres A, Pawlak D, Handa JT, Brownlee M, Najaraj R, Taylor A. Glycation-altered proteolysis as a pathobiological mechanism thatlinks dietary glycemic index, aging, and age-related disease (in nondiabetics) Aging Cell. 2012;11:1–13. doi: 10.1111/j.1474-9726.2011.00752.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uno K, Prow TW, Bhutto IA, Yerrapureddy A, McLeod DS, Yamamoto M, Reddy SP, Lutty GA. Role of Nrf2 in retinal vascular development and the vaso-obliterative phase of oxygen-induced retinopathy. Exp Eye Res. 90(4):493–500. doi: 10.1016/j.exer.2009.12.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vine AK, Stader J, Branham K, Musch DC, Swaroop A. Biomarkers of cardiovascular disease as risk factors for age-related macular degeneration. Ophthalmology. 2005;112 (12):2076–2080. doi: 10.1016/j.ophtha.2005.07.004. [DOI] [PubMed] [Google Scholar]
- Vogt SD, Curcio CA, Wang L, Li CM, McGwin G, Jr, Medeiros NE, Philp NJ, Kimble JA, Read RW. Retinal pigment epithelial expression of complement regulator CD46 is altered early in the course of geographic atrophy. Exp Eye Res. 2011;93 (4):413–423. doi: 10.1016/j.exer.2011.06.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wada M, Gelfman CM, Matsunaga H, Alizadeh M, Morse L, Handa JT, Hjelmeland LM. Density-dependent expression of FGF-2 in response to oxidative stress in RPE cells in vitro. Curr Eye Res. 2001;23 (3):226–231. doi: 10.1076/ceyr.23.3.226.5467. [DOI] [PubMed] [Google Scholar]
- Wajer SD, Taomoto M, McLeod M, McCally RL, Fabry ME, Nagel RL, Lutty GA. Velocity measurements of normal and sickle red blood cells in the rat retinal and choroidal vasculatures. Microvascular Res. 2000;60:281–293. doi: 10.1006/mvre.2000.2270. [DOI] [PubMed] [Google Scholar]
- Walport MJ. Complement. Second of two parts. N Engl J Med. 2001;344 (15):1140–1144. doi: 10.1056/NEJM200104123441506. [DOI] [PubMed] [Google Scholar]
- Walsh N, Valter K, Stone J. Cellular and subcellular patterns of expression of bFGF and CNTF in the normal and light stressed adult rat retina. Exp Eye Res. 2001;72 (5):495–501. doi: 10.1006/exer.2000.0984. [DOI] [PubMed] [Google Scholar]
- Wangsa-Wirawan ND, Linsenmeier RA. Retinal oxygen. Fundamental and clinical aspects. Arch Ophthalmol. 2003;121:547–557. doi: 10.1001/archopht.121.4.547. [DOI] [PubMed] [Google Scholar]
- Weikel KA, Fitzgerald P, Shang F, Caceres MA, Bian Q, Handa JT, Stitt AW, Taylor A. Natural History of Age-Related Retinal Lesions that Precede AMD in Mice Fed High or Low Glycemic Index Diets. Invest Ophthalmol Vis Sci. 2011 doi: 10.1167/iovs.11-8545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weismann D, Hartvigsen K, Lauer N, Bennett KL, Scholl HP, Charbel Issa P, Cano M, Brandstatter H, Tsimikas S, Skerka C, Superti-Furga G, Handa JT, Zipfel PF, Witztum JL, Binder CJ. Complement factor H binds malondialdehyde epitopes and protects from oxidative stress. Nature. 2011;478 (7367):76–81. doi: 10.1038/nature10449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wenkel H, Streilein JW. Evidence that retinal pigment epithelium functions as an immune-privileged tissue. Invest Ophthalmol Vis Sci. 2000;41 (11):3467–3473. [PubMed] [Google Scholar]
- Wood A, Binns A, Margrain T, Drexler W, Povazay B, Esmaeelpour M, Sheen N. Retinal and choroidal thickness in early age-related macular degeneration. Am J Ophthalmol. 2011;152(6):1030–1038. e1032. doi: 10.1016/j.ajo.2011.05.021. [DOI] [PubMed] [Google Scholar]
- Xu GZ, Li WW, Tso MO. Apoptosis in human retinal degenerations. Trans Am Ophthalmol Soc. 1996;94:411–430. discussion 430–411. [PMC free article] [PubMed] [Google Scholar]
- Xu H, Chen M, Forrester JV. Para-inflammation in the aging retina. Prog Retin Eye Res. 2009;28 (5):348–368. doi: 10.1016/j.preteyeres.2009.06.001. [DOI] [PubMed] [Google Scholar]
- Yang Z, Camp NJ, Sun H, Tong Z, Gibbs D, Cameron DJ, Chen H, Zhao Y, Pearson E, Li X, Chien J, Dewan A, Harmon J, Bernstein PS, Shridhar V, Zabriskie NA, Hoh J, Howes K, Zhang K. A variant of the HTRA1 gene increases susceptibility to age-related macular degeneration. Science. 2006;314 (5801):992–993. doi: 10.1126/science.1133811. [DOI] [PubMed] [Google Scholar]
- Yuan X, Gu X, Crabb JS, Yue X, Shadrach K, Hollyfield JG, Crabb JW. Quantitative proteomics: comparison of the macular Bruch membrane/choroid complex from age-related macular degeneration and normal eyes. Mol Cell Proteomics. 2010;9 (6):1031–1046. doi: 10.1074/mcp.M900523-MCP200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou J, Jang YP, Kim SR, Sparrow JR. Complement activation by photooxidation products of A2E, a lipofuscin constituent of the retinal pigment epithelium. Proc Natl Acad Sci U S A. 2006;103 (44):16182–16187. doi: 10.1073/pnas.0604255103. [DOI] [PMC free article] [PubMed] [Google Scholar]